

Abstract
Background
Chronic heart failure (CHF) is associated with skeletal muscle abnormalities contributing to exercise intolerance, muscle loss, and negative impact on patient prognosis. A primary role has been proposed for mitochondrial dysfunction, which may be induced by systemic and tissue inflammation and further contribute to low insulin signalling. The acylated form of the gastric hormone ghrelin (AG) may improve mitochondrial oxidative capacity and insulin signalling in both healthy and diseased rodent models.
Methods
We investigated the impact of AG continuous subcutaneous administration (AG) by osmotic minipump (50 nmol/kg/day for 28 days) compared with placebo (P) on skeletal muscle mitochondrial enzyme activities, mitochondrial biogenesis regulators transcriptional expression and insulin signalling in a rodent post‐myocardial infarction CHF model.
Results
No statistically significant differences (NS) were observed among the three group in cumulative food intake. Compared with sham‐operated, P had low mitochondrial enzyme activities, mitochondrial biogenesis regulators transcripts, and insulin signalling activation at AKT level (P < 0.05), associated with activating nuclear translocation of pro‐inflammatory transcription factor nuclear factor‐κB. AG completely normalized all alterations (P < 0.05 vs P, P = NS vs sham‐operated). Direct AG activities were strongly supported by in vitro C2C12 myotubes experiments showing AG‐dependent stimulation of mitochondrial enzyme activities. No changes in mitochondrial parameters and insulin signalling were observed in the liver in any group.
Conclusions
Sustained peripheral AG treatment with preserved food intake normalizes a CHF‐induced tissue‐specific cluster of skeletal muscle mitochondrial dysfunction, pro‐inflammatory changes, and reduced insulin signalling. AG is therefore a potential treatment for CHF‐associated muscle catabolic alterations, with potential positive impact on patient outcome.
Keywords: Ghrelin, Skeletal muscle, Mitochondria, Insulin signalling
Introduction
Incidence and prevalence of chronic heart failure (CHF) are increasing worldwide, and CHF‐associated morbidity and mortality are a major health problem.1, 2 CHF commonly causes skeletal muscle (SM) metabolic and functional alterations that lead to exercise intolerance and ultimately muscle loss with strong negative impact on patient outcome.3, 4, 5, 6, 7, 8, 9, 10 Altered energy metabolism with low mitochondrial oxidative capacity has been proposed to play a primary role in CHF‐induced SM changes.3, 11, 12 Cardiac and systemic pro‐inflammatory signalling activation has been conversely hypothesized to contribute to low tissue mitochondrial function,13, 14 and skeletal muscle inflammation and mitochondrial dysfunction could further impair insulin signalling15, 16, 17, 18 thereby collectively enhancing muscle catabolism. Effective therapeutic strategies to counteract CHF‐induced SM mitochondrial dysfunction and its potential metabolic consequences are therefore urgently needed.
The gastric hormone acylated ghrelin (AG) has orexigenic effects and complex metabolic activities that may include stimulation of SM mitochondrial oxidative capacity and insulin signalling in both healthy and diseased rodent models.19, 20, 21, 22 Despite potential negative effects on circulating glucose reported in human and experimental models due to potential glucogenic activities,19, 20, 23 AG has been therefore extensively studied and increasingly suggested as potential anti‐catabolic treatment under chronic disease conditions associated with loss of body and muscle mass.8, 22, 24, 25, 26 We and others have shown beneficial effects of AG on body weight, muscle mass, and systemic inflammation in cancer and renal cachexia models.21, 27, 28 In experimental chronic kidney disease, we specifically demonstrated that sustained peripheral AG administration enhances skeletal muscle mitochondrial function also independently of food intake.21 No information is, however, available on potential effects of AG administration on skeletal muscle mitochondrial function and insulin signalling in CHF. In addition, no studies investigated potential AG effects on skeletal muscle inflammation as potential mediator of AG metabolic activities in cachexia‐inducing diseases in vivo.
In the current study, we therefore investigated the impact of continuous 28 day AG subcutaneous administration on SM mitochondrial enzyme activities, transcriptional expression of mitochondrial biogenesis regulators as well as insulin signalling and pro‐inflammatory pathways in a rodent post‐myocardial infarction (IMA) CHF model. Direct AG effects on mitochondrial function and enzyme activities were further verified in vitro in C2C12 myotubes, whereas animals were studied in vivo at AG doses reported to prevent global alterations in food intake through the whole experimental period.29 Because AG was previously reported to reduce systemic inflammation markers in different chronic disease models27 and systemic inflammation may be activated in CHF with potential negative impact on SM inflammatory pathways and mitochondrial and metabolic dysfunction,13, 14 we further examined whether activating nuclear translocation of the master pro‐inflammatory transcription factor nuclear factor‐κB (NFκB) is altered in CHF and normalized by AG. The same parameters were finally investigated in the liver to determine potential tissue distribution and specificity of AG activities because differential acylated ghrelin effects were reported at hepatic level with potential to negatively affect plasma metabolic profile.19, 20, 23
Materials and methods
Experimental protocol and measurements
Animal surgery and treatments
All procedures were approved by the competent ethics committee (Ladesamt fur Gesundheit und Soziales Berlin, Germany; permit number G 0116/05). The study was designed to determine the effects of ghrelin or ghrelin analogues on body weight and composition in chronic heart failure, and part of the study results have been published elsewhere.29, 30 215–230 g male Sprague Dawley rats (Harlan‐Winkelman, Borchen, Germany) underwent surgical procedures with thoracotomy and either coronary artery ligation (IMA) or sham operation with thoracotomy and cardiac exposure without ligation of the coronary artery (Control).29, 30 All rats were treated with buprenorphine for 2 days following surgery for analgesia; 24 h mortality was 31%. Surviving animals were caged individually and fed standard chow under a 12 h light cycle; all rats were treated with furosemide starting 14 days after surgery.29, 30 After two additional weeks, IMA animals were further randomized (n = 18 each) to receive either placebo (IMA‐P) or human ghrelin at 50 nmol/kg/day (IPSEN Pharmaceuticals; IMA‐AG) via osmotic minipumps implanted subcutaneously on the back (Charles River, Sulzfeld, Germany); control animals received placebo via the same route (n = 14). After 14 day infusions, minipumps were replaced on Day 42 from surgery; animals were then sacrificed at Day 56 from surgery after identical additional 14 day infusions. Animal body weight and food intake were recorded twice weekly throughout the study period; body weight changes were analysed and compared among groups during the first week of each infusion period, that is, at Days 28–36 and 42–50 after initial surgery. Echocardiography was performed prior to sacrifice, using am 15 MHz probe and an Acuson Sequoia system (Siemens, Erlangen, Germany); animals with infarct size less than 25% of left ventricle were excluded from analyses (n = 5).
Cytochrome c oxidase and citrate synthase activities and RNA analysis
Mitochondrial cytochrome c oxidase and citrate synthase enzyme activities were measured spectrophotometrically from muscle homogenates as referenced.31 Total RNA was isolated from 40–50 mg of gastrocnemius muscle (Tri Reagent, MRC, Cincinnati, OH, USA). Transcript levels of regulators of mitochondrial biogenesis and lipid metabolism were measured by real‐time polymerase chain reaction (7900 Sequence Detection System, Applied Biosystems, Applied Biosystems Italia, Monza, Italy) as referenced.21 Briefly, 1 mg of total RNA was reverse‐transcribed (RNA Reverse Transcription KIT, Applied Biosystems) and amplified using primers (300 nM) and probes (50 nM) selected using the Primer Express Software (Applied Biosystems). Primers for PGC‐1α amplification were as follows: forward: AGCAAGCTCTGATGCTCTGAAGGA; reverse: ACCGAAGTGAGGTGCTTATGCAGT. The probe sequence for PGC‐1α detection was TTCCAGAAGTCAGCGGCCTTGTGTCAA. Primers for PPARγ amplification were as follows: forward: ACCAGGGAGTTCCTCAAAAGC; reverse: GCAAACTCAAACTTAGGCTCCATAA. The probe sequence for PPARγ detection was TGCGGAAGCCCTTTGGTGA. The reference gene used was 28S rRNA.19, 21 Target and housekeeping genes were amplified separately, and their final quantitation was achieved using a relative standard curve. Results for each gene were divided by the corresponding 28S rRNA abundance and expressed as arbitrary units.
Nuclear factor‐κB nuclear content, AKT insulin signalling, and plasma biochemical profile
Nuclear protein extracts for measurement of nuclear NFκB were obtained as previously reported.32 Protein concentration in all samples was measured by spectrophotometer (BCA Protein Assay Reagent, Pierce, Rockford, IL, USA). For measurement of tissue p65 NFκB subunit a commercially available kit was used following manufacturer instructions (TransAM, Active Motif North America, Carlsbad, CA, USA). Total (T) and phosphorylated (P: activated) AKT muscle protein contents were determined using western blotting as previously described.20 The P/T ratio was calculated as a marker of tissue AKT activation, and results were expressed as a percentage of sham value. Plasma insulin concentration was measured by enzyme‐linked immunosorbent assay (Ultrasensitive ELISA; DRG, Springfield, NJ). Plasma glucose and triglycerides levels were determined by standard enzymatic colorimetric assays.21
C2C12 myotube experiments
C2C12 myoblasts were differentiated in myotubes as previously reported.33, 34 After 4 day incubation periods with differentiation medium and 18 h starvation, they were treated with AG (0.1 or 1 μmol/L) for 48 h, followed by collection and processing. Adenosine triphosphate (ATP) production from cell preparations was measured by luciferin‐luciferase luminometric assay,33, 34, 35 and cytochrome c oxidase and citrate synthase activities were measured spectrophotometrically as referenced.19
Statistical analysis
All values are expressed as mean ± SEM. Comparison of parameters among the three groups were performed using one‐way analysis of variance followed by the Tukey post‐hoc test. P values <0.05 were considered statistically significant. Weight changes within each group were compared using paired t‐test.
Results
Body weight, food intake, and plasma metabolic profile
No statistically significant differences in body weight among groups were observed at any time, and body weight was comparable at time of surgery in all groups despite a less pronounced body weight gain in the last ghrelin infusion period (Table 1). A general overall decline in food intake was observed for all groups between the first and the second ghrelin infusion period, but cumulative food intake remained comparable among the three groups during the whole infusion periods (Table 2). Plasma insulin, glucose, and triglycerides concentrations were comparable in all study groups (Table 3).
Table 1.
Body weights
| Time | Sham | IMA‐P | IMA‐AG | |
|---|---|---|---|---|
| n = 14 | n = 18 | n = 18 | ||
| Body weight at surgery (g) | T0 | 227.9 ± 2.5 | 225.0 ± 2.1 | 227.4 ± 1.6 |
| Body weight at start of treatment (g) | T28 | 322.5 ± 7.4 | 311.4 ± 5.8 | 316.1 ± 3.7 |
| Final body weight (g) | T56 | 357.8 ± 7.8 | 339.4 ± 7.5 | 352.2 ± 5.4 |
Effects of continuous subcutaneous administration of gastric hormone acylated ghrelin (50 nmol/kg/day for 28 days) by osmotic minipump in a rodent post‐myocardial infarction chronic heart failure model on body weight. No statistical significant differences were observed among groups. Mean ± SEM, n = 14–18/group. IMA‐AG, myocardial infarction with gastric hormone acylated ghrelin; IMA‐P, myocardial infarction with placebo
Table 2.
Food intake
| Time | Sham | IMA‐P | IMA‐AG | |
|---|---|---|---|---|
| n = 14 | n = 18 | n = 18 | ||
| Average food intake (g/100 g/day) | T28‐T56 | 5.43 ± 0.07 | 5.32 ± 0.065 | 5.48 ± 0.5 |
Effects of continuous subcutaneous administration of gastric hormone acylated ghrelin (50 nmol/kg/day for 28 days) by osmotic minipump in a rodent post‐myocardial infarction chronic heart failure model on average daily food intake during treatment. No statistical significant differences were observed among groups. Mean ± SEM, n = 14–18/group. IMA‐AG, myocardial infarction with gastric hormone acylated ghrelin; IMA‐P, myocardial infarction with placebo
Table 3.
Animal characteristics
| Time | Sham | IMA‐P | IMA‐AG | |
|---|---|---|---|---|
| n = 14 | n = 18 | n = 18 | ||
| Plasma glucose (mg/dL) | T56 | 112 ± 5 | 107 ± 9 | 121 ± 6 |
| Plasma insulin (μU/mL) | T56 | 12.3 ± 2.1 | 12.8 ± 0.9 | 11.7 ± 1.3 |
| Plasma triglycerides (mg/dL) | T56 | 0.73 ± 0.08 | 0.61 ± 0.04 | 0.72 ± 0.06 |
Effects of continuous subcutaneous administration of gastric hormone acylated ghrelin (50 nmol/kg/day for 28 days) by osmotic minipump in a rodent post‐myocardial infarction chronic heart failure model on final glucose, insulin, and triglyceride plasma levels. No statistical significant differences were observed among groups. Mean ± SEM, n = 14–18/group. IMA‐AG, myocardial infarction with gastric hormone acylated ghrelin; IMA‐P, myocardial infarction with placebo
Skeletal muscle mitochondrial enzyme activities and mitochondrial biogenesis transcript levels (Figure 1)
Figure 1.
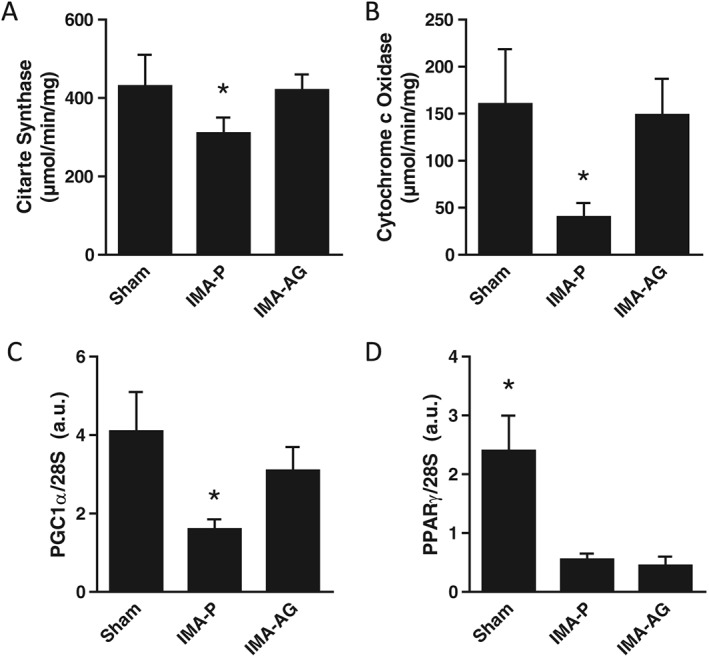
Skeletal muscle mitochondrial enzyme activities and mitochondrial biogenesis transcript levels. Effects of continuous subcutaneous administration of gastric hormone acylated ghrelin (50 nmol/kg/day for 28 days) by osmotic minipump in a rodent post‐myocardial infarction chronic heart failure model on citrate synthase (A) and cytochrome c oxidase (B) activity and on PGC1α (C) and PPARγ (D) gene expression in skeletal muscle. a.u., arbitrary units, *P < 0.05 versus other groups, mean ± SEM, n = 14–18/group. IMA‐P, myocardial infarction with placebo; IMA‐AG, myocardial infarction with gastric hormone acylated ghrelin.
Myocardial infarction with placebo had lower skeletal muscle activities of both cytochrome c oxidase and citrate synthase (P < 0.05 vs control; Figure 1). This alteration was associated with lower transcript levels of master regulators of mitochondrial biogenesis and lipid oxidation PGC‐1a and PPAR‐gamma (Figure 2). Ghrelin treatment was in turn associated with higher activities of both enzymes compared with IMA‐P (P < 0.05 vs IMA‐P, P = NS vs control; Figure 1). Transcript levels of PGC‐1a were also higher after AG treatment than in IMA‐P animals (P < 0.05 vs IMA‐P, P = NS vs control) whereas no effects of AG were observed on PPARγ expression (Figure 2).
Figure 2.
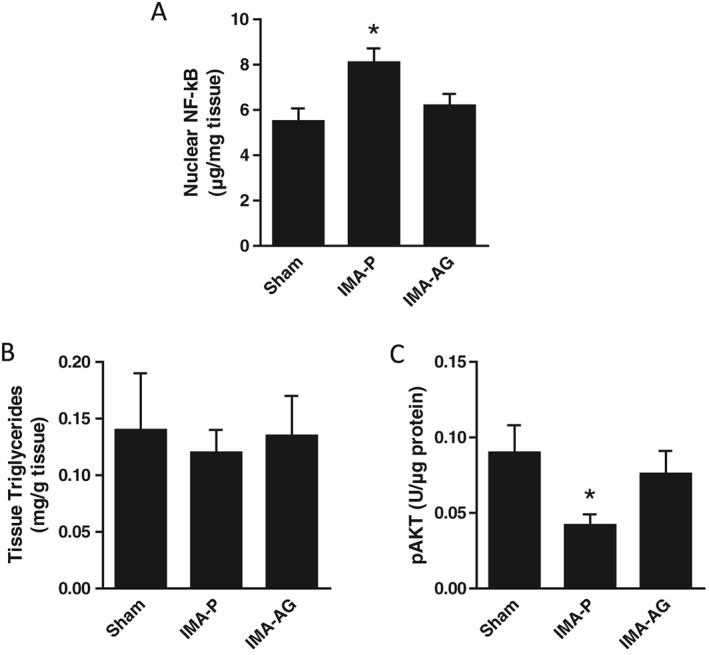
Skeletal muscle nuclear factor‐κB (NFκB) nuclear content and AKT phosphorylation. Effects of continuous subcutaneous administration of AG (50 nmol/kg/day for 28 days) by osmotic minipump in a rodent post‐myocardial infarction chronic heart failure model on nuclear NF‐κB (A), tissue triglycerides (B), and pAKT (C) levels in skeletal muscle. *P < 0.05 vs. other groups, mean ± SEM, n = 14–18/group. IMA‐P, myocardial infarction with placebo; IMA‐AG, myocardial infarction with gastric hormone acylated ghrelin.
Skeletal muscle nuclear factor‐κB nuclear content and AKT phosphorylation (Figure 2)
Higher skeletal muscle nuclear NFκB content and lower AKT phosphorylation were observed in IMA‐P rats compared with control animals. Ghrelin treatment was in turn associated with normalized nuclear NFκB and AKT phosphorylation (both P < 0.05 for IMA‐AG vs IMA‐P; P = NS for IMA‐AG vs control).
Gastric hormone acylated ghrelin activities in vitro in C2C12 myotubes (Figure 3)
Figure 3.
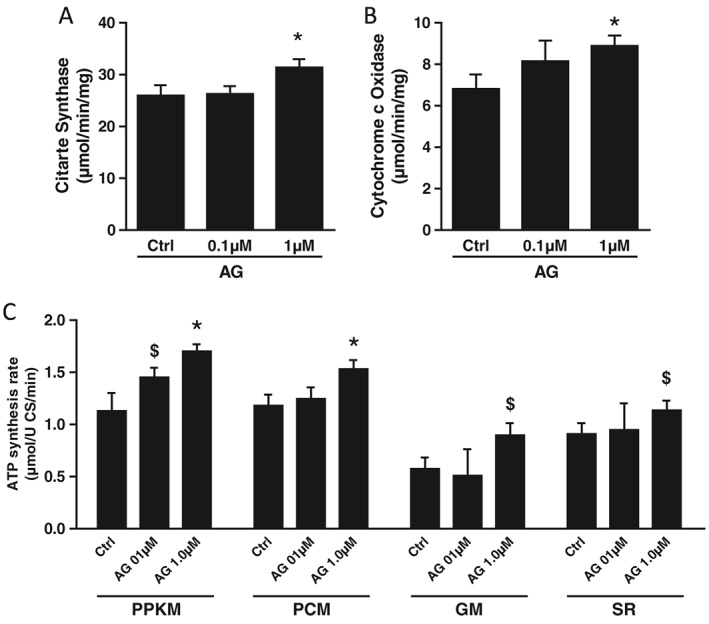
Gastric hormone acylated ghrelin (AG) activities in vitro in C2C12 myotubes. Effects of the incubation with increasing concentrations of AG or unacylated ghrelin versus control (Ct) on citrate synthase (A) and cytochrome c oxidase (B) activity and adenosine triphosphate (ATP) synthesis rate (C) in C2C12 myotubes. PPKM, pyruvate + palmitoyl‐L‐carnintine + α‐ketoglutarate + malate; PCM, palmitoyl‐L‐carnintine + malate; GM, glutamate + malate; SR, succinate + rotenone. *P < 0.05 versus Ctrl, $ P < 0.05 versus AG 0.1 μM, mean ± SEM, n = 14–18/group.
In order to determine potential direct effects of AG in skeletal muscle tissue, we further investigated its mitochondrial activities in vitro in C2C12 myotubes. Consistent with direct mitochondrial effects in skeletal muscle tissue, AG incubation resulted in enhanced mitochondrial ATP production as well as cytochrome c oxidase and citrate synthase activities (P < 0.05).
Liver mitochondrial enzyme activities and mitochondrial biogenesis transcript levels (Figure 4)
Figure 4.
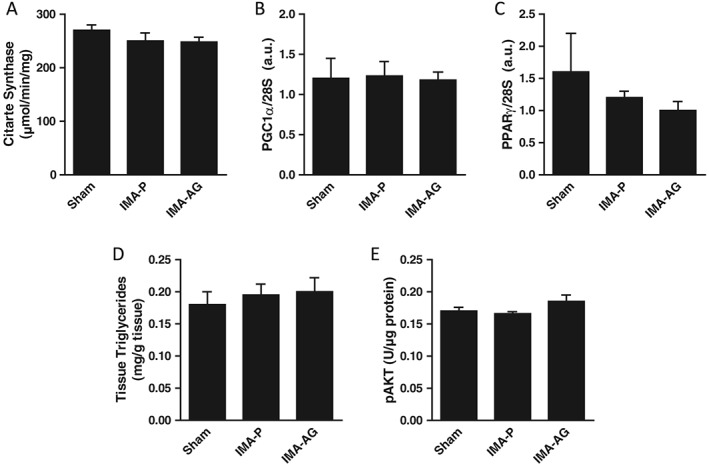
Liver mitochondrial enzyme activities and mitochondrial biogenesis transcript levels. Effects of continuous subcutaneous administration of gastric hormone acylated ghrelin (50 nmol/kg/day for 28 days) by osmotic minipump in a rodent post‐myocardial infarction chronic heart failure model on citrate synthase activity (A), on PGC1α (B), and PPARγ (C) gene expression and on tissue triglycerides (D) and pAKT (E) levels in the liver. Mean ± SEM, n = 14–18/group. IMA‐P, myocardial infarction with placebo; IMA‐AG, myocardial infarction with gastric hormone acylated ghrelin.
At variance with gastrocnemius muscle, liver activities of both cytochrome c oxidase and citrate synthase were not affected by IMA or AG treatment and were comparable in the three study groups. Transcript levels of master regulators of mitochondrial biogenesis and lipid oxidation were similarly comparable in control, IMA‐P, and IMA‐AG animals. Further supporting tissue‐specific CHF and AG activities, no changes were observed in any groups also for NFκB nuclear translocation and AKT phosphorylation.
Discussion
The current study demonstrated that (a) sustained peripheral acylated ghrelin treatment in a rodent model of IMA‐induced chronic heart failure completely normalized a cluster of skeletal muscle mitochondrial dysfunction and tissue pro‐inflammatory and insulin‐desensitizing changes; (b) acylated ghrelin appears to act at least in part directly at skeletal muscle level because its mitochondrial effects are fully reproduced in vitro in muscle cells; (c) effects of chronic heart failure per se as well as those of acylated ghrelin treatment on energy metabolism, inflammation, and insulin signalling are tissue‐specific and do not occur at hepatic level.
Mitochondrial dysfunction, inflammation, and insulin resistance have emerged in recent years as potentially causally related abnormalities with strong negative impact on skeletal muscle metabolism and function in chronic heart failure.3, 10, 11, 12, 13, 14, 15, 17 Comprehensive normalization of CHF‐induced alterations therefore indicates acylated ghrelin as a novel potential treatment for skeletal muscle dysfunction and its negative prognostic impact in CHF patients.4, 6, 7 The current findings are notably in excellent agreement with previous reports of mitochondrial effects of acylated ghrelin with enhanced skeletal muscle oxidative enzyme activities in healthy rodents19 and normalized enzyme activities in experimental chronic kidney disease.21 Anti‐inflammatory and insulin‐sensitizing AG activities were conversely also reported in healthy and obese rodent models20, 27, 36 and in vitro in response to pharmacological induction of muscle cell damage.25 Causal links and vicious cycles likely contribute to associations between muscle mitochondrial dysfunction, inflammation, and low insulin signalling in CHF. In particular, systemic pro‐inflammatory changes are reported to occur in CHF, and they could lead to both mitochondrial and insulin signalling abnormalities in skeletal muscle.13, 14 Mitochondrial dysfunction is, however, in turn also a potential primary cause of tissue inflammation and insulin resistance through enhanced reactive oxygen species generation by damaged organelles.17, 18, 34 A primary role of improved mitochondrial function in global tissue AG activities in the current model is strongly supported by AG‐induced in vitro enhancement of mitochondrial ATP production and oxidative capacity of both Krebs cycle and respiratory chain enzymes. In the current study, we notably also observed a CHF‐induced transcriptional suppression of master mitochondrial biogenesis and oxidative metabolism regulators; PGC1‐α but not PPARγ expression was selectively normalized by AG treatment, and this finding is in agreement with a direct involvement of mitochondrial biogenesis in the observed stimulation of mitochondrial parameters. Further studies should directly confirm this hypothesis.
In the current study, we investigated acylated ghrelin effects in the established IMA‐induced CHF model, and all studied animals showed evidence of impaired left‐ventricular function. Under the current conditions, all groups had comparable body weight and food intake at different experimental stages. Consistent with CHF‐induced catabolism, P‐treated animals, however, had less pronounced growth and total weight gain during the second part of the acylated ghrelin infusion treatment (Day 42–56 from surgery; not shown) despite comparable food intake. Less pronounced growth was conversely not observed in AG‐treated animals, consistent with potential muscle mass preservation through the observed muscle anti‐catabolic effects. Skeletal muscle weight was not available in the current study, and further experiments should directly aim at dissecting potential differential contributions of lean and fat tissues in acylated ghrelin anabolic activities.
Assessment of potential tissue‐specificity of CHF‐induced metabolic alterations as well as differential acylated ghrelin effects in different tissues was an additional aim of the current study. At variance with skeletal muscle, CHF had no relevant impact on hepatic mitochondrial, inflammatory, and insulin signalling parameters. This finding is consistent with available reports of clinically relevant hepatic abnormalities following acute heart failure and hemodynamic changes37 while chronic alterations in heart function may only result in liver metabolic alterations at late disease stages. Lack of AG activities in liver tissue is also consistent with tissue‐specific metabolic effects of AG on skeletal muscle and liver,19, 20 with potentially negative effects on hepatic insulin signalling and stimulation of hepatic glucogenic pathways in healthy lean rodents.19, 20 Under the current experimental conditions, lack of changes in hepatic metabolic parameters was also associated with no negative impact of AG on plasma glucose, insulin, and triglycerides. These findings are in agreement with lack of effects in chronic kidney disease models,21 thereby collectively supporting the concept that AG administration under various chronic disease conditions is metabolically safe and does not alter plasma biochemical profile as well as pivotal liver metabolic pathways.
In conclusion, we demonstrated that sustained peripheral AG treatment normalizes a CHF‐induced cluster of skeletal muscle mitochondrial dysfunction, pro‐inflammatory changes, and reduced insulin signalling. Acylated ghrelin activities at skeletal muscle level appear to be at least partly direct and independent because mitochondrial effects are fully reproduced in vitro in muscle cells. The metabolic impact of chronic heart failure per se and acylated ghrelin treatment are tissue‐specific and do not occur at hepatic level. These findings collectively indicate AG as a potential novel treatment for CHF‐associated muscle catabolic abnormalities, with potential positive impact on skeletal muscle function and patient outcome.
Conflict of interest
None declared.
Acknowledgements
The authors would like to thank Antonella Falcione and Francesca Bortolotti (International Centre for Genetic Engineering and Biotechnology, Trieste, Italy) for excellent assistance in vitro studies. Clara Ferreira and Alessia Pirulli (Department of Medical Sciences, University of Trieste) are also acknowledged for excellent assistance in sample preparation and handling.
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.38
Barazzoni, R. , Gortan Cappellari, G. , Palus, S. , Vinci, P. , Ruozi, G. , Zanetti, M. , Semolic, A. , Ebner, N. , von Haehling, S. , Sinagra, G. , Giacca, M. , and Springer, J. (2017) Acylated ghrelin treatment normalizes skeletal muscle mitochondrial oxidative capacity and AKT phosphorylation in rat chronic heart failure. Journal of Cachexia, Sarcopenia and Muscle, 8: 991–998. doi: 10.1002/jcsm.12254.
References
- 1. McMurray JJ, Pfeffer MA. Heart failure. Lancet 2005;365:1877–1889. [DOI] [PubMed] [Google Scholar]
- 2. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;18:891–975. [DOI] [PubMed] [Google Scholar]
- 3. Adams V, Reich B, Uhlemann M, Niebauer J. Molecular effects of exercise training in patients with cardiovascular disease: focus on skeletal muscle, endothelium, and myocardium. Am J Physiol Heart Circ Physiol 2017;313:H72–H88. [DOI] [PubMed] [Google Scholar]
- 4. Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb‐Peploe KM, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet 1997;349:1050–1053. [DOI] [PubMed] [Google Scholar]
- 5. Harrington D, Anker SD, Chua TP, Webb‐Peploe KM, Ponikowski PP, Poole‐Wilson PA, et al. Skeletal muscle function and its relation to exercise tolerance in chronic heart failure. J Am Coll Cardiol 1997;30:1758–1764. [DOI] [PubMed] [Google Scholar]
- 6. von Haehling S, Ebner N, Dos Santos MR, Springer J, Anker SD. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol 2017;14:323–341. [DOI] [PubMed] [Google Scholar]
- 7. von Haehling S, Lainscak M, Springer J, Anker SD. Cardiac cachexia: a systematic overview. Pharmacol Ther 2009;121:227–252. [DOI] [PubMed] [Google Scholar]
- 8. Ebner N, Sliziuk V, Scherbakov N, Sandek A. Muscle wasting in ageing and chronic illness. ESC Heart Fail 2015;2:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Konishi M, Ishida J, von Haehling S, Anker SD, Springer J. Nutrition in cachexia: from bench to bedside. J Cachexia Sarcopenia Muscle 2016;7:107–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loncar G, Springer J, Anker M, Doehner W, Lainscak M. Cardiac cachexia: hic et nunc. J Cachexia Sarcopenia Muscle 2016;7:246–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murray AJ, Edwards LM, Clarke K. Mitochondria and heart failure. Curr Opin Clin Nutr Metab Care 2007;10:704–711. [DOI] [PubMed] [Google Scholar]
- 12. Rosca MG, Hoppel CL. Mitochondrial dysfunction in heart failure. Heart Fail Rev 2013;18:607–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jahng JW, Song E, Sweeney G. Crosstalk between the heart and peripheral organs in heart failure. Exp Mol Med 2016;48:e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Remels AH, Gosker HR, Bakker J, Guttridge DC, Schols AM, Langen RC. Regulation of skeletal muscle oxidative phenotype by classical NF‐kappaB signalling. Biochim Biophys Acta 2013;1832:1313–1325. [DOI] [PubMed] [Google Scholar]
- 15. Kemppainen J, Tsuchida H, Stolen K, Karlsson H, Bjornholm M, Heinonen OJ, et al. Insulin signalling and resistance in patients with chronic heart failure. J Physiol 2003;550:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006;55:S9–S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ohta Y, Kinugawa S, Matsushima S, Ono T, Sobirin MA, Inoue N, et al. Oxidative stress impairs insulin signal in skeletal muscle and causes insulin resistance in postinfarct heart failure. Am J Physiol Heart Circ Physiol 2011;300:H1637–H1644. [DOI] [PubMed] [Google Scholar]
- 18. Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 2008;118:2992–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barazzoni R, Bosutti A, Stebel M, Cattin MR, Roder E, Visintin L, et al. Ghrelin regulates mitochondrial‐lipid metabolism gene expression and tissue fat distribution in liver and skeletal muscle. Am J Physiol Endocrinol Metab 2005;288:E228–E235. [DOI] [PubMed] [Google Scholar]
- 20. Barazzoni R, Zanetti M, Cattin MR, Visintin L, Vinci P, Cattin L, et al. Ghrelin enhances in vivo skeletal muscle but not liver AKT signaling in rats. Obesity 2007;15:2614–2623. [DOI] [PubMed] [Google Scholar]
- 21. Barazzoni R, Zhu X, Deboer M, Datta R, Culler MD, Zanetti M, et al. Combined effects of ghrelin and higher food intake enhance skeletal muscle mitochondrial oxidative capacity and AKT phosphorylation in rats with chronic kidney disease. Kidney Int 2010;77:23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anker SD, Coats AJ, Morley JE. Evidence for partial pharmaceutical reversal of the cancer anorexia‐cachexia syndrome: the case of anamorelin. J Cachexia Sarcopenia Muscle 2015;6:275–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gauna C, Meyler FM, Janssen JA, Delhanty PJ, Abribat T, van Koetsveld P, et al. Administration of acylated ghrelin reduces insulin sensitivity, whereas the combination of acylated plus unacylated ghrelin strongly improves insulin sensitivity. J Clin Endocrinol Metab 2004;89:5035–5042. [DOI] [PubMed] [Google Scholar]
- 24. Trippel TD, Holzendorf V, Halle M, Gelbrich G, Nolte K, Duvinage A, et al. Ghrelin and hormonal markers under exercise training in patients with heart failure with preserved ejection fraction: results from the Ex‐DHF pilot study. ESC Heart Fail 2017;4:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen JA, Splenser A, Guillory B, Luo J, Mendiratta M, Belinova B, et al. Ghrelin prevents tumour‐ and cisplatin‐induced muscle wasting: characterization of multiple mechanisms involved. J Cachexia Sarcopenia Muscle 2015;6:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hatanaka M, Konishi M, Ishida J, Saito M, Springer J. Novel mechanism of ghrelin therapy for cachexia. J Cachexia Sarcopenia Muscle 2015;6:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deboer MD, Zhu X, Levasseur PR, Inui A, Hu Z, Han G, et al. Ghrelin treatment of chronic kidney disease: improvements in lean body mass and cytokine profile. Endocrinology 2008;149:827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeBoer MD, Zhu XX, Levasseur P, Meguid MM, Suzuki S, Inui A, et al. Ghrelin treatment causes increased food intake and retention of lean body mass in a rat model of cancer cachexia. Endocrinology 2007;148:3004–3012. [DOI] [PubMed] [Google Scholar]
- 29. Palus S, Schur R, Akashi YJ, Bockmeyer B, Datta R, Halem H, et al. Ghrelin and its analogues, BIM‐28131 and BIM‐28125, improve body weight and regulate the expression of MuRF‐1 and MAFbx in a rat heart failure model. PLoS One 2011;6:e26865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lenk K, Palus S, Schur R, Datta R, Dong J, Culler MD, et al. Effect of ghrelin and its analogues, BIM‐28131 and BIM‐28125, on the expression of myostatin in a rat heart failure model. J Cachexia Sarcopenia Muscle 2013;4:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barazzoni R, Zanetti M, Bosutti A, Biolo G, Vitali‐Serdoz L, Stebel M, et al. Moderate caloric restriction, but not physiological hyperleptinemia per se, enhances mitochondrial oxidative capacity in rat liver and skeletal muscle‐‐tissue‐specific impact on tissue triglyceride content and AKT activation. Endocrinology 2005;146:2098–2106. [DOI] [PubMed] [Google Scholar]
- 32. Barazzoni R, Zanetti M, Semolic A, Cattin MR, Pirulli A, Cattin L, et al. High‐fat diet with acyl‐ghrelin treatment leads to weight gain with low inflammation, high oxidative capacity and normal triglycerides in rat muscle. PLoS One 2011;6:e26224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gortan Cappellari G, Semolic A, Ruozi G, Vinci P, Guarnieri G, Bortolotti F, et al. Unacylated ghrelin normalizes skeletal muscle oxidative stress and prevents muscle catabolism by enhancing tissue mitophagy in experimental chronic kidney disease. FASEB J: Official Publication of the Federation of American Societies for Experimental Biology 2017;https://doi.org/10.1096/fj.201700126R. [DOI] [PubMed] [Google Scholar]
- 34. Gortan Cappellari G, Zanetti M, Semolic A, Vinci P, Ruozi G, Falcione A, et al. Unacylated ghrelin reduces skeletal muscle reactive oxygen species generation and inflammation and prevents high‐fat diet‐induced hyperglycemia and whole‐body insulin resistance in rodents. Diabetes 2016;65:874–886. [DOI] [PubMed] [Google Scholar]
- 35. Barazzoni R, Zanetti M, Gortan Cappellari G, Semolic A, Boschelle M, Codarin E, et al. Fatty acids acutely enhance insulin‐induced oxidative stress and cause insulin resistance by increasing mitochondrial reactive oxygen species (ROS) generation and nuclear factor‐kappaB inhibitor (IkappaB)‐nuclear factor‐kappaB (NFkappaB) activation in rat muscle, in the absence of mitochondrial dysfunction. Diabetologia 2012;55:773–782. [DOI] [PubMed] [Google Scholar]
- 36. Porporato PE, Filigheddu N, Reano S, Ferrara M, Angelino E, Gnocchi VF, et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest 2013;123:611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harjola VP, Mullens W, Banaszewski M, Bauersachs J, Brunner‐La Rocca HP, Chioncel O, et al. Organ dysfunction, injury and failure in acute heart failure: from pathophysiology to diagnosis and management. A review on behalf of the Acute Heart Failure Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur J Heart Fail 2017;19:821–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]