

ABSTRACT
Bacterial persisters are a quasidormant subpopulation of cells that are tolerant to antibiotic treatment. The combination of the aminoglycoside tobramycin with fumarate as an antibacterial potentiator utilizes an antipersister strategy that is aimed at reducing recurrent Pseudomonas aeruginosa infections by enhancing the killing of P. aeruginosa persisters. Stationary-phase cultures of P. aeruginosa were used to generate persister cells. A range of tobramycin concentrations was tested with a range of metabolite concentrations to determine the potentiation effect of the metabolite under a variety of conditions, including a range of pH values and in the presence of azithromycin or cystic fibrosis (CF) patient sputum. In addition, 96-well dish biofilm and colony biofilm assays were performed, and the cytotoxicity of the tobramycin-fumarate combination was determined utilizing a lactate dehydrogenase (LDH) assay. Enhanced killing of up to 6 orders of magnitude of P. aeruginosa persisters over a range of CF isolates, including mucoid and nonmucoid strains, was observed for the tobramycin-fumarate combination compared to killing with tobramycin alone. Furthermore, significant fumarate-mediated potentiation was seen in the presence of azithromycin or CF patient sputum. Fumarate also reduced the cytotoxicity of tobramycin-treated P. aeruginosa to human epithelial airway cells. Finally, in mucoid and nonmucoid CF isolates, complete eradication of P. aeruginosa biofilm was observed in the colony biofilm assay due to fumarate potentiation. These data suggest that a combination of tobramycin with fumarate as an antibacterial potentiator may be an attractive therapeutic for eliminating recurrent P. aeruginosa infections in CF patients through the eradication of bacterial persisters.
KEYWORDS: Pseudomonas aeruginosa, aminoglycoside, bacterial metabolite, bacterial persistence, cystic fibrosis, fumarate, persistent infection, persisters, potentiator, tobramycin
INTRODUCTION
In response to various stimuli, including antibiotic treatment, bacteria can enter into a persister state. In this metabolically dormant state, bacteria become tolerant or unresponsive to antibiotics, which can lead to chronic recurrent infections (1). Persistent, or “tolerant,” bacteria are genetically identical to the bacterial population from which they emerge. This subpopulation of nongrowing bacteria can remain in a state of metabolic hibernation for weeks or months in persistently infected patients, only to later “awaken,” often in a more aggressive form (2). Persistent infections have been described in many serious lung diseases, including cystic fibrosis (CF) (3), non-CF bronchiectasis (4, 5), chronic obstructive pulmonary disease (COPD) (6, 7), and nontuberculous mycobacterial pulmonary infections (8, 9). Recurrent infections that are attributable to bacterial persisters occur in a wide variety of biofilm-related infections (e.g., lung, urinary tract, device associated, and wound infections) caused by an array of bacterial pathogens (10–12).
As previously shown by Collins and coworkers, metabolite stimulation of the proton motive force (PMF) enables the uptake of aminoglycoside antibiotics in Escherichia coli and Staphylococcus aureus, effectively eradicating bacterial persisters and clearing an infection (13). In this earlier work, persister populations were selected in planktonic culture by exposure to ofloxacin, and the effects of different carbon sources (normalized to 60 mM carbon concentration) on the sensitivity of the persister population to subsequent exposure to gentamicin were tested. Notably, combining mannitol with the aminoglycoside antibiotic gentamicin significantly enhanced the killing of persistent E. coli in vitro. In addition, in vivo studies in a mouse model of chronic urinary tract infection with a mannitol-gentamicin combination reduced the bacterial load on the catheters by 1.5 orders of magnitude and inhibited the spread of the bacterial infection to the kidneys compared to gentamicin alone (13). Similarly, screening of various bacterial metabolites using the PAO1 strain of Pseudomonas aeruginosa showed that specific carbon sources, such as fumarate (at 15 mM, since it has four carbons), which is involved in the tricarboxylic acid (TCA) cycle (as well as others involved in glycolysis), increase the efficacy of tobramycin (TOB) against PAO1 persisters by four orders of magnitude (14).
Over the course of the disease, up 80% of people with CF become chronically infected with P. aeruginosa. In one study, for example, 98% (39/40) of infant patients had serological or culture evidence of P. aeruginosa infection by age three (15). These infections are associated with accelerated lung function decline and earlier mortality, with 80 to 90% of CF deaths attributable to progressive lung disease (16). Bacterial persisters lead to drug tolerance, which can result in a failure to eradicate the infection, as schematically depicted in Fig. 1 (2). Inhaled TOB has long been known to modulate but not eradicate P. aeruginosa infections (17, 18). Genetic resistance to TOB does not account for the lessening reduction in sputum bacterial density (as measured in CFU) observed with subsequent on-off cycles of treatment. Instead, the decreasing efficacy of TOB over treatment time is consistent with an increase in the numbers of persisters that are tolerant to antibiotic treatment.
FIG 1.
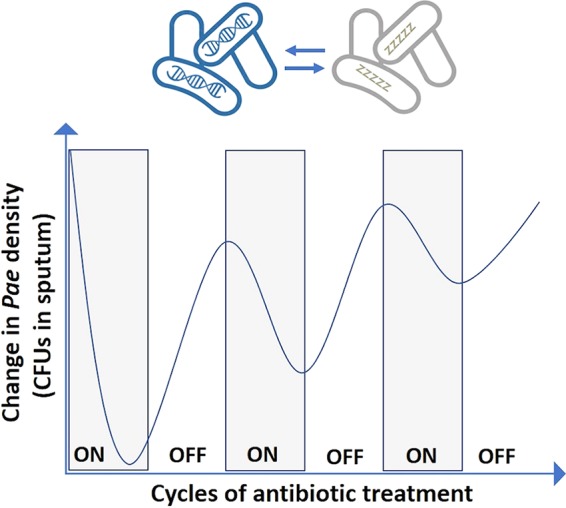
Bacterial persisters lead to antibiotic tolerance and failure to eradicate the infection. Schematic generalized based on data presented by Ramsey et al. (17). Pae, P. aeruginosa.
New disease-modifying agents, such as the cystic fibrosis transmembrane conductance regulator (CFTR) modulators, hold great promise (19, 20) for the treatment of CF patients (19, 20). However, CFTR modulators may ameliorate but are unlikely to reverse long-standing bronchiectasis (21). As such, the prevalence of chronic airway infection is unlikely to decrease among CF patients with existing bronchiectasis. The data from the G551D ObservationAL (GOAL) study has, for example, shown that patients on ivacaftor remain colonized with P. aeruginosa (22). Therefore, anti-infectives that can effectively treat chronic P. aeruginosa CF airway infections will remain an important clinical need that is largely unmet at present. If clinical use of CFTR modulators results in stabilization of CF lung disease, a large fraction of the CF population will nonetheless continue to suffer from chronic airway infections for the next 30 to 50 years (22).
A potential strategy for enhancing the susceptibility of persister cells to antibiotics, such as TOB, is to utilize specific metabolites that sensitize “transiently resistant” or persistent bacteria to antibiotics, thereby potentially enabling the effective treatment of debilitating chronic infections. Potentiating the activity of TOB is an attractive strategy, since it is currently the front-line therapy for treating CF lung infections, and genetic resistance to inhaled TOB has not increased markedly (23–25). Fumarate was selected from a small set of potential metabolite potentiators because it exhibited the highest level of potentiation in P. aeruginosa, and it has been used as an excipient in several inhaled drug products. In the present work, we specifically study the combination of TOB with fumarate in a variety of in vitro models that aim to mimic the environment of the CF lung. In so doing, we further probe the action of the TOB-fumarate combination and its potential utility as a therapeutic for treating P. aeruginosa infections in CF and other lung diseases. Most notably, the potentiation effect of fumarate on TOB in killing P. aeruginosa persisters was examined over a diverse set of CF clinical isolates, across a range pH values relevant to the CF lung, in the presence of CF patient sputum, in combination with succinate, and in two different biofilm models. Since clinically azithromycin is often given concomitantly with tobramycin, the potentiation of tobramycin by fumarate was also examined in the presence of azithromycin. In addition, the cytotoxicity of the combination of TOB plus fumarate was assessed.
RESULTS
Potentiation in CF clinical isolates.
To simulate the treatment of persisters in chronic infection, a diverse set of P. aeruginosa CF isolates, including both mucoid and nonmucoid isolates representing a range of TOB susceptibilities, were tested in the planktonic stationary phase (PSP) time-kill potentiation assay, in which persister cells were generated by allowing the cultures to grow to stationary phase. Potentiation of TOB activity (here referred to simply as potentiation) by fumarate ranging from 3.5 to over 6 orders of magnitude was observed for approximately 90% of the TOB-sensitive CF P. aeruginosa clinical isolates (see Table 1). Mucoid and nonmucoid isolates displayed similar patterns of fumarate-enhanced killing. Representative assays for a nonmucoid (isolate 10004) and a mucoid (isolate 10028) strain are shown in Fig. 2. While the MICs for these strains range from 0.25 to 1 μg/ml, potentiation was observed starting at TOB concentrations of ∼8 μg/ml, which generally leveled off by ∼16 to 32 μg/ml. In fact, data for isolate 10004 were extended out to 1,024 μg/ml TOB and showed that the potentiation effect is maintained (see Fig. S2 in the supplemental material). As expected, fumarate alone had no effect (see Fig. S3 for a replot of the no-TOB data for further clarity). A significant potentiation effect by fumarate was also observed at similar TOB concentrations in the PAO1 and PA14 P. aeruginosa laboratory strains, both of which have an MIC of 0.8 μg/ml (Fig. S4). These results are consistent with previously published results (13, 14) and suggest that potentiation by fumarate is a consequence of fumarate metabolism by the TCA cycle and the generation of PMF. Also consistent with the postulated mechanism of PMF-enhanced killing of persisters, three TOB-resistant strains tested (10022, 10039, and 10084 with MICs of 8, 64, and 128 μg/ml, respectively) were not potentiated (Table S2 and Fig. S5). TOB-sensitive CF isolates 10020 and 10077 did not show potentiation (Fig. S6) despite having low MICs of 0.25 and 0.5 μg/ml, respectively (Table 1). It should be noted that in the PSP potentiation assays for these two strains, after growing the cells overnight for 16 h in lysogeny broth (LB) medium (the step expected to take the cells to stationary phase), the cultures were not as turbid as the cultures for the other P. aeruginosa strains tested. Moreover, the colonies formed by these two strains on LB-agar plates were very small. At the end of the assay, however, after spot plating and counting, the CFU counts for the no-TOB/no-potentiator controls were comparable for these two strains (mean, ∼1.5 × 109 CFU/ml) and the 17 TOB-susceptible strains for which potentiation was observed (mean, ∼3.9 × 109 CFU/ml). Given that isolates 10020 and 10077 did not appear to reach saturation in the PSP assay, we initially extended the overnight culture from 16 to 20 h for these two strains but obtained similar results.
TABLE 1.
Most TOB-susceptible P. aeruginosa CF clinical isolates are potentiated by fumarate
| Strain | Mucoid | MIC (μg/ml)a | Potentiation (log10) |
|---|---|---|---|
| 10028 | Yes | 1 | 5 |
| 10048 | Yes | 0.25 | 4.5 |
| 10005 | Yes | 0.25 | 5 |
| 14003 | Yes | 0.5 | 6 |
| 10140 | Yes | 0.5 | 5 |
| 10063-Sb | Yes | 0.5 | 4.5 |
| 10063-Lb | No | 0.25 | 4.5 |
| 10013 | No | 0.25 | 4 |
| 10019 | No | 0.25 | 5 |
| 10002 | No | 0.25 | 4 |
| 10001 | No | 0.25 | 5 |
| 10008 | No | 0.25 | 4.5 |
| 10004 | No | 0.25 | 4.5 |
| 10045 | No | 0.25 | 3 |
| 10054 | No | 0.5 | 5 |
| 14001 | No | 0.5 | 3.5 |
| 10015 | No | 4 | 2 |
| 10077 | Yes | 0.25 | No |
| 10020 | No | 0.5 | No |
MICs are for TOB alone in an in vitro assay in MHB under standard CLSI conditions, as described in Materials and Methods under “Characterization of bacterial strains.” The addition of fumarate does not affect the values (see Table S1).
S, small colonies; L, large colonies.
FIG 2.

Dose-dependent potentiation effect of fumarate on TOB sensitivity in P. aeruginosa CF clinical isolates. Planktonic stationary-phase (PSP) cultures were used to generate P. aeruginosa persister cells (56). Plots of percent survival versus TOB concentration in the PSP assay are shown for the nonmucoid 10004 isolate (A) and for the mucoid 10028 isolate (B). Values of percent survival depict the mean ± standard deviation (SD) (n = 2). (C and D) Data for the 15 mM fumarate curves are replotted as bar graphs for isolates 10004 (C) and 10028 (D), and statistically significant differences in a one-way analysis of variance (ANOVA) with Dunnett's multiple-comparison test are indicated (**, P < 0.01; ***, P < 0.001; ns, not significant).
To probe the possibility that the lack of potentiation for isolates 10020 and 10077 is due to metabolic and/or TCA cycle impairment through an inability to metabolize fumarate efficiently, and therefore an inability to generate sufficient PMF to facilitate TOB uptake, we generated growth curves under a variety of conditions (see supplemental Materials and Methods). Specifically, we observed delayed growth of 10077 for at least 5 h and a lack of any significant growth of 10020 on fumarate, succinate, or pyruvate as sole carbon sources under diluted conditions (1:30) (Fig. S7). PAO1 under the same conditions grew relatively rapidly on fumarate and succinate but showed negligible growth at 4 h on pyruvate as a sole carbon source (Fig. S7C). Interestingly, pyruvate also potentiated TOB-mediated killing of PAO1 but at a slightly higher concentration of TOB than fumarate (∼5 log at 32 μg/ml TOB with pyruvate versus ∼5.5 log at 16 μg/ml TOB with fumarate; compare Fig. S7D and S4A).
Potentiation in COPD clinical isolates.
Several P. aeruginosa isolates from patients with chronic obstructive pulmonary disease (COPD) were also tested in the PSP assay to assess potentiation by fumarate. Similar levels of enhanced persister killing were observed as for the CF P. aeruginosa isolates, with 3.5 to 5 log of enhanced killing compared to TOB alone (Table S3). Figure 3 shows the results for genetically identical early and late COPD isolates with respect to disease progression (isolates 5072829 and 919193, both with MICs of 0.25 μg/ml). The potentiation effect appears to be greater at a lower TOB and potentiator concentration with the later isolate, although many additional pairs of patient isolates will need to be tested to see if this trend holds.
FIG 3.

Dose-dependent potentiation effect of fumarate on TOB sensitivity in P. aeruginosa COPD isolates. Early and late patient isolates with respect to disease progression were obtained from A. Oliver (6). Plots of percent survival versus TOB concentration in the PSP assay are shown for an early isolate from a patient (5072829) (A) and for a late isolate from the same patient (919193) (B). Strains 5072829 and 919193 are genetically identical (as determined by pulsed-field gel electrophoresis) and have a TOB MIC of 0.25 μg/ml. Values of percent survival depict the mean ± SD (n = 2).
Potentiation across a range of pH values.
In the lungs of CF patients, airway pH can vary between 5.6 and 7.8, as measured in the trachea, the mainstem bronchi, and the smaller secondary and tertiary bronchi (26). In addition, the activity of TOB on planktonic bacteria versus biofilms may be modulated by pH (27). Over 4 orders of magnitude of enhanced killing relative to TOB alone was observed at the two lower pHs tested (5.6 and 7), and 1.5 log enhanced killing was observed at the highest pH (8.5), which is close to the maximum pH for P. aeruginosa growth (Fig. S8). These results indicate that the potentiation effect is robust across a range of pH levels expected to be relevant in the CF lung.
Fumarate in combination with succinate.
Since the production of succinate directly precedes that of fumarate in the TCA cycle, the ability of succinate in combination with fumarate to potentiate the killing of P. aeruginosa persisters was also examined in the PSP assay for P. aeruginosa strain PA14. As expected, based on the work of Meylan et al. (14), succinate also showed significant potentiation of TOB. In addition, we observed an additive (but not synergistic) potentiation effect when fumarate and succinate were combined with TOB; i.e., similar levels of potentiation were observed with 15 mM fumarate, 15 mM succinate, and 7.5 mM each (Fig. 4).
FIG 4.
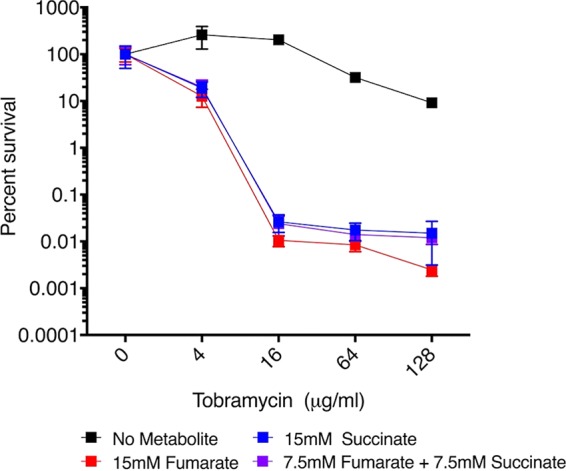
Potentiation effect of succinate alone and in combination with fumarate on TOB for P. aeruginosa strain PA14. A plot of percent survival versus TOB concentration in the PSP assay is shown for strain PA14. Values of percent survival depict the mean ± SD (n = 2), and the results are representative of two repeated experiments.
Potentiation in naive P. aeruginosa biofilm models.
While there is some debate about the precise form of the biofilms involved in CF infections, it is clear that biofilms are present in the CF lung and that they are a form of growth that is highly enriched in persisters (12). Importantly, we saw significant fumarate potentiation of TOB killing of P. aeruginosa biofilm cells in two different biofilm models. Notably, in these experiments, there was no preselection of persisters. In the 96-well plate biofilm assay (28), the extent of P. aeruginosa PA14 biofilm formation and the effect of fumarate on the reduction of biofilm were measured using the dye crystal violet (CV) by quantifying absorbance at 550 nm. Significant reduction in biofilm relative to the TOB-alone control (optical density at 550 nm [OD550], 0.22 ± 0.01) was observed for TOB with increasing concentrations of fumarate (Fig. 5A). The reduction in CV staining was 39% with 7.5 mM fumarate (OD550, 0.13 ± 0.01) and 52% with 15 mM fumarate (OD550, 0.1 ± 0.02). Similar results were observed for the effect of TOB plus fumarate on P. aeruginosa PAO1 biofilm formation (Fig. S9). The PAO1 biofilm biomass relative to the TOB-alone control (OD550, 0.74 ± 0.09) was reduced by 69% with 7.5 mM fumarate (OD550, 0.23 ± 0.04) or 15 mM fumarate (OD550, 0.23 ± 0.02) and 87% with 30 mM fumarate (OD550, 0.09 ± 0.001). Fumarate alone at 30 mM did not reduce the PA14 biofilm (Fig. S10).
FIG 5.

Fumarate potentiation of TOB observed in two different biofilm models. P. aeruginosa strain PA14 was tested in the 96-well plate biofilm assay (A) and in the colony biofilm assay (B). For the 96-well plate biofilm assay, the effect of TOB plus fumarate on the reduction in biofilm was measured using crystal violet dye by quantifying the absorbance at 550 nm. The height of each bar represents the average OD550 values of 4 replicate wells from one representative experiment, and error bars indicate standard deviations. Each experiment was carried out twice, yielding similar results. The difference in OD550 between the TOB-only control and each set of the other wells was statistically significant in a one-way ANOVA (***, P < 0.001). For the colony biofilm assay, error bars represent two biological replicates, and percent survival is normalized to the no-TOB/no-fumarate value; statistically significant differences in a two-way ANOVA are indicated (***, P < 0.001).
In the colony biofilm assay (29–31), PA14 P. aeruginosa biofilm cells were reduced by 2.5 log at 4 μg/ml TOB and 3.5 log at 8 μg/ml TOB with the addition of fumarate (Fig. 5B). Preexposure and postexposure CFU for the no-TOB/no-fumarate control are ∼107 (Fig. S11 gives the CFU for the various conditions). A nonmucoid and a mucoid CF patient strain were also tested in the colony biofilm assay, and a dramatic reduction in the biofilm colonies was observed (Fig. 6). With the nonmucoid strain, there was complete eradication of the P. aeruginosa biofilm at 8 μg/ml TOB plus 15 mM fumarate, while with the mucoid strain this occurred at an even lower TOB concentration of 4 μg/ml TOB plus the fumarate.
FIG 6.

Eradication of P. aeruginosa in colony biofilms from CF clinical isolates. Plots of percent survival versus TOB concentration are shown for the nonmucoid isolate 10004 (A) and for the mucoid isolate 10005 (B). Blue arrows indicate complete eradication with no colonies remaining. Two biological replicates are shown for each condition tested, and the results are representative of two repeated experiments. Percent survival is normalized to the no-TOB/no-fumarate value for the strain, which can result in values greater than 100%. Statistically significant differences in a two-way ANOVA are indicated (***, P < 0.001).
Potentiation observed in the presence of CF sputum.
In a further attempt to mimic as closely as possible the environment of the CF lung, we also tested TOB with fumarate in the PSP assay in the presence of artificial sputum media and CF patient sputum. In each of two artificial sputum medium formulations (32, 33), the TOB-fumarate combination showed enhanced killing of bacterial persisters by approximately 4 orders of magnitude relative to TOB alone (see supplemental Materials and Methods and Fig. S12). Sputum was collected from two different patients, and for patient 2, early (pre-antibiotic treatment) and late (post-antibiotic treatment) samples were collected. All three samples were tested with P. aeruginosa strain PAO1, and the late sputum sample from the second patient was tested with nonmucoid CF isolate 10004. In all cases, in the presence of these CF sputum samples, approximately 3 log enhanced killing was observed in the PSP assay (Fig. 7).
FIG 7.

Fumarate-mediated potentiation observed in the presence of CF patient sputum. The PAO1 laboratory and the CF patient isolate 10004 were tested. Plots of percent survival versus TOB concentration in the PSP assay in the presence of 10% sputum purified through filtration are shown for PAO1 with early sputum from patient 1 (A), for PAO1 with early sputum from patient 2 (B), for PAO1 with late sputum from patient 2 (C), and for clinical isolate 10004 with late sputum from patient 2 (D). Early sputum refers to pre-antibiotic treatment for the most recent exacerbation, while late refers to posttreatment. Values of percent survival depict the mean ± SD (n = 2).
Fumarate potentiation in the presence of azithromycin.
Reducing airway inflammation in a chronic lung disease, such as CF, is desirable, and in fact, azithromycin is often given concurrently with TOB primarily for its anti-inflammatory effect (34, 35), even though it appears clinically to antagonize/reduce the anti-infective effect of TOB (36, 37). Indeed, we found that azithromycin does show a diminished effect of fumarate-mediated potentiation of TOB against P. aeruginosa persisters (compare the red triangle curve to the red square curve in Fig. 8). Nevertheless, >3 log potentiation was still achieved with TOB plus fumarate in the presence of azithromycin (comparing the black curves to the red triangle curve in Fig. 8).
FIG 8.
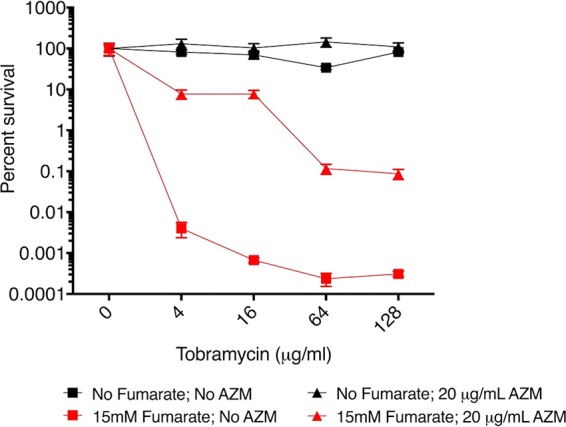
Greater than 3 log of TOB potentiation in the presence of azithromycin. A plot of percent survival versus TOB concentration in the PSP assay is shown for strain PAO1; AZM, azithromycin. Values of percent survival depict the mean ± SD (n = 2).
The TOB-fumarate combination exhibits a cytoprotective effect on airway cells.
Fumarate also had a protective effect on human airway epithelial cells (Fig. 9). For example, for the mucoid CF isolate 14003, the relative cytotoxicity was reduced in a dose-dependent manner from 1.0 to below 0.3 upon the addition of fumarate. This decrease in cytotoxicity of P. aeruginosa is not the result of potentiation by fumarate. Under the conditions of this assay, that is, in the presence of 2 mM glutamine, which is necessary to maintain the human airway epithelial cells, there was no additional potentiation (beyond what was observed with glutamine) with the addition of fumarate (Fig. S13). Furthermore, fumarate alone was tested at the highest concentration (30 mM) with two strains of P. aeruginosa and had no effect on cytotoxicity (Fig. S14). Thus, the data indicate that fumarate plus TOB reduces the cytotoxicity of P. aeruginosa on human airway epithelial cells, suggesting that the TOB-fumarate combination may have the potential to decrease airway inflammation.
FIG 9.

Cytoprotective effect on human epithelial cells. Cytotoxicity was measured in a colorimetric assay that quantitates the amount of lactate dehydrogenase (LDH) released into the media from damaged cells (by recording the absorbance at 490 nm) as a biomarker for cellular cytotoxicity. Reduced cytotoxicity of the human airway epithelial cells was observed in a dose-dependent manner with the addition of fumarate to TOB in the presence of P. aeruginosa strains PAO1, PA14, the nonmucoid CF isolate 14001, and the mucoid CF isolate 14003. Under the conditions of this assay, in the presence of 2 mM glutamine, fumarate does not yield additional potentiation (see Fig. S10). Cytotoxicity values are normalized to the cytotoxicity of the human airway epithelial cells in response to PAO1 P. aeruginosa untreated with TOB or fumarate. Means ± SD (n = 3) are depicted, and statistically significant differences in a one-way ANOVA are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
DISCUSSION
This work demonstrates that the combination of TOB and fumarate can enhance killing in stationary-phase planktonic cultures and biofilms of P. aeruginosa by up to six orders of magnitude compared to TOB alone. These effects occurred with a variety of mucoid and nonmucoid CF and COPD isolates, suggesting that formulations consisting of an aminoglycoside with a metabolic potentiator might have broad therapeutic application. For most TOB-susceptible P. aeruginosa strains tested, a significant potentiation effect of fumarate was seen.
The concentrations of TOB needed for killing of P. aeruginosa persisters in the presence of potentiator in the PSP experiments (Table 1) were higher than the TOB MICs typically observed for log-phase cultures. One explanation for these relatively high TOB concentrations compared to standard MICs is that although fumarate may be enhancing uptake of TOB through the generation of PMF (14), the concentration of TOB in the persisters may be lower than it is in growing cells. Furthermore, an inoculum effect may account for this difference, in that in standard MIC assays, the inoculum is approximately 105 cells, whereas in the PSP assay, it is approximately 109 cells. In any case, potentiation occurs at TOB concentrations that are likely to be relevant to the concentrations present in the lungs of treated CF patients; e.g., the mean concentration of tobramycin in the lung epithelial lining fluid was measured at 90 μg/ml after a single 300-mg inhaled dose of tobramycin (38) and, in a separate study, in lung sputum at 1,237 μg/ml in CF patients (38, 39).
For two strains, no potentiation was observed, perhaps attributable to a metabolic defect or significant growth delay when switched to fumarate as the sole carbon source. Certain strains may have impaired persister cell formation as a consequence of mutations in genes related to quorum sensing, heat shock, toxin-antitoxin systems, or other stress responses (2). Alternatively, strains that display TOB tolerance in stationary phase yet do not display potentiation by fumarate may have mutations in TCA cycle genes as the basis for impaired potentiation.
Thus, one plausible explanation for the lack of potentiation for isolates 10020 and 10077 is that they exhibit an inability to metabolize fumarate efficiently and are therefore not able to generate sufficient PMF to facilitate TOB uptake. Previous studies in E. coli showing that while metabolic stimuli do not induce rapid growth resumption in the PSP assay over an 8-h period, the addition of the potentiator to a diluted culture (1:100) does stimulate growth (13). That is, carbon sources at concentrations per persister 100-fold higher than required for metabolite-enabled persister eradication can stimulate persister growth. Moreover, the carbon sources that caused the highest level of potentiation were the ones that caused the quickest revival of persisters, suggesting that these carbon sources are most efficiently metabolized by persisters. The observed delayed growth of 10077 and the lack of growth of 10020 on fumarate, succinate, or pyruvate as sole carbon sources under diluted conditions, as well as the lack of potentiation observed for these two strains, is consistent with these earlier studies in E. coli.
Persisters derived from TOB-resistant strains tested were not potentiated by fumarate, which is expected if one assumes that the mechanisms underlying their TOB resistance are not related to defects in TOB uptake. If TOB resistance is due to enzymatic inactivation of the drug itself or mutations to the drug target (40) that prevent the drug once inside the bacteria from binding to its target, the potentiator is unlikely to have an effect. If on the other hand, the resistance is due to defects in uptake of the drug, the potentiator might be able to compensate. Genetic resistance to TOB, as opposed to persister-mediated tolerance to TOB during antibiotic therapy, has not increased markedly as the use of inhaled TOB has become widespread among CF patients (23–25). Thus, metabolite enhancement of TOB efficacy would not be expected to drive the selection and dissemination of TOB-resistant P. aeruginosa strains.
The COPD P. aeruginosa clinical isolates tested in the PSP assay also showed a high level of potentiation of TOB by fumarate, suggesting that the combination or similar formulations consisting of an aminoglycoside plus a metabolic potentiator could have broad therapeutic applications. While providing another clinically relevant example of how these findings could translate therapeutically, the sample sizes are small. In general, clinical relevance will need to be probed further through subsequent in-human studies.
The data indicating that the potentiation effects of fumarate and succinate on TOB are additive suggest that it may be possible in future therapeutic applications to combine more than one potentiator molecule with TOB or another aminoglycoside. On the other hand, although combining fumarate and succinate with TOB, as an example, would not be expected to increase the drug load and may help limit the development of resistance to a single potentiator, it might complicate the development path, since three substances would be involved. The combination of fumarate and succinate may be attractive in light of the fact that bacteria have different mechanisms for the uptake of these metabolites (41).
In both biofilm assays (which involve naive biofilms and no selection of persisters per se), a significant potentiation effect was observed. In fact, the concentrations of TOB needed to reduce biofilm formation in the 96-well plate biofilm assay and eradicate the biofilm in the colony biofilm assay are significantly lower than what is required to yield the maximum potentiation effect in the PSP assay. There are a number of differences between the colony biofilm and PSP assays that might account for this effect. Experiments were initiated with ∼107 cells for the colony biofilm assay compared to ∼109 for the PSP assay. A lower cell count was utilized for the colony biofilm assay to avoid diffusion limitations with respect to oxygen, antibiotic, and fumarate that arise from growing the bacteria on solid media. Since the PSP assay was performed in broth with vigorously mixed cultures, the diffusion of the oxygen/antibiotic/metabolite is not a concern even at high cell counts. In addition, the exposure to each antibiotic-metabolite combination was performed for 20 h in the colony biofilm assay compared to 4 h in the PSP assay. The longer incubation time in the colony biofilm assay may allow for enhanced persister killing at a lower tobramycin concentration.
In addition, the observed cytoprotective effect of fumarate on human airway epithelial cells indicates that the potentiator may have the potential to reduce airway inflammation. It is possible that fumarate may reduce the level of cytotoxicity of P. aeruginosa to the airway cells by tuning down virulence factors and influencing the level of toxins secreted by P. aeruginosa. It is known, for example, that at least certain strains of P. aeruginosa utilize the type III secretion system (T3SS) to deliver effector proteins that are cytotoxic to mammalian cells during infection to avoid innate immune clearance (42, 43). As a further indication of the potential importance of the observed cytoprotective effect of fumarate, azithromycin is often given concurrently with TOB primarily for its anti-inflammatory effect (and possible inhibition of bacterial communication through quorum sensing) (34), even though it appears clinically to antagonize/reduce the anti-infective effect of TOB (36). Our data combining azithromycin with TOB plus fumarate in the PSP assay confirm in vitro the antagonistic effect of azithromycin on TOB while showing that significant potentiation by fumarate is maintained.
We observed a significant potentiation effect by fumarate under a variety of conditions designed to mimic the CF lung. These data include enhanced killing of P. aeruginosa persisters across a wide range of pH values (5.5 to 8.5) in the PSP assay, potentiation in the presence of CF patient sputum, and efficacy in two relevant biofilm models of P. aeruginosa infection. The CF sputum results suggest that even in the presence of nutrients available in the CF lung environment, the potentiation of TOB by fumarate is likely to be significant. In addition, within biofilms and under conditions likely to promote persister formation, the nutrient environment of the lung may be somewhat reduced. The biofilm results in general are noteworthy because in those assays, there is no preselection of bacterial persisters, although as discussed, biofilms are expected to be enriched in persisters. The colony biofilm assay results are particularly significant in that complete eradication of the biofilm was observed for the nonmucoid as well as the mucoid strain, and perhaps somewhat surprisingly, the effect is observed at a somewhat lower TOB concentration for the mucoid strain. Mucoid strains are thought to be more tolerant to antibiotics (44), and moreover, CF P. aeruginosa strains convert to mucoidy as the disease progresses (45, 46).
TOB plus fumarate presumably targets persistence and the development of antibiotic tolerance in contrast to heritable resistance. The precise and often stochastic mechanism by which bacteria naturally enter and emerge from a persistent state is not fully known (2, 47, 48). We suspect that if treatment with the TOB-fumarate combination eliminates persisters as they form, not just when they “reawaken” during a course of antibiotic treatment, the TOB-fumarate combination may show an effect on acute infections as well as diminish or eliminate recurrent infections. This hypothesis is supported by data in an acute mouse model of catheter-associated urinary tract infection for a related combination (gentamicin plus mannitol) (13). As such, it may be possible to reduce the dose of TOB in the combination, limiting potential toxicities, thereby making TOB a more effective antibiotic.
If the strategy of enhancing the efficacy of inhaled TOB through the addition of fumarate and/or other metabolites were to enable the eradication of P. aeruginosa persisters in the clinical setting, this innovation could represent a major therapeutic breakthrough in the treatment of CF airway infection, potentially leading to reduced rates of pulmonary exacerbations and disease progression (49). Furthermore, given that fumarate and any other mechanistically similar potentiators enhance the killing of only nongrowing persister cells, the development of resistance to the potentiator is expected to be low. Bacterial persisters, however, are thought to serve as an evolutionary reservoir from which resistant organisms can emerge (2, 50–53). Others have hypothesized that persistence is an actively maintained state that is caused and supported by changes in gene expression patterns in a network of intracellular stress responses. These same stress responses can accelerate processes of adaptive evolution leading to the development of resistance. Thus, eliminating bacterial persisters as early as possible may be key to limiting further resistance and thereby prolonging the lifetimes of clinically important anti-infective agents.
MATERIALS AND METHODS
Antibiotics and chemicals.
TOB (T4014-500MG) and sodium fumarate dibasic (F1506-100G) were obtained from Sigma-Aldrich.
Medium preparation.
LB medium (M1245-500G, lot no. 0000215110; HiMedia) was prepared and sterilized for 15 min by autoclaving. Mueller-Hinton broth (MHB) medium (Fluka 70192) was prepared as per the manufacturer's instructions and sterilized for 25 min by autoclaving. M9 salts (catalog no. 30621015-2, lot no. L14051600; bioWORLD) were prepared as a 5× stock solution. For all PSP experiments, a 1× M9 medium solution was used. The final 1× M9 solution was created using 200 ml of 5× M9, 2 ml of sterile 1 M MgSO4 (final concentration, 2 mM), and 100 μl of sterile 1 M CaCl2 (final concentration, 0.1 mM) and supplemented to 1 liter with sterile water.
Characterization of bacterial strains.
P. aeruginosa CF clinical isolates were collected over a period of 2 years at two CF centers in the United States (54). In an initial round of characterization, 93 isolates were prescreened for mucoidy and TOB resistance by inoculating from frozen stock cultures into 50 μl of LB broth and streaking LB-agar plates supplemented with 0, 4, 8, 16, 32, and 64 μg/ml TOB. The streaked plates were incubated for 24 h at 37°C, followed by 48 h of incubation at room temperature. The plates without TOB were used to estimate the growth and mucoidy of the CF isolates. The plates with various TOB concentrations were used to estimate the level of resistance for each isolate. Out of 93 strains tested, we selected 21 isolates, including seven mucoid strains, with no major growth defects in a second round of characterization. We determined the TOB MICs for these isolates by broth microdilution using the Clinical and Laboratory Standards Institute (CLSI) recommendations (55), with P. aeruginosa ATCC 27853 and E. coli ATCC 25922 as controls. Colonies of each clinical isolate were suspended in 1 ml of MHB and diluted to a density of 5 × 105 CFU/ml into 2-fold serial dilutions of TOB (0 to 64 μg/ml final concentration). The plates were incubated at 37°C for up to 48 h.
Isolates were considered susceptible if the TOB MIC was ≤4 μg/ml, intermediate if their MIC was >4 μg/ml and <16 μg/ml, and resistant if their MIC was ≥16 μg/ml. A total of 18 isolates, 7 mucoid and 11 nonmucoid, were TOB sensitive; one nonmucoid strain was TOB intermediate, and three were TOB resistant.
PSP time-kill potentiation assay.
PSP cultures are expected to have higher numbers of persisters and, in terms of antibiotic tolerance, are considered more similar than exponential-phase cultures to bacterial biofilms (56). The PSP time-kill potentiation assay was utilized to assess TOB-potentiator combinations with respect to in vitro activity and efficacy. In this assay, P. aeruginosa bacterial cultures in planktonic stationary phase were used to enrich for bacterial persisters. Bacterial strains were evaluated using the time-kill method (CLSI M26), using various concentrations of TOB and potentiator.
Specifically, bacterial cells were streaked from frozen stock on LB-agar (Miller formulation) plates and individual colonies picked, inoculated into 3 ml of LB broth, and incubated at 37°C with shaking at 200 rpm to an optical density at 600 nm (OD600) of 0.25 to 0.3. Cultures were then diluted 1:1,000 in 40 ml of LB medium overnight and grown in 250-ml shake flasks for 16 h at 37°C at 200 rpm. After 16 h, cells were harvested by centrifugation at 5,000 × g for 15 min and were washed by resuspending the pellet in 10 ml of 1× phosphate-buffered saline (PBS). This step was followed by centrifugation of the cells at 5,000 × g for 15 min and resuspension of the washed cells in 20 ml of 1× M9 medium supplemented with 2 mM MgSO4 and 0.1 mM CaCl2.
Stock solutions of 1 M fumarate (and/or other carbon sources being tested as potentiators) were prepared for addition to the media. Each TOB and fumarate concentration was prepared at 2 times the final desired concentration in 0.5 ml of medium. Half a milliliter of the 2× fumarate plus TOB combination was mixed with 0.5 ml of resuspended cells in Eppendorf tubes. All combinations were incubated for 4 h at 37°C at 200 rpm. After 4 h, cells were centrifuged at 5,000 × g for 5 min and washed by resuspending each pellet in 1 ml of 1× PBS. The cells were centrifuged again at 5,000 × g for 5 min and resuspended in 1× M9 medium. Aliquots were taken from each combination and serially diluted in a 96-well plate (catalog no. 3370; Costar). Five-microliter aliquots were plated for each dilution on LB-agar plates. The plates were incubated overnight at 37°C, and CFU were enumerated the next day. See Fig. S1 for a summary of the PSP assay.
In the standard PSP assay described above, the pH at the start of the incubation was between 7.0 and 7.5. The potentiation effect of fumarate on TOB was also specifically explored in vitro in the PSP assay at pH values of 5.6, 7, and 8.5 (see supplemental Materials and Methods for details). These pH values are close to the minimum (5.6), optimal (6.6 to 7), and maximum (8) pH for P. aeruginosa growth (57).
Ninety-six-well plate biofilm assay.
In preparation for the 96-well plate biofilm assay (28), P. aeruginosa PA14 was grown in liquid culture in rich LB medium. After overnight incubation at 37°C, the culture was diluted 1:100 into fresh M63 minimal medium supplemented with 0.4% (wt/vol) arginine. M63 minimal medium, a standard biofilm assay medium for P. aeruginosa, was supplemented with arginine as the sole carbon and energy source to promote less planktonic growth and a more robust biofilm formation in this assay (28).
For each condition tested, a final volume of 100 μl was added to each well of a 96-well plate in quadruplicate. Plates were incubated for 8 h at 37°C, and the biofilms were stained and quantified immediately afterwards as described below. Planktonic cells were removed by turning the plate over and shaking out the liquid. Plates were gently submerged in a small tub of water, and the water was subsequently shaken out. To lower the background in the assay, the process was repeated twice to remove unattached cells and medium components that stain with CV. After the washes, 125 μl of a 0.1% solution of CV (Sigma) was added in water, and the plates were incubated at room temperature for 20 min. The plates were subsequently washed 3 to 4 times with water, the water was shaken out, and the plates were blotted on a stack of paper towels. Then, they were turned upside down and dried for 2 to 3 h. To quantitate the amount of biofilm in each well, 200 μl of 20% acetic acid was added to each well. Plates were left to sit for 15 min, and 100 μl was transferred to a new flat-bottom microtiter plate. The plate was read at an absorbance of 550 nm using 20% acetic acid in water as a blank.
In this assay, the biofilm was allowed to form in the presence of increasing concentrations of TOB to identify the concentration of TOB that inhibits the formation of biofilm by approximately 50%. A TOB concentration of 2.5 μg/ml was therefore utilized for subsequent assays involving TOB to assess the effect of TOB with fumarate on biofilm formation. The following conditions were then tested: no exposure, TOB alone, 30 mM fumarate alone, TOB plus 7.5 mM fumarate, TOB plus 15 mM fumarate, and TOB plus 30 mM fumarate.
Colony biofilm assay.
Colony biofilms of each strain were grown as previously described, with minor modifications (29–31). Briefly, overnight cultures of each strain in LB were diluted to 1 × 108 CFU/ml in PBS. Single 5-μl droplets (5 × 105 cells) of the diluted culture were placed on individual autoclave-sterilized 0.2-μm-pore-size polycarbonate filter membranes (GTBP 02500; Millipore) placed on top of M9-agar plates. The plates were inverted and incubated for 20 h at 37°C. The colony biofilms that formed on the filter membranes were moved onto fresh M9-agar plates with or without TOB and the potentiator and incubated for 20 h at 37°C. Biofilms were harvested by resuspension in 9 ml of 1× PBS, vigorously vortexed, and then serially diluted. Serial dilutions were plated on LB-agar and incubated overnight at 37°C for CFU enumeration the next day.
CF patient sputum collection and preparation.
CF sputum samples were collected at the Massachusetts General Hospital (MGH) Cystic Fibrosis Center (institutional review board [IRB] no. 2011P000620) by having each patient expectorate into a sterile cup. The collected sputum was weighed, fast-frozen on dry ice, and stored at −80°C until needed. At the time of the experiment, the sputum samples were thawed on ice and diluted 1:1 (wt/vol) in water, followed by sonication on ice for 4 constant cycles of 30 s using a microtip probe. After sonication, the insoluble debris fraction was removed by centrifugation at 18,000 × g for 10 min. The soluble fraction was filter sterilized (0.22-μm pore size) to generate a 50% sputum stock kept at 4°C for up to 1 week. For the PSP assay, the sputum stock was diluted with 1× M9 medium to a final concentration of 10%.
Cytotoxicity assay.
Twenty-four-well tissue culture plates were seeded with 2 × 105 cystic fibrosis bronchial epithelial (CFBE) cells in 0.5 ml of feeding medium per well (58). The medium was replaced every 2 to 3 days. One to two days before bacterial inoculation, the feeding medium was replaced with minimal essential medium (MEM; without phenol red), supplemented with 10% fetal bovine serum (FBS) and 2 mM glutamine. The plates were ready to use 7 to 10 days after the initial seeding.
Epithelial cell cytotoxicity was determined by first inoculation of CFBE cells with overnight cultures of P. aeruginosa at a multiplicity of infection (MOI) of 30:1 in 0.5 ml of assay medium, 0.4% arginine, 40 μg/ml TOB (or 8 μg/ml for PA14, since higher concentrations eliminated this more sensitive strain), and either no fumarate, 7.5 mM, 15 mM, or 30 mM fumarate. Cocultures were incubated for 16 h at 37°C and 5% CO2 before epithelial cell cytotoxicity was assessed. The following strains were tested: PAO1, PA14, nonmucoid clinical strain 14001, and mucoid clinical strain 14003.
For cytotoxicity measurements, 300 μl of supernatant was harvested prior to washing the wells with 1× PBS. The supernatant was centrifuged at 16,000 × g in a microcentrifuge for 2 min, and 50 μl was assayed in triplicate for lactate dehydrogenase (LDH) release using the CytoTox96 nonradioactive cytotoxicity assay kit (catalog no. G1780; Promega, Madison, WI), according to the manufacturer's instructions (58). Cytotoxicity was determined by measuring spontaneous release of LDH from CFBE cells (untreated). To assay the toxicity of TOB or fumarate alone, CFBE cells, prepared as described above, were incubated with 40 μg/ml TOB (or 8 μg/ml for PA14) or 30 mM fumarate for 16 h at 37°C in the presence of 5% CO2.
Supplementary Material
ACKNOWLEDGMENTS
We thank Daniel J. Ferullo and Jeffrey A. Radding from EnBiotix and James J. Collins from MIT for helpful discussions, and Antonio Oliver from the Instituto de Investigación Sanitaria de Palma for providing pairs of COPD patient isolates.
We thank the Massachusetts Life Science Center for a Cooperative Research Matching Grant supporting this work.
M.K., A.D.G., J.D.W., F.M.A., S.M.M., and D.J.-M. conceived and designed the experiments. M.K., A.D.G., W.H., and L.M.Y. performed the experiments. M.K., A.D.G., G.A.O., F.M.A., and D.J.-M. analyzed the data. L.M.Y., G.A.O., and S.M.M. contributed reagents/materials/analysis tools. M.K., A.D.G., L.M.Y., G.A.O., F.M.A., S.M.M., and D.J.-M. drafted and edited the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00987-17.
REFERENCES
- 1.Allison KR, Brynildsen MP, Collins JJ. 2011. Heterogeneous bacterial persisters and engineering approaches to eliminate them. Curr Opin Microbiol 14:593–598. doi: 10.1016/j.mib.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen NR, Lobritz MA, Collins JJ. 2013. Microbial persistence and the road to drug resistance. Cell Host Microbe 13:632–642. doi: 10.1016/j.chom.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mulcahy LR, Burns JL, Lory S, Lewis K. 2010. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J Bacteriol 192:6191–6199. doi: 10.1128/JB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McShane PJ, Naureckas ET, Tino G, Strek ME. 2013. Non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med 188:647–656. doi: 10.1164/rccm.201303-0411CI. [DOI] [PubMed] [Google Scholar]
- 5.White L, Mirrani G, Grover M, Rollason J, Malin A, Suntharalingam J. 2012. Outcomes of Pseudomonas eradication therapy in patients with non-cystic fibrosis bronchiectasis. Respir Med 106:356–360. doi: 10.1016/j.rmed.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 6.Martínez-Solano L, Macia MD, Fajardo A, Oliver A, Martinez JL. 2008. Chronic Pseudomonas aeruginosa infection in chronic obstructive pulmonary disease. Clin Infect Dis 47:1526–1533. doi: 10.1086/593186. [DOI] [PubMed] [Google Scholar]
- 7.Murphy TF. 2008. The many faces of Pseudomonas aeruginosa in chronic obstructive pulmonary disease. Clin Infect Dis 47:1534–1536. doi: 10.1086/593187. [DOI] [PubMed] [Google Scholar]
- 8.Cortesia C, Lopez GJ, de Waard JH, Takiff HE. 2010. The use of quaternary ammonium disinfectants selects for persisters at high frequency from some species of non-tuberculous mycobacteria and may be associated with outbreaks of soft tissue infections. J Antimicrob Chemother 65:2574–2581. doi: 10.1093/jac/dkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson MM, Odell JA. 2014. Nontuberculous mycobacterial pulmonary infections. J Thorac Dis 6:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fauvart M, De Groote VN, Michiels J. 2011. Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J Med Microbiol 60:699–709. doi: 10.1099/jmm.0.030932-0. [DOI] [PubMed] [Google Scholar]
- 11.Michiels JE, Van den Bergh B, Verstraeten N, Fauvart M, Michiels J. 2016. In vitro emergence of high persistence upon periodic aminoglycoside challenge in the ESKAPE pathogens. Antimicrob Agents Chemother 60:4630–4637. doi: 10.1128/AAC.00757-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebeaux D, Ghigo JM, Beloin C. 2014. Biofilm-related infections: bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol Mol Biol Rev 78:510–543. doi: 10.1128/MMBR.00013-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allison KR, Brynildsen MP, Collins JJ. 2011. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meylan S, Porter CBM, Yang JH, Belenky P, Gutierrez A, Lobritz MA, Park J, Kim SH, Moskowitz SM, Collins JJ. 2017. Carbon sources tune antibiotic susceptibility in Pseudomonas aeruginosa via tricarboxylic acid cycle control. Cell Chem Biol 23:195–206. doi: 10.1016/j.chembiol.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, Rosenfeld M, Hiatt P, McCoy K, Castile R, Smith AL, Ramsey BW. 2001. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis 183:444–452. doi: 10.1086/318075. [DOI] [PubMed] [Google Scholar]
- 16.O'Sullivan BP, Freedman SD. 2009. Cystic fibrosis. Lancet 373:1891–1904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 17.Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, Vasiljev KM, Borowitz D, Bowman CM, Marshall BC, Marshall S, Smith AL. 1999. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 340:23–30. [DOI] [PubMed] [Google Scholar]
- 18.Gibson RL, Burns JL, Ramsey BW. 2003. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168:918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 19.Derichs N. 2013. Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Eur Respir Rev 22:58–65. doi: 10.1183/09059180.00008412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ong T, Ramsey BW. 2013. Modifying disease in cystic fibrosis: current and future therapies on the horizon. Curr Opin Pulm Med 19:645–651. doi: 10.1097/MCP.0b013e328365ab5f. [DOI] [PubMed] [Google Scholar]
- 21.O'Reilly R, Elphick HE. 2013. Development, clinical utility, and place of ivacaftor in the treatment of cystic fibrosis. Drug Des Devel Ther 7:929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heltshe SL, Mayer-Hamblett N, Burns JL, Khan U, Baines A, Ramsey BW, Rowe SM, GOAL (the G551D Observation-AL) Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. 2015. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis 60:703–712. doi: 10.1093/cid/ciu944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burns JL, Emerson J, Stapp JR, Yim DL, Krzewinski J, Louden L, Ramsey BW, Clausen CR. 1998. Microbiology of sputum from patients at cystic fibrosis centers in the United States. Clin Infect Dis 27:158–163. doi: 10.1086/514631. [DOI] [PubMed] [Google Scholar]
- 24.Emerson J, McNamara S, Buccat AM, Worrell K, Burns JL. 2010. Changes in cystic fibrosis sputum microbiology in the United States between 1995 and 2008. Pediatr Pulmonol 45:363–370. [DOI] [PubMed] [Google Scholar]
- 25.Moskowitz SM, Silva SJ, Mayer-Hamblett N, Pasta DJ, Mink DR, Mabie JA, Konstan MW, Wagener JS, Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis (ESCF). 2008. Shifting patterns of inhaled antibiotic use in cystic fibrosis. Pediatr Pulmonol 43:874–881. doi: 10.1002/ppul.20873. [DOI] [PubMed] [Google Scholar]
- 26.McShane D, Davies JC, Davies MG, Bush A, Geddes DM, Alton EW. 2003. Airway surface pH in subjects with cystic fibrosis. Eur Respir J 21:37–42. doi: 10.1183/09031936.03.00027603. [DOI] [PubMed] [Google Scholar]
- 27.Moriarty TF, Elborn JS, Tunney MM. 2007. Effect of pH on the antimicrobial susceptibility of planktonic and biofilm-grown clinical Pseudomonas aeruginosa isolates. Br J Biomed Sci 64:101–104. doi: 10.1080/09674845.2007.11732766. [DOI] [PubMed] [Google Scholar]
- 28.O'Toole GA. 2011. Microtiter dish biofilm formation assay. J Vis Exp 47:e2437. doi: 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderl JN, Franklin MJ, Stewart PS. 2000. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob Agents Chemother 44:1818–1824. doi: 10.1128/AAC.44.7.1818-1824.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, Britigan BE, Singh PK. 2011. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334:982–986. doi: 10.1126/science.1211037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walters MC III, Roe F, Bugnicourt A, Franklin MJ, Stewart PS. 2003. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob Agents Chemother 47:317–323. doi: 10.1128/AAC.47.1.317-323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diraviam Dinesh S. 2010. Artificial sputum medium. Protoc Exch doi: 10.1038/protex.2010.212. [DOI] [Google Scholar]
- 33.Sriramulu DD, Lunsdorf H, Lam JS, Romling U. 2005. Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J Med Microbiol 54:667–676. doi: 10.1099/jmm.0.45969-0. [DOI] [PubMed] [Google Scholar]
- 34.Høiby N. 2011. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med 9:32. doi: 10.1186/1741-7015-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yousef AA, Jaffe A. 2010. The role of azithromycin in patients with cystic fibrosis. Paediatr Respir Rev 11:108–114. [DOI] [PubMed] [Google Scholar]
- 36.Nick JA, Moskowitz SM, Chmiel JF, Forssen AV, Kim SH, Saavedra MT, Saiman L, Taylor-Cousar JL, Nichols DP. 2014. Azithromycin may antagonize inhaled tobramycin when targeting Pseudomonas aeruginosa in cystic fibrosis. Ann Am Thorac Soc 11:342–350. doi: 10.1513/AnnalsATS.201310-352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nichols DP, Happoldt CL, Bratcher PE, Caceres SM, Chmiel JF, Malcolm KC, Saavedra MT, Saiman L, Taylor-Cousar JL, Nick JA. 2016. Impact of azithromycin on the clinical and antimicrobial effectiveness of tobramycin in the treatment of cystic fibrosis. J Cyst Fibros 16:358–366. doi: 10.1016/j.jcf.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruddy J, Emerson J, Moss R, Genatossio A, McNamara S, Burns JL, Anderson G, Rosenfeld M. 2013. Sputum tobramycin concentrations in cystic fibrosis patients with repeated administration of inhaled tobramycin. J Aerosol Med Pulm Drug Deliv 26:69–75. doi: 10.1089/jamp.2011.0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geller DE, Pitlick WH, Nardella PA, Tracewell WG, Ramsey BW. 2002. Pharmacokinetics and bioavailability of aerosolized tobramycin in cystic fibrosis. Chest 122:219–226. doi: 10.1378/chest.122.1.219. [DOI] [PubMed] [Google Scholar]
- 40.Cag Y, Caskurlu H, Fan Y, Cao B, Vahaboglu H. 2016. Resistance mechanisms. Ann Transl Med 4:326. doi: 10.21037/atm.2016.09.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valentini M, Storelli N, Lapouge K. 2011. Identification of C4-dicarboxylate transport systems in Pseudomonas aeruginosa PAO1. J Bacteriol 193:4307–4316. doi: 10.1128/JB.05074-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rolsma SL, Frank DW. 2014. In vitro assays to monitor the activity of Pseudomonas aeruginosa type III secreted proteins. Methods Mol Biol 1149:171–184. doi: 10.1007/978-1-4939-0473-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sato H, Frank DW. 2004. ExoU is a potent intracellular phospholipase. Mol Microbiol 53:1279–1290. doi: 10.1111/j.1365-2958.2004.04194.x. [DOI] [PubMed] [Google Scholar]
- 44.Alkawash MA, Soothill JS, Schiller NL. 2006. Alginate lyase enhances antibiotic killing of mucoid Pseudomonas aeruginosa in biofilms. APMIS 114:131–138. doi: 10.1111/j.1600-0463.2006.apm_356.x. [DOI] [PubMed] [Google Scholar]
- 45.Pritt B, O'Brien L, Winn W. 2007. Mucoid Pseudomonas in cystic fibrosis. Am J Clin Pathol 128:32–34. doi: 10.1309/KJRPC7DD5TR9NTDM. [DOI] [PubMed] [Google Scholar]
- 46.Li Z, Kosorok MR, Farrell PM, Laxova A, West SE, Green CG, Collins J, Rock MJ, Splaingard ML. 2005. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 293:581–588. doi: 10.1001/jama.293.5.581. [DOI] [PubMed] [Google Scholar]
- 47.Epstein SS. 2009. Microbial awakenings. Nature 457:1083. doi: 10.1038/4571083a. [DOI] [PubMed] [Google Scholar]
- 48.Harms A, Maisonneuve E, Gerdes K. 2016. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 354. [DOI] [PubMed] [Google Scholar]
- 49.Sanders DB. 2015. The epidemiology of poor outcomes after pulmonary exacerbations. J Cyst Fibros 14:679–680. doi: 10.1016/j.jcf.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 50.Vogwill T, Comfort AC, Furio V, MacLean RC. 2016. Persistence and resistance as complementary bacterial adaptations to antibiotics. J Evol Biol 29:1223–1233. doi: 10.1111/jeb.12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Levin BR, Rozen DE. 2006. Non-inherited antibiotic resistance. Nat Rev Microbiol 4:556–562. doi: 10.1038/nrmicro1445. [DOI] [PubMed] [Google Scholar]
- 52.Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ. 2017. Antibiotic tolerance facilitates the evolution of resistance. Science 355:826–830. doi: 10.1126/science.aaj2191. [DOI] [PubMed] [Google Scholar]
- 53.Lewis K, Shan Y. 2017. Why tolerance invites resistance. Science 355:796. doi: 10.1126/science.aam7926. [DOI] [PubMed] [Google Scholar]
- 54.Ernst RK, Moskowitz SM, Emerson JC, Kraig GM, Adams KN, Harvey MD, Ramsey B, Speert DP, Burns JL, Miller SI. 2007. Unique lipid a modifications in Pseudomonas aeruginosa isolated from the airways of patients with cystic fibrosis. J Infect Dis 196:1088–1092. doi: 10.1086/521367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.CLSI. 2015. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 10th ed Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 56.Spoering AL, Lewis K. 2001. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J Bacteriol 183:6746–6751. doi: 10.1128/JB.183.23.6746-6751.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Todar K. 2006. Todar's online textbook of bacteriology. University of Wisconsin-Madison Department of Bacteriology, Madison, WI. [Google Scholar]
- 58.Anderson GG, Moreau-Marquis S, Stanton BA, O'Toole GA. 2008. In vitro analysis of tobramycin-treated Pseudomonas aeruginosa biofilms on cystic fibrosis-derived airway epithelial cells. Infect Immun 76:1423–1433. doi: 10.1128/IAI.01373-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.