

Abstract
The endoplasmic reticulum (ER) stress is a cellular process that occurs as a consequence of several stress circumstances, such as the accumulation of unfolded proteins in the lumen of the ER or distinct insults that disturb the ER normal function. Different conditions in the tumor microenvironment (TME), including hypoxia, nutrient deprivation, and the elevated production of reactive oxygen and nitrogen species destabilize the loading and dispatching of the newly synthesized proteins, triggering ER stress in cancer cells and tumor-infiltrating leukocytes. In order to cope with TME-induced ER stress, tumor and stromal cells initiate an adaptive response process that aims to resolve ER stress and to restore cellular homeostasis, which is referred as the unfolded protein responses (UPR). Paradoxically, the UPR can also induce cell death under severe and/or permanent ER stress. The UPR is started through three mediators, the activation of the inositol-requiring enzyme-1α, the pancreatic ER kinase-like ER kinase, and the activating transcription factor 6. In this minireview, we will discuss the pro- and anti-tumorigenic role of the UPR in cancer cells. In addition, we will describe the effects of the TME-induced ER stress in the immunosuppressive activity of tumor-infiltrating myeloid cells. Also, we will review the results of emerging therapeutic interventions that target ER stress and the UPR mediators in cancer. We postulate that the inhibition of ER stress or the UPR-related elements could represent a significant approach to increase the efficacy of various forms of cancer immunotherapy.
Keywords: Tumor microenvironment, Myeloid-derived suppressor cells, Regulatory myeloid suppressor cells, T cells, Endoplasmic reticulum stress, Unfolded protein responses
Introduction
The endoplasmic reticulum (ER) is the cellular organelle where production, folding, and proper modifications of proteins occur [1]. It also functions as a major quality control step for several protein assembly processes. Properly folded proteins are usually secreted outside the ER via defined pathways. On the other hand, unfolded or improperly folded proteins undergo ER-associated degradation (ERAD) to ensure their elimination [2]. The balance between the entry of nascent peptides and the secretion of correctly folded and modified proteins is maintained by the ER under normal physiological conditions. Due to the harsh environmental cues that are associated with infections, injuries or tumorigenesis, the ER homeostatic balance cannot be maintained, leading to ER stress [3]. Cellular ER stress is sensed through the dissociation of the inactive form of the HSP70-type BiP/GRP78 [4], leading to the activation of three intraluminal ER sensors; the inositol-requiring enzyme-1α (IRE-1α), the protein kinase RNA-(PKR) like ER kinase (PERK), and the activating transcription factor 6 (ATF6) [3]. When these mediators are activated, several related signaling pathways are initiated and referred as the unfolded protein responses (UPR). The ultimate physiological goal of the UPR is to restore the functions of the ER back to homeostatic conditions, which ensures cellular survival. However, under unresolved ER stress conditions, the same UPR elements may ultimately trigger cell death [5]. In this minireview, we will summarize the physiological and pathological role of the UPR in cancer cells and immune populations such as myeloid cells and T lymphocytes (Fig. 1). Also, we will illustrate some approaches that target ER stress or the specific UPR mediators and that we postulate could serve as novel platforms for therapies against cancer.
Fig. 1.
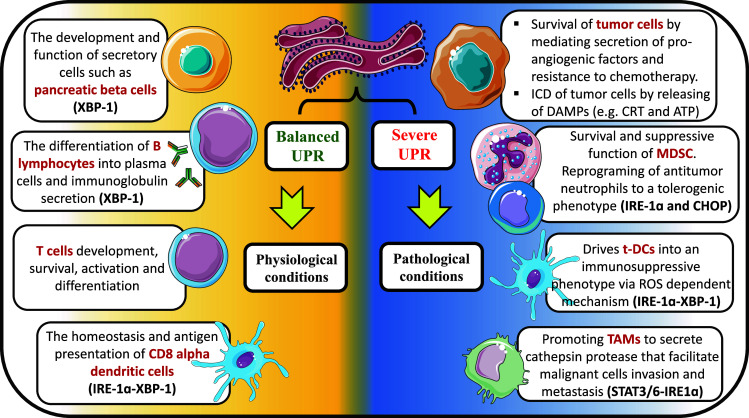
UPR mediators have versatile roles in different cell types either in the presence of balanced or unresolved ER stress. Under physiological conditions, the balanced UPR is crucial for the development of cells with secretory functions as pancreatic β islets, hepatocytes, and B lymphocytes. Also, the maturation and the antigen-presenting capability of CD8α DCs require moderated activation of UPR that is mediated though IRE1α-XBP-1 pathway. Furthermore, the effect of the balanced UPR in the T cell development and function is emerging. An exaggerated form of UPR can be triggered by different stress factors associated with pathological conditions as infections, autoimmunity, and cancer. Tumors cells either survive or undergo ICD depending on the degree of UPR severity. Tumor-infiltrating myeloid cells (t-DC, TAM, and MDSC) are characterized by sustained and amplified UPR activity which converts them into immunosuppressive cells that promote tumor growth and invasion
UPR mediators
At steady state, the three UPR mediators, IRE-1α, PERK, and ATF6, are sequestered in an inactive form by binding to the chaperone BiP [4]. However, because BiP exhibits a higher affinity for misfolded proteins, the induction of ER stress causes the dissociation of BiP from the associated UPR elements, leading to their activation and subsequent initiation of the UPR signaling pathway. Following is a brief description of how those mediators initiate UPR to cope with stress conditions and promote cellular survival or apoptosis.
IRE-1α
A type I transmembrane protein that contains both kinase and RNase domains. Once the luminal side senses the unfolded proteins, IRE-1α undergoes oligomerization, which activates its RNase domain that subsequently cleaves the X-box binding protein-1 (XBP-1) mRNA in the cytosol [6]. The cleaved fragments are then ligated by RNA ligase to yield the active form, XBP-1s [6]. XBP-1s functions as a transcription factor that activates several genes encoding for ER stress chaperones, lipids biosynthesis enzymes, and ERAD mediators [7, 8]. IRE-1α can also attenuate protein synthesis during ER stress by degrading various mRNAs through a mechanism known as IRE-1-dependent decay of mRNA (RIDD). Besides the RNase function, IRE-1α also autophosphorylates via the activity of its own kinase domain, which creates docking sites for the adaptor protein TNF receptor-associated factor 2 (TRAF2) [9]. The formed IRE-1α-TRAF2 complex enables the inhibition of various BCL2 family members through ER stress-induced JNK kinases, thereby leading to cellular apoptosis.
PERK
A type I transmembrane kinase that promotes protein translation arrests under ER stress through the phosphorylation of the eukaryotic initiation factor 2α (eIF2α) [10]. Phosphorylation of eIF2α inhibits the initiation of de novo protein synthesis, which reduces the protein loading in the ER and therefore alleviates ER stress. Some transcripts such as the activation transcription factor 4 (ATF4) escape the translation block induced by phospho-eIF2α, thereby controlling the final effects induced by this pathway [10]. As such, ATF4 induces the expression of the pro-apoptotic factor, CAAT/enhancer binding protein (C/EBP) homologous protein (CHOP), which leads to programmed cell death [10]. Paradoxically, PERK-induced ATF4 also promotes survival of ER-stressed cells through the transient expression of the microRNA miR-211, which degrades/downregulates CHOP mRNA [11]. Thus, PERK plays a dual role in deciding the fate of cells depending on the strength and duration of the ER stress signal.
ATF6
A type II transmembrane protein with a bZIP transcription factor in its N terminal end. In response to ER stress, ATF6 is translocated via COPII-coated vesicles to the Golgi apparatus where it undergoes sequential cleavages. That process leads to the release of the ATF6 N terminal domain, which translocates to the nucleus to initiate the transcription of several UPR downstream genes involved in ER membrane expansion and ERAD [12]. In contrast to IRE-1α, systemic deletion of ATF6 is not embryonic lethal [9]. However, ATF6 deficiency affects cell survival under chemically induced ER stress, suggesting an interplaying role between ATF6 and the other UPR-related elements [13].
The intrinsic role of the UPR in malignant cell physiology
Several reports have provided evidence of the increased activation of UPR mediators in patients with different cancer types and their overexpression usually correlated with poor prognosis and/or resistance to various therapies. As such, injection of mice with malignant cells lacking IRE-1α, XBP-1, PERK, or ATF6; or treatment of tumor-bearing mice with specific inhibitors of the UPR mediators resulted in reduced tumor growth, which was attributed to low angiogenesis, increased sensitivity to hypoxia, and elevated production of reactive oxygen species (ROS) [14]. Furthermore, the upregulation of the UPR sensors or their master regulator BiP in cancer cells induced an elevated tumor growth and resistance to chemotherapy [15]. Also, XBP-1 mediated the aggressiveness and chemoresistance of triple-negative breast cancer (TNBC) by assembling a transcriptional complex with the hypoxia-inducible factor 1 alpha (HIF-1α) and promoting the expression of HIF-1α target genes [16]. Although the severe and/or prolonged activation of the PERK and ATF6 arms of the UPR are known to result in pro-apoptotic effects through CHOP induction, the stimulation of these mediators after tolerable ER stress has also been shown to promote survival of the malignant cells. In fact, transient activation of PERK in tumor cells blocked the expression of CHOP through the induction of miR-211 [11]. In addition, NF-E2-related factor-2 (Nrf2) phosphorylation and activation through PERK was found to protect stressed cells by maintaining glutathione levels, thereby blocking the accumulation of deleterious ROS [17]. ATF6 promoted the survival of tumors and their adaptation to chemotherapy treatments by activating mTOR signaling [18]. Furthermore, a recent study demonstrated that the activation of Notch1 pathway by ATF6 confers resistance of human glioblastoma cells to gamma radiation [19]. These results suggest the dual effect of the UPR mediators depending on the severity and time of exposure to the ER stress insult.
It is well known that growing cancer cells produce pro-angiogenic factors as a response to nutrients and oxygen deprivation to initiate the formation of new blood vessels. The IRE-1α-XBP-1 pathway promoted the transcription of the angiogenic factor, vascular endothelial growth factor A (VEGF-A), through binding of XBP-1 to VEGF-A promoter [20]. Congruently, knockdown of XBP-1 in fibrosarcoma cells turned them to form avascular tumors compared to the wild-type counterparts [21]. Moreover, K-Ras oncogene-transformed mouse embryonic fibroblasts lacking PERK showed a decreased angiogenic potential and smaller tumors after injection into mice [22]. Thus, the activation of the UPR mediators in tumor cells promotes their angiogenic potential, and resistance to chemotherapy, supporting the rationale for the combination of regular chemotherapy or radiotherapy approaches with UPR inhibitors as treatments for cancer.
The UPR regulates homeostatic functions of subsets of antigen-presenting cells
The role of UPR in the immune system was the first documented in the differentiation of B cells to plasma cells. XBP-1-deficient B lymphocytes displayed a severe defect in their differentiation into immunoglobulin-secreting plasma cells [23]. Additionally, the IRE-1α-XBP-1 pathway was found to regulate the phenotype and function of CD8α conventional dendritic cells (cDCs) [24]. XBP-1-deficient CD8α cDCs failed to cross prime CD8+ T cells, suggesting the activating role of XBP-1 in the antigen presentation capacity of CD8α cDCs. Interestingly, the UPR mediators have been shown to synergize with some pathogen recognition receptors (PRR) like toll-like receptors (TLRs). Lymphoid-deficient XBP-1 mice showed decreased expansion and survival of conventional and plasmacytoid DCs after TLR stimulation [25]. Moreover, TLR2 and TLR4 activation stimulates IRE-1α-XBP-1 and regulates the optimal production of pro-inflammatory cytokines in macrophages [26]. The canonical ER stress inducer, thapsigargin, was found to synergize with TLR4 agonist to induce the production of IL-23 in DCs [27]. That effect was mediated by CHOP, a major transcription factor downstream of PERK. These observations suggest that under physiological conditions, the UPR is necessary for the maturation, function, and activation of various specific populations of antigen-presenting cells (Fig. 1).
The UPR mediators reprogram tumor-associated myeloid cells
Myeloid cells represent the major compartment of immune cells in the TME. The aggressive and hostile conditions, soluble factors, and cytokines within the TME trigger ER stress and the UPR in myeloid cells, thereby promoting their immunosuppressive phenotype (Fig. 2). The specific effect of the UPR mediators in regulating the function of tumor-infiltrating myeloid cells has been recently studied by different groups. Seminal work by Cubillos-Ruiz et al. showed that over-activation of the IRE-1α-XBP-1 drives the tolerogenic activity of DCs infiltrating ovarian carcinoma tumors [28]. Mechanistically, the ROS within the TME-induced lipid peroxidation in tumor-infiltrating DCs (t-DC) resulting in ER stress and activation of IRE-1α-XBP-1. Deletion or silencing of XBP-1 or treatment with the antioxidant vitamin E transformed immunosuppressive t-DC into myeloid cells that triggered immunogenic anti-tumor responses. That study introduced a new molecular mechanism that can be a potential target for resolving the dominant immune tolerance mediated by t-DC in the TME [29].
Fig. 2.
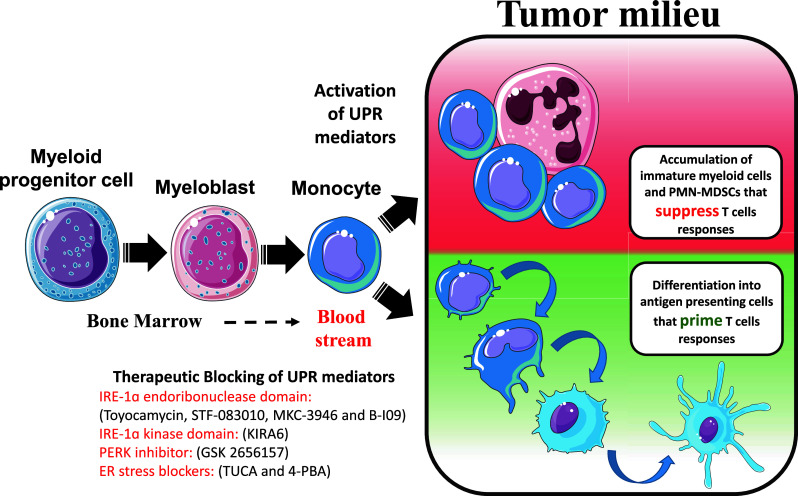
Myeloid cells originate from precursors in the bone marrow and transfer through blood stream until they reach tumor sites. Stress conditions in the tumor microenvironment activate UPR pathways which either retain tumor-infiltrating myeloid cells in an immature stage or switch them to tolerogenic populations that can blunt T cells. Therapeutic approaches to block UPR mediators can reprogram immature tumor-infiltrating myeloid cells into professional antigen-presenting cells rather than immunosuppressive phenotypes. As such, the inhibition of the UPR mediators could serve as a major opportunity for cancer treatments
Myeloid-derived suppressive cells (MDSC) are a heterogeneous population of immature cells that accumulate in individuals with tumors and block-protective anti-tumor T cell responses. In mice, MDSC can be divided into monocytic MDSC (M-MDSC, CD11b+ Ly6G− Ly6Chigh) and granulocytic MDSC (PMN-MDSC, CD11b+ Ly6G+ Ly6Clow). A recent work from Condamine et al. showed the role of ER stress in MDSC survival. Results showed that ER stress occurred primarily in tumor-infiltrating MDSC and promoted MDSC apoptosis through the TNF-related apoptosis-induced ligand receptor 2 (DR5) and caspase 8 activation [30]. Latest results from the same investigators showed that pharmacological targeting of IRE-1α-XBP-1 reprogrammed the low-density immunosuppressive G-MDSC into high-density anti-tumor neutrophils [31]. The upregulation of the lectin-type oxidized LDL receptor-1 (LOX-1) upon ER stress also enabled the discrimination between suppressive PMN-MDSC and non-tolerogenic neutrophils. We also studied the role of the ER stress downstream target CHOP in tumor MDSC. When MDSC were isolated from tumors of CHOP-deficient mice, they showed lower ability to impair T cell responses compared to their wild-type counterparts [32]. This correlated with a heightened expression of MHC-II and co-stimulatory molecules, and a higher ability to increase T cell responses after DC vaccination. This effect appears to be mediated through a decreased C/EBPβ and IL-6/p-STAT-3 signaling, which are known to regulate the function and expansion of MDSC [33]. Accordingly, CHOP levels controlled the development of “alternatively activated” myeloid cells that produced cytokines known to impair T cell responses [11, 27], suggesting a potential master role of ER stress mediators and CHOP in the immunosuppressive function of tumor myeloid cells.
A recent report has pointed on the role of the UPR mediator IRE-1α on the secretory function of tumor-associated macrophages (TAM) [34]. Type 2 T helper cells (Th2) cytokines like IL-4, IL-6 and IL-10 synergize to instigate ER stress and activate IRE-1α in TAM, which enhanced the production of cathepsin protease, thereby facilitating invasion of tumor cells. Importantly, deletion of the Th2 signalizing or inhibition of IRE-1α in TAM was sufficient to reduce tumor growth and invasion. This suggests the importance of the UPR in TAM in the regulation of cancer cell invasion.
Collectively, all those studies outlined the effect of the ER stress on triggering myeloid cell dysfunction within the TME (Fig. 2). Also, they emphasized that the UPR mediators can be promising targets to either inhibit the suppressive capacity of myeloid cells in tumors or to induce their reprogramming into highly immunogenic antigen-presenting cells. Because the accumulation of myeloid cells in tumors is a major obstacle to current forms of immunotherapy, the development of strategies to reprogram tumor-associated myeloid cells into immunogenic populations is expected to significantly increase the efficacy of these forms of cancer treatments.
A cross talk between UPR-undergoing tumor cells and myeloid cells
The UPR mediators induce multiple intracellular changes but also lead to specific extrinsic effects. Several reports showed that tumor cells experiencing ER stress promote tolerogenic signals in the myeloid cells [35]. Macrophages and MDSC cultured in a conditioned medium of ER-stressed tumor cells upregulated the ER stress response genes BiP, Chop and Xbp-1s, increased the production of pro-inflammatory cytokines IL-6, TNF-α, and IL-23, and elevated the expression of the immunosuppressive enzyme arginase I and the pro-angiogenic molecule VEGF, all of which are known to promote tumor growth and metastasis [36]. TLR4 in macrophages was crucial to mediate that response. These results suggest that ER stress in tumor cells can potentially alter the behavior of myeloid cells within the TME. However, it remains unknown the factors and molecular mechanisms by which the ER-stressed tumor cells triggered this immunotolerogenic effect in myeloid cells.
The role of UPR in T cells under physiological and pathological conditions
Seminal studies have provided evidence on the role of the UPR signaling in T cell differentiation and activation. Through applying a XBP-1-GFP reporter system, researchers revealed that the IRE-1α-XBP-1 branch is active in CD4+CD8+ T cells from the thymus, indicating a role of the UPR in T cell development [37]. Another study reported that ablation of Rpl22 (RP ribosomal protein L22), which restrains ER stress, blocked the development of αβ, but not γδ T cell progenitors [38]. On the other hand, T cells containing a point mutation in the Kdelr1 gene (encoding ER chaperone KDEL receptor 1) have higher levels of phospho-eIF2α and impaired T cell survival [39]. Similar to this finding, Skfn2 null T cells undergo chronic ER stress, which leads to proliferation inhibition and low viability. Interestingly, this developmental abnormality was reversed after deleting CHOP or XBP-1 [40]. Those studies suggest that evoking UPR is necessary for T cell development, but elevated stress will lead to impaired T cell function and survival.
T cells are activated by TCR-peptide-MHC recognition plus a secondary stimulatory interaction with APCs, which can be mimicked by anti-CD3/CD28 antibodies or the combination of the PKC activator PMA and the Ca2+ ionophore ionomycin. Elevated ER chaperones components were found after T cell priming with anti-CD3/CD28 [41, 42]. In the process of CD4+ T cell polarization into Th2 cells, high levels of BiP, ATF4, and CHOP were detected [43]. Furthermore, spliced XBP-1 mRNA is increased during CD8+ T cell activation and the overexpression of XBP-1 spliced isoform enhances CD8+ T cell cytotoxic function [44]. Those discoveries indicate that elevated UPR is necessary during T cell activation. Besides this, UPR also regulates T cell apoptosis. In Gimap5 null hosts, which have spontaneous apoptosis in T cells, knocking down of CHOP inhibited T cell apoptosis [45]. In addition, a recent study revealed that deletion of Hrd1, an E3 ligase involved in UPR-induced ERAD, led to reduced T cell numbers and attenuated T cell polarization into Th1, Th2, and Th17 [46]. Therefore, moderate UPR promoted T cell function, whereas overwhelming UPR signaling will lead to impaired T cell responses and cell death.
Abnormal UPR amplitudes or dysregulated expression of core UPR components are related to pathological disorders, giving a possibility that UPR mediators can be a therapeutic target in such diseases. XBP-1-mediated UPR is critical for CD8+ T cell differentiation during acute bacterial or viral infection [44]. Increased XBP-1 splicing, but not PERK or ATF6 activation, along with high expression of BiP and CHOP was found in acute necrotizing enterocolitis (A-NEC) patients who also had lower Th17 and Treg numbers [47]. In the experimental autoimmune encephalomyelitis (EAE) model, downregulating UPR via weakening ERAD leads to low disease progress, indicating that targeting UPR pathways may be a promising therapeutic option for multiple sclerosis [46]. Currently, whether UPR regulates T cell viability and function in the TME remains mostly elusive.
Therapeutic approaches targeting the UPR mediators
Severe ER stress incites immunogenic cell death in tumor cells
Malignant cells killed by intrinsic or extrinsic stress factors within the TME generate danger-associated molecular patterns (DAMPs), including HMGB-1, ROS, IL-1α, ATP, and plasma membrane exposure of the ER resident protein calreticulin (CRT) that evoke inflammation and immunogenic responses [48]. This phenomenon is known as immunogenic cell death (ICD). Based on this, ICD inducers have evolved as potential anticancer agents and were classified into two categories based on the mechanisms through which they target the ER and induce apoptosis of tumor cells [49]. Type I ICD inducers, as cyclophosmamide and gamma radiation, stimulate immunogenic apoptosis via secondary ER stress effects, while type II ICD triggers, as hypericin-based photodynamic therapy, activate immunogenicity by directly disrupting ER homeostasis leading to UPR. The relationship between ER stress and ICD is more likely to be bidirectional and regulated throughout highly complexed mechanisms. The release of HMGB1 upon ICD and its binding to RAGE receptor was found to activate UPR mediators in bystander cells [50]. On the other hand, cytotoxin-induced ICD was found to be mediated through PERK activation and calcium flux [51]. Accordingly, the PERK-eIF2α pathway was shown to be involved in CRT exposure following anthracyclines and oxaliplatin treatment [52], and the phosphorylation of eIF2α was documented to be a key parameter correlated with ICD [53]. The combination of cisplatin with ER stress inducers like, tunicamycin or thapsigargin, was found to be therapeutically effective in some tumors rather than using cisplatin alone [54]. Those findings indicate that the activation of the UPR arms mediates the production of ICD-associated factors and represents a mechanistic prerequisite for the clinical outcome of various ICD-inducing agents in cancer.
The increased production and accumulation of ROS in cells have been related with ICD after chemotherapy. The inflammatory responses associated with ICD were reduced in the presence of antioxidants [55]. As such, ER stress synergizes with ROS production to promote ICD by increasing the levels of DAMPs. However, it is still unknown if ICD can also occur in immune cells infiltrating tumors and whether that can have any impact on cancer therapy. In addition, a clear understanding of the mechanisms regulating the immunogenic vs. immunosuppressive effects induced by the UPR mediators in the context of ICD remains to be determined.
Blocking UPR mediators as cancer therapy
It is an interesting fact that ER stress is crucial for the survival of malignant cells, but also could drive cancer cells toward ICD and recognition by the immune system. That can be explained in terms of the severity of the ER stress. In other words, cancer cells undergoing moderate ER stress activate the UPR mediators as a response to adapt to the aggressive tumor milieu, while cancer cells exposed to severe or permanent ER stress activate UPR to mediate cell death and potentially ICD. In agreement with the therapeutic potential of blocking moderate ER stress in cancer cells, pharmacological inhibition of the UPR arms in malignant cells resulted in tumoricidal effects in vivo. A class of compounds that includes toyocamycin, STF-083010, MKC-3946, and B-I09 can interfere with the endoribonuclease domain of IRE-1α and showed therapeutic benefits in multiple myeloma and chronic lymphocytic leukemia models [56, 57]. Also, oral administration of the PERK inhibitor, GSK2656157, delayed tumor growth of xenografts in immunocompromised mice [58]. Similar results were obtained after treatment of tumor-bearing mice with the ER stress scavengers, tauroursodeoxycholic acid or 4-phenylbutyrate. Furthermore, induction of severe ER stress using tunicamycin or thapsigargin has shown to impair tumor growth and increase the effects of ICD inducers [54].
Although the systemic inhibition of UPR mediators showed to induce anti-tumor effects [56], it can also trigger side effects such as the pancreatic toxicity associated with the inhibition of PERK [59]. Therefore, specific targeting of UPR mediators in myeloid cells will remain a big challenge to the field. Based on their unique phagocytic properties, t-DC, MDSC, and TAM can be considered as candidates for nanoparticle therapies. Targeting t-DC with siRNA-loaded nanoparticles against XBP-1 transformed them into immunogenic cells that primed anti-tumor T cell responses [28]. Knocking down IRE-1α by siRNA in bone marrow-derived macrophages blocked their ability to express cathepsin in response to Th2 cytokines, thereby, preventing macrophage-mediated cancer cells invasion [34]. Specific blocking of UPR mediators in tumor-associated myeloid cells can be a potential approach to either reprogram them into effectors or deprive them from producing pro-tumorigenic factors. Also, they might improve the therapeutic effect when combined with checkpoint blockades, adoptive cell therapy, or tumor vaccines.
Closing remarks and future directions
The role of myeloid cells in the inhibition of anti-tumor immune surveillance and as a major obstacle for the successful development of effective therapies against cancer is subject of intense interest in the field. To date, however, the approaches to inhibit the accumulation and function of myeloid cells in tumors are limited to myelosuppressive agents or multi-tyrosine kinase inhibitors that are only partially effective and secondarily enhance populations such as MDSC as the bone marrow recovers. We propose that the fundamental problem is that the limited understanding of the central signaling pathways governing the accumulation and function of myeloid cells in tumors has prevented the development of successful therapies. The results discussed here support the central role of the UPR mediators in the overall behavior of myeloid cells in tumors. We postulate that therapeutic inhibition of ER stress or the UPR elements in tumors could reprogram immunosuppressive myeloid subsets into immunogenic cells being capable to induce protective immunity (Fig. 2), thereby enhancing the effect of chemotherapy, radiotherapy, and immunotherapy. This hypothesis remains to be explored in animal models and clinical studies.
Acknowledgements
This work was partially supported by the National Cancer Institute, NIH Grant No. CA18485 to Paulo C. Rodriguez, PhD.
Abbreviations
- A-NEC
Acute necrotizing enterocolitis
- ATF4
Activation transcription factor 4
- ATF6
Activating transcription factor 6
- C/EBP
CAAT/enhancer binding protein
- cDCs
Conventional dendritic cells
- CHOP
CAAT/enhancer binding protein (C/EBP) homologous protein
- CRT
Calreticulin
- DAMPs
Danger-associated molecular patterns
- DR5
TNF-related apoptosis-induced ligand receptor 2
- EAE
Autoimmune encephalomyelitis
- eIF2α
Eukaryotic initiation factor 2α
- ER stress
Endoplasmic reticulum stress
- ERAD
ER-associated degradation
- HIF-1α
Hypoxia-inducible factor 1 alpha
- ICD
Immunogenic cell death
- IRE-1α
Inositol-requiring enzyme-1α
- Kdelr1
KDEL receptor 1
- LOX-1
Lectin-type oxidized LDL receptor-1
- M-MDSC
Monocytic MDSC
- MDSC
Myeloid-derived suppressor cells
- Nrf2
NF-E2-related factor-2
- PERK
Pancreatic ER kinase (PKR)-like ER kinase
- PKR
Protein kinase RNA
- PMN-MDSC
Granulocytic MDSC
- PRR
Pathogen recognition receptors
- RIDD
IRE-1-dependent decay of mRNA
- ROS
Reactive oxygen species
- Rpl22
RP ribosomal protein L22
- t-DC
Tumor-infiltrating dendritic cells
- TAM
Tumor-associated macrophages
- Th2
Type 2 T helper cells
- TLRs
Toll-like receptors
- TME
Tumor microenvironment
- TNBC
Triple-negative breast cancer
- TRAF2
TNF receptor-associated factor 2
- UPR
Unfolded protein responses
- VEGF-A
Vascular endothelial growth factor A
- XBP-1
X-box binding protein-1
Compliance with ethical standards
Conflict of interest
The authors do not have conflict of interests to disclose.
Footnotes
This paper is a Focussed Research Review based on a presentation given at the conference Regulatory Myeloid Suppressor Cells: From Basic Discovery to Therapeutic Application which was hosted by the Wistar Institute in Philadelphia, PA, USA, 16th–19th June, 2016. It is part of a Cancer Immunology, Immunotherapy series of Focussed Research Reviews.
Contributor Information
Eslam Mohamed, Phone: 706-721-9285, Email: emohamed@augusta.edu.
Paulo C. Rodriguez, Phone: 706-446-5808, Email: paurodriguez@augusta.edu
References
- 1.Janssens S, Pulendran B, Lambrecht BN. Emerging functions of the unfolded protein response in immunity. Nat Immunol. 2014;15(10):910–919. doi: 10.1038/ni.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334(6059):1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. 2015;33:107–138. doi: 10.1146/annurev-immunol-032414-112116. [DOI] [PubMed] [Google Scholar]
- 4.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 5.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 7.Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR, 3rd, Su AI, Kelly JW, Wiseman RL. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013;3(4):1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167(1):35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 10.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5(5):897–904. doi: 10.1016/S1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 11.Chitnis NS, Pytel D, Bobrovnikova-Marjon E, Pant D, Zheng H, Maas NL, Frederick B, Kushner JA, Chodosh LA, Koumenis C, Fuchs SY, Diehl JA. miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol Cell. 2012;48(3):353–364. doi: 10.1016/j.molcel.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33(1):75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- 13.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7(9):880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahonen TJ, Xie J, LeBaron MJ, Zhu J, Nurmi M, Alanen K, Rui H, Nevalainen MT. Inhibition of transcription factor Stat5 induces cell death of human prostate cancer cells. J Biol Chem. 2003;278(29):27287–27292. doi: 10.1074/jbc.M304307200. [DOI] [PubMed] [Google Scholar]
- 15.Daneshmand S, Quek ML, Lin E, Lee C, Cote RJ, Hawes D, Cai J, Groshen S, Lieskovsky G, Skinner DG, Lee AS, Pinski J. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum Pathol. 2007;38(10):1547–1552. doi: 10.1016/j.humpath.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, Mai J, Shen H, Hu DZ, Adoro S, Hu B, Song M, Tan C, Landis MD, Ferrari M, Shin SJ, Brown M, Chang JC, Liu XS, Glimcher LH. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508(7494):103–107. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279(19):20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 18.Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA. 2008;105(30):10519–10524. doi: 10.1073/pnas.0800939105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dadey DY, Kapoor V, Khudanyan A, Urano F, Kim AH, Thotala D, Hallahan DE. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget. 2016;7(2):2080–2092. doi: 10.18632/oncotarget.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pereira ER, Liao N, Neale GA, Hendershot LM. Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS One. 2010 doi: 10.1371/journal.pone.0012521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romero-Ramirez L, Cao H, Regalado MP, Kambham N, Siemann D, Kim JJ, Le QT, Koong AC. X box-binding protein 1 regulates angiogenesis in human pancreatic adenocarcinomas. Transl Oncol. 2009;2(1):31–38. doi: 10.1593/tlo.08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, Koumenis C, Harding HP, Ron D, Holcik M, Bell JC. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol. 2006;26(24):9517–9532. doi: 10.1128/MCB.01145-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412(6844):300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 24.Osorio F, Tavernier SJ, Hoffmann E, Saeys Y, Martens L, Vetters J, Delrue I, De RR, Parthoens E, Pouliot P, Iwawaki T, Janssens S, Lambrecht BN. The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha+ dendritic cells. Nat Immunol. 2014;15(3):248–257. doi: 10.1038/ni.2808. [DOI] [PubMed] [Google Scholar]
- 25.Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. 2007;204(10):2267–2275. doi: 10.1084/jem.20070525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11(5):411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodall JC, Wu C, Zhang Y, McNeill L, Ellis L, Saudek V, Gaston JS. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci USA. 2010;107(41):17698–17703. doi: 10.1073/pnas.1011736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, Ellenson LH, Caputo T, Lee AH, Conejo-Garcia JR, Glimcher LH. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527–1538. doi: 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168(4):692–706. doi: 10.1016/j.cell.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, El-Deiry WS, Winograd R, Vonderheide RH, English NR, Knight SC, Yagita H, McCaffrey JC, Antonia S, Hockstein N, Witt R, Masters G, Bauer T, Gabrilovich DI. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. 2014;124(6):2626–2639. doi: 10.1172/JCI74056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, Tcyganov E, Hashimoto A, Nefedova Y, Lin C, Partlova S, Garfall A, Vogl DT, Xu X, Knight SC, Malietzis G, Lee GH, Eruslanov E, Albelda SM, Wang X, Mehta JL, Bewtra M, Rustgi A, Hockstein N, Witt R, Masters G, Nam B, Smirnov D, Sepulveda MA, Gabrilovich DI. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016 doi: 10.1126/sciimmunol.aaf8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, Ochoa AC, Cui Y, Del VL, Rodriguez PC. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity. 2014;41(3):389–401. doi: 10.1016/j.immuni.2014.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sonda N, Chioda M, Zilio S, Simonato F, Bronte V. Transcription factors in myeloid-derived suppressor cell recruitment and function. Curr Opin Immunol. 2011;23(2):279–285. doi: 10.1016/j.coi.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Yan D, Wang HW, Bowman RL, Joyce JA. STAT3 and STAT6 signaling pathways synergize to promote cathepsin secretion from macrophages via IRE1alpha activation. Cell Rep. 2016;16(11):2914–2927. doi: 10.1016/j.celrep.2016.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci USA. 2011;108(16):6561–6566. doi: 10.1073/pnas.1008942108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zanetti M, Rodvold JJ, Mahadevan NR. The evolving paradigm of cell-nonautonomous UPR-based regulation of immunity by cancer cells. Oncogene. 2016;35(3):269–278. doi: 10.1038/onc.2015.108. [DOI] [PubMed] [Google Scholar]
- 37.Brunsing R, Omori SA, Weber F, Bicknell A, Friend L, Rickert R, Niwa M. B- and T-cell development both involve activity of the unfolded protein response pathway. J Biol Chem. 2008;283(26):17954–17961. doi: 10.1074/jbc.M801395200. [DOI] [PubMed] [Google Scholar]
- 38.Solanki NR, Stadanlick JE, Zhang Y, Duc AC, Lee SY, Lauritsen JP, Zhang Z, Wiest DL. Rpl22 loss selectively impairs alphabeta T cell development by dysregulating endoplasmic reticulum stress signaling. J Immunol. 2016;197(6):2280–2289. doi: 10.4049/jimmunol.1600815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamimura D, Katsunuma K, Arima Y, Atsumi T, Jiang JJ, Bando H, Meng J, Sabharwal L, Stofkova A, Nishikawa N, Suzuki H, Ogura H, Ueda N, Tsuruoka M, Harada M, Kobayashi J, Hasegawa T, Yoshida H, Koseki H, Miura I, Wakana S, Nishida K, Kitamura H, Fukada T, Hirano T, Murakami M. KDEL receptor 1 regulates T-cell homeostasis via PP1 that is a key phosphatase for ISR. Nat Commun. 2015;6:7474. doi: 10.1038/ncomms8474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Omar I, Lapenna A, Cohen-Daniel L, Tirosh B, Berger M. Schlafen2 mutation unravels a role for chronic ER stress in the loss of T cell quiescence. Oncotarget. 2016;7(26):39396–39407. doi: 10.18632/oncotarget.9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takano S, Ando T, Hiramatsu N, Kanayama A, Maekawa S, Ohnuma Y, Enomoto N, Ogawa H, Paton AW, Paton JC, Kitamura M, Nakao A. T cell receptor-mediated signaling induces GRP78 expression in T cells: the implications in maintaining T cell viability. Biochem Biophys Res Commun. 2008;371(4):762–766. doi: 10.1016/j.bbrc.2008.04.132. [DOI] [PubMed] [Google Scholar]
- 42.Pino SC, O’Sullivan-Murphy B, Lidstone EA, Thornley TB, Jurczyk A, Urano F, Greiner DL, Mordes JP, Rossini AA, Bortell R. Protein kinase C signaling during T cell activation induces the endoplasmic reticulum stress response. Cell Stress Chaperones. 2008;13(4):421–434. doi: 10.1007/s12192-008-0038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheu S, Stetson DB, Reinhardt RL, Leber JH, Mohrs M, Locksley RM. Activation of the integrated stress response during T helper cell differentiation. Nat Immunol. 2006;7(6):644–651. doi: 10.1038/ni1338. [DOI] [PubMed] [Google Scholar]
- 44.Kamimura D, Bevan MJ. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol. 2008;181(8):5433–5441. doi: 10.4049/jimmunol.181.8.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pino SC, O’Sullivan-Murphy B, Lidstone EA, Yang C, Lipson KL, Jurczyk A, diIorio P, Brehm MA, Mordes JP, Greiner DL, Rossini AA, Bortell R. CHOP mediates endoplasmic reticulum stress-induced apoptosis in Gimap5-deficient T cells. PLoS One. 2009;4(5):e5468. doi: 10.1371/journal.pone.0005468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Y, Zhao F, Qiu Q, Chen K, Wei J, Kong Q, Gao B, Melo-Cardenas J, Zhang B, Zhang J, Song J, Zhang DD, Zhang J, Fan Y, Li H, Fang D. The ER membrane-anchored ubiquitin ligase Hrd1 is a positive regulator of T-cell immunity. Nat Commun. 2016;7:12073. doi: 10.1038/ncomms12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu P, Struijs MC, Mei J, Witte-Bouma J, Korteland-van Male AM, de Bruijn AC, van Goudoever JB, Renes IB. Endoplasmic reticulum stress, unfolded protein response and altered T cell differentiation in necrotizing enterocolitis. PLoS One. 2013;8(10):e78491. doi: 10.1371/journal.pone.0078491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8(4):279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 50.Luo Y, Li SJ, Yang J, Qiu YZ, Chen FP. HMGB1 induces an inflammatory response in endothelial cells via the RAGE-dependent endoplasmic reticulum stress pathway. Biochem Biophys Res Commun. 2013;438(4):732–738. doi: 10.1016/j.bbrc.2013.07.098. [DOI] [PubMed] [Google Scholar]
- 51.Tufi R, Panaretakis T, Bianchi K, Criollo A, Fazi B, Di Sano F, Tesniere A, Kepp O, Paterlini-Brechot P, Zitvogel L, Piacentini M, Szabadkai G, Kroemer G. Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin. Cell Death Differ. 2008;15(2):274–282. doi: 10.1038/sj.cdd.4402275. [DOI] [PubMed] [Google Scholar]
- 52.Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P, Yuan J, Zitvogel L, Madeo F, Williams DB, Kroemer G. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28(5):578–590. doi: 10.1038/emboj.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kepp O, Semeraro M, Bravo-San Pedro JM, Bloy N, Buque A, Huang X, Zhou H, Senovilla L, Kroemer G, Galluzzi L. eIF2alpha phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol. 2015;33:86–92. doi: 10.1016/j.semcancer.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 54.Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, Michaud M, Menger L, Gdoura A, Tajeddine N, Tesniere A, Zitvogel L, Kroemer G. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30(10):1147–1158. doi: 10.1038/onc.2010.500. [DOI] [PubMed] [Google Scholar]
- 55.Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G, Schlemmer F, Sulpice E, Locher C, Gidrol X, Ghiringhelli F, Modjtahedi N, Galluzzi L, Andre F, Zitvogel L, Kepp O, Kroemer G. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012;4(143):143ra199. doi: 10.1126/scitranslmed.3003807. [DOI] [PubMed] [Google Scholar]
- 56.Tang CH, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, Del Valle JR, Hu CC. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest. 2014;124(6):2585–2598. doi: 10.1172/JCI73448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ri M, Tashiro E, Oikawa D, Shinjo S, Tokuda M, Yokouchi Y, Narita T, Masaki A, Ito A, Ding J, Kusumoto S, Ishida T, Komatsu H, Shiotsu Y, Ueda R, Iwawaki T, Imoto M, Iida S. Identification of toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012;2(7):e79. doi: 10.1038/bcj.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N, Choudhry AE, Alsaid H, Jucker BM, Axten JM, Kumar R. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013;73(6):1993–2002. doi: 10.1158/0008-5472.CAN-12-3109. [DOI] [PubMed] [Google Scholar]
- 59.Yu Q, Zhao B, Gui J, Katlinski KV, Brice A, Gao Y, Li C, Kushner JA, Koumenis C, Diehl JA, Fuchs SY. Type I interferons mediate pancreatic toxicities of PERK inhibition. Proc Natl Acad Sci USA. 2015;112(50):15420–15425. doi: 10.1073/pnas.1516362112. [DOI] [PMC free article] [PubMed] [Google Scholar]