

Tackling the multifactorial nature of virulence and antifungal drug resistance in A. fumigatus requires the mechanistic interrogation of a multitude of genes, sometimes across multiple genetic backgrounds. Classical fungal gene replacement systems can be laborious and time-consuming and, in wild-type isolates, are impeded by low rates of homologous recombination. Our simple and universal CRISPR-Cas9 system for gene manipulation generates efficient gene targeting across different genetic backgrounds of A. fumigatus. We anticipate that our system will simplify genome editing in A. fumigatus, allowing for the generation of single- and multigene knockout libraries. In addition, our system will facilitate the delineation of virulence factors and antifungal drug resistance genes in different genetic backgrounds of A. fumigatus.
KEYWORDS: Aspergillus fumigatus, CRISPR-Cas9, gene deletion, genome editing, in vitro assembly, pksP gene
ABSTRACT
CRISPR (clustered regularly interspaced short palindromic repeat)-Cas9 is a novel genome-editing system that has been successfully established in Aspergillus fumigatus. However, the current state of the technology relies heavily on DNA-based expression cassettes for delivering Cas9 and the guide RNA (gRNA) to the cell. Therefore, the power of the technology is limited to strains that are engineered to express Cas9 and gRNA. To overcome such limitations, we developed a simple and universal CRISPR-Cas9 system for gene deletion that works across different genetic backgrounds of A. fumigatus. The system employs in vitro assembly of dual Cas9 ribonucleoproteins (RNPs) for targeted gene deletion. Additionally, our CRISPR-Cas9 system utilizes 35 to 50 bp of flanking regions for mediating homologous recombination at Cas9 double-strand breaks (DSBs). As a proof of concept, we first tested our system in the ΔakuB (ΔakuBku80) laboratory strain and generated high rates (97%) of gene deletion using 2 µg of the repair template flanked by homology regions as short as 35 bp. Next, we inspected the portability of our system across other genetic backgrounds of A. fumigatus, namely, the wild-type strain Af293 and a clinical isolate, A. fumigatus DI15-102. In the Af293 strain, 2 µg of the repair template flanked by 35 and 50 bp of homology resulted in highly efficient gene deletion (46% and 74%, respectively) in comparison to classical gene replacement systems. Similar deletion efficiencies were also obtained in the clinical isolate DI15-102. Taken together, our data show that in vitro-assembled Cas9 RNPs coupled with microhomology repair templates are an efficient and universal system for gene manipulation in A. fumigatus.
IMPORTANCE Tackling the multifactorial nature of virulence and antifungal drug resistance in A. fumigatus requires the mechanistic interrogation of a multitude of genes, sometimes across multiple genetic backgrounds. Classical fungal gene replacement systems can be laborious and time-consuming and, in wild-type isolates, are impeded by low rates of homologous recombination. Our simple and universal CRISPR-Cas9 system for gene manipulation generates efficient gene targeting across different genetic backgrounds of A. fumigatus. We anticipate that our system will simplify genome editing in A. fumigatus, allowing for the generation of single- and multigene knockout libraries. In addition, our system will facilitate the delineation of virulence factors and antifungal drug resistance genes in different genetic backgrounds of A. fumigatus.
INTRODUCTION
CRISPR (clustered regularly interspaced short palindromic repeat)-Cas9 is an emerging tool for programmable genome editing in prokaryotes and eukaryotes (for reviews, refer to references 1 and 2. In this technique, which was originally discovered in bacteria as a mechanism of acquired resistance against viral infections, the Cas9 DNA nuclease recognizes and cleaves specific DNA sequences after forming a ribonucleoprotein (RNP) complex with a guide RNA (gRNA) (3–5). This gRNA is a duplex that is composed of a CRISPR RNA (crRNA) and a transactivating CRISPR RNA (tracrRNA) (1, 2). The crRNA contains a 20-base stretch designated the “protospacer,” which facilitates specific DNA cleavage by binding to complementary sequences in the target site (6). However, Cas9-mediated DNA cleavage occurs only if the protospacer is followed by a protospacer-adjacent motif (PAM) in the target sequence. For the commonly used Cas9 from Streptococcus pyogenes, the PAM site is normally an NGG (2, 7). Additionally, the crRNA contains a 22-base stretch for binding the tracrRNA (refer to Fig. 1 in reference 8. Although the tracrRNA does not directly mediate DNA binding processes, it is crucial for regulating crRNA maturation and DNA cleavage by crRNA-bound Cas9 (9, 10). In addition to the naturally occurring crRNA/tracrRNA duplex, single-guide-RNA (sgRNA) systems have been engineered to incorporate crRNA and tracrRNA into a single chimeric RNA (2, 9).
FIG 1 .

Overview of microhomology-mediated gene deletion coupled with in vitro-assembled dual Cas9 RNP cleavage. (A) (1) Cas9, tracrRNA, and dual crRNAs that cleave upstream and downstream of the pksP gene were purchased from a commercial vendor. The assembly of dual gRNA duplexes was performed by separately mixing each crRNA with equimolar amounts of tracrRNA to a final concentration of 33 μM. The two mixtures were boiled at 95°C for 5 min and then cooled to room temperature (20 to 25°C) for 10 to 15 min to allow hybridization of the crRNA to the tracrRNA. (2) For generation of dual Cas9 RNPs, each gRNA duplex was separately mixed with Cas9 (1 µg/µl) and incubated at room temperature for 5 min to allow for the formation of RNP complexes. (3) For generation of the repair template, the HygR cassette was PCR amplified using primer sets that insert 35 bp or 50 bp of flanking microhomology regions for targeting the pksP gene locus. The resulting PCR fragments were purified and utilized as repair templates. (4) The two RNP reaction mixtures were mixed with the HygR repair template and then added to A. fumigatus protoplast suspension (5 × 107 conidia/ml). The protoplasts were then transformed according to a standard protocol. (B) Inside the protoplasts, the dual Cas9 RNPs cleave upstream and downstream of pksP, resulting in complete removal of the pksP coding sequence. In the presence of the HygR repair template, the cleaved pksP gene is replaced by the HygR repair template mediated by 35 to 50 bp of microhomology regions. Deletion mutants of the pksP gene exhibit white conidia, which allow for simple assessment of gene deletion based on the conidial color of the colonies.
The DNA double-stranded break (DSB) introduced by Cas9 is primarily repaired by three pathways: nonhomologous end joining (NHEJ), homology-directed repair (HDR), and microhomology-mediated end joining (MMEJ). The NHEJ pathway (also termed classical nonhomologous end joining [C-NHEJ]) is used to introduce indels (i.e., insertion or deletions) with variable length at the DSB site, resulting in frameshift mutations. In contrast, the HDR pathway integrates an endogenous sister chromatid or an exogenous repair template containing large regions of DNA homologous to sequences flanking the DSB site. Therefore, the HDR pathway introduces mutations ranging from a 1-bp point mutation up to the integration of several coding sequences at the DSB site (2, 11). In the MMEJ pathway, which is a subset of the alternative nonhomologous end joining pathway (A-NHEJ), short homology regions (i.e., microhomology) flanking the upstream and downstream regions of the DSB site are recombined to restore DNA integrity (see Fig. 1 in references 12 and 13. With the widespread use of CRISPR-Cas9 for genome editing, the MMEJ pathway has also now been employed for Cas9-mediated manipulations by using exogenous repair templates containing microhomology regions flanking the DSB site (11).
Although the HDR and MMEJ repair machineries are both activated by a DSB and require a linear DNA fragment for DSB repair, each pathway retains distinct mechanisms for processing the resected ends of the DSB (for reviews on the mechanisms of HDR and MMEJ repair pathways, refer to reference 14. HDR is an error-free repair pathway which is predominantly active during the late S phase and the entire G2 phase of the cell cycle. In contrast, MMEJ is an error-prone pathway that is activated during G1 and early S phases of the cell cycle (12, 14–16). Another major difference between HDR and MMEJ is the size of the homology regions harbored within the linear DNA fragment. Whereas HDR requires long homology flanking regions (~500 to 5,000 bp) for precise insertion of the repair template, MMEJ-based integration of the repair template is mediated by short microhomology regions (2 to 40 bp) (12, 17–19).
Aspergillus fumigatus is the most common etiologic agent of invasive aspergillosis (IA), an often-fatal infection in immunocompromised patients (20, 21). Although current antifungals display anti-Aspergillus activity, treatment of IA infections is being challenged by the emergence of antifungal drug resistance (22). Therefore, there is an increasing need for the development of efficient genome-editing platforms to identify virulence factors and antifungal drug resistance factors across genetically distinct strains of A. fumigatus. Several CRISPR-Cas9 platforms have been established successfully to edit the genome of A. fumigatus (23–26). However, the current state of the technology relies heavily on DNA-based systems for delivering Cas9 and the gRNA to the nucleus. In these systems, Cas9 and gRNA expression cassettes are delivered on linear DNA fragments or circular plasmids to the nucleus. After integration in the genome, Cas9 and the gRNA are produced to form a Cas9 RNP complex (23–26). Alternatively, transient CRISPR-Cas9 expression systems that utilize autonomously replicating plasmids were also established in A. fumigatus (24). The major limitation to such DNA-based systems is that they require the construction of expression vectors and fungal strains for their use, restricting studies to these strains only. Additionally, DNA-based systems require the construction of species-specific expression cassettes. However, this might challenge the portability of CRISPR-Cas9 to other fungi that lack well-established genetic tools (27).
One method to overcome these drawbacks is to use in vitro-assembled Cas9 RNPs. In this system, Cas9 and gRNA expression cassettes are replaced by purified Cas9 enzyme and synthetic gRNA that are either purchased from commercial vendors or produced in the laboratory. Cas9 and the gRNA are mixed in vitro to allow the formation of functional RNPs, which are then mixed with a repair template and delivered into the host cell using traditional transformation procedures. In vitro assembly of Cas9 RNPs has been implemented previously for genome editing of Penicillium chrysogenum and several Candida species (27, 28). The major advantages of this system are simplicity (i.e., it does not require strain construction) and portability (it can be used across different species of the same genus or different strains of the same species). Accordingly, we sought in this work to utilize the advantages of the in vitro assembly of Cas9 RNPs to develop a universal system for gene deletion that works across different genetic backgrounds of A. fumigatus. Our system utilizes dual Cas9 RNP complexes that cleave upstream and downstream of the target gene, allowing for complete deletion of the target gene. Deleted genes are then replaced by a repair template (i.e., an antibiotic selection cassette) that is flanked by 35 to 50 bp of microhomology regions adjacent to Cas9 cutting sites (Fig. 1). Our results suggest that this system is a simple yet efficient tool for manipulating genomes of different A. fumigatus laboratory strains and clinical isolates.
RESULTS
Selection of the crRNA protospacer sequences for complete deletion of the pksP gene.
Our goal was to establish a simple and universal system for complete gene deletion that coupled in vitro assembly of dual Cas9 RNPs with a repair template that is flanked by microhomology regions adjacent to the target gene. Gene replacement efficiency was evaluated by using the polyketide synthase gene (pksP, Afu2g17600) as a target gene throughout this study. PksP mediates the biosynthesis of dihydroxynaphthalene (DHN)-melanin, a secondary metabolite that accounts for the gray-green color of conidia in A. fumigatus (29). Deletion of the pksP gene in A. fumigatus results in white conidia, allowing for simple assessment of gene deletion efficacy based on the conidial color of the resulting transformants. The pksP gene has been previously used as a reporter gene to assess the efficacy of several CRISPR-Cas9 platforms in A. fumigatus (23, 24).
To delete the entire coding sequence of pksP, we designed two crRNAs (designated cr5′pksP and cr3′pksP) that direct Cas9 cleavage within the 5′ untranslated region (UTR) and 3′ UTR of pksP, respectively (Fig. 2B and C; Table 1). We used the well-annotated reference strain Af293 to design the crRNAs. We first identified all PAM sites (NGG) located from −30 to +4 bp upstream and from −30 to 0 bp downstream of the pksP start and stop codons, respectively. Two PAM sites were found in the pksP upstream region: one located within the first 4 nucleotides of the pksP coding sequence and the second located 18 bp upstream of the pksP start codon. To limit the deletion of non-pksP coding sequences, we chose the cr5′pksP protospacer with the closest proximity to the start codon (Fig. 2B). The 30-bp region downstream of the pksP gene was noted to be G rich, and thus, several PAM sites were identified (Fig. 2C). The cr3′pksP protospacer was chosen because it precedes a multiple-PAM-site region (Fig. 2C) and such regions have been shown to increase Cas9 cleavage specificity (30).
FIG 2 .

Selection of dual crRNA protospacer sequences. (A) Schematic diagram of pksP coding sequence and the flanking regions that are targeted by dual in vitro-assembled Cas9 RNPs. Designing the protospacers is described in Results. (B and C) Sequence alignment of pksP upstream (B) and downstream (C) regions of three distinct genetic backgrounds of A. fumigatus. The consensus sequence (highlighted in orange) was manually generated based on sequence alignment. The 5′ crRNA and 3′ crRNA protospacer sequences are marked by red open boxes. The protospacer-adjacent motif (PAM) sequences are marked by blue open boxes. Start and stop codons are highlighted in gray. The additional adenine in the pksP downstream region of the ΔakuB strain and the clinical isolate DI15-102 is shown in red font. Cas9 DSB sites (i.e., 3 nucleotides upstream of the PAM site [1, 9, 57]) are marked by a vertical line in the sequence. The sequences of the 35-bp and 50-bp regions that are used for microhomology-mediated integration are marked by a line above the sequence.
TABLE 1 .
Oligonucleotides and crRNAs used for this study
| Type, purpose, and name | Sequencea |
|---|---|
| Oligonucleotides for | |
| Cloning HygR cassette into pCR-Blunt II-TOPO |
|
| gpdA(p)-For | CGAGCTCCCAAATCTGTCC |
| trpC(t)-Rev | AGCTTGCATGCCTGCAGG |
| Amplification of HygR cassette with 35-bp microhomology |
|
| 35 bp-pksP-HygR-F | ACTCCTTGCAGTCACGGCCGCGCAAACCACTCGCCCGAGCTCCCAAATCTGTCCA |
| 35 bp-pksP-HygR-R | AGAAACCAATTTAGGTATGCACAACTCCACCCCCAAGCTTGCATGCCTGCAGGTC |
|
Amplification of HygR cassette with 50-bp microhomology |
|
| 50 bp-pksP-HygR-F | TCCAGGATAATCACTACTCCTTGCAGTCACGGCCGCGCAAACCACTCGCCCGAGCTCCCAAATCTGTCCA |
| 50 bp-pksP-HygR-R | CAAGGAGAAAACAAAAGAAACCAATTTAGGTATGCACAACTCCACCCCCAAGCTTGCATGCCTGCAGGTC |
| Amplification of Southern blot probe | |
| pksP-Probe-Forward | GGCACGCTGTGCAGGACG |
| pksP-Probe-Reverse | GCCCGTCATCCCACTTGC |
| Sequencing DI15-102 isolate | |
| pksP(p)-159-for | TGAAGAGGTTGGACCCCAGTCGT |
| pksP-141-Rev | TTCCTGACGCAGTGCATGGAAGC |
| pksP-6521-For | CCAAACAAATGGGATACTTTGGTGGGCC |
| pksP(t)-159-Rev | ATTAGATACTCCAAACGCAACTAACTTCGGGTTCA |
| Seq-pksP(p)-126-For | GCACTTAAAGACTCCTCTTTTCAA |
| Seq-pksP-6606_For | CGGGACAGAAGGCGAA |
| crRNA sequences | |
| cr5′pksP | GCCGCGCAAACCACTCGCCA |
| cr3′pksP | GTGGGGTGAGTTCCTAGGTT |
Underlined sequences are the microhomology arms; bold sequences are for the amplification of the HygR cassette.
The chosen protospacers were then examined for sequence conservation in the ΔakuBku80 strain (henceforth referred to as the ΔakuB strain) and DI15-102. Because the genome sequence of the DI15-102 clinical isolate is not available, we used Sanger technology to sequence the 50-bp and 69-bp regions that are located upstream and downstream of the pksP start and stop codons, respectively. Next, we manually carried out sequence alignment for the 50-bp and 69-bp regions among Af293, the ΔakuB strain, and the clinical isolate. With the exception of an adenine located 9 bases downstream of the stop codon in the ΔakuB strain, our data demonstrate that the two regions are conserved among all three strains of A. fumigatus (Fig. 2A to C). Although the target sequence of the cr3′pksP protospacer contains an additional adenine in the ΔakuB strain, this additional adenine is located outside the last 6 bp of the protospacer, known as the seed region (Fig. 2C). While mismatches in the seed region have been shown to abrogate Cas9 cleavage activities, mismatches outside this region do not significantly reduce Cas9 cleavage activities (31).
Finally, each protospacer sequence was utilized to perform a BLAST search in the A. fumigatus Af293 genome (http://www.aspgd.org). To minimize Cas9 off-target cleavage, protospacers that displayed greater than 15 bp of identity to off-target (yet PAM-adjacent) genomic loci should be omitted (32). Our BLAST analysis demonstrated that neither of the designed crRNAs displayed 15-bp (or more) off-target identity in the A. fumigatus genome (data not shown). Additionally, off-target sequences with high homology to either of the designed crRNAs were lacking adjacent PAM sites, which are essential for Cas9 recognition of target sequences (9, 33–35). Accordingly, we decided to exploit these two regions for Cas9 cleavage and gene replacement events.
In vitro-assembled dual Cas9 RNPs coupled with microhomology repair templates result in efficient gene targeting in various A. fumigatus genetic backgrounds. (i) The A. fumigatus ΔakuB strain, a laboratory strain with a defective NHEJ pathway.
As a proof of concept, our initial experimentation focused on deletion of pksP in the ΔakuB strain. This strain is deficient in NHEJ repair and thus exhibits high rates of homologous recombination (36). This strain has been an important tool for A. fumigatus genetics research as a method for circumventing the typically low level of homologous recombination in wild-type strains. The repair template employed for gene replacement included a hygromycin resistance cassette (HygR), allowing selection of transformants resistant to hygromycin. To analyze the importance of the length of the homology arms flanking the HygR cassette, we investigated the effect of two sizes of microhomology regions on the efficiency of gene replacement. Thus, oligonucleotides designed for amplification of the HygR repair template included two components: (i) a 20-base region of homology to the HygR cassette for template annealing during PCR amplification and (ii) a region of microhomology of either 35 or 50 bp in length that was targeted to the genomic DNA sequence flanking the Cas9-induced DSBs at the pksP locus (Table 1; Fig. 2B and C). Finally, we also examined the effect of repair template concentration on gene replacement efficiency by performing separate transformation experiments with two different concentrations of repair template DNA (i.e., 2 µg or 10 µg). The in vitro assembly of Cas9 RNPs and subsequent transformation of A. fumigatus were both performed as described in the legend to Fig. 1 and in Materials and Methods. Gene replacement efficiency, calculated as the percentage of white colonies among the total number of transformants, was close to 100% in all four experiments using the ΔakuB strain as the parent strain. The gene replacement efficiency was approximately 97% using 2 µg of either 35-bp or 50-bp microhomology repair templates (Fig. 3A). When we increased the amount of the repair template to 10 µg, we did not observe a significant change in gene replacement efficiency (Fig. 3A). Moreover, the additional adenine in the pksP downstream region of the ΔakuB strain (Fig. 2C) did not seem to affect cr3′pksP binding activities and Cas9 cleavage since pksP deletion efficiencies in this strain were close to 100% (Fig. 3A). Taken together, our results demonstrate that in vitro-assembled Cas9 RNPs coupled with repair templates harboring as little as 35 bp of sequence homology are sufficient to generate high rates of gene deletion in the NHEJ-deficient strain. In this genetic background, the concentration of the HygR repair template did not appear to affect homologous recombination rates.
FIG 3 .

High efficiency of gene deletion in all tested genetic backgrounds of A. fumigatus. In vitro-assembled Cas9 RNPs coupled with microhomology-mediated integration of the HygR cassette were tested in ΔakuB (A), Af293 (B), and DI15-102 (C) strains. (Above) Representative transformation plates are shown for each strain using 2 µg of the HygR repair template that is flanked by 35-bp microhomology arms. (Below) The assessment of pksP deletion efficiency across different strains is plotted as the number of ΔpksP mutants out of the total number of transformation colonies. Deletion efficiencies were assessed based on the color of conidia. The ΔpksP mutant produces white colonies, while ectopic integrations result in green colonies. Deletion efficiencies represent the average from at least three independent transformations. Error bars represent the standard deviation calculated for each combination of strain, the size of HygR microhomology arms, and concentration of the HygR repair template for all experimental replicates.
(ii) A. fumigatus strain Af293, the reference genome strain.
Interstrain variability of virulence is a recently recognized and important phenomenon among environmental fungal pathogens like A. fumigatus. In fact, recent studies have highlighted important differences in virulence attributes between the two major laboratory strains of A. fumigatus, Af293 and CEA10 (37). Therefore, it is important to develop tools that may facilitate molecular studies across multiple A. fumigatus strains. Because the ΔakuB mutant was generated in the CEA10 genetic background (36), we sought to test our system in the A. fumigatus reference strain Af293. In contrast to the ΔakuB strain, the Af293 wild-type strain displays low levels of homologous integration using standard transformation protocols. Nevertheless, using our portable CRISPR-Cas9 system, we were able to increase gene deletion frequencies to 40 to 74% in Af293. When transformations were performed with 2 µg of the HygR repair template and 35-bp-homology flanking regions, 46% (±4.8%) pksP gene replacement efficiencies were observed (Fig. 3B). When the length of the homology flanks was increased to 50 bp, pksP gene deletion efficiencies increased to 74% (±10.5%) (Fig. 3B). However, when the amount of the repair template was increased to 10 µg, gene deletion efficiencies dropped for 35-bp- and 50-bp-flanked HygR cassettes to 40% (±1.5%) and 54% (±6.9%), respectively (Fig. 3B). These data suggested that increasing the concentration of the repair template in a strain with an intact NHEJ pathway resulted in an increased number of ectopic integration events. We reasoned that, because Af293 has a functional NHEJ pathway, there was an increased chance of generating unwanted ectopic integrations even in the transformants counted as successful homologous recombination events (all white colonies). Therefore, we performed a Southern blot analysis using six randomly chosen white colonies from the Af293 transformations utilizing 2 µg of the transforming repair template and 35-bp microhomology. Our analysis demonstrated that five colonies displayed a single integration event with the expected pksP deletion band size (~3.8 kb) (Fig. 4A and B). One transformant, however, displayed a band at twice the predicted pksP deletion band size (~7.6 kb) while not producing the proper 3.8-kb pksP deletion band (Fig. 4A and B). In addition, this transformant did not produce a band matching the expected wild-type banding pattern (~1.8 kb). For these reasons, we presume that this transformant is likely to contain a tandem integration of the HygR repair template at the pksP locus. Interestingly, Southern blot analysis of six randomly chosen transformants from the ΔakuB strain experiment yielded the exact same results (data not shown). Although the downstream region of the pksP gene completely matches the cr3′pksP sequence (Fig. 2C), deletion efficiencies in the Af293 strain were lower than those obtained in the ΔakuB strain (Fig. 3A and B), which contains an additional adenine in the same region. These results suggest a major role for the DNA repair pathways in modulating repair template integration. In contrast, mutations outside the protospacer seed region exhibit minor effects on the efficiencies of gene deletion. Taken together, our data suggest that the typically low levels of site-specific integration in the Af293 wild-type strain can be overcome by coupling Cas9-mediated DSBs with a microhomology repair template. However, a small portion of the resulting transformants may carry tandem integrations at the target site.
FIG 4 .
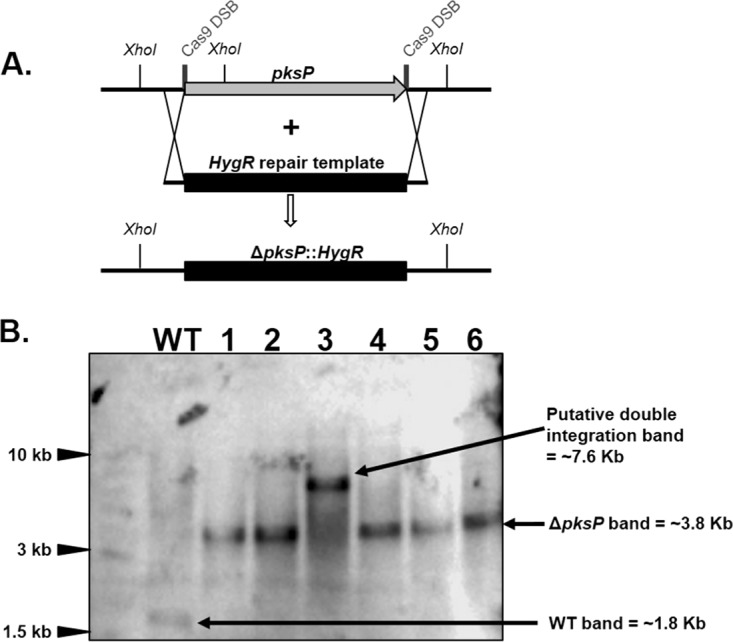
Southern blot analysis of ΔpksP mutant generated in the Af293 background. (A) Schematic representation of the genomic locus of the Af293 and ΔpksP strains. Deletion of the pksP gene was carried out using the HygR cassette. The cleavage sites of the dual in vitro-assembled Cas9 RNPs are marked by thick vertical lines. XhoI cutting sites are indicated in the pksP locus of the wild-type and ΔpksP strains. (B) Southern blot analysis of 6 arbitrarily selected colonies after digesting genomic DNA with the XhoI restriction enzyme. The wild type (WT) produced a 1.8-kb band that matches the expected wild-type banding pattern. Lanes 1, 2, 4, 5, and 6 displayed a 3.8-kb band which matches the expected pksP deletion banding pattern. The colony in lane 3 displayed a 7.6-kb band, likely containing a tandem integration of the HygR repair template at the pksP locus.
(iii) A. fumigatus DI15-102, a clinical isolate with an unknown genotype.
With the emergence of antifungal drug resistance in A. fumigatus, direct manipulation of clinical isolates is currently receiving increased attention to delineate specific resistance mechanisms in diverse clinical isolate backgrounds (38). However, like the Af293 strain employed here, mutational analyses of A. fumigatus clinical isolates are typically plagued by the same low rates of homologous recombination. Therefore, we sought to test the portability of our system into an A. fumigatus clinical isolate with a largely uncharacterized genetic background. The A. fumigatus clinical isolate DI15-102 was employed for this purpose. DI15-102 has previously been confirmed as A. fumigatus by sequence analysis of the β-tubulin gene and has been characterized as triazole resistant (39). Employing DI15-102 as the parental strain, transformation with 2 µg of a HygR repair template that includes 35 bp of microhomology resulted in 50% (±11%) deletion efficiency of pksP (Fig. 3C). Increasing the length of the homology regions to 50 bp improved gene targeting rates to 90% (±2.7%). Similarly to the Af293 reference strain, increasing the concentration of the HygR repair template to 10 µg resulted in lower efficiency of gene targeting (Fig. 3C). Our data again suggested that increasing the concentration of the repair template from 2 µg to 10 µg resulted in an elevated number of nonspecific, ectopic integrations. Additionally, pksP deletion efficiencies in DI15-102 were comparable to or higher than those obtained in Af293 (Fig. 3B and C). Both strains exhibit a functional NHEJ repair pathway, and the pksP downstream region completely matches the cr3′pksP sequence (Fig. 2C). These results suggest that Cas9-mediated gene deletion is primarily controlled by DNA repair pathways and that the effect of mutations outside the protospacer seed region is minor. It is noteworthy that transformation efficiencies of the DI15-102 clinical isolate were significantly lower than those of the ΔakuB and Af293 strains, as significantly lower total numbers of transformants were obtained. However, variability in the sequence identity of upstream and downstream regions of homology is not the reason for decreased efficiency in the clinical isolate, as these areas are identical (Fig. 2B and C). Collectively, our data demonstrate that in vitro-assembled Cas9 RNPs coupled with microhomology repair templates are a simple and universal system that works in different genetic backgrounds of A. fumigatus.
Concentration of Cas9 directly correlates with the efficiency of gene deletion.
In the previous experiments, we analyzed the effects of the concentration of the repair template, the size of the flanking homology regions, and the genetic background of A. fumigatus. We found that the efficiencies of gene deletion are primarily controlled by DNA repair pathways. Additionally, our data demonstrate that increasing the length of the homology arms to 50 bp improved gene targeting rates in Af293 and DI15-102. In contrast, increasing the concentration of the HygR repair template to 10 µg reduced deletion efficiencies in Af293 and DI15-102, as increased numbers of ectopic integrations are obtained. Throughout these initial studies, we used Cas9 nuclease at a high concentration of 1 µg/µl. Therefore, we sought here to inspect the effects of Cas9 concentration on gene deletion efficiency. We analyzed gene targeting efficiencies at two additional concentrations of Cas9. Using 2 µg of the transforming HygR repair template with 35-bp microhomology regions, gene replacement efficacies were compared between 1-µg/µl, 0.5-µg/µl, and 0.2-µg/µl Cas9 concentrations in the Af293 wild-type strain. Transformations with 1 µg/µl of Cas9 resulted in 52% (±4.8%) white colonies (Fig. 5), a percentage that is comparable to our previous results with Af293 under the same conditions (Fig. 3B). Interestingly, when the Cas9 concentration was decreased to 0.5 µg/µl, pksP deletion efficiency was not significantly affected (Fig. 5); however, the overall transformation efficiency (i.e., total number of hygromycin-resistant transformants) decreased by ~60% (data not shown). In contrast, dilution of Cas9 to 0.2 µg/µl resulted in moderately reduced pksP deletion efficiency to 28% (±5.3%) and overall transformation efficacy by ~66%, respectively. Taken together, our data demonstrate that the concentration of Cas9 correlates with transformation and gene deletion efficiencies.
FIG 5 .
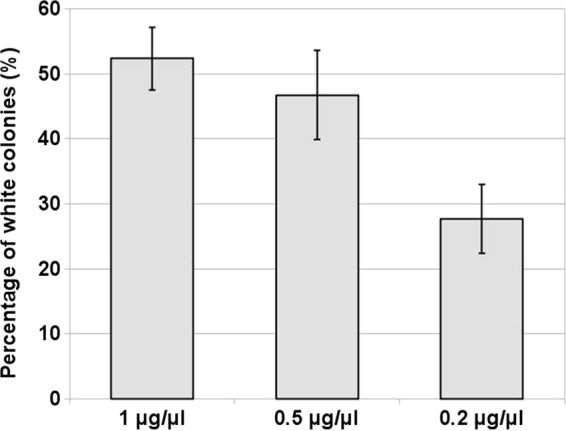
The concentration of Cas9 directly correlates with the efficiency of gene deletion. The analysis was carried out in the Af293 strain using 2 µg of the HygR repair template flanked by 35 bp of microhomology regions. Dilution of Cas9 is described in Materials and Methods. The effect of Cas9 concentration on pksP deletion rates was assessed based on the color of conidia. Deletion efficiencies represent the average from at least three independent transformations. Error bars represent the standard deviation calculated for each concentration of Cas9.
DISCUSSION
The novelty of this work lies in establishing a universal platform for site-specific gene targeting that can be employed across various genetic backgrounds of A. fumigatus. This technique offers multiple advantages over the classical technologies, which rely on homologous recombination for DNA integration. First, our system is simple as it relies only on the in vitro assembly of commercially available Cas9 proteins with the appropriate crRNA and tracrRNA (also produced commercially) to form the necessary ribonucleoprotein complexes for gene targeting. Another major advantage of our platform is that it requires only microhomology regions (35 to 50 bp) for efficient gene deletion. Currently, classical gene deletion systems in A. fumigatus wild-type strains are dependent on laborious and time-consuming construction of selectable markers with large flanking homology regions (~750 to 1,500 bp) (40). We were able to omit the standard construct building steps of subcloning DNA fragments and/or overlap extension PCR and replace them with a single PCR that amplifies a selectable marker with incorporated microhomology regions. This repair template is then mixed with in vitro-assembled Cas9 RNPs and added to protoplasts according to standard transformation protocols to generate mutant strains. A third major advantage of our system is that it results in higher gene targeting rates than the classical systems. In the Af293 wild-type strain, we achieved 46% and 74% gene deletion rates using 35-bp and 50-bp homology regions, respectively. In contrast, classical gene manipulation systems normally result in very low (~5 to 20%) gene targeting rates in the Af293 strain.
Using our current protocol with commercially supplied components, we calculate an average cost of ~$165/transformation. One possibility to reduce the cost of in vitro-assembled Cas9 RNP systems is to produce purified Cas9 and gRNA components in the laboratory. Further, as with other innovations, we would expect the cost of commercially prepared Cas9 protein and the synthesis of tracrRNA and crRNA to drop over time. CRISPR-Cas9 systems have been previously established in A. fumigatus using DNA-based expression cassettes and are promoted as cheaper ways to employ CRISPR-mediated mutagenesis (23–25). However, employing DNA-based systems for genome editing has several disadvantages compared with the purified Cas9 RNP systems. First, the DNA-based systems can be laborious and time-consuming since they require the construction of vectors with different gRNAs for targeting different genes. Second, the portability of DNA-based systems can be challenging, as many fungal species lack well-established genetic tools (27). Third, there are risks that Cas9 and gRNA expression cassettes might integrate randomly in the genome or at the DSB site (23). Finally, DNA-based delivery systems can be associated with higher rates of off-target cleavage (discussed below).
To minimize the potential for off-site targeting in our system, we performed a BLAST analysis for each of the designed gRNAs. In order for Cas9 to bind and cleave off-target sites, two gRNA criteria must be met. The first criterion is the presence of ≤5 mismatches between the chosen gRNA sequence and the off-target sequence. Cas9 normally tolerates mismatches that are located at the 5′ end of the gRNA, outside the seed region (31). This is supported in our studies, as we found that a single nucleotide mutation located outside the crRNA seed region did not affect the portability of our in vitro-assembled Cas9 RNP system. The second criterion is that the off-target sequences must precede a PAM site (9, 33–35). Although our analysis identified several sequences that display 15 bases or less of identity to either of the gRNAs, these sequences were not followed by a PAM site and therefore were not considered potential Cas9 off-target sites. Further, the delivery of a purified Cas9 RNP complex has been associated with minimal off-target cleavage events (41–43). In contrast, DNA-based delivery systems could be complicated by the potential for higher numbers of off-target effects from Cas9 expression in the cell (44). For example, the use of regulatable promoter systems could lead to low levels of constitutively expressed Cas9 nuclease if induction or repression of expression is not tightly regulated. This, in turn, could lead to increased off-target DSBs and result in undesired phenotypes. Because our system relies on only transient presence of the Cas9 protein immediately following transformation, we believe that our in vitro-assembled Cas9-gRNA-based gene manipulation in A. fumigatus carries a relatively low risk for off-site mutations compared to DNA-based systems. Our laboratory is currently working to test this hypothesis.
Our study is the first report to apply an in vitro assembly of a CRISPR-Cas9 system for complete gene deletion in A. fumigatus. To achieve the highest possible gene deletion efficiencies, we implemented dual gRNAs to target both upstream and downstream PAM sites of the pksP gene. In contrast, previous studies have used only one gRNA for site-specific gene disruption via targeting sequences with close proximity to the annotated ATG (23, 24). However, such approaches have two major drawbacks. First, inaccuracies in genome annotation might cause targeting of a nearby upstream noncoding ATG instead of the actual start codon of the target gene (45, 46). Such an error might lead to the disruption of noncoding sequences instead of genes of interest. Second, alternative transcription and splicing are common in eukaryotes and produce multiple isoforms from the same gene. These various isoforms can then have different cellular functions (47). Gene disruption by insertion of selectable markers in a single site breaks the open reading frame of some gene isoforms but not others. To overcome these risks, complete gene deletion remains the most powerful tool for the analysis of gene function. Apart from this, our work also shows that targeting two separate PAM sites in a single transformation is an efficient process with in vitro-assembled Cas9 RNPs. This might open the door for several applications. For example, targeting two separate genes with fluorophore tags could be accomplished in a single experiment. Additionally, delineating multifactorial virulence factors in fungi requires the deletion of multiple genes in one fungal strain. Accordingly, our system might be useful for marker recycling. This process, which is designated CRISPR-Cas9-induced marker excision (CRIME), has been established successfully in Candida albicans using DNA-based CRISPR-Cas9 expression cassettes (48). CRIME is also feasible using in vitro-assembled dual Cas9 RNPs.
One prerequisite for our system is the availability of a PAM site on both ends of the target gene. This should be achievable in fungi with relatively high genomic GC content such as A. fumigatus (GC content is 50% [49]). However, the identification of two PAM sites flanking the target gene in close proximity to the start and stop codons remains a challenge in fungi like Candida albicans, which has a low genomic GC content (33.5%) (50). In these organisms, two alternative solutions can be applied. The first solution is to use the Cas9 homologue Cpf1, which targets AT-rich PAM sites. Cpf1 is a type V-A endonuclease of the CRISPR-Cas class 2 that recognizes a TTN PAM sequence, and therefore, it is suitable for editing genomes with a low GC content (51, 52). The second alternative is to generate oligonucleotides with large flanking regions that would, in effect, reincorporate areas of DNA removed by inducing DSBs at PAM sites distal to the stop or start codons.
In summary, we have developed a simple and universal gene manipulation system for A. fumigatus by combining the techniques of Cas9-gRNA in vitro assembly with a microhomology repair template and adapting them for Aspergillus transformation. This system allows for efficient gene targeting in multiple genetic backgrounds, including clinical isolates. Future studies will evaluate the potential for off-target DNA cleavage caused by our in vitro RNP system.
MATERIALS AND METHODS
Strains and preparation of conidia.
The wild-type strain A. fumigatus Af293, the clinical isolate DI15-102, and the genetically engineered ΔakuB (ΔakuBku80) strain, which is deficient in the NHEJ pathway, were used in this study (36, 39, 53). For harvesting of conidia, all strains were cultivated on glucose minimal medium (GMM) agar plates (54) for 3 days at 37°C. Conidia were harvested from GMM agar plates using sterile water. After being washed three times with sterile water, conidia were counted and stored at 4°C until used.
Construction and amplification of the HygR repair template.
A hygromycin B phosphotransferase expression cassette, referred to as here as HygR, was used as the selectable marker for Cas9-mediated gene deletion throughout this work. A 2,890-bp region, which spans 1,053 bp of the gpdA promoter, 1,020 bp of hygromycin B phosphotransferase (hph), and 817 bp of the trpC terminator, was PCR amplified from plasmid pUCGH (55) using the primers gpdA(p)-For and trpC(t)-Rev (Table 1). The resulting HygR cassette was cloned into pCR-Blunt II-TOPO using the Zero Blunt TOPO PCR cloning kit (Invitrogen) according to the manufacturer’s instructions. Positive clones were Sanger sequenced to confirm the absence of mutations, and the resulting plasmid was designated pCR-HygR. For generation of the repair templates needed for Cas9-mediated gene deletion, the HygR cassette was PCR amplified from plasmid pCR-HygR using either primer set 35 bp-pksP-HygR-F and 35 bp-pksP-HygR-R or primer set 50 bp-pksP-HygR-F and 50 bp-pksP-HygR-R. The resulting PCR fragments were purified using the GeneJET gel extraction kit (Thermo Scientific) and eluted using nuclease-free water. These purified PCR products were utilized as the completed repair templates and were composed of a 2,890-bp HygR cassette flanked by either 35 bp or 50 bp of microhomology regions targeting the pksP gene locus (Afu2g17600).
In vitro assembly of Cas9-gRNA ribonucleoprotein complexes.
Cas9 ribonucleoproteins (RNPs), composed of crRNA, tracrRNA, and the Cas9 protein, were assembled in vitro using commercially available Alt-R-CRISPR-Cas9 components from Integrated DNA Technologies (IDT). For preparation of the synthesized RNA components, 100 μM stock solutions of lyophilized crRNA and tracrRNA were prepared using nuclease-free duplex buffer, supplied by IDT, and stored at −20°C until use. The Cas9 enzyme, delivered at a concentration of 10 µg/µl, was diluted 1:10 to a final concentration of 1 µg/µl using nuclease-free Cas9 working buffer (20 mM HEPES, 150 mM KCl, pH 7.5) and stored at −20°C until use.
To delete the entire coding sequence of the pksP gene, a dual Cas9-gRNA system was implemented. To do this, two separate crRNAs were designed to target selected protospacer sequences in the 5′ UTR and 3′ UTR of the pksP gene. These crRNAs were designated cr5′pksP and cr3′pksP, respectively (Table 1). Selection of the protospacer sequences is described in Results (Fig. 2). For generation of the gRNA, each crRNA was separately hybridized to the tracrRNA by mixing equal molar amounts of crRNA and tracrRNA in nuclease-free duplex buffer to a final concentration of 33 μM (Fig. 1). The two mixtures were boiled at 95°C for 5 min and then cooled to room temperature (20 to 25°C) for 10 to 15 min. The resulting gRNAs were stored on ice for up to 5 h preceding transformation or at −20°C for long-term storage. For generation of the Cas9 RNP complexes, 1.5 µl of each gRNA was separately combined with 0.75 µl of Cas9 (1 µg/µl) and 11 µl of nuclease-free Cas9 working buffer in a sterile 1.5-ml tube (final volume, 13.25 µl). These mixtures were incubated at room temperature for 5 min to allow for the formation of dual RNP complexes (Fig. 1). The two RNP reaction mixtures were mixed to a final volume of 26.5 µl and then used for A. fumigatus protoplast transformation, as described below.
Transformation of A. fumigatus.
Transformation of A. fumigatus protoplasts was carried out according to a standard protocol (56), with minor modifications. Briefly, conidia were inoculated into 100 to 150 ml of liquid YG (5 g/liter yeast extract and 20 g/liter d-glucose) at a final concentration of 1 × 106 conidia/ml and cultivated for 16 h at 30°C with shaking at 250 rpm. Mycelia were harvested by filtration, washed with sterile water, resuspended in protoplasting buffer (5% [wt/vol] VinoTastePro lytic enzyme mix, 1.2 M MgSO4⋅7H2O, 10 mM sodium phosphate buffer, pH 6.5), and incubated at 32°C with shaking at 75 rpm until protoplasts were produced (approximately 2 to 5 h, depending on the strain). Protoplasts were separated from mycelial debris by overlaying the protoplast mixture with trapping buffer (0.6 M sorbitol, 100 mM Tris-HCl, pH 7.0) and centrifuging for 15 min at 3,500 rpm and 4°C. The protoplast layer was transferred to a new tube, washed with 3 volumes of STC buffer (1.2 M sorbitol, 7.55 mM CaCl2⋅H2O, 10 mM Tris-HCl, pH 7.5), and centrifuged for 10 min at 3,500 rpm and 4°C. The protoplast pellet was then resuspended in STC buffer to a final concentration of 5 × 107 protoplasts/ml. To proceed with Cas9-mediated transformation, 200 µl of protoplasts was transferred to a sterile 15-ml tube containing the full 26.5-µl reaction mixture of the dual RNPs, prepared as described above (Fig. 1). Immediately, 2 or 10 µg of the purified repair template (described above) and 25 µl of polyethylene glycol (PEG)-CaCl2 buffer (60% [wt/vol] PEG 3350, 50 mM CaCl2⋅H2O, 450 mM Tris-HCl, pH 7.5) were added, and the mixture was incubated on ice for 50 min. Afterward, 1.25 ml PEG-CaCl2 buffer was added, and the mixture was incubated at room temperature for 20 min. Subsequently, the mixture was diluted to a total volume of 3 ml with STC buffer and plated on SMM agar plates (GMM supplemented with 1.2 M sorbitol and 1.5% [wt/vol] agar). The plates were incubated overnight at room temperature to allow regeneration of the fungal cell wall. Finally, all transformation plates were overlaid with SMM top agar (GMM supplemented with 1.2 M sorbitol and 0.7% [wt/vol] agar) containing hygromycin (final concentration of 150 µg/ml), and the plates were incubated at 37°C for 3 days.
Southern blot analysis of ΔpksP mutants.
A pksP-specific probe was amplified from genomic DNA of Af293 by using the primer set pksP-Probe-Forward and pksP-Probe-Reverse (Table 1). The resulting 504-bp PCR fragment was biotinylated with the North2South biotin random primer labeling kit (Thermo Scientific), according to the manufacturer’s protocols. The genomic DNA of 6 arbitrarily selected white colonies, which were transformed with 2 µg of the 35-bp-flanked HygR repair template, was isolated using a standard phenol-chloroform extraction protocol. Following quantification by a NanoDrop spectrophotometer and integrity verification by agarose gel electrophoresis, the genomic DNA was digested overnight at 37°C using the restriction enzyme XhoI and was separated on an agarose gel before being transferred to a Biodyne B modified nylon membrane (Thermo Scientific). Membrane-bound, digested genomic DNA was hybridized with the biotinylated probe and then labeled with horseradish peroxidase-conjugated streptavidin (Thermo Scientific). Finally, the membrane was visualized by using the North2South chemiluminescent hybridization and detection kit (Thermo Scientific).
Sequencing of clinical isolate DI15-102.
The genomic DNA of the clinical isolate DI15-102 was extracted using a standard phenol-chloroform extraction protocol. For each of the pksP dual crRNAs, a 300-bp fragment that spans 140 bp upstream and downstream of the crRNA binding site was PCR amplified. The primer pair pksP(p)-159-For and pksP-141-Rev was used to amplify the 300-bp region flanking the binding site of pksP 5′ crRNA. Similarly, pksP-6521-For and pksP(t)-159-Rev were used to amplify the 300-bp region flanking the binding site of pksP 3′ crRNA. Both fragments were purified using the GeneJET gel extraction kit (Thermo Scientific) and eluted with nuclease-free water. The purified DNA fragments were mixed with the sequencing primers Seq-pksP(p)-126-For and Seq-pksP-6606-For, which bind 126 bases and 53 bases upstream of pksP start and stop codons, respectively. Mixtures were sequenced by Genewiz Inc. using Sanger sequencing technology.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01AI106925 to J.R.F.
We thank Nathan P. Weiderhold, University of Texas Health Science Center at San Antonio, for the gift of clinical isolate DI15-102.
REFERENCES
- 1.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sander JD, Joung JK. 2014. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horvath P, Barrangou R. 2010. CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 4.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 5.Hsu PD, Lander ES, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah SA, Erdmann S, Mojica FJM, Garrett RA. 2013. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol 10:891–899. doi: 10.4161/rna.23764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karvelis T, Gasiunas G, Young J, Bigelyte G, Silanskas A, Cigan M, Siksnys V. 2015. Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biol 16:253. doi: 10.1186/s13059-015-0818-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahdar M, McMahon MA, Prakash TP, Swayze EE, Bennett CF, Cleveland DW. 2015. Synthetic CRISPR RNA-Cas9–guided genome editing in human cells. Proc Natl Acad Sci U S A 112:E7110–E7117. doi: 10.1073/pnas.1520883112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakuma T, Nakade S, Sakane Y, Suzuki K-IT, Yamamoto T. 2016. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat Protoc 11:118–133. doi: 10.1038/nprot.2015.140. [DOI] [PubMed] [Google Scholar]
- 12.Seol JH, Shim EY, Lee SE. 2017. Microhomology-mediated end joining: good, bad and ugly. Mutat Res doi: 10.1016/j.mrfmmm.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Xu X. 2017. Microhomology-mediated end joining: new players join the team. Cell Biosci 7:6. doi: 10.1186/s13578-017-0136-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ceccaldi R, Rondinelli B, D’Andrea AD. 2016. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taleei R, Nikjoo H. 2013. Biochemical DSB-repair model for mammalian cells in G1 and early S phases of the cell cycle. Mutat Res 756:206–212. doi: 10.1016/j.mrgentox.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 16.González-Huici V, Wang B, Gartner A. 2017. A role for the nonsense-mediated mRNA decay pathway in maintaining genome stability in Caenorhabditis elegans. Genetics 206:1853–1864. doi: 10.1534/genetics.117.203414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z, Liang Y, Ang EL, Zhao H. 2017. A new era of genome integration—simply cut and paste! ACS Synth Biol 6:601–609. doi: 10.1021/acssynbio.6b00331. [DOI] [PubMed] [Google Scholar]
- 18.Yao X, Wang X, Hu X, Liu Z, Liu J, Zhou H, Shen X, Wei Y, Huang Z, Ying W, Wang Y, Nie YH, Zhang CC, Li S, Cheng L, Wang Q, Wu Y, Huang P, Sun Q, Shi L, Yang H. 2017. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res 27:801–814. doi: 10.1038/cr.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamae K, Nishimura Y, Takenaga M, Nakade S, Sakamoto N, Ide H, Sakuma T, Yamamoto T. 2017. Establishment of expanded and streamlined pipeline of PITCh knock-in—a web-based design tool for MMEJ-mediated gene knock-in, PITCh designer, and the variations of PITCh, PITCh-TG and PITCh-KIKO. Bioengineered 8:302–308. doi: 10.1080/21655979.2017.1313645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Latgé JP. 1999. Aspergillus fumigatus and aspergillosis. Clin Microbiol Rev 12:310–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin SJ, Schranz J, Teutsch SM. 2001. Aspergillosis case-fatality rate: systematic review of the literature. Clin Infect Dis 32:358–366. doi: 10.1086/318483. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Effron G, Dilger A, Alcazar-Fuoli L, Park S, Mellado E, Perlin DS. 2008. Rapid detection of triazole antifungal resistance in Aspergillus fumigatus. J Clin Microbiol 46:1200–1206. doi: 10.1128/JCM.02330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuller KK, Chen S, Loros JJ, Dunlap JC. 2015. Development of the CRISPR/Cas9 system for targeted gene disruption in Aspergillus fumigatus. Eukaryot Cell 14:1073–1080. doi: 10.1128/EC.00107-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang C, Meng X, Wei X, Lu L. 2016. Highly efficient CRISPR mutagenesis by microhomology-mediated end joining in Aspergillus fumigatus. Fungal Genet Biol 86:47–57. doi: 10.1016/j.fgb.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 25.Weber J, Valiante V, Nødvig CS, Mattern DJ, Slotkowski RA, Mortensen UH, Brakhage AA. 2017. Functional reconstitution of a fungal natural product gene cluster by advanced genome editing. Synth Biol 6:62–68. doi: 10.1021/acssynbio.6b00203. [DOI] [PubMed] [Google Scholar]
- 26.Zhang C, Lu L. 2017. Precise and efficient in-frame integration of an exogenous GFP tag in Aspergillus fumigatus by a CRISPR system. Methods Mol Biol 1625:249–258. doi: 10.1007/978-1-4939-7104-6_17. [DOI] [PubMed] [Google Scholar]
- 27.Grahl N, Demers EG, Crocker AW, Hogan DA. 2017. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere 2:e00218-17. doi: 10.1128/mSphere.00218-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pohl C, Kiel JAKW, Driessen AJM, Bovenberg RAL, Nygård Y. 2016. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. Synth Biol 5:754–764. doi: 10.1021/acssynbio.6b00082. [DOI] [PubMed] [Google Scholar]
- 29.Heinekamp T, Thywissen A, Macheleidt J, Keller S, Valiante V, Brakhage AA. 2013. Aspergillus fumigatus melanins: interference with the host endocytosis pathway and impact on virulence. Front Microbiol 3:440. doi: 10.3389/fmicb.2012.00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. 2014. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng T, Hou Y, Zhang P, Zhang Z, Xu Y, Zhang L, Niu L, Yang Y, Liang D, Yi F, Peng W, Feng W, Yang Y, Chen J, Zhu YY, Zhang LH, Du Q. 2017. Profiling single-guide RNA specificity reveals a mismatch sensitive core sequence. Sci Rep 7:40638. doi: 10.1038/srep40638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. 2014. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X, Kriz AJ, Sharp PA. 2014. Target specificity of the CRISPR-Cas9 system. Quant Biol 2:59–70. doi: 10.1007/s40484-014-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai SQ, Joung JK. 2016. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat Rev Genet 17:300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferreira ME, da S, Kress MRVZ, Savoldi M, Goldman MHS, Härtl A, Heinekamp T, Brakhage AA, Goldman GH. 2006. The akuBKU80 mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus. Eukaryot Cell 5:207–211. doi: 10.1128/EC.5.1.207-211.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kowalski CH, Beattie SR, Fuller KK, McGurk EA, Tang YW, Hohl TM, Obar JJ, Cramer RA. 2016. Heterogeneity among isolates reveals that fitness in low oxygen correlates with Aspergillus fumigatus virulence. mBio 7:e01515-16. doi: 10.1128/mBio.01515-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fraczek MG, Bromley M, Buied A, Moore CB, Rajendran R, Rautemaa R, Ramage G, Denning DW, Bowyer P. 2013. The cdr1B efflux transporter is associated with non-cyp51a-mediated itraconazole resistance in Aspergillus fumigatus. J Antimicrob Chemother 68:1486–1496. doi: 10.1093/jac/dkt075. [DOI] [PubMed] [Google Scholar]
- 39.Wiederhold NP, Gil VG, Gutierrez F, Lindner JR, Albataineh MT, McCarthy DI, Sanders C, Fan H, Fothergill AW, Sutton DA. 2016. First detection of TR34 L98H and TR46 Y121F T289A Cyp51 mutations in Aspergillus fumigatus isolates in the United States. J Clin Microbiol 54:168–171. doi: 10.1128/JCM.02478-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brakhage A, Jahn B, Schmidt A. 1999. Aspergillus fumigatus: biology, clinical aspects and molecular approaches to pathogenicity. Karger, Basel, Switzerland. [Google Scholar]
- 41.Kim S, Kim D, Cho SW, Kim J, Kim JS. 2014. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, Carte J, Chen W, Roark N, Ranganathan S, Ravinder N, Chesnut JD. 2015. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol 208:44–53. doi: 10.1016/j.jbiotec.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 43.Shin SE, Lim JM, Koh HG, Kim EK, Kang NK, Jeon S, Kwon S, Shin WS, Lee B, Hwangbo K, Kim J, Ye SH, Yun JY, Seo H, Oh HM, Kim KJ, Kim JS, Jeong WJ, Chang YK, Jeong BR. 2016. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Sci Rep 6:27810. doi: 10.1038/srep27810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Meng X, Pan J, Jiang N, Zhou C, Wu Z, Gong Z. 2017. CRISPR/Cas9-mediated noncoding RNA editing in human cancers. RNA Biol doi: 10.1080/15476286.2017.1391443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koonin EV, Galperin MY. 2003. Sequence—evolution—function: computational approaches in comparative genomics. Kluwer Academic, Boston, MA. [PubMed] [Google Scholar]
- 46.Poptsova MS, Gogarten JP. 2010. Using comparative genome analysis to identify problems in annotated microbial genomes. Microbiology 156:1909–1917. doi: 10.1099/mic.0.033811-0. [DOI] [PubMed] [Google Scholar]
- 47.Dapas M, Kandpal M, Bi Y, Davuluri RV. 2017. Comparative evaluation of isoform-level gene expression estimation algorithms for RNA-seq and exon-array platforms. Brief Bioinform 18:260–269. doi: 10.1093/bib/bbw016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang MY, Mitchell AP. 2017. Marker recycling in Candida albicans through CRISPR-Cas9-Induced marker excision. mSphere 2:e00050-17. doi: 10.1128/mSphere.00050-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ, Crabtree J, Silva JC, Badger JH, Albarraq A, Angiuoli S, Bussey H, Bowyer P, Cotty PJ, Dyer PS, Egan A, Galens K, Fraser-Liggett CM, Haas BJ, Inman JM, Kent R, Lemieux S, Malavazi I, Orvis J, Roemer T, Ronning CM, Sundaram JP, Sutton G, Turner G, Venter JC, White OR, Whitty BR, Youngman P, Wolfe KH, Goldman GH, Wortman JR, Jiang B, Denning DW, Nierman WC. 2008. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet 4:e1000046. doi: 10.1371/journal.pgen.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Butler G, Rasmussen MD, Lin MF, Santos MAS, Sakthikumar S, Munro CA, Rheinbay E, Grabherr M, Forche A, Reedy JL, Agrafioti I, Arnaud MB, Bates S, Brown AJP, Brunke S, Costanzo MC, Fitzpatrick DA, de Groot PWJ, Harris D, Hoyer LL, Hube B, Klis FM, Kodira C, Lennard N, Logue ME, Martin R, Neiman AM, Nikolaou E, Quail MA, Quinn J, Santos MC, Schmitzberger FF, Sherlock G, Shah P, Silverstein KAT, Skrzypek MS, Soll D, Staggs R, Stansfield I, Stumpf MPH, Sudbery PE, Srikantha T, Zeng Q, Berman J, Berriman M, Heitman J, Gow NAR, Lorenz MC, Birren BW, Kellis M, Cuomo CA. 2009. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459:657–662. doi: 10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fonfara I, Richter H, Bratovič M, Le Rhun A, Charpentier E. 2016. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532:517–521. doi: 10.1038/nature17945. [DOI] [PubMed] [Google Scholar]
- 52.Nishimasu H, Yamano T, Gao L, Zhang F, Ishitani R, Nureki O. 2017. Structural basis for the altered PAM recognition by engineered CRISPR-Cpf1. Mol Cell 67:139–147.e2. doi: 10.1016/j.molcel.2017.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C, Bennett J, Bowyer P, Chen D, Collins M, Coulsen R, Davies R, Dyer PS, Farman M, Fedorova N, Fedorova N, Feldblyum TV, Fischer R, Fosker N, Fraser A, García JL, García MJ, Goble A, Goldman GH, Gomi K, Griffith-Jones S, Gwilliam R, Haas B, Haas H, Harris D, Horiuchi H, Huang J, Humphray S, Jiménez J, Keller N, Khouri H, Kitamoto K, Kobayashi T, Konzack S, Kulkarni R, Kumagai T, Lafton A, Latgé J-P, Li W, Lord A, Lu C, Majoros WH, May GS, Miller BL, Mohamoud Y, Molina M, Monod M, Mouyna I, Mulligan S, Murphy L, O’Neil S, Paulsen I, Peñalva MA, Pertea M, Price C, Pritchard BL, Quail MA, Rabbinowitsch E, Rawlins N, Rajandream M-A, Reichard U, Renauld H, Robson GD, de Córdoba SR, Rodríguez-Peña JM, Ronning CM, Rutter S, Salzberg SL, Sanchez M, Sánchez-Ferrero JC, Saunders D, Seeger K, Squares R, Squares S, Takeuchi M, Tekaia F, Turner G, de Aldana CRV, Weidman J, White O, Woodward J, Yu J-H, Fraser C, Galagan JE, Asai K, Machida M, Hall N, Barrell B, Denning DW. 2005. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438:1151–1156. doi: 10.1038/nature04332. [DOI] [PubMed] [Google Scholar]
- 54.Shimizu K, Keller NP. 2001. Genetic involvement of a cAMP-dependent protein kinase in a G protein signaling pathway regulating morphological and chemical transitions in Aspergillus nidulans. Genetics 157:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langfelder K, Philippe B, Jahn B, Latgé JP, Brakhage AA. 2001. Differential expression of the Aspergillus fumigatus pksP gene detected in vitro and in vivo with green fluorescent protein. Infect Immun 69:6411–6418. doi: 10.1128/IAI.69.10.6411-6418.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yelton MM, Hamer JE, Timberlake WE. 1984. Transformation of Aspergillus nidulans by using a trpC plasmid. Proc Natl Acad Sci U S A 81:1470–1474. doi: 10.1073/pnas.81.5.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gasiunas G, Barrangou R, Horvath P, Siksnys V. 2012. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]