

Abstract
Background
Muscle ring finger 1 (MuRF1) is a muscle‐specific ubiquitin E3 ligase activated during clinical conditions associated with skeletal muscle wasting. Yet, there remains a paucity of therapeutic interventions that directly inhibit MuRF1 function, particularly in vivo. The current study, therefore, developed a novel compound targeting the central coiled coil domain of MuRF1 to inhibit muscle wasting in cardiac cachexia.
Methods
We identified small molecules that interfere with the MuRF1–titin interaction from a 130 000 compound screen based on Alpha Technology. A subset of nine prioritized compounds were synthesized and administrated during conditions of muscle wasting, that is, to C2C12 muscle cells treated with dexamethasone and to mice treated with monocrotaline to induce cardiac cachexia.
Results
The nine selected compounds inhibited MuRF1–titin complexation with IC50 values <25 μM, of which three were found to also inhibit MuRF1 E3 ligase activity, with one further showing low toxicity on cultured myotubes. This last compound, EMBL chemical core ID#704946, also prevented atrophy in myotubes induced by dexamethasone and attenuated fibre atrophy and contractile dysfunction in mice during cardiac cachexia. Proteomic and western blot analyses showed that stress pathways were attenuated by ID#704946 treatment, including down‐regulation of MuRF1 and normalization of proteins associated with apoptosis (BAX) and protein synthesis (elF2B‐delta). Furthermore, actin ubiquitinylation and proteasome activity was attenuated.
Conclusions
We identified a novel compound directed to MuRF1's central myofibrillar protein recognition domain. This compound attenuated in vivo muscle wasting and contractile dysfunction in cardiac cachexia by protecting de novo protein synthesis and by down‐regulating apoptosis and ubiquitin‐proteasome‐dependent proteolysis.
Keywords: Muscle wasting, Chemical biology, Diaphragm, Myofibrillar proteins, Protein synthesis, Ubiquitin‐proteasome system
Introduction
Skeletal muscle wasting is manifested under a variety of clinical conditions, including chronic heart failure, pulmonary hypertension, mechanical ventilation, cancer, immobilization, and sepsis. The loss of muscle mass (i.e. atrophy), which occurs in both limb and respiratory muscles, plays a critical role in skeletal muscle weakness and respiratory complications observed in many patients, which, in turn, exacerbates symptoms and prognosis.1 The underlying mechanisms of muscle atrophy include the activation of so‐called atrogins, leading to an enhanced degradation of proteins via the autophagosome and ubiquitin‐proteome system2, 3, 4, 5 with muscle ring finger 1 (MuRF1) believed to provide a key step in the transfer of multi‐ubiquitinated muscle proteins to the latter system for degradation, respectively.6 Consistent with the view that MuRF1 acts as an atrogin, its expression is intimately associated with muscle wasting in numerous clinical conditions (as reviewed in Bodine and Baehr2) or following exposure to pharmacological treatments (e.g. glucocorticoids, inflammatory cytokines, reactive oxygen species7, 8, 9, 10), while its gene inactivation confers partial resistance to muscle wasting conditions.7, 8, 11, 12 Therefore, several approaches have attempted to down‐regulate MuRF1 activity therapeutically, either directly in vitro via targeted inhibition of MuRF1 by a muscle‐specific small molecule13 and adenoviral knockdowns14, 15 or indirectly in vivo via exercise training16 or antioxidant administration17, which resulted in a significant reduction in muscle wasting.
Here, we performed a high‐throughput screen based on Alpha Technology to identify compounds that inhibit the previously documented interaction between MuRF1 and titin.18 Our earlier studies assigned MuRF1's interaction with muscle proteins to a central coiled coil domain (‘MuRF1 central’).19, 20 Therefore, we hypothesized that it might be beneficial to inhibit the recognition of muscle proteins by this MuRF1 central domain, thereby maintaining its basal expression level and E3 ligase activity that resides in MuRF1's N‐terminal ring finger domain. This approach identified almost 100 compounds from the EMBL in‐house library that inhibit the titin–MuRF1 complex formation in vitro (for a prototype of this screening chemistry, refer to Labeit et al. 21). From the total of about 100 hits of the high‐throughput screen, one compound has been characterized here in detail because of its low toxicity on cultured cells and its ability to influence muscle atrophy and dysfunction in an animal model of cardiac cachexia. Overall, our data suggest that this novel compound EMBL ID#704946 directed to MuRF1 central attenuated in vivo muscle wasting and contractile dysfunction in cardiac cachexia by mechanisms that involved a protection of muscle protein de novo synthesis and a down‐regulation in apoptosis.
Methods and materials
Identification of MuRF1 central‐directed compounds
Small‐molecule screens were performed as previously described,21 as based upon our previous MuRF1 and titin interaction studies that identified the MuRF1 coiled coil domain to interact with titin A168‐170.19, 20 These interacting fragments were expressed as GST and His6 fusion proteins so that complex formation could be monitored with glutathione donor and nickel chelate (Ni‐NTA) acceptor beads, respectively, in an AlphaScreen assay.22 For this, 125 nM GST‐Murf1 and 250 nM His‐tagged titin were combined in screening buffer (PBS, 1 mM DTT, 0.05% Tween 20) and pre‐incubated for 60 min in the presence of compounds or under mock control conditions [dimethyl sulfoxide (DMSO)]. Afterwards, 5 μg/mL glutathione‐coated donor and Ni‐NTA‐coated acceptor beads were added. Following 60 min of incubation, alpha signals were recorded using an EnVision™ plate reader (PerkinElmer Life Sciences). In the primary screen, compounds from the 130 000 compound encompassing EMBL library were tested at a final concentration of 40 μM. Reordered compounds were verified by a concentration‐response experiment using an 11‐fold 1:1 serial dilution starting at 200 μM. The serially diluted compounds were also tested as an additional technical control in a de‐selection assay with 20 nM GSTHis, a fusion protein containing both an N‐terminal GST tag and a C‐terminal His tag. Taken together, our survey of the 130 000 compound‐encompassing in‐house EMBL library identified a total of 79 molecules with IC50 < 25 μmol/L for the MuRF1–titin interaction. Compounds were next assessed for effects on MuRF1 E3 ligase activity directed to titin or to MuRF1 itself (self‐ubiquitination). For this, respective compounds were added to an assay containing 75 nmol/L UBE1 (Boston Biochem), 1 μmol/L UbcH5c (Boston Biochem), 100 μmol/L ubiquitin, 4 mmol/L ATP, 100 nmol/L titin A168‐170. Reactions were started by the addition of 220 nmol/L MuRF1, followed by 1 h at 37°C SDS PAGE and western blot analysis with MuRF1 and titin‐specific antibodies. All reactions also included 5% DMSO. Three compounds were then selected based on ubiquitination patterns and thereafter synthesized at a 10 g scale by Enamine (Kiev, Ukraine). For subsequent experiments in cells or mice, compounds were either dissolved at 10 mmol/L in DMSO and added to the culture medium 0.01 to 50 μmol/L or added at 0.1% w/w to standard rodent diet (Sniff Spezialdiäten, Germany), respectively.
Cell culture experiments
Murine C2C12 myoblasts (CRL‐1772, ATCC) were cultured in DMEM (Lonza; Basel, Switzerland) supplemented with 10% foetal calf serum (Gibco® Invitrogen, Carlsbad, CA). For induction of differentiation into myotubes, subconfluent cultures were switched to DMEM containing 2% horse serum (Sigma‐Aldrich; Seelze, Germany). Thereafter, myotubes first underwent a single 2 h pre‐treatment with increasing compound concentrations (0.1 to 10 μmol/L, dissolved in DMSO) or with an equal volume of DMSO, which was followed by the addition of dexamethasone (10 μmol/L; DEX; Sigma‐Aldrich; Seelze, Germany) to the cell culture media for 24 h. Myotube diameter was then evaluated by image analysis software (Analysis 3.0, Olympus Soft Imaging Solutions GmbH, Münster, Germany). To determine cytotoxicity of selected compounds, myoblasts or myotubes were incubated with increasing concentrations for 24 h. Subsequently, the concentration of lactate dehydrogenase activity was quantified in the cell culture supernatant as a measure for cell destruction as previously described.23
Animal experimentation
The animal experiments were approved by the Regierungspräsidium Karlsruhe (35‐9185.81/G‐141/13) and the Regierungspräsidium Leipzig (TVV 40/16). Three groups of mice were included in this study, including (1) saline‐treated (sham; n = 20), (2) monocrotaline (MCT)‐treated fed a normal chow (MCT; n = 27), and (3) MCT‐treated fed a MuRF1 inhibitor chow (MCT + compound; n = 27). Briefly, C57BL/6 mice (aged 8 weeks) were subcutaneously injected weekly with either MCT (600 mg/kg) or a matched volume of saline for 6 weeks—a time period where MCT is known to induce cardiac cachexia due to pulmonary hypertension and subsequent RV dysfunction rather than anorexia.24 Mice were exposed to identical conditions under a 12:12 h light/dark cycle with food and water provided ad libitum. The MCT + compound group started receiving the inhibitor chow 1 week prior to the MCT injections, whereas the sham and MCT groups were fed an identical chow but without the addition of the selected compound. Body weight was recorded every week for each mouse. Mice were sacrificed following deep anesthetization with i.p. administration of fentanyl (0.05 mg/kg), medetomidine (0.5 mg/kg), midazolam (5 mg/kg), and ketamine (100 mg/kg). At sacrifice, the heart and lungs were dissected, cleaned, blotted dry, and weighed, with the heart fixed in 4% PBS‐buffered formalin. The left tibialis anterior (TA), soleus, extensor digitorum longus (EDL), and section of costal diaphragm were also dissected, weighed, and fixed in 4% PBS‐buffered formalin, while the remaining muscle portions were immediately frozen in liquid N2 for molecular analysis. Muscle wet weights were normalized to tibia length, which allowed a fair comparison of relative changes in muscle mass between all groups due to differences in body weight.
Contractile function
The left costal diaphragm muscle was prepared in a Krebs–Hanseleit buffer solution (120.5 NaCl, 4.8 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 20.4 NaHCO3, 1.6 CaCl2, 10 dextrose, 1 pyruvate; in mmol/L at a pH of ~7.4) at room temperature equilibrated with 95% O2–5% CO2 for contractile measurements. Briefly, a muscle bundle connected from rib to central tendon was dissected, attached to silk sutures (4‐0) at either end, and mounted vertically in a buffer‐filled organ bath (~22°C) for in vitro contractile function to be assessed using a length‐controlled lever system (301B, Aurora Scientific Inc., Aurora, Canada), as previously described.25 The muscle bundle was set at optimal length and after a 15 min thermo‐equilibration period was stimulated over a force–frequency protocol between 1 and 300 Hz (600 mA; 500 ms train duration; 0.25 ms pulse width). The muscle then underwent a force–velocity protocol whereby the muscle was allowed to shorten against external loads (80–10% of the maximal tetanic force; each separated by 1 min) after being stimulated at 150 Hz for 300 ms. Shortening velocity was determined 10 ms after the first change in length and on the linear section of the transient (DMA software, Aurora Scientific). Force (N) was normalized to muscle cross‐sectional area (CSA; cm2) by dividing muscle mass (g) by the product of L o (cm) and estimated muscle density (1.06), which allowed specific force in N/cm2 to be calculated. Shortening velocity was normalized to optimal muscle length (in L o/s), while power was calculated for each load as the product of shortening velocity and specific force (in W/cm2). A total of 16 mice per group were assessed.
Tissue analyses
RNA isolation and quantification of mRNA expression
Total RNA was isolated from C2C12 cells and reverse transcribed into cDNA using random hexamers and Sensiscript Reverse Transcriptase (Qiagen, Hilden, Germany). An aliquot of the cDNA was used for quantitative RT‐PCR, applying the Light Cycler system (Roche Diagnostics, Mannheim, Germany). The expression of specific genes was normalized to the expression of hypoxanthin phosphoribosyltransferase mRNA. For quantification of MuRF1 expression, fluorescence resonance energy transfer technology was applied using the following primers (TIB MolBiol, Berlin, Germany) and conditions: hypoxanthin phosphoribosyltransferase: 5′‐CTCATggACTgATTATggACAggAC‐3′ and 5′‐gCAggTCAgCAAAgAACTTATAgCC‐3′, 60°C annealing; MuRF1: 5′‐gATgTgCAAggAACACgAA‐3′, 5′‐CCTTCACCTggTggCTATTC‐3′, LC640‐gCACAAggAgCAAgTAggCACCTCAC‐PH, 5′‐gCCTggTgAgCCCCAAACACCT‐FL, annealing 58°C.
Histology
The heart (medial section) and TA muscle were embedded in paraffin; 3 μm sections were obtained, which where mounted on slides and stained with haematoxylin and eosin. Sections were than captured as images on a computer connected to a microscope and subsequently evaluated using Analysis software (Analysis 3.0, Olympus Soft Imaging Solutions GmbH, Münster, Germany). RV wall thickness (in μm) was determined from the mean of 10 individual measurements distributed along the free ventricular wall, while mean fibre CSA (in μm2) of the TA was evaluated after assessment of approximately 300–500 fibres per animal.
Proteomic and western blot analysis
Proteins from frozen diaphragm samples in sham, MCT, and MCT + compound mice (n = 3 per group) were powdered under liquid N2. Mass spectrometry was then performed at the DZHK mass spectrometry core facility at Bad Nauheim (for a detailed description, please refer to Konzer et al. 26). The relative ratios for MCT/sham and MCT + compound/MCT were determined, with western blot used to further study hits deemed as differentially expressed and highly significant (P < 0.01). The western blot analyses consisted of frozen TA muscle samples being homogenized in relax buffer (90 mmol/L HEPES, 126 mmol/L KCl, 36 mmol/L NaCl, 1 mmol/L MgCl, 50 mmol/L EGTA, 8 mmol/L ATP, 10 mmol/L creatine phosphate; pH 7.4) containing a protease inhibitor mix (inhibitor mix M, Serva, Heidelberg, Germany) and sonicated. Protein concentration was determined (bicinchoninic acid assay, Pierce, Bonn, Germany), and aliquots (5–20 μg) were separated by SDS‐polyacrylamide gel electrophoresis. Proteins were transferred to a polyvinylidene fluoride membrane and incubated overnight at 4°C with the following primary antibodies: MAFbx (1/2000, Eurogentec, Seraing, Belgium), MuRF1 (1/1000, Myomedix Ltd., Neckargemünd, Germany), CARP (1/500, Myomedix Ltd., Neckargemünd, Germany), BAX (1/1000; Abcam, Cambridge, UK), eIF2B‐delta (1/200; Santa Cruz Biotechnolgy, Santa Cruz, USA), myosin heavy chain (1/1000; Sigma‐Aldrich, Taufkirchen, Germany), myosin light chain (1/1000; Sigma‐Aldrich, Taufkirchen, Germany), sarcomeric actin (1/500; Sigma‐Aldrich, Taufkirchen, Germany), and ubiquitin (linkage‐K48; 1/1000; Abcam, Cambridge, UK). Membranes were subsequently incubated with a horseradish peroxidase‐conjugated secondary antibody and specific bands visualized by enzymatic chemiluminescence (Super Signal West Pico, Thermo Fisher Scientific Inc., Bonn, Germany) and densitometry quantified using a 1D scan software package (Scanalytics Inc., Rockville, USA). Blots were then normalized to the loading control GAPDH (1/30 000; HyTest Ltd., Turku, Finland). For proteasome activity in the cytosolic fraction, muscle samples were homogenized in a buffer containing 19 mM Tris (pH 7.5), 1 mM EDTA, 2 mM ATP, 20% glycerol, and 4 mM DTT. Briefly, chymotrypsin‐like activity was assayed using the fluorogenic peptide Suc‐LLVY‐7‐amino‐4‐methylcoumarine (Biomol, Hamburg, Germany). Proteins (20 μg) were incubated with reaction buffer (0.05 mol/L Tris–HCl, pH 8.0, 0.5 mmol/L EDTA) and the labelled peptide (40 μmol/L), with the kinetics of the reaction recorded using a spectrofluorometer (Tecan Safir 2, Tecan, Crailsheim, Germany) at an excitation of 380 nm and emission at 440 nm. Only the proportion of the reaction that could be inhibited by MG132 (20 μmol/L, Sigma, Taufkirchen, Germany) was regarded as chymotrypsin‐like activity. For the calculation of enzymatic activity, a calibration curve of free amino‐4‐methylcoumarine (Sigma, Taufkirchen, Germany) was recorded and values then determined as milliunits per milligram protein. All data are presented as fold change relative to sham.
Statistical analyses
Data are presented as mean ± SEM. One‐way analysis of variance (ANOVA) followed by Bonferroni post hoc was used to compare conditions in cell culture and animal experiments, while two‐way repeated measures ANOVA followed by Bonferroni post hoc was used to assess muscle contractile function in mice (GraphPad Prism). Significance was accepted as P < 0.05.
Results
In vitro selection of small molecules perturbing MuRF1
Figure 1A provides a summary of the compound selection scheme, which started with in vitro screenings for compounds that inhibit MuRF1 central titin binding functions. This scheme resulted in the compound EMBL ID#704946 being selected for our subsequent cell and mouse studies. Briefly, the in‐house library of the Chemical Biology Core Facility comprising 130 000 compounds from four commercial suppliers was screened for inhibitors in the GST‐MuRF1/His6‐titin AlphaScreen shown in Figure 1A–C. In total, 393 compounds with >35% inhibition at 40 μmol/L were selected and tested in triplicate to confirm their activity in the original MuRF1 AlphaScreen and also the absence of activity in an AlphaScreen with GSTHis as a negative control for specificity. From these, 79 compounds with confirmed MuRF1 activity were reordered as fresh powder from the suppliers in 1 mg scale to perform concentration–response titration experiments. Using as selection criteria IC50 < 25 μmol/L, no or low activity in the GSTHis counter assay, and availability from the suppliers at 1 mg scale, we established a final priority list of nine compounds (‘top 9’). Top 9 compounds fulfilled the following criteria: inhibition of MuRF1 central to titin A168‐170 interaction with IC50 < 25 μmol/L and the absence in 12 other screens carried out by the EMBL core facility. Next, we studied the effect of these identified nine compounds on the E3 ligase activity of full‐length MuRF1 constructs: The N‐terminal ring finger domain in MuRF1 catalyses in vitro the ubiquitin transfer to titin A168‐A170 (data not shown), or to itself (Figure 1D). Several compounds decreased E3 ligase activity, both to titin and to MuRF1, while others had little or no effect. In summary, we established from a 130 000 compound screen a prioritized top 9 compound list with high MuRF1–titin specificity. Of these, three compounds also inhibited MuRF1's E3 ligase activities (Figure 1D and 1E), and one compound (lane ID#704946) had a low cytotoxicity towards C2C12 myoblasts or myotubes (Figure 1F and 1G): At 25 μmol/L, the compound ID#704946 showed no apparent toxicity in C2C12 cells, whereas two structurally related analogues (analogues #704946‐5 and #704946‐8, respectively) were toxic for cells at lower concentrations. Therefore, subsequent cell and animal experiments in this study were conducted with compound ID#704946.
Figure 1.
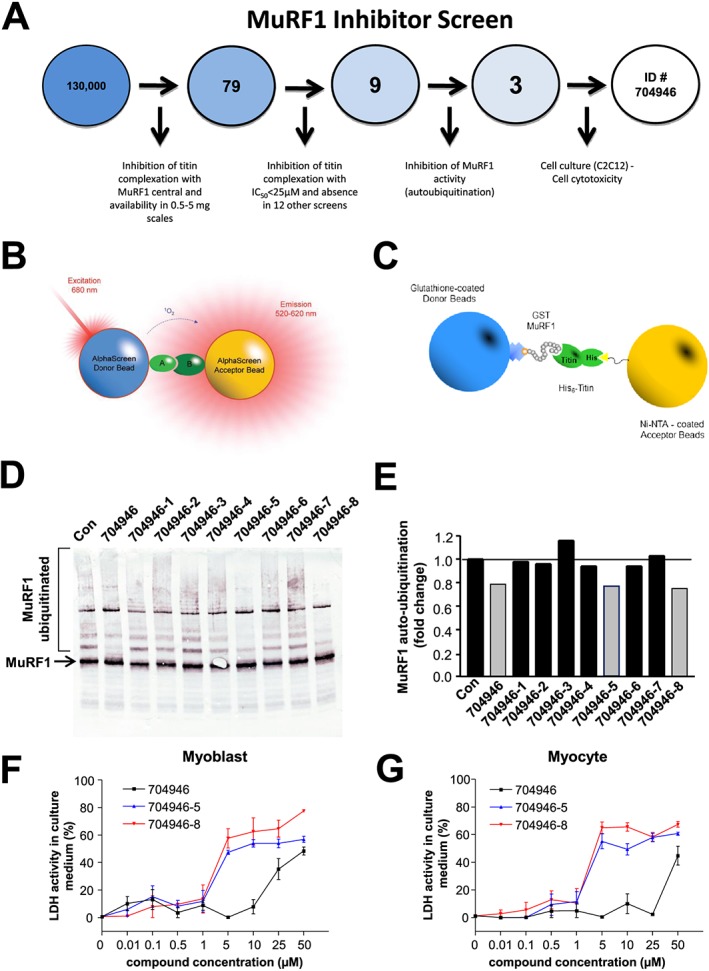
An overview of the screening process that finally prioritized the compound EMBL ID#704946 in this study (A). An AlphaScreen of 130 000 compounds was first performed, which resulted in 79 hits based on their ability to inhibit the interaction between titin and MuRF1's ‘central domain’ (B–C). These interacting fragments were expressed as GST and His‐tagged fusion proteins so that complex formation could be monitored in an AlphaScreen assay with glutathione donor and Ni‐NTA acceptor beads, respectively (C). Of these 79 compounds, a total of 9 were further synthesized and tested for inhibition towards MuRF1 autoubiquitination (D), with 3 compounds (#704946, #704946‐5, #704946‐8) showing the greatest inhibition (E). These final three compounds were then tested for cytotoxicity in myoblasts (F) and myocytes (G), with increasing concentrations of ID#704946 clearly showing a lower activity in lactate dehydrogenase. Refer to text for further details. Data are presented as mean ± SEM.
In vitro effects of the MuRF1 inhibitor ID#704946 in dexamethasone‐treated myotubes
Dexamethasone treatment induced an approximately 2‐fold increase in MuRF1 mRNA expression relative to untreated cells (P < 0.05; Figure 2A). The addition of 10 μmol/L #704946 to the culture medium attenuated this DEX‐induced increase in MuRF1 expression (for 10 μmol/L, P < 0.05; for 0.1 μmol/L, no significant effect; Figure 2A). Myotube diameter was also reduced in DEX‐treated cells by 23% (P < 0.05; Figure 2B), and while pre‐incubation of myotubes with 0.1 μmol/L #704946 had no effect, a concentration of 10 μmol/L completely abolished fibre atrophy (P < 0.05; Figure 2B). Overall, therefore, our data indicated that #704946 compound at 10 μmol/L in the culture medium attenuated DEX‐induced myotube atrophy, possibly due to attenuating MuRF1 expression.
Figure 2.

Expression of MuRF1 at the mRNA level (A) and atrophy (B) was elevated in myotubes following 24 h incubation with dexamethasone (DEX; 10 μmol/L). In contrast, pre‐treatment for 2 h with ID#704946 at 10 μmol/L (but not 0.1 μmol/L) reduced MuRF1 mRNA levels and prevented fibre atrophy. Data are presented as mean ± SEM. *P < 0.05 vs. CON. § P < 0.05 vs. DEX.
Mouse model of cardiac cachexia
Consistent with the literature,24 MCT injections over the 6 week period resulted in cardiac cachexia: Sham mice increased body weight by 7.9 ± 0.8% compared with MCT mice where a 0.7 ± 0.6% loss was observed (Figure 3A). In addition, an increase in pulmonary congestion (Figure 3B), heart weight (Figure 3C), and RV hypertrophy (Figure 3D and 3E) was also present (all P < 0.05), which is consistent with the development of pulmonary hypertension and subsequent RV dysfunction. The observed weight loss was due, at least in part, to a reduction in skeletal muscle mass, as wet weights for the EDL (Figure 4A), soleus (Figure 4B), and TA (Figure 4C) were all lower in MCT mice compared with shams. Specifically, MCT mice had a reduction in TA muscle weight of ~10% (Figure 4C), and this was associated with a lower fibre CSA of ~20% (P < 0.05; Figure 4D and 4E). In addition, skeletal muscle contractile function was also impaired in MCT mice: While no significant effects were seen in isometric forces (Figure 5A), the more clinically relevant measures of shortening velocity (Figure 5B) and power (Figure 5C) were impaired in MCT compared with sham mice, with peak values for both reduced by ~20% (P < 0.05).
Figure 3.
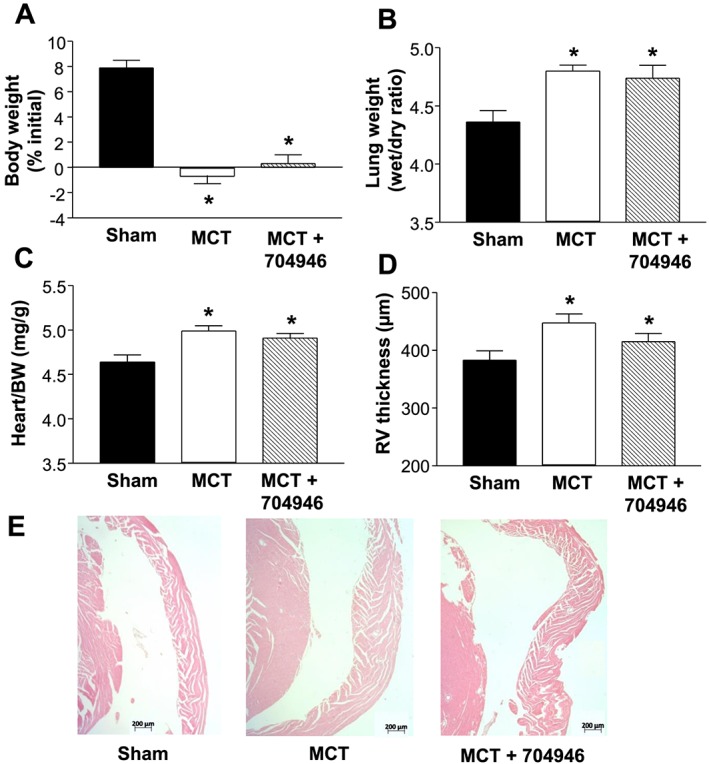
Physical characteristics of sham and mice treated with monocrotaline fed either normal chow (MCT) or EMBL ID#704946 compound (MCT + 704946). Data confirmed that MCT treatment induced cardiac cachexia independent of the chow administrated, as demonstrated by an impaired weight gain (A), increased pulmonary congestion (B), increased heart weight (C), and right ventricular (RV) hypertrophy (D), the latter visualized by representative haematoxylin and eosin‐stained sections (E). Data are presented as mean ± SEM, with n = 20 (sham), n = 27 (MCT), and n = 27 (MCT + 704946) per group. *P < 0.01 vs. sham.
Figure 4.

Skeletal muscle wet weights (normalized to tibia length) for the EDL (A), soleus (B), and tibialis anterior (TA) (C). In addition, the fibre cross‐sectional area for the TA muscle is also presented (D) and representative histological sections (E). Data are presented as mean ± SEM, with n = 20 (sham), 27 (MCT), and 27 (MCT + 704946) per group. *P < 0.05 vs. sham; § P < 0.01 vs. sham and MCT + ID#704946.
Figure 5.
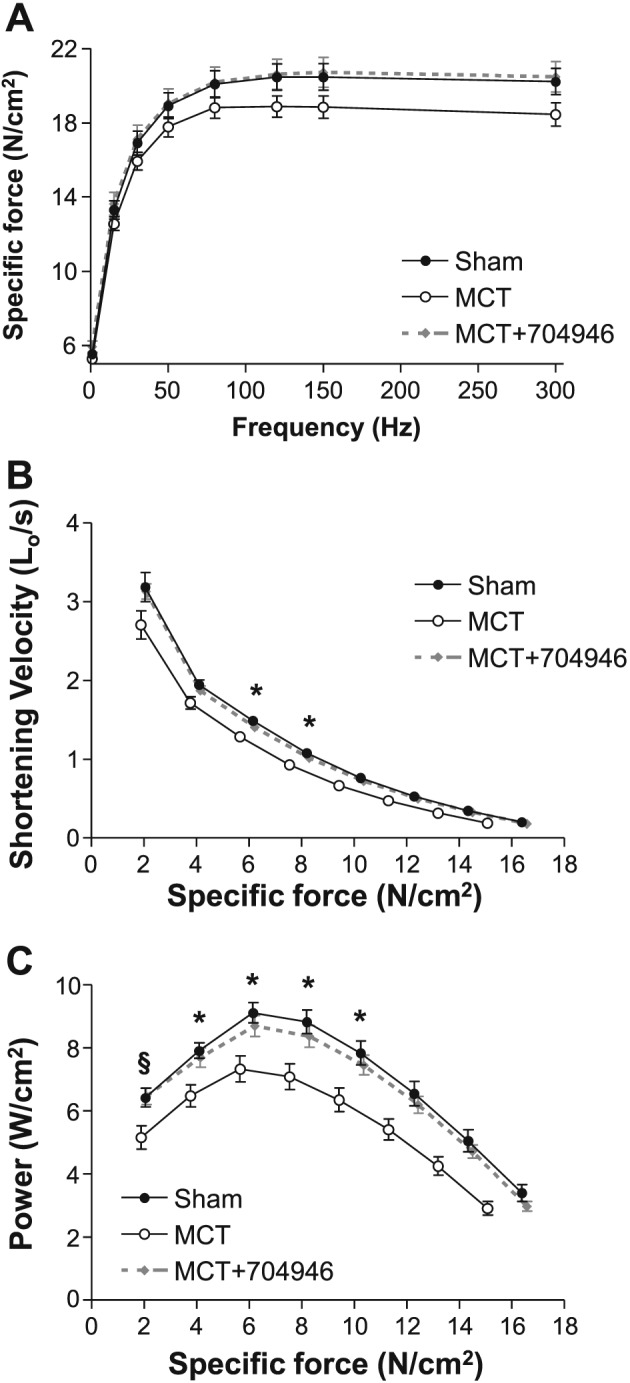
In vitro skeletal muscle contractile function, as assessed during isometric contractions (A) and also isotonic contractions, whereby shortening velocity (B) and power (C) were determined. MCT‐treated mice demonstrated impairments in shortening velocity and power by around 20% compared with shams, but this was essentially prevented in MCT mice fed the ID#704946 compound. Data are presented as mean ± SEM, with n = 16 per group. *P < 0.05 vs. sham; § P < 0.01 vs. sham and MCT + ID#704946.
In vivo effects of the MuRF1 inhibitor in cardiac cachexia
As our in vitro studies identified ID#704946 as having the greatest inhibition of ubiquitin transfer (Figure 1E) in combination with the lowest toxicity (Figure 1F and 1G), while further being able to lower MuRF1 expression and myotube atrophy in DEX‐treated C2C12 cells, we next tested the effects of this compound in mice with cardiac cachexia induced by MCT. MCT‐treated mice fed with ID#704946 underwent the same pathophysiological alterations of cardiac cachexia to that observed in the MCT mice on normal chow: Pulmonary congestion was accompanied by right ventricular hypertrophy, while weight gain was absent (Figure 3). In marked contrast to MCT mice on normal chow, however, ID#704946 feeding rescued TA from atrophy both with respect to total muscle weight (Figure 4C) and fibre CSA (Figure 4D and 4E). We also observed effects in other skeletal muscles in the ID#704946‐fed mice, such that wet weights of the EDL and soleus muscles in MCT + ID#704946 mice were not statistically different to shams (Figure 4A and 4B). Importantly, ID#704946 feeding in MCT‐treated mice was also able to restore muscle contractile function, both with regards to shortening velocity and power when compared with sham mice (P < 0.05; Figure 5B and 5C). In summary, therefore, our animal feeding experiments indicated that ID#704946 attenuated skeletal muscle wasting and dysfunction in cardiac cachexia, with the most obvious effects noted in the TA and diaphragm. As such, we performed subsequent molecular analyses on these muscle groups.
Effects of compound treatment on signalling and contractile proteins
To gain insights into the apparent protective mechanisms of ID#704946, we next analysed the proteomes from muscle tissue in mice from each experimental group (Figure 6A and 6B). Comparison of proteome data sets revealed 51 proteins from the MCT group and 70 proteins from the MCT + compound group significantly differentially expressed (P < 0.01; Figure 6C), out of which 5 were normalized by compound treatment and identified as positive hits (P < 0.01; Figure 6C and 6D). Two of the identified hits were suggestive of a protective mechanism: Reversal of MCT‐induced reduction in eIF2B (subunit δ) implies that ID#704946 prevented dysregulation in protein translation, while reversal of the MCT‐induced increase in BAX by this substance implies a protection from augmented apoptosis in this cardiac cachexia model. To verify our mass spectrometry screening data, we next performed western blots with specific antibodies. This independent method confirmed that both eIF2B and BAX were normalized in compound‐fed animals: eIF2Bδ expression was significantly reduced by 25% in MCT mice compared with shams, but this was prevented in mice fed by the compound, while conversely, BAX expression was significantly increased by 103% in MCT mice compared with shams, but this again was not observed in MCT + compound mice (Figure 6E–G). While mass spectrometry provides global unbiased overviews, low abundant proteins are not displayed in whole proteome data sets. Therefore, we also analysed protein expression of a number of key signalling genes by western blots, including MuRF1 as well as MAFbx (another key E3 ligase), in order to confirm that the ID#704946 compound was specific to MuRF1, while also quantifying CARP (a binding target of MuRF127). In support of our cell culture experiments, the immunoblots revealed that the compound was able to down‐regulate the increased MuRF1 expression induced by cardiac cachexia (P < 0.05; Figure 7A), whereas the expression of MAFbx remained unchanged between groups (P > 0.05; Figure 7B). In addition, the expression of the MuRF1 target CARP was also normalized to sham values following treatment with the compound (P > 0.05; Figure 7C).
Figure 6.
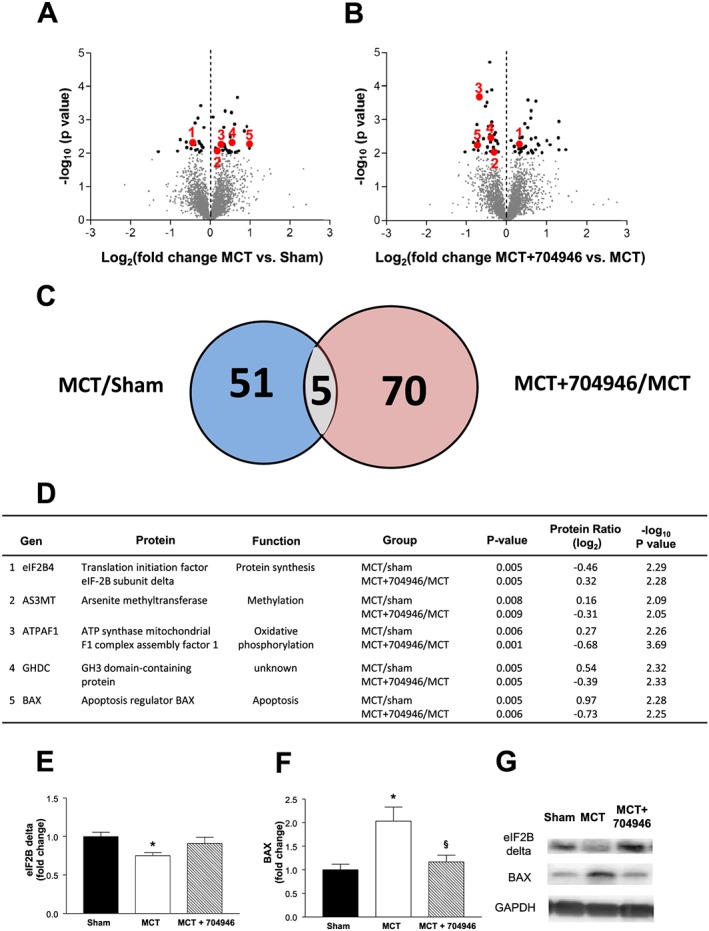
The proteomes expressed in TA muscle samples were analysed using mass spectrometry to identify differentially expressed protein in the different groups (n = 3 per group). (A, B) Differentially expressed proteins found in MCT/sham mice (A) and in MCT + ID#704946/MCT mice (B) are shown by volcano plots. (C) The set of 51 differentially expressed proteins from group A and of 70 proteins from group B share a group of 5 proteins that are differentially expressed in both sets with high significance. We therefore consider them as candidates to be modulated by the ID#704946 compound during MCT stress: These five proteins are detailed in the table (D). Of these, two proteins were further investigated by western blot (n = 9 per group) including eIF2B subunit‐delta (E) and BAX (F), with representative blots also presented (G). Data are presented as mean ± SEM. *P < 0.05 vs. sham; § P < 0.01 vs. MCT.
Figure 7.
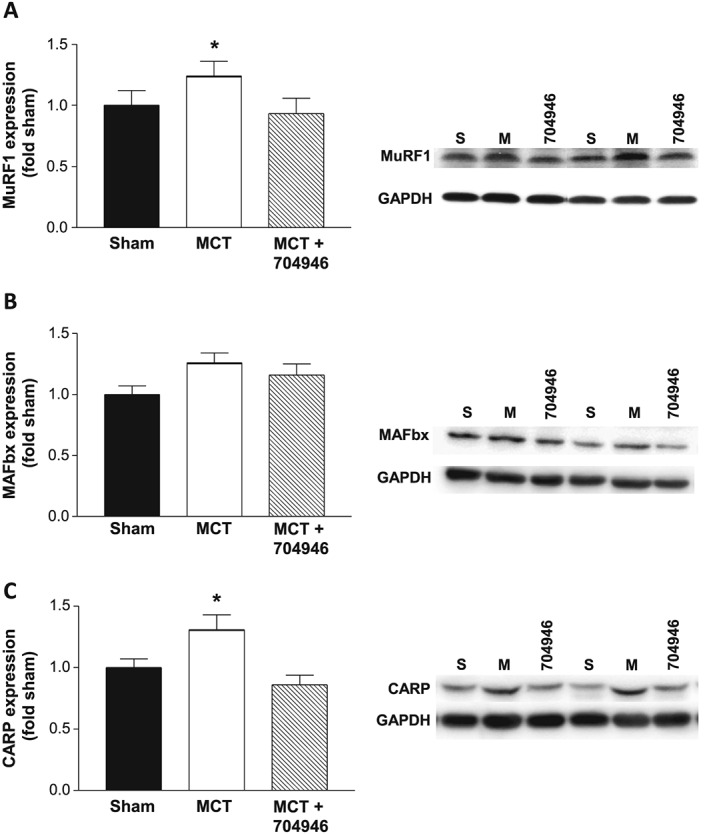
Protein expression and representative blots for MuRF1 (A), MAFBx (B), and CARP (C) for sham, MCT, and MCT + ID#704946 mice. Our data revealed that MCT treatment leads to an increase in MuRF1 and CARP expression, but this was attenuated in mice fed the #704946 compound. No changes were detected in the levels of MAFBx. Data are presented as mean ± SEM, with n = 12–16 per group. *P < 0.05 vs. sham and MCT + ID#704946.
We finally assessed also contractile proteins, because the compound screen was designed in the first place to perturb recognition of myofibrillar proteins by MuRF1: While the expression of myosin heavy and light chains were not different between groups (P > 0.05; Figure 8A and 8B), we found sarcomeric actin to be reduced in MCT mice compared with shams by 14% (P < 0.05; Figure 8C). Depletion of sarcomeric actin by MCT stress was prevented in mice treated with the compound ID704946 (P > 0.05; Figure 8C). Consistent with the idea that this compound can attenuate stress‐induced E3 ligase activities towards myofibrillar proteins, we found ubiquitinated actin to be elevated in MCT mice by 19% (P < 0.05; Figure 8D), whereas mice treated with MCT and the compound had only a 10% increase as compared with shams (P > 0.05; Figure 8D). In line with these findings, proteasome activity was shown to be up‐regulated in the MCT mice compared with both sham and MCT + compound mice by ~30% (Figure 8F). Collectively, therefore, the combined proteome, immunoblot, and activity data are consistent with the notion that ID#704946 acts as a MuRF1 antagonist that blunts ubiquitination and subsequent proteasome activity.
Figure 8.
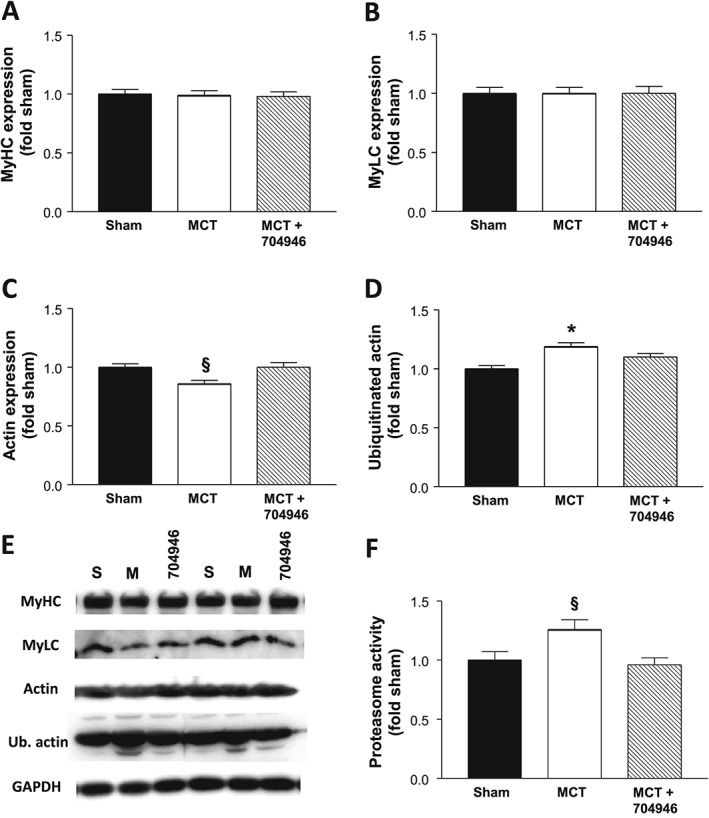
Protein expression for the contractile proteins myosin heavy (A) and light chains (B), sarcomeric actin (C), as well as ubiquitinated actin (D), with the respective representative blots also presented (E) for sham, MCT, and MCT + ID#704946 mice. In addition, proteasome activity was also measured in each group (F). Data are presented as mean ± SEM, with n = 12–16 per group. *P < 0.05 vs. sham and MCT + ID#704946.
Discussion
Development of the MuRF1 inhibitor compound
Previous studies have reported that small‐molecule knockdowns of MuRF1 can prevent atrophy ex vivo in myotubes.13, 14, 15 However, to our knowledge, this concept had yet to be translated to an in vivo setting, such as animal models that mimic clinically relevant disease conditions. It is likely that a complete inhibition of MuRF1, such as blockade of its ring finger functions or by gene inactivation, may be detrimental as MuRF1 is suggested to have additional functions beyond simply that of an atrogin.28 For example, MuRF1 knockout (KO) mice are much more susceptible to cardiac hypertrophy,28 and patients with null mutations in the MuRF1 gene also suffer a similar fate,29 which indicates that basal MuRF1 expression is required for myocyte homeostasis. In addition, small molecules directed to the ubiquitin transfer catalysing ring finger of MuRF1 might provide toxicity by lack of target specificity and thus leading to protein aggregates.18, 27, 30 Based on these considerations, we hypothesized that small molecules inhibiting the MuRF1 interaction with the giant filament titin may provide an alternative approach towards perturbing the action of MuRF1 on myofibrillar proteins and myofibril atrophy, but may leave MuRF1 basal expression in place. The MuRF1–titin compound screens used in the present study were developed along this rationale and were able to identify nine compounds that fulfilled our in vitro selection criteria (Figure 1). These novel identified compounds (i.e. not FDA approved) were first assessed for toxicity prior to our animal experiments, with the compound ID#704946 evidencing both the lowest toxicity in cultured myotubes (Figure 1G) and also a rapid bioavailability in the serum [i.e. 2 h after i.p. injection in a linear dose–serum concentration relationship, as determined by liquid chromatography coupled with mass spectrometry; data not shown]. Taken together, therefore, the selected compound fulfilled the entry criteria for our animal experiments, which included inhibiting the MuRF1–titin interaction, inhibiting MuRF1 E3 ligase activity, showing least apparent toxicity in cultured myotube assays, and also protecting myotubes from DEX‐induced atrophy.
Effects of in vivo MuRF1 inhibition on cardiac cachexia
For in vivo studies with the ID#704946 compound, we stressed mice with the alkaloid MCT such that weekly i.p. injections induced pulmonary hypertension, right ventricular dysfunction, and subsequent cardiac cachexia over a 6 week period.24 Clinical indices (i.e. pulmonary congestion, heart weight, RV hypertrophy, and weight gain) were near identical between the MCT and MCT + compound‐fed mice, which suggest that disease severity progressed similarly in both groups independent of compound feeding. Importantly, however, while MCT‐treated mice showed a progressive loss of skeletal muscle mass, the MCT + compound‐fed mice did not follow this trend and were protected with the most obvious effect seen in the TA muscle. Interestingly, despite muscle atrophy being attenuated in the MCT mice treated with compound compared with normal chow, body weights between groups remained similar. This is likely explained by body weight (which was ~21 g in our mice) providing a gross measure that is simply too insensitive to detect subtle changes (in mg) of muscle mass, or alternatively, it may be related to reductions in muscle mass being masked by concomitant increases in fat mass and/or fluid retention—a point the present study cannot confirm. To provide a direct functional assessment, we also measured contractility in diaphragm muscle fibres and observed that contractile dysfunction (in terms of shortening velocity and power) induced by cardiac cachexia was essentially prevented in mice fed the ID#704946 compound. Collectively, therefore, these two findings suggest that the selective inhibition of MuRF1 by ID#704946 mediated a benefit to both skeletal muscle quantity (i.e. mass) and quality (i.e. contractile function) in cardiac cachexia.
Signalling pathways underlying the MuRF1 inhibitor compound
Clearly, further studies are warranted to identify the mechanism(s) in detail by which the compound ID#704946 mediated its benefits in the MCT‐stressed mice (i.e. on maintaining contractile function and muscle mass). Here, for an initial overview, the present study included an unbiased proteome profiling survey. The expression levels of five proteins were found to specifically respond to the compound, as indicated by comparison of the MCT and MCT + ID#704946 proteomes (Figure 6). This included an up‐regulation of eIF2B (subunit δ) and down‐regulation of BAX, which we later confirmed by immunoblotting. The eIF2B pathway is a known translational regulator of protein synthesis, with evidence showing that its content can directly influence fibre size.31 The direct targeting of elF2B by MuRF1 appears plausible because our previous yeast two‐hybrid screens identified elF2B as a MuRF1 interacting factor, linking MuRF1 as a negative regulator of protein synthesis.32 In line with a direct regulation of protein translation by MuRF1, injection of D5‐phenylalanine into MuRF1 KO mice demonstrated an elevated muscle protein translation,27 while MuRF1 KO mice are protected against a reduction in protein synthesis following DEX treatment.11 As such, we suggest that MuRF1 mediated a depletion of the translation initiation factor elF2B under MCT stress, and this was relieved by the compound. Whether this functionally accounts for the maintenance of muscle mass remains to be determined, but this obviously warrants further investigation. In addition, the compound also modulated the pro‐apoptotic regulator BAX, with this protein up‐regulated in MCT mice compared with shams and normalized in MCT + ID#704946 mice. In human patients with chronic heart failure, apoptosis is increased in skeletal muscle, and this closely correlates to the degree of atrophy.33, 34 Indeed, it has been noted before that BAX is elevated in cardiac cachexia and associated with an increased MuRF1 expression.35, 36 Therefore, future studies are warranted to determine if MuRF1 inhibition by ID#704946 provides anti‐apoptotic signals in a muscle‐specific fashion.
In line with our in vitro studies, we also confirmed that the increased protein expression of MuRF1 in MCT mice was prevented by the compound, while no effects were noted for the other apparent key atrogin MAFBx. Therefore, the underlying mechanism of the compound appears to be MuRF1 specific, which would be expected based on our preliminary in vitro studies. However, it still remains unclear whether the main effects of the compound in vivo are exerted by the inhibition of MuRF1's enzyme activity or rather its down‐regulation in protein expression, although our in vitro and in vivo data suggest that a combination of both is likely playing a role. MuRF1 is also known to interact with numerous substrates, with one in particular being CARP (a member of the muscle ankyrin repeat protein family20, 37), which is a purported nuclear‐based and sarcomere (titin)‐based protein with transcriptional functions.20, 37 CARP is known to be up‐regulated in stress‐related conditions and is associated with contractile dysfunction and muscle atrophy.20, 38, 39 In line with such evidence, we also observed an increase in CARP expression in MCT‐stressed mice, but this effect was abolished in the ID#704946‐fed mice. This suggests that this compound may blunt CARP expression via its inhibition on MuRF1, which, in turn, may contribute to the maintenance of muscle mass and function. Importantly, additional experiments also provided evidence that the compound was able to prevent the reduction in both the content and ubiquitination of the key sarcomeric protein actin and a corresponding significant reduction in the activity of the ubiquitin proteasome pathway. Interestingly, while myosin heavy chain is generally suggested to be the key target of MuRF1‐mediated degradation,6, 15 other studies have also found that actin is similarly susceptible40: In vitro and in vivo data have demonstrated lower protein content in line with a greater ubiquitination of actin in catabolic conditions, with MuRF1 found to interact with and polyubiquitinate actin. As such, our data provide strong support that the developed compound was able to perturb the myofibril‐directed MuRF1 atrophy axis, which, in turn, inhibited actin ubiquitination and degradation and thus overall muscle proteolysis.
Clinical considerations
The treatment of cardiac cachectic patients using compounds naturally requires long‐term interventions, raising concerns on their potential cardiac and general toxicity. In this study, we used high doses (about 3 mg per day per mouse) over a 6 week period. Therefore, it is next mandatory to further evaluate in detail in vivo toxicities. Currently, we are monitoring closely ID#704946 and other structurally related compounds for their effects on cardiac remodelling and for bioavailability by liquid chromatography coupled with mass spectrometry. Clearly, this study provides only a starting point because all of the top 9 compounds are novel and uncharacterized with regards to their toxicity and pharmacology. Importantly, all steps involved in their selection can be adapted to high‐throughput approaches. Therefore, future studies will aim to further expand the MuRF1 central–titin inhibitor compound line and in parallel to optimize individual hits such as ID#704946, to test whether such compounds can be translated to treat stress‐induced myopathic pathologies.
Conclusions
Our data show that a novel compound (ID#704946) selected for its inhibition of the muscle‐specific E3 ligase MuRF1 was able to attenuate in vivo skeletal muscle atrophy and contractile dysfunction in a mouse model of cardiac cachexia. These findings were further supported by our in vitro experiments, whereby myotube atrophy induced by the synthetic glucocorticoid DEX was abolished following treatment with the ID#704946 compound. The compound‐induced attenuation of muscle wasting both in vivo and in vitro was associated with reduced levels of MuRF1 expression, while additional proteome analyses revealed a protection of de novo protein synthesis and a down‐regulation in apoptosis.
Conflict of interest
Volker Adams, Joe Lewis, Dittmar Labeit, Siegfried Labeit: A patent is pending for ID#704946. Dittmar Labeit and Siegfried Labeit: The given distributor for anti‐MuRF1 antibodies Myomedix is owned by DL, SL.
Acknowledgements
We thank Sylvia Jeratsch and Anne Konzer from the DZHK Proteome Core Facility in Bad Nauheim and for support by the DZHK column B project ‘titin mass spec’. In addition, we thank Dr Lee Sweeney and Dr David Hammers for discussions and advice. The authors certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia, and Muscle.41 This research was supported by the Fondation Leducq (TNE 13CVD04) and the EU network ‘Muscle Stress Relief’.
Bowen, T. S. , Adams, V. , Werner, S. , Fischer, T. , Vinke, P. , Brogger, M. N. , Mangner, N. , Linke, A. , Sehr, P. , Lewis, J. , Labeit, D. , Gasch, A. , and Labeit, S. (2017) Small‐molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. Journal of Cachexia, Sarcopenia and Muscle, 8: 939–953. doi: 10.1002/jcsm.12233.
Contributor Information
Volker Adams, Email: adav@medizin.uni-leipzig.de.
Siegfried Labeit, Email: labeit@medma.de.
References
- 1. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 2015;14:58–74. [DOI] [PubMed] [Google Scholar]
- 2. Bodine SC, Baehr LM. Skeletal muscle atrophy and the e3 ubiquitin ligases murf1 and mafbx/atrogin‐1. Am J Physiol Endocrinol Metab 2014;307:E469–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin‐proteasome pathway in normal and disease states. J Am Soc Nephrol 2006;17:1807–1819. [DOI] [PubMed] [Google Scholar]
- 4. Sandri M. Protein breakdown in muscle wasting: role of autophagy‐lysosome and ubiquitin‐proteasome. Int J Biochem Cell Biol 2013;45:2121–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Willis MS, Bevilacqua A, Pulinilkunnil T, Kienesberger P, Tannu M, Patterson C. The role of ubiquitin ligases in cardiac disease. J Mol Cell Cardiol 2014;71:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1‐dependent ubiquitylation. J Cell Biol 2009;185:1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adams V, Mangner N, Gasch A, Krohne C, Gielen S, Hirner S, Thierse HJ, Witt CC, Linke A, Schuler G, Labeit S. Induction of MuRF1 is essential for tnf‐alpha‐induced loss of muscle function in mice. J Mol Biol 2008;384:48–59. [DOI] [PubMed] [Google Scholar]
- 8. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704–1708. [DOI] [PubMed] [Google Scholar]
- 9. Li W, Moylan JS, Chambers MA, Smith J, Reid MB. Interleukin‐1 stimulates catabolism in c2c12 myotubes. Am J Physiol Cell Physiol 2009;297:C706–C714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li YP, Chen Y, Li AS, Reid MB. Hydrogen peroxide stimulates ubiquitin‐conjugating activity and expression of genes for specific e2 and e3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol 2003;285:C806–C812. [DOI] [PubMed] [Google Scholar]
- 11. Baehr LM, Furlow JD, Bodine SC. Muscle sparing in muscle ring finger 1 null mice: response to synthetic glucocorticoids. J Physiol 2011;589:4759–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hooijman PE, Beishuizen A, Witt CC, de Waard MC, Girbes AR, Spoelstra‐de Man AM, Niessen HW, Manders E, van Hees HW, van den Brom CE, Silderhuis V, Lawlor MW, Labeit S, Stienen GJ, Hartemink KJ, Paul MA, Heunks LM, Ottenheijm CA. Diaphragm muscle fiber weakness and ubiquitin‐proteasome activation in critically ill patients. Am J Respir Crit Care Med 2015;191:1126–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eddins MJ, Marblestone JG, Suresh Kumar KG, Leach CA, Sterner DE, Mattern MR, Nicholson B. Targeting the ubiquitin e3 ligase MuRF1 to inhibit muscle atrophy. Cell Biochem Biophys 2011;60:113–118. [DOI] [PubMed] [Google Scholar]
- 14. Castillero E, Alamdari N, Lecker SH, Hasselgren PO. Suppression of atrogin‐1 and murf1 prevents dexamethasone‐induced atrophy of cultured myotubes. Metabolism 2013;62:1495–1502. [DOI] [PubMed] [Google Scholar]
- 15. Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, Rakhilin SV, Stitt TN, Patterson C, Latres E, Glass DJ. The e3 ligase murf1 degrades myosin heavy chain protein in dexamethasone‐treated skeletal muscle. Cell Metab 2007;6:376–385. [DOI] [PubMed] [Google Scholar]
- 16. Gielen S, Sandri M, Kozarez I, Kratzsch J, Teupser D, Thiery J, Erbs S, Mangner N, Lenk K, Hambrecht R, Schuler G, Adams V. Exercise training attenuates murf‐1 expression in the skeletal muscle of patients with chronic heart failure independent of age: the randomized leipzig exercise intervention in chronic heart failure and aging catabolism study. Circulation 2012;125:2716–2727. [DOI] [PubMed] [Google Scholar]
- 17. Servais S, Letexier D, Favier R, Duchamp C, Desplanches D. Prevention of unloading‐induced atrophy by vitamin e supplementation: links between oxidative stress and soleus muscle proteolysis? Free Radic Biol Med 2007;42:627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol 2001;306:717–726. [DOI] [PubMed] [Google Scholar]
- 19. Mrosek M, Labeit D, Witt S, Heerklotz H, von Castelmur E, Labeit S, Mayans O. Molecular determinants for the recruitment of the ubiquitin‐ligase MuRF‐1 onto M‐line titin. FASEB J 2007;21:1383–1392. [DOI] [PubMed] [Google Scholar]
- 20. Witt CC, Ono Y, Puschmann E, McNabb M, Wu Y, Gotthardt M, Witt SH, Haak M, Labeit D, Gregorio CC, Sorimachi H, Granzier H, Labeit S. Induction and myofibrillar targeting of carp, and suppression of the nkx2.5 pathway in the mdm mouse with impaired titin‐based signaling. J Mol Biol 2004;336:145–154. [DOI] [PubMed] [Google Scholar]
- 21. Labeit S, Witt CC, Hirner S, Labeit D. A host cell deficient for MuRF1 and MuRF2. World International Property Organisation: International Bureau WO2009/077618. European Patent (LAB65256EP). 2009.
- 22. Sehr P, Pawlita M, Lewis J. Evaluation of different glutathione s‐transferase‐tagged protein captures for screening e6/e6ap interaction inhibitors using alphascreen. J Biomol Screen 2007;12:560–567. [DOI] [PubMed] [Google Scholar]
- 23. Bellocci M, Ronzitti G, Milandri A, Melchiorre N, Grillo C, Poletti R, Yasumoto T, Rossini GP. A cytolytic assay for the measurement of palytoxin based on a cultured monolayer cell line. Anal Biochem 2008;374:48–55. [DOI] [PubMed] [Google Scholar]
- 24. Ahn B, Empinado HM, Al‐Rajhi M, Judge AR, Ferreira LF. Diaphragm atrophy and contractile dysfunction in a murine model of pulmonary hypertension. PLoS One 2013;8: e62702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bowen TSES, Drobner J, Fischer T, Werner S, Linke A, Mangner N, Schuler G, Adams V. High‐intensity interval training prevents oxidant‐mediated diaphragm muscle weakness in hypertensive mice. FASEB J 2017;31:60–71. [DOI] [PubMed] [Google Scholar]
- 26. Konzer A, Ruhs A, Braun T, Kruger M. Global protein quantification of mouse heart tissue based on the silac mouse. Methods Mol Biol 2013;1005:39–52. [DOI] [PubMed] [Google Scholar]
- 27. Witt CC, Witt SH, Lerche S, Labeit D, Back W, Labeit S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J 2008;27:350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Willis MS, Zungu M, Patterson C. Cardiac muscle ring finger‐1—friend or foe? Trends Cardiovasc Med 2010;20:12–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen SN, Czernuszewicz G, Tan Y, Lombardi R, Jin J, Willerson JT, Marian AJ. Human molecular genetic and functional studies identify trim63, encoding muscle ring finger protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ Res 2012;111:907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ, Richardson JA, Bassel‐Duby R, Olson EN. Myosin accumulation and striated muscle myopathy result from the loss of muscle ring finger 1 and 3. J Clin Invest 2007;117:2486–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mayhew DL, Hornberger TA, Lincoln HC, Bamman MM. Eukaryotic initiation factor 2b epsilon induces cap‐dependent translation and skeletal muscle hypertrophy. J Physiol 2011;589:3023–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koyama S, Hata S, Witt CC, Ono Y, Lerche S, Ojima K, Chiba T, Doi N, Kitamura F, Tanaka K, Abe K, Witt SH, Rybin V, Gasch A, Franz T, Labeit S, Sorimachi H. Muscle ring‐finger protein‐1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J Mol Biol 2008;376:1224–1236. [DOI] [PubMed] [Google Scholar]
- 33. Adams V, Jiang H, Yu J, Mobius‐Winkler S, Fiehn E, Linke A, Weigl C, Schuler G, Hambrecht R. Apoptosis in skeletal myocytes of patients with chronic heart failure is associated with exercise intolerance. J Am Coll Cardiol 1999;33:959–965. [DOI] [PubMed] [Google Scholar]
- 34. Vescovo G, Volterrani M, Zennaro R, Sandri M, Ceconi C, Lorusso R, Ferrari R, Ambrosio GB, Dalla LL. Apoptosis in the skeletal muscle of patients with heart failure: Investigation of clinical and biochemical changes. Heart 2000;84:431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dalla Libera L, Ravara B, Volterrani M, Gobbo V, Della Barbera M, Angelini A, Danieli Betto D, Germinario E, Vescovo G. Beneficial effects of gh/igf‐1 on skeletal muscle atrophy and function in experimental heart failure. Am J Physiol Cell Physiol 2004;286:C138–C144. [DOI] [PubMed] [Google Scholar]
- 36. Rezk BM, Yoshida T, Semprun‐Prieto L, Higashi Y, Sukhanov S, Delafontaine P. Angiotensin ii infusion induces marked diaphragmatic skeletal muscle atrophy. PLoS One 2012;7: e30276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller MK, Bang ML, Witt CC, Labeit D, Trombitas C, Watanabe K, Granzier H, McElhinny AS, Gregorio CC, Labeit S. The muscle ankyrin repeat proteins: Carp, ankrd2/arpp and darp as a family of titin filament‐based stress response molecules. J Mol Biol 2003;333:951–964. [DOI] [PubMed] [Google Scholar]
- 38. Laure L, Suel L, Roudaut C, Bourg N, Ouali A, Bartoli M, Richard I, Daniele N. Cardiac ankyrin repeat protein is a marker of skeletal muscle pathological remodelling. FEBS J 2009;276:669–684. [DOI] [PubMed] [Google Scholar]
- 39. Moulik M, Vatta M, Witt SH, Arola AM, Murphy RT, McKenna WJ, Boriek AM, Oka K, Labeit S, Bowles NE, Arimura T, Kimura A, Towbin JA. Ankrd1, the gene encoding cardiac ankyrin repeat protein, is a novel dilated cardiomyopathy gene. J Am Coll Cardiol 2009;54:325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Polge C, Heng AE, Jarzaguet M, Ventadour S, Claustre A, Combaret L, Béchet D, Matondo M, Uttenweiler‐Joseph S, Monsarrat B, Attaix D, Taillandier D. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J 25:3790–3802. [DOI] [PubMed] [Google Scholar]
- 41. von Haehling S, Morley JE, Coats AJ, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]