

Abstract
Over the past 20 years, a diverse group of ligands targeting surface biomarkers or receptors has been identified with several investigated to target siRNA to tumors. Many approaches to developing tumor-homing peptides, RNA and DNA aptamers, and single-chain variable fragment antibodies by using phage display, in vitro evolution, and recombinant antibody methods could not have been imagined by researchers in the 1980s. Despite these many scientific advances, there is no reason to expect that the ligand field will not continue to evolve. From development of ligands based on novel or existing biomarkers to linking ligands to drugs and gene and antisense delivery systems, several fields have coalesced to facilitate ligand-directed siRNA therapeutics. In this review, we discuss the major categories of ligand-targeted siRNA therapeutics for tumors, as well as the different strategies to identify new ligands.
1. Introduction
In tumor-bearing mice, nanoparticles (NPs) greater than 10 nm in diameter have a proclivity to accumulate in the tumor. Accumulation of these NPs in tumors, however, is modest and only about 20 to 40% greater than its accumulation by normal tissues [1]. The preferential uptake of NPs by tumors is due to the enhanced permeation and retention (EPR) effect. The EPR effect is thought to result from a combination of leakiness of tumor blood vessels resulting in flux of NPs from the blood into the tumor tissue and reduced numbers of lymphatic vessels in tumors associated with decreased drainage of NPs and occurs despite a higher interstitial pressure within the tumor. Notably, several rarely used therapies, such as nitroglycerin, may enhance the EPR effect and augment accumulation of NPs within tumors [2, 3]. When NPs have diameters less than ~10 nm, they are rapidly secreted by the kidneys and the effect of EPR is greatly reduced [4]. Moreover, NPs with very short half-life and/or with their nonspecific binding may accumulate within the tumor to a greater extent, if the EPR effect is enhanced by pegylation of particles. By prolonging blood circulation (plasma half-life) of the NP and reducing nonspecific binding, pegylation may also enable accumulation of ligand-nanoparticle conjugates in tumors above the EPR effect.
Building on accumulation of NPs in tumors from the EPR effect, researchers have sought to increase their tumor delivery by coating the particles with tumor-localizing ligands. The mechanism by which ligands increase the antitumor efficacy of their cargo (in our case siRNA) is somewhat controversial. Most investigators have determined that increased efficacy of targeted ligand-siRNA NPs is due to enhanced binding to the tumor surface marker and accumulation of NPs in the tumor compared to that in nontargeted tissues. Some investigators, however, have found that accumulation of targeted and nontargeted NPs within tumors was similar and found that increased efficacy of the targeted NP was due to enhanced receptor-mediated endocytosis and increased intracellular localization of the siRNA therapeutic [5]. Most likely, both mechanisms have important roles in ligand- targeted therapy, improving efficacy, and depend on the delivery vehicle, the target of the ligand, and strategy used in making the ligand (i.e., aptamer, peptide, or antibody).
In this review, we describe various strategies that have been developed for ligand-siRNA therapeutics to increase their selectivity toward tumors (Figure 1). “Decorating” the NP with the ligand together with PEG shell, however, does not adequately describe how ligand molecules may affect stability of the core particle. As investigators have reported, ligand molecules and their specific linkages to the NP may significantly influence release of siRNA and their efficacy [6].
Figure 1.

Schematic overview of the different ligands and core particles that target tumors. An array of core particles and ligands has been used to carry siRNA which inhibit oncogenes or induce apoptosis of tumor cells.
In addition, the efficacy (or lack thereof) of the siRNA-NP may interfere with the independent evaluation of ligand-directed therapies. It is our goal in this review not only to cover the array of ligand-targeted siRNA NPs, but also to indicate possible flaws in the particular study and alert the reader to the potential of the ligand, independent of the efficacy of the NP. This determination will be particularly important in cases in which there has been reduced antitumor efficacy with the nanoparticle.
2. Ligands Targeting Tumor Cells and Vessels
Ligands targeting tumor cells and their angiogenic vessels have primarily been peptides isolated by the phage display method (Table 1) (Figure 2) [7–15] (see review by [16]). Since tumor cells and angiogenic blood vessels often have similar cell surface receptors, ligands can have dual targeting capabilities for both tumor vasculature and tumor cells. When this is the case, siRNA therapeutic agents that have an inhibitory effect on both cells may be preferred for use with these targeting ligands, as recently reported with a miRNA mimetic [17].
Table 1.
Ligands targeting endothelial and tumor cells.
| Ligand/target1 | Carrier | siRNA and/or active moiety | Cells | Results | Other comments | References |
|---|---|---|---|---|---|---|
| cRGD2/αvβ3/β5 | Polymer (PEI)3 | siVEGFR2 | N2A neuroblastoma allografts, implanted SC | Marked inhibition of tumor growth (versus nontargeted NP) | May target physiologic angiogenesis |
[22] |
| Blocked copolymers micelles | siVEGF/siVEGFR2 | HeLa xenografts, SC |
Marked inhibition of tumor growth | Silencing both VEGFR2/VEGF more effective than either alone | [27] | |
|
| ||||||
| APRPG/VEGFR-1 | Polymer (PEI) | siVEGF | MCF7 xenografts, SC | Targeted and nontargeted PEG-NP inhibited tumor similarly | Reduction of intratumoral VEGF and mRNA greater with targeted NP | [34] |
| Liposome | si-mTOR | B16F10, IV, tumor-laden lungs | Targeted and nontargeted NP reduced tumor burden similarly | Wild-type PTEN expressing tumors | [35] | |
|
| ||||||
| cNGR/CD13 | Liposome | sic-MYC/doxorubicin | HT-1080 (fibrosarcoma) xenografts, SC | Tumor uptake and inhibition greater with targeted NP | HT-1080, a CD13+ cell | [38] |
|
| ||||||
| F3/nucleolin | Silk-polylysine-copolymer NP | Plasmid-based luciferase expression | MDA-MB-231 xenografts, SC |
Targeted NP increased gene expression in tumors | Only study in vivo with F3 ligand; F3 more effective than CGKRK in vitro | [44] |
| Liposomes | siPLK1 | PC3 (in vitro study) |
Uptake and silencing greater with targeted therapy | Cells pretreated with siPLK1 were more sensitive to the inhibitory effects of paclitaxel | [42] | |
|
| ||||||
| CGKRK/p32 | Nanoworms | Mitochondria-targeting peptide | Orthotopic breast cancer model (MCF10CA1a) | Targeted NP gave greater tumor inhibition than nontargeted NP | Addition of iRGD increased tumor inhibition of targeted NP | [45] |
|
| ||||||
| iRGD | PEI micelles | Shsurvivin/paclitaxel | A549 lung cancer xenografts, SC |
Greater tumor size inhibition with targeted therapy | Plasmid-based expression of shRNA; dose-response inhibition; no siRNA studies yet | [51] |
1Selected representatives of targeted nanoparticles. 2See text for amino acid sequences of ligands; 3PEI, polyethylenimine; NP, nanoparticle; siVEGFR2, small inhibitory RNA targeting VEGFR2; PLK1, polo-like kinase 1; SC, subcutaneous; shRNA, small hairpin RNA expressed by plasmids; siRNA, small inhibitory RNA.
Figure 2.
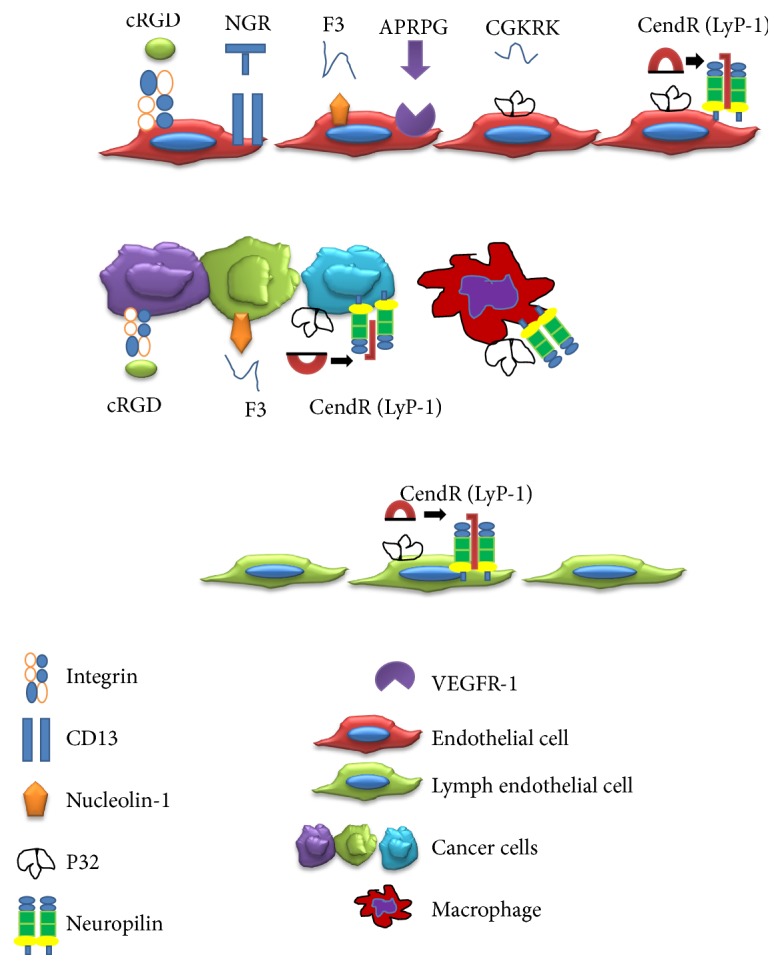
Peptide ligands targeting tumor endothelial cells and tumor cells. Ligands and their receptors are shown with associated cells.
Of note, a few promising targeting peptides discussed in this section have not been tested for their efficacy with siRNA therapy although they have shown their ability to target tumors with cytotoxic peptides and chemotherapeutic drugs. Moreover, except for cRGD-targeted therapies, the remaining siRNA studies are limited and additional studies, particularly with CendR and NGR targeting peptides, are required to confirm the encouraging in vivo results.
2.1. Cyclic(c) RGD
cRGD, isolated from an in vivo tumor phage selection display, was found to bind to αvβ3 and αvβ5 integrins. These two integrins are highly expressed on angiogenic endothelial cells of tumors as well as on a number of tumor cells (about 10–15%). Interestingly, αvβ3 and αvβ5 both have important roles in tumor angiogenesis, yet they control different aspects of angiogenesis. Whereas αvβ3 has an important role in the bFGF-mediated angiogenic pathway, αvβ5 has a more critical role with the VEGF-mediated pathway [18]. The functions of these integrins have not been clearly defined in tumor cells, yet they do modulate apoptosis.
The cRGD ligand has been extensively used for targeted therapy against an array of cancer models. cRGD binds to integrins at least 100-fold more tightly than linear RGD [19]. Although the initial cRGD required oxidation of cysteines to achieve its cyclic form (A[CRGDMFGC]A), the use of D-phenylalanine enabled cyclization of the peptide (cRGDfV) without the unwanted cys-cys reactions [19, 20]. An important paper demonstrating the efficacy of cRGD therapy was published by Hood and colleagues in 2002 [21]. This plasmid-based approach with cRGD liposomal carriers of dominant-negative mutants of Raf-1 not only inhibited tumor xenografts, but also resulted in their regression. This study provided the stimulus for siRNA therapy targeting RAF-1, VEGF, VEGFR2, ERG, FAK, and PLXDC1 and several other oncogenes [22–28].
Antiangiogenic therapy with siRNAs was first demonstrated with cRGD-containing PEI siRNA-NP targeting VEGF (siVEGF) to inhibit the growth of N2A murine neuroblastomas [22]. Coadministered free excess cyclic RGD markedly decreased the efficacy of the targeted therapy, demonstrating the specificity of cRGD-NP. Later, a cRGD functionalized chitosan NP targeting PLXDC1 (upregulated in the tumor vasculature) inhibited ovarian tumor growth by about 90%; the cRGD-NP was about 60% more effective than the untargeted NP [23]. Since the A2780 tumor cell did not express αvβ3 integrins, the authors concluded that the efficacy of the therapy was its direct effect on the endothelial cells. In contrast, Chou et al. utilized an αvβ3-expressing melanoma cell line in their murine model and directed their therapy toward genes expressed in the tumor cells [24, 29]. Chou and colleagues reduced luciferase expression by more than 80% in MDA-MB-435 tumor xenografts with their RGD-biodegradable NP carrying luciferase siRNA. Moreover, when the Raf-1 siRNA RGD-NP targets human RAF-1 in the melanoma cells, but not the mouse Raf-1 (in blood vessels), the tumor was inhibited by more than 60% [29]. When cRGD-PEG-NPs were compared with PEG-NPs for reduction in tumor luciferase expression, the targeting cRGD moiety was responsible for 50% of reduction of their target gene. Christie et al. targeted both VEGF and VEGFR2 with their cRGD micelle-siRNA particle, which was stabilized with 2-iminothiolane; compared to untreated tumors, these particles reduced tumor growth by more than 80% [27]. Christie et al. noted that the combination of VEGFR2 and VEGF siRNAs was more effective than administration of VEGF siRNA alone. Targeting VEGFR2 located on endothelial cells by the cRGD-NP is likely to be very effective because of its ready accessibility, whereas targeting secreted VEGF from both tumor and endothelial cells may be less effective, due to reduced penetration of the tumor by the NP.
2.2. APRPG
APRPG peptide, first found from a phage-displayed peptide library in 2002, is an angiogenic vessel-homing peptide [11], which binds to VEGFR-1 [30]. There have been several studies with APRPG-conjugated NPs including investigations that incorporated antiangiogenic or chemotherapeutic agents within targeted liposomes. Doxorubicin, TNP-470, and the tyrosine-kinase VEGFR2 inhibitor-SU1498 which were encapsulated within APRPG-liposomes, showed significant antitumor activity in vivo, including colon and ovarian cancers [31–33]. Compared to the nontargeted NP, the targeted NP showed greater efficacy at downregulating its target and in most cases, they showed significantly greater antitumor activity.
In addition to chemotherapy and tyrosine-kinase inhibitors, siRNA [34, 35] and miRNA [36] have been incorporated within APRPG-NPs. Although APRPG primarily targets angiogenic vessels, Lu et al. interestingly found that APRPG targeted PEI in complex with VEGF siRNA inhibited the growth of breast cancer MCF-7 cells in vitro more effectively than untargeted PEI-polyplexes [34]. Inhibition of tumor growth, however, with the targeted PEI complex was no greater than the nonligand NP. Nonetheless, the targeted PEI polyplex reduced intratumoral VEGF mRNA and VEGF significantly more than the nontargeted polyplex. In a lung tumor model in which wild-type PTEN was expressed, liposomes which were decorated with APRPG and carried si-mTOR markedly reduced the tumor burden compared to the phosphate-buffered saline-treated group [35]. Although the targeted ligand NP accumulated in the lung tumors significantly more than nontargeted NPs, inhibition of the tumor was similar to the targeted and untargeted therapies. The authors speculated that the discrepancy between accumulation and the antitumor efficacy may be due to differences in the distribution of the liposomes within the tumor. Whereas the targeted NP may be primarily located in the tumor vasculature, the nontargeted NPs may be found throughout the tumor interstitium.
2.3. NGR
Cyclic NGR (i.e., CNGRCVSGCAGRC) is a ligand for CD13 [8], which is upregulated primarily in the tumor vasculature and in a few cancers such as fibrosarcomas. Although the cyclic NGR binds with higher affinity than linear NGR, and drug conjugates with the cyclic peptide have 10-fold greater antitumor efficacy, the linear NGR has been the preferred ligand on the surface of nucleic acid carriers because it avoids cysteine bridges that may occur between different carrier molecules. In addition, biodistribution and targeting with linear NGR conjugated to liposome surfaces have been reported to be quite good, probably because of the multivalence presentation of the ligand on the NP. Recently, Negussie and colleagues developed a cyclic NGR (cKNGRE) without disulfide bonds, which had about 3.5-fold greater affinity (with and without liposomes) toward CD13+ cells compared to the linear NGR (KNGRG) [37]. Furthermore, the linear and cyclic peptides when conjugated to liposomes both had a 10-fold greater affinity for CD13+ cells compared to their respective free peptides. With an estimated ~4200 peptides per liposome, it is not clear nor has it demonstrated that the cyclic NGR conjugated to liposomes would improve their biodistribution or antitumor activity compared to noncyclic NGR liposomes. With NPs (or proteins) containing a limited number of NGR peptides on their surface, the cyclic peptide may have a greater role in enhancing binding and selectivity.
To the best of our knowledge, there has only been a single NP-siRNA study that used NGR. In this report, the linear NGR (GNGRGGVRSSSRTPSDKY) was attached to the PEG on a liposomal polycationic DNA (LPD) carrier of siRNA. The NP delivered si-cMYC to the cytoplasm and downregulated the target gene in the CD13+ HT 1080 cells but not CD13− HT-29 cells in vitro [38]. Notably, the NGR si-cMYC NP reduced tumor size of HT-1080 xenografts by about 30% more than untargeted NP. When doxorubicin was in complex with DNA, the targeted NP containing doxorubicin/DNA and si-cMYC reduced the size of tumors by a further 40% compared to the particle containing si-cMYC alone [38].
2.4. F3 Peptide
The F3 peptide (KDEPQRRSARLSAKPAPPKPEPKPKKAPAKK), a 31-amino acid fragment of HMGN2 protein, binds to nucleolin which is on the cell surface of tumor and angiogenic endothelial cells [39]. After the F3 peptide binds to nucleolin at the cell surface, they are rapidly internalized and transported to the nucleus [40]. Compared to untargeted liposomes, a F3 peptide-modified liposomal carrier of siRNA enhanced internalization by 10-fold in both breast cancer cells and angiogenic blood endothelial cells and this uptake correlated with more effective gene silencing [41]. Moreover, these investigators showed that the F3 ligand conjugated to liposomes containing siPLK1 (polo-like kinase-1) reduced cell viability by 40% and sensitized the PC3 prostate cancer cells to paclitaxel (IC50 reduced by about 45%) [42]. Similar synergistic results with an apoptotic siRNA (AllStars Death-siRNA, Qiagen) and paclitaxel were observed for ovarian cancer cells with a F3 targeted multifunctional NP [43]. Numata and colleagues were the only group to demonstrate the efficacy of F3-targeted NPs to tumors in vivo, using a plasmid-based gene delivery system [44]. Compared to the vascular homing (CGKRK) particles, the F3-targeted silk particles enhanced luciferase expression in the two malignant cells (MDA-MB-435 and MDA-MB-231) in vitro. Neither targeted peptide NPs (CGKRK or F3) resulted in measurable luciferase activity in a nonmalignant cell. For the in vivo study, the F3-nanoparticles (injected iv twice a week) markedly enhanced luciferase expression after the first week in MDA-MB-231 derived xenografts but F3-targeted particles were not compared with non-F3 particles as carriers of luciferase. Notably, utility of the F3 peptide conjugated to siRNA NPs in vivo has not yet been examined.
2.5. CGKRK
CGKRK targets primarily the angiogenic vessels of tumors. Similar to LyP-1 peptide (see below, Section 2.6), it binds with high affinity to the p32 receptor which aids in the transport of the conjugate or NP into the cytosol of the angiogenic vessels. Unlike LyP-1, the CGKRK does not contain a CendR motif with tumor-penetrating properties, and consequently, NPs containing CGKRK primarily targets endothelial cells of the tumor. Based on targeting of angiogenic vessels, the CGKRK targeted a dimer of the proapoptotic peptide, klaklak (D-amino acids) to brain glioma xenografts, resulting in its enhanced accumulation in the tumor vasculature and prolonged survival of mice [45]. To date, there have not been any reports of siRNA NPs with the CGKRK targeting ligand in vitro or in vivo.
2.6. Tumor-Penetrating Peptides with the CendR Motif
In contrast to the tumor-homing peptides discussed above (i.e., cRGD and NGR), tumor-penetrating peptides and their associated NPs penetrate deeply and pervasively throughout the tumor parenchyma. There have been three tumor-penetrating peptides identified thus far: LyP-1, iRGD, and iNGR. Whereas LyP-1 and iRGD were the first two tumor-penetrating peptides to be identified and were isolated by in vivo phage display, iNGR was the result of de novo synthesis gained from knowledge of the other two peptides [7, 12, 14, 15, 46, 47]. Prototypical sequences of LyP-1, iRGD, and iNGR are CGNKRTRGC, CRGDKGPDC, CRNGRGPDC, respectively, with the CendR motif underlined. For extravasation of NPs into tumors, the peptide requires two motifs, one for tumor homing and the other for tumor penetration. With the exception of LyP-1, the peptides must be cyclic for optimal binding and specificity for tumor homing. Moreover, for tumor homing of the NP, the LyP-1 peptide binds to p32 receptors on tumor cells, tumor blood vessels and lymphatics, and tumor macrophages, whereas the iRGD and iNGR peptides initially bind to αvβ3 integrins and CD13 receptors, respectively, on the angiogenic vessels of tumors. After the tumor-specific components of the penetrating peptide bind to the specific receptor in the tumor, proteolytic degradation exposes the CendR sequence (R/KXXR/K-OH, where R/K represents either arginine or lysine and X represents any amino acid; see underlined sequences above). Exposure of the cryptic CendR activates the neuropilin-1 and neuropilin-2 pathways, enhancing extravasation of the NP into the tumor. At least part of the extravasation of the NP is thought to be due to transcytosis [48] (Figure 3).
Figure 3.
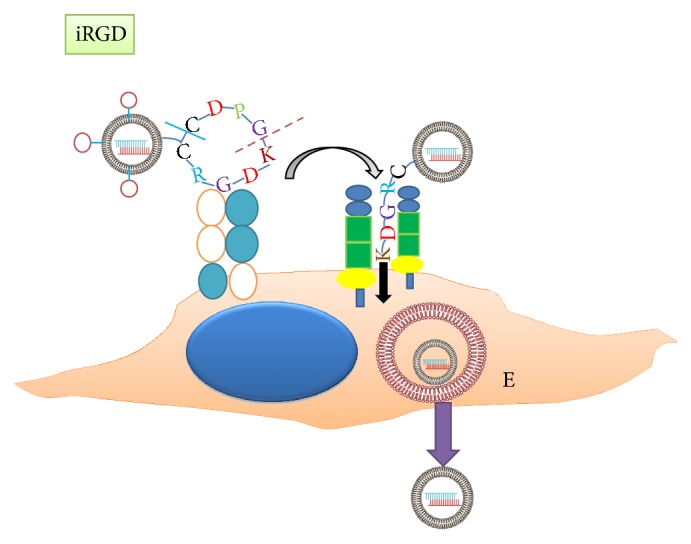
Proposed mechanism of CendR tumor-peptides to transport NPs into tumor matrix. After a CendR peptide such as iRGD binds to av integrins, furin-like enzymes cleave the cyclic peptide (dash line) on the carboxyl side of the lysine group. With reduction of the cystine linkage (solid line), the peptide, KDGR, binds to the neuropilin receptor and activates the transcytosis pathway. The peptide together with the NP is then endocytosed and transported through the endothelial cell to the tumor milieu. E, endosome.
Although several studies show that tumor-penetrating peptides improve antitumor efficacy of drugs or NPs incorporating drugs [12, 47, 49, 50], there have been few studies using tumor-penetrating peptides with siRNA (or shRNA) nanoparticles [10, 51]. Ren and colleagues demonstrated that LyP-1 siRNA NPs accumulated 3-fold more in tumors compared to untargeted NPs [10]. Moreover, location of the targeted NP within the tumor was extravascular in contrast to the intravascular location of the untargeted NP. With a LyP-1 containing NP carrying an siIDF (inhibitor of DNA binding 4), ovarian cancer xenografts were reduced by nearly 80% compared to tumors treated with the siGFP control group. A similar study was done with iRGD-NPs carrying both paclitaxel and a plasmid-based shRNA targeting survivin [51]. The size of the tumors treated with iRGD targeted NPs remained unchanged during the course of therapy (~280 mm3), whereas size of tumors treated with untargeted NP or saline grew to about 900 and 2200 mm3, respectively.
In contrast to cRGD peptides-NPs that do not contain cysteines, iRGD-NPs with cysteines may have reduced stability in solution when reconstituted due to intermolecular bridge formation between NPs from reduced cysteines. The tendency of reduced cysteines to form these side reactions may limit the clinical utility of cyclic iRGD/iNGR conjugated to NPs. Importantly, not only administration of iRGD-NPs but also coadministration of iRGD and NPs significantly enhanced tumor penetration and antitumor efficacy of the NP [14].
3. Targeting Tumor Cells
The diverse approaches and ligands to target tumors cells have indeed been numerous. Whereas ligands targeting angiogenic vessels were peptides as discussed previously, the ligands targeting markers/receptors on tumor cells have ranged from single-chain variable fragment antibodies to RNA aptamers and from low molecular weight ligands such as folate to high molecular weight ligands such as transferrin and hyaluronic acid (Table 2). The ligands in this section primarily target tumor cells although they may target other tumor-associated cells to a lesser extent.
Table 2.
Ligands targeting tumor cells.
| Ligand/target1 | Carrier | siRNA and/or active moiety | Cells | Results | Other comments | References |
|---|---|---|---|---|---|---|
| High MW endogenous ligands | ||||||
|
| ||||||
| Transferrin (Tfr)/TfR receptor | Polymer (cyclodextrin) | siRRM2 | Neuro2A, murine neuroblastoma, implanted SC | Greater tumor inhibition with targeted than nontargeted NP | Dose-related efficacy using transferrin ligand | [59] |
|
| ||||||
| Hyaluronic acid/CD44 | Liposomes | siPLK1 | U87MG, human glioblastoma, orthotopic | Prolonged survival, reduced PLK1 mRNA by 80% in tumor xenografts versus control siRNA | CD44 increased on cell surfaces of many cancers | [63] |
|
| ||||||
| ApoA1/SR-B1 | Liposomes | siVEGF | MCF-7, SC | Inhibited tumor size and prolonged survival | Hepatocellular carcinomas have been targeted | [72] |
|
| ||||||
| Aptamers | ||||||
|
| ||||||
| Aptamer/PSMA2 | Aptamer-siRNA chimera | siPLK1 | PSMA+ (22Rv) versus PSMA− (PC-3) cells, SC | Tumor regression of 22Rv xenografts treated with apta-mer-PLK1 siRNA | PSMA upregulated in prostate cancer | [78] |
| Aptamer/PSMA | Aptamer- dsRNA binding domain- polyhistidine chimera | siGFP | PSMA+ (LNCaP) versus PSMA− (PC-3) cells, in vitro study only | Silenced GFP efficiently in LNCaP cells | dsRNA-binding domain binds to siRNA; chimera may not efficiently lyse endosomes in vivo | [80] |
|
| ||||||
| Aptamer/PSMA | Polymer (atelocollagen) | (miR15a and miR-16-1) | Bone metastatic prostate model-LNCap cells | Prolonged mouse survival versus nontargeted NP | Reduced expression of Bcl-2, cyclin D1, and Wnt3a in vitro with targeted NP | [86] |
|
| ||||||
| Antibodies | ||||||
|
| ||||||
| scFv/Her2+ | scFv-protamine-conjugate | siPLK1 | Her2+ versus Her2− breast cancer, orthotopic | Marked tumor inhibition on Her2+ tumors in vivo versus no/little effect on Her2− tumors | Ionic interaction between siRNA and protamine; siRNA cocktail targeting PLK1, CCND1, and AKT more effective than PLK1 alone | [89] |
|
| ||||||
| scFv/Her2+ | Polymer (PLA) | siPLK1 | Her2+ (BT474) versus Her 2− (MCF7) breast cancer, orthotopic | Targeted NP inhibited both Her2+/- xenografts but showed the most efficacy toward Her2+ tumors | Dose-dependent antitumor response of the targeted NP was observed | [95] |
| Small molecule nonpeptide ligands | ||||||
|
| ||||||
| Folate/folate receptor | Low MW PEI cross-linked with by cyclodextrin | siVEGF | HeLa xenografts, SC | Targeted NP reduced tumor size more than nontargeted | Folate receptor is widely distributed in epithelial cancers | [109] |
| Tetrahedral DNA | siGFP siLuciferase |
KB expressing GFP (in vitro) or luciferase, SC | Folate NP, given IV or IT, reduced tumor luciferase expression by 60% | 3 ligands on same face of tetrahedron to maximize uptake; well-defined NPs | [119] | |
| Polymer (oligoamine amides) | siEG5 | KB xenografts, SC | Methotrexate/siEG5 NP enhanced survival | Methotrexate targets folate receptor, small NPs (~6 nm) delivered IT | [120] | |
|
| ||||||
| Anisamide/sigma-1, -2 receptors | Liposome | siMDM2, sic-MYC, and siVEGF | B16F10 murine melanoma, SC, and lung metastases | Reduced primary and metastatic tumor burden and prolonged survival | Receptor widely distributed in tumors and proliferating cells; sigma-2 ligands induce apoptosis in tumors | [125, 144] |
|
| ||||||
| Galactose/asialoglycoprotein receptors | Multilayered NP (mesoporous silica NP, polymers) | siVEGF/doxorubicin | QGY-7703, a hepatocarcinoma, SC | Marked inhibition with targeted NP (91.3%) versus nontargeted NP (75.2%) | Ligand targets primarily liver cells | [130] |
|
| ||||||
| Peptide ligands | ||||||
|
| ||||||
| T73/transferrin receptor | Cholesterol-grafted polymer/lipid hybrid | siEGFR | MC7 xenografts, SC | About 50% greater reduction in tumor size with targeted than nontargeted NPs | Binding of T7 to receptor is independent of endogenous transferrin concentration | [134] |
|
| ||||||
| MMP2-cleavable peptide | PEI-lipid hybrid micelle | Fluorescently-labeled siRNA and paclitaxel | A549 lung cancer xenografts, SC | Enhanced tumor uptake by 2.4-fold with MMP2-micelle | MMP2-triggered PEG deshielding | [135] |
|
| ||||||
| CP15/unknown | Polymer (chitosan) | siPLK1, biotin-labeled RNA | SW480 colon cancer cells, SC | Modest increase in tumor uptake/specificity of targeted NP (versus nontargeted NP) | Identified by phage display; primarily targets colon cancer; study requires validation | [136] |
|
| ||||||
| LHRH/LHRH receptor | Polymer (PPI) | siCD44/paclitaxel | Ovarian cancer xenograft, SC | Targeted therapy significantly more effective than nontargeted therapy | Combination of paclitaxel and siCD44 NP most effective in reducing tumor size | [139] |
|
| ||||||
| GRP/BB2 receptor | Conjugate | Fluorescently-labeled siRNA, siSurvivin | MDA-MB-231 xenografts (in vitro) | Competitive inhibition indicates specific uptake of GRP-siRNA conjugates into cells; marked reduction of survivin with targeted NP | Requires in vivo validation; BB2 reported to be on breast, prostate, lung, and colon cancers | [140] |
|
| ||||||
| RVG/acetylcholine receptor | Polymer (cyclodextrin) | siGADPH | U87 glioblastoma, HeLa (RVG- control), (in vitro) | Targeted NP reduced GADPH mRNA about 25% more than nontargeted NP in U87 | Requires in vivo validation; in vitro efficacy is modest | [142] |
1Selected representatives of targeted nanoparticles. 2PSMA, prostate specific membrane antigen; scFv, single-chain variable fragment antibody; MMP2, metalloproteinase-2; LHRH, luteinizing-hormone releasing hormone; GRP, gastrin-releasing peptide; BB2, a subtype of the bombesin receptor; RVG, rabies virus glycoprotein; dsRNA, double-stranded RNA binding domain; PLA, poly(D,L-lactide); PEI, polyethylenimine; PPI, polypropylenimine; RRM2, ribonucleoside-diphosphate reductase subunit M2; PLK1, polo-like kinase-1; EG5, eglin 5; GFP, green fluorescent protein; EGFR, epidermal growth factor receptor; GADPH, glyceraldehyde-3 phosphate dehydrogenase; SC, subcutaneous; NP, nanoparticle; IT, intratumoral. 3See text for amino acid sequences of ligands.
3.1. High MW Endogenous Ligands
3.1.1. Transferrin
To deliver the growth mediator iron intracellularly, the transferrin receptor (TfR) is markedly upregulated in many tumor cells, as much as severalfold higher than in normal cells [52]. When iron binds to its receptor, they are endocytosed and the iron is released in the acidic endosomal compartment and the receptor is recycled to the surface [53]. Transferrin has been evaluated extensively as a ligand to enhance the delivery of drugs and NPs to tumors [5, 54–58]. The most extensively studied transferrin-targeted siRNA targeting tumors was based on the cyclodextrin carrier. Marked suppression of the oncogene (M2 subunit of ribonucleotide reductase) [59] or exogenous marker (luciferase) [60] was reported with intravenous delivery of the transferrin-NP. Thus far, this has been the only targeted ligand-siRNA NP (CALAA-01) tested in a cancer clinical trial (this therapy was discontinued after phase 1 due in part to adverse clinical events) [61]. Other investigators have found that transferrin coupled to NP enhanced the specificity and potency of the NP within the tumor [56]. For example, Liu et al. found that transferrin-PEI-plasmid shRNA (targeting HIF-α) accumulated in a time-dependent manner in tumors expressing high levels of the TfR [56], reducing the growth rate by 8-fold compared to control groups. Interestingly, biodistribution studies did not detect a difference in accumulation within the tumor between untargeted and transferrin-containing NPs, despite their having marked differences in their antitumor efficacies [5]. The authors indicated that transferrin was essential for delivery of the NP and siRNA intracellularly. Studies with DNAzymes carried by the transferrin-cyclodextrin also support the notion that internalization within the tumor cell and not accumulation and localization within the tumor is the important factor [55].
Notably, the cyclodextrin studies from Davis group also underscored that the method to form the targeted nanoparticle was important [62]. Whereas grafting of PEG-ligand onto preformed polyplexes may augment transfection of PEI-polyplexes, polyplexes formed with the low molecular weight cyclodextrin may be destabilized by this approach, particularly in the presence of salt. When cyclodextrin, nucleic acids, and adamantine-PEG-ligand were mixed together, polyplexes were stable in different salt concentrations and maintained their ability to target cells. Although this approach was initially used for cyclodextrin DNA polyplexes, this strategy was later used to form stable targeted cyclodextrin-siRNA polyplexes. In many cases, optimal targeting is dependent on the method used to form the nanoparticle.
3.1.2. Hyaluronic Acid
Hyaluronic acid (HA), a negatively charged natural polymer, has recently been investigated as a targeting agent for drug delivery systems. HA binds to the surface CD44 receptors, which are overexpressed in primary and metastatic tumor cells, and consequently, several studies have utilized HA-coated NPs in which siRNA has been incorporated to enhance antitumor efficacy. After determining that HA-grafted liposomal carriers of siPLK1 reduced its target gene expression in vitro by about 90%, Cohen and colleagues investigated the efficacy of these HA-grafted liposomes in vivo [63]. In a glioblastoma orthotopic mouse model, the HA-grafted liposomal carrier of siPLK1 administered by local injections markedly prolonged mouse survival. Compared to the untreated and siLuc control groups in which all mice had died prior to day 40, six of the ten mice in the HA-liposomal-siPLK1 group survived to day 95. In addition to liposomes, a chitosan-siRNA-NP, which was coated with HA, showed enhanced uptake, particularly in cells expressing high levels of CD44 [64].
Other targeting ligands have been added to the surface of HA-coated NPs to further enhance delivery to tumors of nucleic acids, although this approach has not been used for siRNA therapeutics yet. For example, Yu's group determined that transferrin and HA-coated liposomes enhanced plasmid-based gene expression (EGFP) to A549 lung adenocarcinoma xenografts by about 40% compared to either single-ligand liposomal NP [65]. Similarly, micelles coated with HA and folate increased accumulation of paclitaxel by about 42% in MCF7 xenografts compared to HA-only coated micelles [66]. On the bases of these studies, the approach of dual-ligand targeting for siRNA delivery has a great deal of promise for hyaluronic acid and the other ligands discussed in this review.
Besides coating preform NPs with HA, alternative strategies have been used to conjugate (or complex) cationic polymers or cholesterol with HA. Discrete and limited conjugation of cationic polymers, such as PEI, polyarginine, and polylysine, with HA has been done to form positive and negative surfaces [67, 68]. Whereas the positive surface of the modified HA binds to the siRNA, the negatively charged HA coating the NP can bind to CD44-positive tumor cells. Choi and colleagues have also conjugated cholesterol with HA to form micelle-like NPs. To incorporate siRNA within these NPs, Choi et al. utilized the viral protein, 2b, which specifically binds siRNA in a pH-dependent manner [69]. Although non-HA micelles were not used as controls, the HA-cholesterol micelles were more effective in silencing red-fluorescent protein (about 80%) in vitro compared to Lipofectamine 2000 (about 64%).
3.1.3. ApoA1
Apo-A1-HDL (rHDL) particles bind tightly to the scavenger receptor class B-type 1 (SR-B1), which is found predominantly on the surface of hepatocytes. Not surprisingly, rHDL particles incorporating cholesterol siRNA conjugates have been injected intravenously at low dosages (≤2 mg/kg) to silence genes in a hepatitis B mouse model, resulting in a marked decrease of the hepatitis surface antigens [70]. Moreover, it has been recognized that the SR-B1 receptor is upregulated in several tumors, ranging from hepatocellular to breast cancers. With an siRNA conjugate encapsulated within rHDL-coated particles, Ding et al. demonstrated the utility of this delivery method by targeting an oncogene (VEGF, Pokemon, or BCL-2) with tumor uptake and tumor inhibitory studies [71, 72]. Accumulation of the rHDL particle was significantly higher in MCF 7 xenografts (5.5-fold higher) which expressed increased levels of SR-B1 compared to the HT1080 tumor xenografts [72]. Moreover, mice with MCF-7 tumors treated with rHDL-siVEGF had a further 70% reduction in their size compared to nontargeted NPs or to untreated mice. Concomitant with the decrease in tumor size, marked reduction in VEGF mRNA levels and numbers of microvessels in the tumor were also observed in the rHDL-siVEGF-treated group compared to untargeted NPs and untreated groups.
3.2. Aptamer
Aptamers are small molecular weight (8–13 KDa) single-stranded RNA or DNA molecules with low nanomolar binding affinities toward their targets and with low immunogenicity (for supplementary reviews, see [73, 74]). These nucleic acid ligands are isolated from combinatorial libraries by a method known as SELEX (Systematic Evolution of Ligands by exponential enrichment). Large quantities of aptamers can readily be synthesized and degradation of aptamers by nucleases may be avoided through chemical modification. By binding with high affinity to target molecules, investigators have found a number of biological applications for aptamers, including target validation, inhibitors of receptors or enzymes, carriers for nucleic acids and chemotherapy agents, and ligands conjugated to NPs. Recently, clinical trials with aptamers alone as antitumor agents have been initiated [75]. For siRNA studies targeting tumors, we will provide an overview on aptamer-siRNA chimeras and aptamer-siRNA NPs (Figure 4).
Figure 4.

Aptamer mediated delivery of siRNA. (a) Aptamers and siRNAs have been conjugated with one another to form a chimera (upper). To enhance lysis of endosomes and minimize formation of polyplexes, aptamer-siRNA conjugates have been complexed to double stranded DNA domain- (DSD-) polyhistidine conjugates (lower). Upon entry into acidic endosomes, the polyhistidine component becomes protonated which aids in the lysis of endosomes. (b) Alternatively, aptamers have been conjugated to the surface of core particles (i.e., liposomes, polyplexes) that have incorporated siRNA.
3.2.1. Aptamer-siRNA Conjugates
Initially most investigators focused on aptamers-siRNA conjugates (Figure 4(a), upper). With aptamers-siRNA conjugates, aptamers serve two purposes: one as a carrier of siRNA and the other as a ligand targeting a tumor-associated antigen. With these conjugates, the aptamer and siRNA could be joined by either a linker or directly as a chimera.
Chu et al. developed a first-generation aptamer that targeted prostate-specific membrane antigen (PSMA), which is highly expressed on LNCaP prostate cancer cells [76]. Through a streptavidin-biotin linkage, a laminin A/C siRNA was linked to the aptamers with the conjugate reducing its target gene levels by about 70%. Moreover, LNCaP tumor xenografts regressed by 2.21-fold from days 6 to 21 after ten intratumoral injections (every other day) of the anti-PSMA aptamer-siPLK chimera; in contrast, the tumors in the control group increased by 3.63-fold [77]. The chimera had no effect on the size of PC3 prostate tumors, which do not express PSMA.
Building on these findings, Dassie et al. made several modifications to a PSMA-siPLK chimera that resulted in 22Rv tumor regression when the chimera was administered systemically (one injection per day for 10 days) [78]. Indeed, 70% of the tumors regressed completely from a pretreatment size of about 400 mm3. This is indeed quite remarkable, although it has been noted that high dosages of chimera were used, suggesting that significant amounts of the chimera were entrapped in the lysosomal pathway. In contrast to the 22Rv tumors, the chimera had no effect on the PC3 tumors. Particularly relevant modifications of the chimera that augmented in vivo activity included pegylation to increase its half-life, addition of 2′-fluoropyrimidines on the passenger strand of the siRNA to decrease nuclease activity, 2 nucleotide 3′ overheads, and swapping the position of the guide and passenger strands. These modifications significantly improved the ability of the chimera to reduce tumor size and/or to decrease PLK1 expression. For instance, pegylation of the chimera silenced PLK1 mRNA in the tumor by about 45% 5 days after a single injection, whereas the nonpegylated form had no effect. Consistent with this finding, pegylation of the chimera had an increased half-life to more than 30 hours, whereas the nonpegylated chimera had a half-life of less than 35 minutes. A similar strategy but with different targets for the siRNA and aptamer was used by Subramanian et al. [79]. By targeting EpCAM with the aptamer as well as with a siRNA of EpCAM, the systemically delivered chimera resulted in the regression of tumor xenografts derived from MCF-7.
Since most aptamer-siRNA chimeras are degraded in the lysosomal pathway, efforts have been made to increase endosomal lysis, enabling the escape of the siRNA intracellularly. One such strategy is a fusion PSMA aptamer-dsRNA binding domain-polyhistidine dual block protein, in which 1 or 2 siRNA docked with the binding domain and the polyhistidine enabled endosomal lysis (Figure 4(a), lower) [80]. Addition of an 18-mer of polyhistidine was optimal in augmenting endosomal escape and increased gene silencing (GFP) by about 5-fold in LNCaP cells. Interestingly, addition of histidine-tags of 24 or 30 amino acids inhibited siRNA binding to the RNA-binding domain. Although the authors state that these chimeras remain discrete in solution, we would be surprised if the polyhistidine tag did not form aggregates at least to some degree, based on the proclivity of histidines to interact with each other through hydrogen bonding [25].
3.2.2. Aptamer-siRNA-Nanoparticles
Several aptamers, including those targeting EpCAM, TfR (CD71 or the transferrin receptor), CD30, PSMA, and a T-cell marker, have been conjugated to NPs (Figure 4(b)) [81–86]. The potential advantage of aptamer-targeting NPs is that endosomal lysis properties can be readily incorporated within the particles to release the siRNA into the cytosol, potentially a significant limitation with the aptamer-siRNA chimeras. Nevertheless, there are potential disadvantages of aptamer-targeting NPs such as greater size, limited tumor penetration, and increased complexity in their preparation compared to the aptamer alone or the aptamer-siRNA chimeras. With one exception utilizing miRNA, the aptamer-siRNA-NP has been limited to studies in vitro.
In 2010, a sub-10 nm aptamer conjugated to the polyamidoamine (PAMAM) dendrimer nanoplex was developed by Zhou et al. [87]. Although no RNAi cargo was incorporated, the group demonstrated that the targeted nanoplex had high affinity for a surface receptor on the T-lymphocytic lymphoma cell line. The DNA aptamer, sgc8c, which binds to the leukemia biomarker PTK7, was identified by a cell-SELEX method. Bagalkot and Gao then demonstrated that a PSMA aptamer-siRNA conjugated to PEI which coated a quantum dot significantly enhanced uptake with concomitant greater silencing of green fluorescent protein (GFP) in prostate cancer cells compared to nontargeted controls [83]. Critical to enhancement of this targeted nanoparticle was a 2-step method of preparation, thereby preserving the targeting function of the aptamer. Zhao and colleagues targeted the CD30 surface biomarker found primarily on anaplastic large cell lymphomas (ALCL) [82]. The CD30-aptamer PEI siRNA nanoplex bound selectively to Karpas 299 cells (an ALCL line), whereas this targeted nanoplex showed little binding to Jurkat cells, a cell line that does not have the CD30 surface marker. Not surprisingly, selective gene silencing for cells with C30 surface marker was demonstrated with the targeted nanoplexes. In Karpas 299 cells expressing GFP, there was a 71% reduction in those treated with the targeted nanoplex where in contrast, there was no reduction in GFP in Jurkat cells treated with nanoplexes. Similar results were observed with the targeted nanoplexes silencing the ALK gene with greater than a 60% reduction in cell number of Karpas 299 cells compared with the untreated and nontargeted control groups, whereas the targeted nanoplexes and control groups had no effect on the proliferation of non-CD30 expressing Jurkat cells. More recently, Subramanian et al. have developed a polymeric-nanoplex containing an EpCAM siRNA and aptamer targeting the EpCAM adhesion molecule on tumor cells [85]. The EpCAM surface protein and mRNA were efficiently downregulated by the targeted nanoplex compared to the nontargeted nanoplex. Moreover, whereas the scrambled siRNA nanoplex had negligible effect on the proliferation of the cells, the targeted nanoplex reduced cell number by about 70%.
Although aptamers have been primarily conjugated to polymeric carriers, Wilner and colleagues conjugated an anti-TfR aptamer (C2) to the SNALP liposomal carrier of siRNA [84]. Compared to the high affinity form of transferrin for its receptor, the C2 aptamer had greater affinity (see Tables 2 and 3). Both uptake and silencing activities of the targeted NPs were reduced by addition of transferrin to the medium, indicating the important role for TfR-mediated endocytosis. Moreover, the targeted and untargeted siEGFR2 NPs reduced EGFR2 mRNA by about 75% and 55%, respectively, compared to untreated HeLa cells.
Table 3.
Multiple strategies for a single target.
| Target | Ligand | Carrier | siRNA target | Cell targets | Results of targeted ligand | Reference |
|---|---|---|---|---|---|---|
| Transferrin receptor | Transferrin | Liposome | RRM2 | MV4-11, an AML line | Improved tumor accumulation (versus nontargeted NP) and enhanced silencing (versus control siRNA NP) | [54] |
| T7 peptide | Liposome-polymer hybrid | EGFR | MCF-7 | Marked tumor size reduction (versus nontargeted NP) | [134] | |
| Aptamer | SNALP liposome | GFP or LamA/C | HeLa-EGFP cells (in vitro) | Enhanced uptake and silencing (versus transferrin-NP) | [84] | |
| scFv1 | scFv-polylysine conjugate | Survivin | U87 glioma cells | Enhanced silencing and improved survival of mice with orthotopic tumors (versus nontargeted NP) | [91] | |
| scFv | Liposome | (Fluorescently-labeled siRNA)3 | Pancreatic, prostate, and melanoma cell lines | Specific delivery to orthotopic and metastatic tumors in vivo | [145] | |
|
| ||||||
| CD44 | HA | Liposome | PLK1 | U87MG glioblastoma cells | Decrease PLK1 by 80%; prolonged survival in orthotopic model | [63] |
| scFv | Polymer | KRAS | PANC-1, pancreatic cancer | Enhanced in vitro/in vivo silencing and efficacy (versus nontargeted NP) | [94] | |
| scFv | Polymer- (PEI-) iron oxide | Nontargeting siRNA | High (SGC-7901) versus low (A375) CD44 expressing cells | Increased uptake of targeted NP via magnetic resonance in SCC-7901 compared to A375 | [146] | |
| Aptamer | Liposome | — | CD44+ (AS49, MDA-MB-231) versus CD44− (NIH/3T3) (in vitro) | Increased accumulation of targeted-NP in CD44+ cells | [147] | |
|
| ||||||
| p32 | Peptide (LyP-1) | Polymeric micelles | ID4 | OVCAR-4, 8 | Marked tumor reduction in vivo, administered IP | [10] |
| scFv, trimerbody | — | — | MDA-MB-231 | Widespread distribution of labeled antibody in tumor xenografts, particularly with the trimerbody | [148] | |
|
| ||||||
| VEGFR2 | VRBP1 peptide2 | — | — | HUVEC, H460 | VRBP1 reduced size of tumor xenografts | [149] |
| Aptamer | Magnetic particle | — | PAEC, U87MG glioblastoma | Increased MRI signal in tumors in vivo (versus nontargeted NP) | [150] | |
1scFv, single-chain variable fragment antibody; HA, hyaluronic acid; SNALP, stable nucleic acid lipid particles; PEI, polyethylenimine; RRM2, ribonucleoside-diphosphate reductase subunit M2; EGFR, epidermal growth factor receptor; GFP, green fluorescent protein; LamA/C, lamin A/C; PLK1, polo-like kinase 1; ID4, inhibitor of DNA binding 4; AML, acute myeloid cell leukemia; HUVEC, human umbilical vein endothelial cells; PAEC, porcine aortic endothelial cells; CPP, cell penetration peptide; NP, nanoparticle. 2Sequence of VRBP1 peptide - YDGNSFYEMVVGVKPASES; 3nontargeting siRNA.
While there have not been any targeted aptamer-NP studies using siRNA in vivo, there was an interesting report with microRNA [86]. By using a bone metastatic model of prostate cancer, Hao and colleagues demonstrated that a PSMA-functionalized atelocollagen carrier of miRNA (miR-15-A and miR-16-1) prolonged the survival of mice significantly (p < 0.05) (mean survival: 57 days) compared to saline-treated (mean survival: 27.2 days), targeted control miR (mean survival: 28.2 days), or untargeted miR-15-A/16-1 (mean survival: 38 days) NP groups [86]. These in vivo results were consistent with the in vitro studies in which the targeted NP had about a 4.4-fold lower IC50 for the LNCaP prostate cancer cells compared to the untargeted NP. The miR-15-A/miR-16-1 downregulated a number of proteins important for tumor invasion, proliferation, and survival, indicating the utility of these tumor suppressor microRNAs for targeted therapy. We expect that more in vivo studies will follow based on these in vitro and in vivo studies.
3.3. Antibody
In contrast to small molecule toxins, siRNA conjugated to antibodies have shown reduced antitumor efficacy and have not advanced to clinical trials. Whereas the efficacy of most radioisotopes- or chemotherapy-antibody conjugates is not usually reduced from enzymatic degradation by lysosomes, the siRNA component of the conjugates is susceptible to enzymatic degradation and must be delivered intact intracellularly to have a biological effect. Thus, the targeted siRNA therapy must escape from acidic endosomes before reaching the lysosomes. Moreover, antibody-toxins have been determined to be more effective against leukemia and lymphomas compared to solid tumors. Penetration of solid tumors may not be effective with the large molecular weight antibodies, and of course, penetration of the antibodies conjugated to NPs could present an even greater challenge. As a result, the trend in the siRNA field is to use scFv or Fab fragments, because of their small size and their ability to deliver silencing activity with their siRNA similar to the much larger parent antibodies. When conjugated directly to a cationic peptide or to NP, the smaller MW Fab antibody fragments or single-chain variable fragment antibodies have a much higher likelihood of better penetration in solid tumors. Single-chain variable fragment antibodies are about 12–15 kDa in size, smaller than the Fab fragments which are 50 kDa, and significantly smaller than the parent antibodies of 150 to 160 kDa. Thus, scFv are about the same size as RNA and DNA aptamers. This section will focus on antibody-siRNA conjugates or antibody-siRNA-NP therapies that enable targeting of tumor cells in vitro and in vivo (Figure 5).
Figure 5.

Antibody mediated delivery of siRNA. (a) siRNA can interact with the cationic proteins such as protamine that has been directly conjugated with the antibody. (b) Similar to aptamers, antibodies may be conjugated to a carrier of siRNA (liposomes, polyplexes). In addition to their direct conjugation to the membrane surface of NP, the antibody may be attached to PEG (far right). The antibody ligand includes not only the parent antibody or its Fab fragment, but as in this case, it may include the single-chain variable fragment form of the antibody.
3.3.1. Antibody-siRNA Conjugates
Unlike aptamers and siRNA, which are similarly negatively charged, there have been concerns about highly negatively charged siRNA interfering with targeting of the antibody modified with siRNA or with cationic polymers. Cuellar and colleagues examined this potential problem and found that the siRNA did not interfere with the binding affinity of the antibody [88]. In this report, seven monomeric antibody-siRNA conjugates that differed in their chemical linkage or utilized several routes of internalization were examined. Although the conjugates demonstrated both targeting and silencing with cells and tumors in vivo, the silencing was at best modest (about 50% in vitro and 33% in the perivascular area of the tumor in vivo). No in vivo tumor efficacy studies were reported. The authors concluded that the two major problems for this approach were that the antibody conjugates clustered near blood vessels within tumors and that escape from the endosomes of tumors was limited. Notably, single-chain variable fragment antibodies were not tested in this study, which likely would improve tumor penetration and gene silencing with this approach.
Most tumor efficacy studies have utilized a fusion antibody-cationic peptide that is mixed with siRNA. Yao and colleagues complexed siPLK1 through ionic interactions with a single-chain variable fragment Her2 antibody-protamine fusion protein. Each fusion fragment-protamine bound to approximately 6 siRNA molecules (Figure 5(a)). With the fusion siRNA construct administered biweekly, there was marked inhibition of Her2+ tumors (80 to 90%), whereas there was no effect of the therapy on the Her2− tumors [89]. PLK1 mRNA in Her2+ tumors was knocked down by about 75% in the siRNA treatment group. In addition to binding siRNA, the cationic protamine likely has a role in lysis of endosomes, releasing siRNA into the cytosol. With a similar strategy, an EGFR antibody-siKras complex reduced its target in vitro and in vivo, resulting in inhibition of the tumors by 75% compared to controls [90]. There have been concerns that these fusion protein-siRNA therapies, which are dependent on electrostatic interactions, might be too heterogeneous for clinical trials [88]. Although more biophysical characterizations are certainly required to understand and minimize the heterogeneity of antibody-siRNA conjugates, the same criticism could be leveled at polyplexes which have a degree of heterogeneity.
Although the TfR is upregulated in many tumors, investigators have also taken advantage of its presence on the blood-brain barrier. For example, Wang and colleagues used a transferrin antibody-attached to a NP to enable its transport across the blood-brain barrier [91]. In this orthotopic glioblastoma model, treatment with a transferrin scFv NP carrier of shsurvivin prolonged survival of the mice compared to those in the control treated group (p < 0.01) [91]. The antibody targeting transferrin not only augmented uptake by the tumor based on in vitro data but also increased transport of the antibody-shRNA complex through the blood-brain barrier. By using streptavidin-biotin linkage between the antibody and siRNA, Xia et al. also demonstrated the utility of a transferrin antibody to deliver siRNA targeting luciferase to orthotopic brain tumors [92]. Luciferase activity was reduced by 70–80% in the treated brain tumors.
While cationic peptides have been fused at the C-terminal end of single-chain antibody fragments through genetic engineering [89], a more flexible site-specific approach has been developed by Lu et al. [93]. After genetically incorporating an unnatural amino acid (p-acetyl L-phenylalanine) at different locations within 2 full-length and 1 Fab fragment antibodies, a cationic polymer was conjugated to this amino acid via oxime bonds. Two of the siRNA conjugates were effectively internalized by the Her2+ cells in vitro and consequently silenced their target gene by more than 70%. The reduced silencing by one conjugate was attributed to the proximity of the basic polymer and the antigen binding sites, interfering with cellular uptake. Thus, this method can be utilized with different types of antibodies, and antibody conjugates may be peptides or nonpeptide polymers.
3.3.2. Antibody-siRNA-Nanoparticles
Antibodies, including the smaller molecular Fab and single-chain fragment variable derivatives, have been conjugated to liposomes and polymers to deliver chemotherapeutic agents, plasmids, and siRNA to tumors (Figure 5(b)) [94–104]. Conjugation of antibodies to NPs has been plagued by reduced binding affinity, heterogeneity of binding, and inadequate density, and consequently, a number of groups have investigated these issues to advance development of these targeted NPs. For instance, Deng and colleagues investigated whether different linkages of the EGFR2 Fab to PEG would affect silencing activity of the NP [6]. The PEG maleimide-derivative linker enhanced the uptake and silencing activity of the NP compared to the PEG-CO2H derivative. It is likely that the amide bond resulted in a PEG-CONH-Fab conjugate which interfered with the binding affinity of the antibody. Similar to what was previously discussed with the cyclodextrin nanoparticle [62], this illustrates that the method to form a nanoparticle is critical in developing the most effective silencing particle.
Dou and colleagues targeted Her2+ xenografts but with a scFv-Ab NP in which the siRNA targeted Polo-like kinase-1 (PLK1) [95]. The core of the NP was composed of a PEG-poly(D,L-lactide) copolymer, a cationic lipid, and siRNA. The targeted NP augmented uptake significantly in Her2+ xenografts, had greater silencing of PLK1, and reduced tumor size to a greater extent than did untargeted NPs. The tumor efficacy of the targeted NP was particularly marked at lower dosages of therapy, underscoring the need for multiple dosages in these studies. In Her2− tumors, there was little difference in the silencing and antitumor activity of targeted and nontargeting NPs. Combination of chemotherapy agents with siRNA has also been examined with targeted therapy. Zhao et al. incorporated docetaxel and siPLK1 within a Herceptin Ab-labeled micelle [105]. The targeted micelle decreased the IC50 by 94.7% in highly expressing Her2+ cells compared to untargeted micelles.
By taking advantage of the overexpression of TfR on the blood-brain barrier, Partridge downregulated EGFR by 90% and prolonged survival of mice with orthotopic tumors treated with TfR-Ab-pegylated liposomes-shEGFR [106]. An alternative strategy for treatment of brain tumors was based on their overexpression of the GD2 ganglioside. Shen et al. targeted neuroblastomas with a GD2 scFv-Ab theranostic iron oxide NP [101]. The GD2 scFv-Ab-NP containing siBcl-2 reduced its target by about 60% and reduced the size of the neuroblastomas by about 50% compared to the nontargeting NP. Further corroboration was provided by MR imaging of the tumors which showed a 35% decrease in the T2 signal intensity in mice treated with the targeted NP compared to the nontargeted NP.
In pancreatic cells and cancers, there is an increased expression of CD44 transmembrane glycoproteins. As a result, Zeng et al. tested whether an NP consisting of CD44 scFV-Ab-PEG-polylysine copolymer in complex with siKRAS would inhibit pancreatic cells and tumors [94]. Although proliferation of a pancreatic cancer cell line (PANC-1) was somewhat inhibited with targeted therapy (versus nontargeted therapy), the number of colonies formed in soft agar was markedly reduced with cells treated with the targeted therapy. With imaging of the tumor and various tissues in vivo, there was an increased uptake of targeted NPs within implanted pancreatic tumors at several time points compared to that of untargeted NPs (p < 0.05). Uptake differences were further confirmed by confocal microscopy, which also demonstrated intracellular localization of the NP. In contrast, there was no difference between targeted and nontargeted therapy in their uptake in normal tissues. Despite the uptake differences, the differences in the tumor sizes were modest between groups of mice treated with the targeted and nontargeted KRAS NP. Lower dosages of the NP may have provided greater distinction between targeted and nontargeted NPs, as observed by Dou et al. [95].
3.4. Small Molecule Ligands
3.4.1. Folate
The most common small molecule ligand that has been conjugated to NPs to target tumors is clearly folate. Folate has been attached to diverse groups of siRNA carriers including liposomes [107], an array of polymers [108–111], polymer-liposome [112] or polymer-micelle combinations [113], nanogels [114], packaging RNA [115], PEG conjugates [116], iron oxide NPs [117], mesoporous silica particles [118], and DNA tetrahedral structures [119]. Folate can bind with high affinity to its receptor, which is overexpressed on numerous solid tumors such as ovarian carcinomas, glioblastomas, and lung adenocarcinomas. Once the folate receptor binds with the folate-containing NP, the NP is rapidly internalized via receptor-mediated endocytosis, releasing its therapeutic content.
With so many publications on conjugation of folate to NPs, we have selected a few as representatives of the field. Li et al. demonstrated that folate coupled to cyclodextrin-PEI core in complex with siVEGF reduced the target mRNA and tumor size of HeLa xenografts by about 50 and 40%, respectively, compared to the nontargeted carrier [109]. In nasopharyngeal cancer xenografts, folate-linked lipid-based carriers of siHer-2 (three injections of 10 μg of siRNA) reduced tumor growth significantly (approximately 35% decrease; p < 0.01), whereas the nonligand carrier reduced tumor size by less than 10% and was not significant [107]. With a tetrahedral DNA-based carrier, Lee and colleagues demonstrated that at least three folates per carrier were essential for optimal silencing with siRNA in vitro. In addition, maximal silencing was achieved when the folate ligands were in close proximity with one another on the tetrahedral structure. Moreover, this DNA-based carrier of siRNA injected intravenously had increased accumulation within the tumor (compared to other organs) and effectively reduced luciferase expression (about 60%) of human KB nasopharyngeal xenografts in vivo, although a nonfolate carrier was not compared [119]. Recently, Wagner's group used methotrexate-labeled NPs to target folate receptors on a KB cervical tumor model [120]. With the dual-functioning methotrexate not only targeting but also inhibiting tumor growth, the sequence-defined NP carrying an siRNA directed toward the oncogene, eglin 5 (EG5), markedly prolonged the survival of KB-bearing mice. The combination of methotrexate and siEG5 incorporated in the NP (methotrexate/siEG5 NP) resulted in the survival of 50% of the tumor-bearing mice for more than 70 days (with no recurrence of the tumor), whereas untargeted siEGF NPs or the folate/siEG5 resulted in all mice euthanized by day 40 (mice euthanized when tumor reached 1000 mm3 in size).
Some normal tissues such as lung and kidney in humans express high levels of folate receptors (primarily the alpha receptor), but these receptors are localized on the apical surfaces of highly polarized epithelial cells [121, 122]. As a result, the folate receptors on normal cells may not be accessible to blood-borne NPs. Indeed, little accumulation in the lungs of humans was observed with a folate-conjugate injected intravenously, indicating that off-target effects by folate-labeled NPs injected intravenously may be minimal. As a result, despite the high expression levels of receptors in lungs of humans compared to those of mice, their apical location likely minimizes differences in biodistribution of intravenously delivered folate-NPs between mice and men [122].
3.4.2. Anisamide
Sigma receptors, overexpressed on the membranes of several malignant tumors and dividing normal cells, bind tightly to haloperidol and analogues, including anisamide. Interestingly, binding of these exogenous ligands to the sigma receptors and in particular the sigma-2 receptor resulted in death of malignant cells (but not normal cells) by apoptotic and nonapoptotic mechanisms. Nevertheless, targeting the sigma receptor of tumors with siRNA NPs enabled their uptake and enhanced their antitumor activity. For example, Huang' s group has published several papers demonstrating that anisamide-containing lipid-based NPs are significantly better carriers of pooled siRNA (targeting VEGF, MDM2, and c-myc) than are the nontargeted NP in different models of lung cancer [123–125]. More recently, combination therapy utilizing photodynamic therapy and anisamide-containing lipid-based carriers of siHIF-1α substantially inhibited the growth of squamous cell carcinomas in mice compared to either therapy alone [126]. In addition, Guo et al. demonstrated that the anisamide-cyclodextrin carrier of siVEGF was about 35% and 60% more effective than the nontargeted and PBS-treated groups, respectively, in reducing the size of prostate cancer xenografts [127]. Thus, in a diverse group of tumors, targeting the sigma receptor increased delivery of the siRNA-NP and holds future promise.
3.4.3. Galactose
NPs containing galactose have been developed to deliver therapeutic agents (chemotherapeutic agents, DNA) to the asialoglycoprotein receptor on liver cells for over 30 years [128, 129]. Han and colleagues revisited and exploited this targeting ligand by developing galactose-containing mesoporous silica NPs, enabling their entry into hepatocarcinoma cells. These systemically delivered NPs sequentially released siVEGF and then doxorubicin into hepatocarcinoma xenografts implanted into nude mice. Compared to controls, there was a 90% reduction in tumor size [130]. Without galactose attached to the surface of the NP, the efficacy of NPs was reduced by about 20%. Han and colleagues also used the oral route to deliver galactose-containing NPs [131]. Specific amounts of galactose conjugated to trimethyl-chitosan markedly enhanced uptake of the siRNA and shRNA by the tumor xenografts. Whereas galactose was important for the uptake and enhanced specificity of the NP, the trimethyl-chitosan was essential for its transport through the intestinal wall. Interestingly, NPs carrying siVEGF and shsurvivin were synergistic in antitumor activity. In both studies, the liver, which expresses elevated amounts of the asialoglycoprotein receptor, did not show toxicity from these NPs. Although these studies show promise, further studies are required to demonstrate that the liver is not a major target of galactose ligand NPs.
3.5. Peptide Ligands Targeting Tumors
3.5.1. T7 Peptide (HAIYPRH)
The T7 peptide was isolated by a phage display approach using cells that express high levels of the TfR [132]. Unlike most peptide ligands, the T7 peptide was reported to have a high affinity (Kd about 10 nM) for its receptor [133] and since it binds to a different site of the receptor than does transferrin, the endogenous ligand does not inhibit uptake of T7-mediated receptor-endocytosis [132, 133]. Although the vast majority of NPs have targeted TfR with transferrin or an antibody, Gao and colleagues targeted the receptor with the T7 peptide [134]. The siEGFR2 NP labeled with the T7 peptide inhibited tumor size about 40% more than did the untargeted NPs, and 70% more than the glucose-treated group. The tumor-inhibitory results of the various treatment groups correlated with degree of downregulation of the EGRF2 protein. Similar to the transferrin ligand which coated the surface of cyclodextrin NP [5], T7 did not appear to increase accumulation of the NP within the tumor. Notably, there was no evidence of toxicity or cytokine induction with the T7 targeted particles.
3.5.2. MMP2-Cleavable Octapeptide (GPLGIAGQ)
This cleavable peptide linker between PEG layer and the remainder of the NP represents a unique method of tumor targeting (Figure 6) [135]. Unlike the vast majority of NPs that target the tumor cells or their stroma, the uptake of these enzyme-sensitive NPs in tumor cells is greatly enhanced by metalloproteinase-2 (MMP2), an enzyme secreted at high levels of many tumors, but found in low levels in most normal tissues. When the peptide substrate (GPLGIAGQ) incorporated on the surface of the NP is cleaved by MMP2, the PEG shield is released from the NP, enabling the multifunctional micelle particle containing siRNA and paclitaxel to interact with tumor cell surface and be imported into the cell. This targeting strategy was validated with a nonsmall cell lung cancer (A549) in vitro and in vivo. Paclitaxel and the siRNA were codelivered with the MMP2-sensitive NP to more than 98% of the cells in vitro, whereas they were codelivered to about 70% of cells with the MMP2-insensitive NP. Similarly, the MMP2 showed improved codelivery of siRNA and paclitaxel to tumor cells in vivo (14.4%, sensitive versus 6% uptake, insensitive).
Figure 6.

Enhanced tumor uptake of MMP2-degraded NP. Several tumors secrete high levels of the MMP2 enzyme into their stroma. By incorporating the substrate, GPLGIAGQ, between PEG and the NP, the MMP2 cleaved the peptide and releases PEG from the NP. This enables the NP to bind to the negatively charged surface of tumor cells with subsequent endocytosis of the NP.
3.5.3. CP15 (VHLGYAT)
The CP15 peptide, identified with a phage display library, is able to target colon cancer cells yet it is not able to recognize normal intestinal epithelial cells. The C15 peptide (VHLGYAT) was conjugated to the PEG-chitosan (CS) copolymer prior to addition of a cross-linker (sodium tripolyphosphate) and siRNA. Silencing of the targeted PLK1 mRNA and protein in human tumor xenografts with the C15-PEG-CS NPs was approximately 50% of that in the control siRNA group [136]. Although silencing differences between the targeted C15-PEG-CS NP and untargeted PEG-CS NP groups were not compared, accumulation in the tumor was modestly greater (about 20%) with the targeted NP compared to the untargeted NP.
3.5.4. FSH (FSH-β, 33–53 Amino Acids, YTRDLVYKDPARPKIQKTCTF); LHRH (QHTSYkcLRP)
Both FSH and LH ligands bind receptors present on the normal ovary as well as on ovarian cancer cells, and peptide fragments of these hormone ligands have been used to target NPs. Hong and colleagues examined the efficacy of NPs composed of FSHβ (33–55)-PEG-PEI and the cytokine siGro-α against ovarian tumor cells [137]. Compared to the untargeted NP in complex to siGro-α, the numbers of cells and migration distance of cells were reduced by about 35% and 75%, respectively. Although these studies with siRNA were in vitro, the same laboratory had previously demonstrated that tumor volume and weight were significantly reduced with the FSH-targeting ligand NPs containing paclitaxel compared to the nontargeting NPs [138].
Similarly, Shah et al. targeted ovarian cancer with a LHRH peptide-polypropylenimine-NP containing paclitaxel and a siRNA that downregulates CD44 [139]. The LHRH sequence, QHTSYkcLRP, was conjugated to PEG through interaction of its thiol group with the maleimide on the PEG. In an in vivo experiment, the LHRH-peptide-NP-Pax/siCD44 prevented growth and indeed caused regression of the ovarian tumor xenografts that were about 400 mm3 prior to initiation of therapy by about 50% (after the 8th treatment). The targeted NP containing paclitaxel or siCD44 reduced tumor growth markedly more than did the untargeted NP groups. Moreover, the CD44 protein in the xenografts was clearly downregulated by the siRNA when the LHRH-NP-paclitaxel and the LHRH-NP-paclitaxel/siCD44 groups were compared.
3.5.5. Gastrin-Releasing Peptides (GRPs) (CGGNHWAVGHLM)
The gastrin-releasing peptide, isolated from the peptide phage library, binds to the BB2 receptor on several malignancies including colon, breast, lung, and prostate cancer cell lines. The authors found that the GRP peptide did bind to the MDA-MB-231 cells but did not bind to endothelial cells [140]. Moreover, the cells treated with GRP-siSurvivin conjugates had survivin mRNA levels that were about 15% of the GRP-scrambled siRNA or siRNA controls.
3.5.6. RVG-Brain Delivery (YTIWMPENPRPGTPCDIFTNSRGKRASNG)
Similar to the transferrin-mediated NPs, the rabies virus glycoprotein- (RVG-) derived peptides may augment transport of NPs across the blood-brain barrier to the central nervous system [141]. To date, there has been only one report (an in vitro study) examining RVG-directed NPs with siRNA targeting cancer [142]. That the acetylcholine receptor for the RVG peptide is found on the surface of glioblastoma cells is indicated by the enhanced uptake of RVD-NPs in U87 cells compared to untargeted NPs. In contrast, there was no difference in the uptake of RVD-NPs and untargeted NPs in the cervical HeLa cell line. Downregulation of GADPH mRNA was about 25% more with the targeted siGADPH NP than with the untargeted NP.
4. Conclusion
Diverse numbers of ligands and approaches to augment siRNA delivery to tumor cells and to tumors in mouse models have been described in this review. More complex ligand targeting NPs, no matter how promising in preclinical trials, must be justified economically and overcome barriers with production scale-up for commercial manufacturing. It is thus not surprising that the less complex ligands (i.e., antibodies and aptamers) are investigated initially in clinical trials. For example, aptamers are being tested in clinical trials and CenR peptides with their recent demonstration of preclinical efficacy may not be far behind. In addition, antibodies directed against Her-2/neu or EGFR (Her-1) tumor-associated receptors or antigens have already demonstrated marked clinical efficacy. Nonetheless, the progressive shift to the use of mAb as targeting ligands for toxins or chemotherapeutics certainly supports development of ligand-targeted forms of siRNA.
Of course, the economically more feasible targeted siRNA therapies (antibody-siRNA or aptamer-siRNA conjugates) could be less effective in some cases than more complex NPs. Multicomponent NPs may have significant advantages due to greater tumor specificity (i.e., multivalency), reduced need to use chemically modified siRNA, and enhanced intracellular siRNA delivery avoiding lysosomal degradation. Nevertheless, because of the challenges with their manufacturing, the more complex multicomponent NPs may not be tested in patients despite their preclinical efficacy. This remains a major challenge in the field for the most promising multicomponent NPs to be tested for clinical efficacy.
The type of ligand to conjugate on the NP may also be challenging. This is illustrated by the number of approaches to identify a ligand targeting the TfR (Table 3). Investigators have used the natural ligand, monoclonal antibodies, single-chain fragment antibodies, aptamers, and even peptides to target the TfR. Which of these strategies is preferred will likely be based on their availability, ease of synthesis and purification, their target affinity, and selectivity as well as the size of the NP. Of these approaches for TfR, the aptamer and the peptide ligand are particularly attractive because of their high affinity for their target, their low molecular weight, and relative ease of synthesis. The greater the size of the NP, the more concern there is about the molecular weight of the ligand. Thus, given a choice between conjugating an antibody or scFv antibody or aptamer to a liposome, it would seem that lower molecular weight ligands would enable greater penetration of the NP within the tumor. Moreover, the peptide ligand differs from the other ligands for TfR in that its binding site is different from the site for transferrin; as a result, the uptake of the T7 NP will not be affected by the relatively high serum levels of endogenous transferrin (25 μM).
An additional layer of complexity is the interaction between the ligand-siRNA therapeutic and the blood components. Although considerable attention has been given to measuring the biophysical parameters of the NP, less has been given to what may occur to the NP in vivo. Silencing experiments done in the presence of medium plus serum reduced the targeting ability of the transferrin-linked NP, by enabling a corona of proteins to surround the NP [57]. This finding will likely extend to other ligands in addition to transferrin, and the presence of the corona may depend on the core particle (i.e., cationic versus neutral liposomes) and the type of ligand (aptamer, endogenous ligand, antibody, small molecule, and others).
In addition to the many promising ligands that have been investigated for targeting siRNA to tumors, many other ligands that have considerable potential have not been tested with siRNA. One example is the urokinase plasminogen activator (uPA) ligand for its receptor, which is upregulated in the majority of pancreatic (about 90%) and breast cancers (60–90%). With a recombinant N-terminal peptide of the uPA, high levels of the NP injected via the tail vein were found in tumors with low levels in normal tissue [143]. Indeed, the number of known ligands that need to be tested and compared in vitro and/or in vivo for enhanced siRNA efficacy against tumors is certainly challenging. Nevertheless, we think it is likely that the many permutations of the ligand, siRNA, and NPs will yield promising drug candidates for improved treatment of a wide range of life-threatening cancers.
Acknowledgments
The authors thank P. Talalay for careful reading and invaluable editing assistance of this manuscript. This work was supported by the National Institutes of Health (R01-CA136938).
Conflicts of Interest
None of the authors of this paper have any financial interest that has influenced the results or interpretation of this paper.
References
- 1.Kobayashi H., Watanabe R., Choyke P. L. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2014;4(1):81–89. doi: 10.7150/thno.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maeda H. Nitroglycerin enhances vascular blood flow and drug delivery in hypoxic tumor tissues: Analogy between angina pectoris and solid tumors and enhancement of the EPR effect. Journal of Controlled Release. 2010;142(3):296–298. doi: 10.1016/j.jconrel.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: Background and future prospects. Bioconjugate Chemistry. 2010;21(5):797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 4.Mehvar R., Robinson M. A., Reynolds J. M. Dose dependency of the kinetics of dextrans in rats: Effects of molecular weight. Journal of Pharmaceutical Sciences. 1995;84(7):815–818. doi: 10.1002/jps.2600840706. [DOI] [PubMed] [Google Scholar]
- 5.Bartlett D. W., Su H., Hildebrandt I. J., Weber W. A., Davis M. E. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proceedings of the National Acadamy of Sciences of the United States of America. 2007;104(39):15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng L., Zhang Y., Ma L., et al. Comparison of anti-EGFR-Fab′ conjugated immunoliposomes modified with two different conjugation linkers for siRNA delivery in SMMC-7721 cells. International Journal of Nanomedicine. 2013;8:3271–3283. doi: 10.2147/IJN.S47597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laakkonen P., Porkka K., Hoffman J. A., Ruoslahti E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nature Medicine. 2002;8(7):751–755. doi: 10.1038/nm720. [DOI] [PubMed] [Google Scholar]
- 8.Pasqualini R., Koivunen E., Kain R., et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Research. 2000;60:722–727. [PMC free article] [PubMed] [Google Scholar]
- 9.Pasqualini R., Koivunen E., Ruoslahti E. |[alpha]| v Integrins as receptors for tumor targeting by circulating ligands. Nature Biotechnology. 1997;15(6):542–546. doi: 10.1038/nbt0697-542. [DOI] [PubMed] [Google Scholar]
- 10.Ren Y., Cheung H. W., Von Maltzhan G., et al. Targeted tumor-penetrating siRNA nanocomplexes for credentialing the ovarian cancer oncogene ID4. Science Translational Medicine. 2012;4(147) doi: 10.1126/scitranslmed.3003778.147ra112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oku N., Asai T., Watanabe K., et al. Anti-neovascular therapy using novel peptides homing to angiogenic vessels. Oncogene. 2002;21(17):2662–2669. doi: 10.1038/sj.onc.1205347. [DOI] [PubMed] [Google Scholar]
- 12.Roth L., Agemy L., Kotamraju V. R., et al. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene. 2012;31(33):3754–3763. doi: 10.1038/onc.2011.537. [DOI] [PubMed] [Google Scholar]
- 13.Ruoslahti E., Bhatia S. N., Sailor M. J. Targeting of drugs and nanoparticles to tumors. The Journal of Cell Biology. 2010;188(6):759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugahara K. N., Teesalu T., Prakash Karmali P., et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328(5981):1031–1035. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teesalu T., Sugahara K. N., Kotamraju V. R., Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proceedings of the National Acadamy of Sciences of the United States of America. 2009;106(38):16157–16162. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teesalu T., Sugahara K. N., Ruoslahti E. Tumor-penetrating peptides. Frontiers in Oncology. 2013;3 doi: 10.3389/fonc.2013.00216.00216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Babae N., Bourajjaj M., Liu Y., et al. Systemic miRNA-7 delivery inhibits tumor angiogenesis and growth in murine xenograft glioblastoma. Oncotarget . 2014;5(16):6687–6700. doi: 10.18632/oncotarget.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hood J. D., Frausto R., Kiosses W. B., Schwartz M. A., Cheresh D. A. Differential αv integrin-mediated Ras-ERK signaling during two pathways of angiogenesis. The Journal of Cell Biology. 2003;162(5):933–943. doi: 10.1083/jcb.200304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurrath M., Muller G., Kessler H., Aumailley M., Timpl R. Conformation/activity studies of rationally designed potent anti-adhesive RGD peptides. European Journal of Biochemistry. 1992;210:911–921. doi: 10.1111/j.1432-1033.1992.tb17495.x. [DOI] [PubMed] [Google Scholar]
- 20.Allman R., Cowburn P., Mason M. In vitro and in vivo effects of a cyclic peptide with affinity for the αvβ3 integrin in human melanoma cells. European Journal of Cancer. 2000;36(3):410–422. doi: 10.1016/S0959-8049(99)00279-8. [DOI] [PubMed] [Google Scholar]
- 21.Hood J. D., Bednarski M., Frausto R., et al. Tumor regression by targeted gene delivery to the neovasculature. Science. 2002;296(5577):2404–2407. doi: 10.1126/science.1070200. [DOI] [PubMed] [Google Scholar]
- 22.Schiffelers R. M., Ansari A., Xu J., et al. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Research. 2004;32:p. e149. doi: 10.1093/nar/gnh140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han H. D., Mangala L. S., Lee J. W., et al. Targeted gene silencing using RGD-labeled chitosan nanoparticles. Clinical Cancer Research. 2010;16(15):3910–3922. doi: 10.1158/1078-0432.CCR-10-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou S.-T., Leng Q., Scaria P., Woodle M., Mixson A. J. Selective modification of HK peptides enhances siRNA silencing of tumor targets in vivo. Cancer Gene Therapy. 2011;18(10):707–716. doi: 10.1038/cgt.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chou S.-T., Hom K., Zhang D., et al. Enhanced silencing and stabilization of siRNA polyplexes by histidine-mediated hydrogen bonds. Biomaterials. 2014;35(2):846–855. doi: 10.1016/j.biomaterials.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li G., Hu Z., Yin H., et al. A novel dendritic nanocarrier of polyamidoamine-polyethylene glycol-cyclic RGD for "smart" small interfering RNA delivery and in vitro antitumor effects by human ether-à-go-go-related gene silencing in anaplastic thyroid carcinoma cells. International Journal of Nanomedicine. 2013;8:1293–1306. doi: 10.2147/IJN.S41555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christie R. J., Matsumoto Y., Miyata K., et al. Targeted polymeric micelles for siRNA treatment of experimental cancer by intravenous injection. ACS Nano. 2012;6(6):5174–5189. doi: 10.1021/nn300942b. [DOI] [PubMed] [Google Scholar]
- 28.Li Y.-H., Shi Q.-S., Du J., et al. Targeted delivery of biodegradable nanoparticles with ultrasound-targeted microbubble destruction-mediated hVEGF-siRNA transfection in human PC-3 cells in vitro. International Journal of Molecular Medicine. 2013;31(1):163–171. doi: 10.3892/ijmm.2012.1175. [DOI] [PubMed] [Google Scholar]
- 29.Chou S.-T., Leng Q., Scaria P., et al. Surface-modified HK:siRNA nanoplexes with enhanced pharmacokinetics and tumor growth inhibition. Biomacromolecules. 2013;14(3):752–760. doi: 10.1021/bm3018356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koide H., Asai T., Furuya K., et al. Inhibition of Akt (ser473) phosphorylation and rapamycin-resistant cell growth by knockdown of mammalian target of rapamycin with small interfering RNA in vascular endothelial growth factor receptor-1-targeting vector. Biological & Pharmaceutical Bulletin. 2011;34(5):602–608. doi: 10.1248/bpb.34.602. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y., Liu P., Duan Y., et al. Specific cell targeting with APRPG conjugated PEG-PLGA nanoparticles for treating ovarian cancer. Biomaterials. 2014;35(3):983–992. doi: 10.1016/j.biomaterials.2013.09.062. [DOI] [PubMed] [Google Scholar]
- 32.Maeda N., Takeuchi Y., Takada M., Sadzuka Y., Namba Y., Oku N. Anti-neovascular therapy by use of tumor neovasculature-targeted long-circulating liposome. Journal of Controlled Release. 2004;100(1):41–52. doi: 10.1016/j.jconrel.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 33.Katanasaka Y., Ida T., Asai T., Maeda N., Oku N. Effective delivery of an angiogenesis inhibitor by neovessel-targeted liposomes. International Journal of Pharmaceutics. 2008;360(1-2):219–224. doi: 10.1016/j.ijpharm.2008.04.046. [DOI] [PubMed] [Google Scholar]
- 34.Lu Z. X., Liu L. T., Qi X. R. Development of small interfering RNA delivery system using PEI-PEG-APRPG polymer for antiangiogenic vascular endothelial growth factor tumor-targeted therapy. International Journal of Nanomedicine. 2011;6:1661–1673. doi: 10.2147/IJN.S22293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koide H., Asai T., Kato H., et al. Susceptibility of PTEN-positive metastatic tumors to small interfering RNA targeting the mammalian target of rapamycin. Nanomedicine: Nanotechnology, Biology and Medicine. 2015;11(1):185–194. doi: 10.1016/j.nano.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Ando H., Asai T., Koide H., et al. Advanced cancer therapy by integrative antitumor actions via systemic administration of miR-499. Journal of Controlled Release. 2014;181(1):32–39. doi: 10.1016/j.jconrel.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 37.Negussie A. H., Miller J. L., Reddy G., Drake S. K., Wood B. J., Dreher M. R. Synthesis and in vitro evaluation of cyclic NGR peptide targeted thermally sensitive liposome. Journal of Controlled Release. 2010;143(2):265–273. doi: 10.1016/j.jconrel.2009.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y., Wu J. J., Huang L. Nanoparticles targeted with NGR motif deliver c-myc siRNA and doxorubicin for anticancer therapy. Molecular Therapy. 2010;18(4):828–834. doi: 10.1038/mt.2009.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porkka K., Laakkonen P., Hoffman J. A., Bernasconi M., Ruoslahti E. A fragment of the HMGN2 protein homes to the nuclei of tumor cells and tumor endothelial cells in vivo. Proceedings of the National Acadamy of Sciences of the United States of America. 2002;99(11):7444–7449. doi: 10.1073/pnas.062189599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christian S., Pilch J., Akerman M. E., Porkka K., Laakkonen P., Ruoslahti E. Nucleolin expressed at the cell surface is a marker of endothelial cells in angiogenic blood vessels. The Journal of Cell Biology. 2003;163(4):871–878. doi: 10.1083/jcb.200304132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomes-Da-Silva L. C., Santos A. O., Bimbo L. M., et al. Toward a siRNA-containing nanoparticle targeted to breast cancer cells and the tumor microenvironment. International Journal of Pharmaceutics. 2012;434(1-2):9–19. doi: 10.1016/j.ijpharm.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 42.Gomes-Da-Silva L. C., Ramalho J. S., Pedroso De Lima M. C., Simões S., Moreira J. N. Impact of anti-PLK1 siRNA-containing F3-targeted liposomes on the viability of both cancer and endothelial cells. European Journal of Pharmaceutics and Biopharmaceutics. 2013;85(3):356–364. doi: 10.1016/j.ejpb.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 43.Elsabahy M., Shrestha R., Clark C., Taylor S., Leonard J., Wooley K. L. Multifunctional hierarchically assembled nanostructures as complex stage-wise dual-delivery systems for coincidental yet differential trafficking of siRNA and paclitaxel. Nano Letters. 2013;13(5):2172–2181. doi: 10.1021/nl4006645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Numata K., Reagan M. R., Goldstein R. H., Rosenblatt M., Kaplan D. L. Spider silk-based gene carriers for tumor cell-specific delivery. Bioconjugate Chemistry. 2011;22(8):1605–1610. doi: 10.1021/bc200170u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agemy L., Kotamraju V. R., Friedmann-Morvinski D., Sharma S., Sugahara K. N., Ruoslahti E. Proapoptotic peptide-mediated cancer therapy targeted to cell surface p32. Molecular Therapy. 2013;21(12):2195–2204. doi: 10.1038/mt.2013.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sugahara K. N., Teesalu T., Karmali P. P., et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16(6):510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alberici L., Roth L., Sugahara K. N., et al. De Novo design of a tumor-penetrating peptide. Cancer Research. 2013;73(2):804–812. doi: 10.1158/0008-5472.CAN-12-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pang H.-B., Braun G. B., Friman T., et al. An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nature Communications. 2014;5, article no. 4904 doi: 10.1038/ncomms5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu Q., Gao X., Kang T., et al. CGKRK-modified nanoparticles for dual-targeting drug delivery to tumor cells and angiogenic blood vessels. Biomaterials. 2013;34(37):9496–9508. doi: 10.1016/j.biomaterials.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 50.Miao D., Jiang M., Liu Z., et al. Co-administration of dual-targeting nanoparticles with penetration enhancement peptide for antiglioblastoma therapy. Molecular Pharmaceutics. 2014;11(1):90–101. doi: 10.1021/mp400189j. [DOI] [PubMed] [Google Scholar]
- 51.Shen J., Meng Q., Sui H., et al. IRGD conjugated TPGS mediates codelivery of paclitaxel and survivin shRNA for the reversal of lung cancer resistance. Molecular Pharmaceutics. 2014;11(8):2579–2591. doi: 10.1021/mp400576f. [DOI] [PubMed] [Google Scholar]
- 52.Walker R. A., Day S. J. Transferrin receptor expression in non-malignant and malignant human breast tissue. Journal of Pathology. 1986;148:217–224. doi: 10.1002/path.1711480305. [DOI] [PubMed] [Google Scholar]
- 53.Gkouvatsos K., Papanikolaou G., Pantopoulos K. Regulation of iron transport and the role of transferrin. Biochimica et Biophysica Acta (BBA) - General Subjects. 2012;1820(3):188–202. doi: 10.1016/j.bbagen.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 54.Yang Z., Yu B., Zhu J., et al. A microfluidic method to synthesize transferrin-lipid nanoparticles loaded with siRNA LOR-1284 for therapy of acute myeloid leukemia. Nanoscale. 2014;6(16):9742–9751. doi: 10.1039/c4nr01510j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pun S. H., Tack F., Bellocq N. C., et al. Targeted delivery of RNA-cleaving DNA enzyme (DNAzyme) to tumor tissue by transferrin-modified, cyclodextrin-based particles. Cancer Biology and Therapy. 2004;3:641–650. doi: 10.4161/cbt.3.7.918. [DOI] [PubMed] [Google Scholar]
- 56.Liu Y., Tao J., Li Y., et al. Targeting hypoxia-inducible factor-1α with Tf-PEI-shRNA complex via transferrin receptor-mediated endocytosis inhibits melanoma growth. Molecular Therapy. 2009;17(2):269–277. doi: 10.1038/mt.2008.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salvati A., Pitek A. S., Monopoli M. P., et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nature Nanotechnology. 2013;8(2):137–143. doi: 10.1038/nnano.2012.237. [DOI] [PubMed] [Google Scholar]
- 58.Hong M., Zhu S., Jiang Y., Tang G., Pei Y. Efficient tumor targeting of hydroxycamptothecin loaded PEGylated niosomes modified with transferrin. Journal of Controlled Release. 2009;133(2):96–102. doi: 10.1016/j.jconrel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 59.Bartlett D. W., Davis M. E. Impact of tumor-specific targeting and dosing schedule on tumor growth inhibition after intravenous administration of siRNA-containing nanoparticles. Biotechnology and Bioengineering. 2008;99(4):975–985. doi: 10.1002/bit.21668. [DOI] [PubMed] [Google Scholar]
- 60.Bartlett D. W., Davis M. E. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Research. 2006;34(1):322–333. doi: 10.1093/nar/gkj439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zuckerman J. E., Davis M. E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nature Reviews Drug Discovery. 2015;14(12):843–856. doi: 10.1038/nrd4685. [DOI] [PubMed] [Google Scholar]
- 62.Suzie H. P., Davis M. E. Development of a nonviral gene delivery vehicle for systemic application. Bioconjugate Chemistry. 2002;13(3):630–639. doi: 10.1021/bc0155768. [DOI] [PubMed] [Google Scholar]
- 63.Cohen Z. R., Ramishetti S., Peshes-Yaloz N., et al. Localized RNAi therapeutics of chemoresistant grade IV glioma using hyaluronan-grafted lipid-based nanoparticles. ACS Nano. 2015;9(2):1581–1591. doi: 10.1021/nn506248s. [DOI] [PubMed] [Google Scholar]
- 64.Almalik A., Day P. J., Tirelli N. HA-coated chitosan nanoparticles for CD44-mediated nucleic acid delivery. Macromolecular Bioscience. 2013;13(12):1671–1680. doi: 10.1002/mabi.201300302. [DOI] [PubMed] [Google Scholar]
- 65.Bin Z., Yueying Z., Yu D. Lung cancer gene therapy: Transferrin and hyaluronic acid dual ligand-decorated novel lipid carriers for targeted gene delivery. Oncology Reports. 2017;37(2):937–944. doi: 10.3892/or.2016.5298. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y., Sun J., Lian H., Cao W., Wang Y., He Z. Folate and CD44 receptors dual-targeting hydrophobized hyaluronic acid paclitaxel-loaded polymeric micelles for overcoming multidrug resistance and improving tumor distribution. Journal of Pharmaceutical Sciences. 2014;103(5):1538–1547. doi: 10.1002/jps.23934. [DOI] [PubMed] [Google Scholar]
- 67.Kim E.-J., Shim G., Kim K., Kwon I. C., Oh Y.-K., Shim C.-K. Hyaluronic acid complexed to biodegradable poly L-arginine for targeted delivery of siRNAs. The Journal of Gene Medicine. 2009;11(9):791–803. doi: 10.1002/jgm.1352. [DOI] [PubMed] [Google Scholar]
- 68.Yang X., Iyer A. K., Singh A., et al. Cluster of differentiation 44 targeted hyaluronic acid based nanoparticles for MDR1 siRNA delivery to overcome drug resistance in ovarian cancer. Pharmaceutical Research. 2014 doi: 10.1007/s11095-014-1602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi K.-M., Jang M., Kim J. H., Ahn H. J. Tumor-specific delivery of siRNA using supramolecular assembly of hyaluronic acid nanoparticles and 2b RNA-binding protein/siRNA complexes. Biomaterials. 2014;35(25):7121–7132. doi: 10.1016/j.biomaterials.2014.04.096. [DOI] [PubMed] [Google Scholar]
- 70.Kim S. I., Shin D., Choi T. H., et al. Systemic and specific delivery of small interfering RNAs to the liver mediated by apolipoprotein A-I. Molecular Therapy. 2007;15(6):1145–1152. doi: 10.1038/sj.mt.6300168. [DOI] [PubMed] [Google Scholar]
- 71.Ding Y., Wang W., Feng M., et al. A biomimetic nanovector-mediated targeted cholesterol-conjugated siRNA delivery for tumor gene therapy. Biomaterials. 2012;33(34):8893–8905. doi: 10.1016/j.biomaterials.2012.08.057. [DOI] [PubMed] [Google Scholar]
- 72.Ding Y., Wang Y., Zhou J., et al. Direct cytosolic siRNA delivery by reconstituted high density lipoprotein for target-specific therapy of tumor angiogenesis. Biomaterials. 2014;35(25):7214–7227. doi: 10.1016/j.biomaterials.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 73.Li X., Zhao Q., Qiu L. Smart ligand: Aptamer-mediated targeted delivery of chemotherapeutic drugs and siRNA for cancer therapy. Journal of Controlled Release. 2013;171(2):152–162. doi: 10.1016/j.jconrel.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 74.Xiang D., Shigdar S., Qiao G., et al. Aptamer-mediated cancer gene therapy. Current Gene Therapy. 2015;15(2):109–119. doi: 10.2174/1566523214666141224095105. [DOI] [PubMed] [Google Scholar]
- 75.Rosenberg J. E., Bambury R. M., Van Allen E. M., et al. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Investigational New Drugs. 2014;32(1):178–187. doi: 10.1007/s10637-013-0045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chu T. C., Twu K. Y., Ellington A. D., Levy M. Aptamer mediated siRNA delivery. Nucleic Acids Research. 2006;34(10, article e73) doi: 10.1093/nar/gkl388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McNamara J. O., II, Andrechek E. R., Wang Y., et al. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nature Biotechnology. 2006;24(8):1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 78.Dassie J. P., Liu X.-Y., Thomas G. S., et al. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nature Biotechnology. 2009;27(9):839–846. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Subramanian N., Kanwar J. R., Kanwar R. K., et al. EpCAM aptamer-siRNA chimera targets and regress epithelial cancer. PLoS ONE. 2015;10(7) doi: 10.1371/journal.pone.0132407.e0132407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu H. Y., Gao X. A universal protein tag for delivery of SiRNA-aptamer chimeras. Scientific Reports. 2013;3, article 3129 doi: 10.1038/srep03129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou J., Rossi J. J. The therapeutic potential of cell-internalizing aptamers. Current Topics in Medicinal Chemistry. 2009;9(12):1144–1157. doi: 10.2174/156802609789630893. [DOI] [PubMed] [Google Scholar]
- 82.Zhao N., Bagaria H. G., Wong M. S., Zu Y. A nanocomplex that is both tumor cell-selective and cancer gene-specific for anaplastic large cell lymphoma. Journal of Nanobiotechnology. 2011;9, article no. 2 doi: 10.1186/1477-3155-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bagalkot V., Gao X. SiRNA-aptamer chimeras on nanoparticles: preserving targeting functionality for effective gene silencing. ACS Nano. 2011;5(10):8131–8139. doi: 10.1021/nn202772p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wilner S. E., Wengerter B., Maier K., et al. An RNA alternative to human transferrin: A new tool for targeting human cells. Molecular Therapy - Nucleic Acids. 2012;1, article no. e21 doi: 10.1038/mtna.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Subramanian N., Kanwar J. R., Athalya P. K., et al. EpCAM aptamer mediated cancer cell specific delivery of EpCAM siRNA using polymeric nanocomplex. Journal of Biomedical Science. 2015;22(1, article no. 4) doi: 10.1186/s12929-014-0108-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hao Z., Fan W., Hao J., et al. Efficient delivery of micro RNA to bone-metastatic prostate tumors by using aptamer-conjugated atelocollagen in vitro and in vivo. Drug Delivery. 2016;23(3):874–881. doi: 10.3109/10717544.2014.920059. [DOI] [PubMed] [Google Scholar]
- 87.Zhou J., Soontornworajit B., Martin J., Sullenger B. A., Gilboa E., Wang Y. A hybrid DNA aptamer-dendrimer nanomaterial for targeted cell labeling. Macromolecular Bioscience. 2009;9(9):831–835. doi: 10.1002/mabi.200900046. [DOI] [PubMed] [Google Scholar]
- 88.Cuellar T. L., Barnes D., Nelson C., et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Research. 2015;43(2):1189–1203. doi: 10.1093/nar/gku1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yao Y.-D., Sun T.-M., Huang S.-Y., et al. Targeted delivery of PLK1-siRNA by ScFv suppresses Her2 + breast cancer growth and metastasis. Science Translational Medicine. 2012;4(130) doi: 10.1126/scitranslmed.3003601.130ra48 [DOI] [PubMed] [Google Scholar]
- 90.Bäumer S., Bäumer N., Appel N., et al. Antibody-mediated delivery of anti-KRAS-siRNA in vivo overcomes therapy resistance in colon cancer. Clinical Cancer Research. 2015;21(6):1383–1394. doi: 10.1158/1078-0432.CCR-13-2017. [DOI] [PubMed] [Google Scholar]
- 91.Wang F., Bai H.-R., Wang J., Bai Y.-Z., Dou C.-W. Glioma growth inhibition in vitro and in vivo by single chain variable fragments of the transferrin receptor conjugated to survivin small interfering RNA. Journal of International Medical Research. 2011;39(5):1701–1712. doi: 10.1177/147323001103900512. [DOI] [PubMed] [Google Scholar]
- 92.Xia C.-F., Zhang Y., Zhang Y., Boado R. J., Pardridge W. M. Intravenous siRNA of brain cancer with receptor targeting and avidin-biotin technology. Pharmaceutical Research. 2007;24(12):2309–2316. doi: 10.1007/s11095-007-9460-8. [DOI] [PubMed] [Google Scholar]
- 93.Lu H., Wang D., Kazane S., et al. Site-specific antibody-polymer conjugates for siRNA delivery. Journal of the American Chemical Society. 2013;135(37):13885–13891. doi: 10.1021/ja4059525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zeng L., Li J., Li J., et al. Effective suppression of the kirsten rat sarcoma viral oncogene in pancreatic tumor cells via targeted small interfering rna delivery using nanoparticles. Pancreas. 2015;44(2):250–259. doi: 10.1097/MPA.0000000000000241. [DOI] [PubMed] [Google Scholar]
- 95.Dou S., Yang X.-Z., Xiong M.-H., et al. ScFv-Decorated PEG-PLA-Based Nanoparticles for Enhanced siRNA Delivery to Her2+ Breast Cancer. Advanced Healthcare Materials. 2014;3(11):1792–1803. doi: 10.1002/adhm.201400037. [DOI] [PubMed] [Google Scholar]
- 96.Chan D. P. Y., Deleavey G. F., Owen S. C., Damha M. J., Shoichet M. S. Click conjugated polymeric immuno-nanoparticles for targeted siRNA and antisense oligonucleotide delivery. Biomaterials. 2013;34(33):8408–8415. doi: 10.1016/j.biomaterials.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y., Liu P., Du J., Sun Y., Li F., Duan Y. Targeted siRNA delivery by anti-HER2 antibody-modified nanoparticles of mPEG-chitosan diblock copolymer. Journal of Biomaterials Science, Polymer Edition. 2013;24(10):1219–1232. doi: 10.1080/09205063.2012.745716. [DOI] [PubMed] [Google Scholar]
- 98.Okamoto A., Asai T., Kato H., et al. Antibody-modified lipid nanoparticles for selective delivery of siRNA to tumors expressing membrane-anchored form of HB-EGF. Biochemical and Biophysical Research Communications. 2014;449(4):460–465. doi: 10.1016/j.bbrc.2014.05.043. [DOI] [PubMed] [Google Scholar]
- 99.Höbel S., Vornicescu D., Bauer M., Fischer D., Keusgen M., Aigner A. A novel method for the assessment of targeted PEI-based nanoparticle binding based on a static surface plasmon resonance system. Analytical Chemistry. 2014;86(14):6827–6835. doi: 10.1021/ac402001q. [DOI] [PubMed] [Google Scholar]
- 100.Mitra M., Kandalam M., Rangasamy J., et al. Novel epithelial cell adhesion molecule antibody conjugated polyethyleneimine-capped gold nanoparticles for enhanced and targeted small interfering RNA delivery to retinoblastoma cells. Molecular Vision. 2013;19:1029–1038. [PMC free article] [PubMed] [Google Scholar]
- 101.Shen M., Gong F., Pang P., et al. An MRI-visible non-viral vector for targeted Bcl-2 siRNA delivery to neuroblastoma. International Journal of Nanomedicine. 2012;7:3319–3332. doi: 10.2147/IJN.S32900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bourseau-Guilmain E., Béjaud J., Griveau A., et al. Development and characterization of immuno-nanocarriers targeting the cancer stem cell marker AC133. International Journal of Pharmaceutics. 2012;423(1):93–101. doi: 10.1016/j.ijpharm.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 103.Felber A. E., Castagner B., Elsabahy M., Deleavey G. F., Damha M. J., Leroux J.-C. SiRNA nanocarriers based on methacrylic acid copolymers. Journal of Controlled Release. 2011;152(1):159–167. doi: 10.1016/j.jconrel.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 104.Chen Y., Zhu X., Zhang X., Liu B., Huang L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Molecular Therapy. 2010;18(9):1650–1656. doi: 10.1038/mt.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao J., Mi Y., Feng S.-S. Targeted co-delivery of docetaxel and siPlk1 by herceptin-conjugated vitamin E TPGS based immunomicelles. Biomaterials. 2013;34(13):3411–3421. doi: 10.1016/j.biomaterials.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 106.Pardridge W. M. shRNA and siRNA delivery to the brain. Advanced Drug Delivery Reviews. 2007;59(2-3):141–152. doi: 10.1016/j.addr.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yoshizawa T., Hattori Y., Hakoshima M., Koga K., Maitani Y. Folate-linked lipid-based nanoparticles for synthetic siRNA delivery in KB tumor xenografts. European Journal of Pharmaceutics and Biopharmaceutics. 2008;70(3):718–725. doi: 10.1016/j.ejpb.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 108.Kim J. S., Oh M. H., Park J. Y., Park T. G., Nam Y. S. Protein-resistant, reductively dissociable polyplexes for in vivo systemic delivery and tumor-targeting of siRNA. Biomaterials. 2013;34(9):2370–2379. doi: 10.1016/j.biomaterials.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 109.Li J.-M., Wang Y.-Y., Zhang W., Su H., Ji L.-N., Mao Z.-W. Low-weight polyethylenimine cross-linked 2-hydroxypopyl-β-cyclodextrin and folic acid as an efficient and nontoxic siRNA carrier for gene silencing and tumor inhibition by VEGF siRNA. International Journal of Nanomedicine. 2013;8:2101–2117. doi: 10.2147/IJN.S42440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Novo L., Takeda K. M., Petteta T., et al. Targeted decationized polyplexes for siRNA delivery. Molecular Pharmaceutics. 2015;12(1):150–161. doi: 10.1021/mp500499x. [DOI] [PubMed] [Google Scholar]
- 111.Dong D., Gao W., Liu Y., Qi X.-R. Therapeutic potential of targeted multifunctional nanocomplex co-delivery of siRNA and low-dose doxorubicin in breast cancer. Cancer Letters. 2015;359(2):178–186. doi: 10.1016/j.canlet.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 112.Lin S.-Y., Zhao W.-Y., Tsai H.-C., Hsu W.-H., Lo C.-L., Hsiue G.-H. Sterically polymer-based liposomal complexes with dual-shell structure for enhancing the siRNA delivery. Biomacromolecules. 2012;13(3):664–675. doi: 10.1021/bm201746t. [DOI] [PubMed] [Google Scholar]
- 113.Li H., Miteva M., Kirkbride K. C., et al. Dual MMP7-proximity-activated and folate receptor-targeted nanoparticles for siRNA delivery. Biomacromolecules. 2015;16(1):192–201. doi: 10.1021/bm501394m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.De Backer L., Braeckmans K., Stuart M. C. A., Demeester J., De Smedt S. C., Raemdonck K. Bio-inspired pulmonary surfactant-modified nanogels: A promising siRNA delivery system. Journal of Controlled Release. 2015;206:177–186. doi: 10.1016/j.jconrel.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 115.Lee T. J., Haque F., Shu D., et al. RNA nanoparticle as a vector for targeted siRNA delivery into glioblastoma mouse model. Oncotarget . 2015;6(17):14766–14776. doi: 10.18632/oncotarget.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dohmen C., Edinger D., Fröhlich T., et al. Nanosized multifunctional polyplexes for receptor-mediated SiRNA delivery. ACS Nano. 2012;6(6):5198–5208. doi: 10.1021/nn300960m. [DOI] [PubMed] [Google Scholar]
- 117.Krais A., Wortmann L., Hermanns L., et al. Targeted uptake of folic acid-functionalized iron oxide nanoparticles by ovarian cancer cells in the presence but not in the absence of serum. Nanomedicine: Nanotechnology, Biology and Medicine. 2014;10(7):1421–1431. doi: 10.1016/j.nano.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 118.Ma X., Zhao Y., Ng K. W., Zhao Y. Integrated hollow mesoporous silica nanoparticles for target drug/siRNA co-delivery. Chemistry - A European Journal. 2013;19(46):15593–15603. doi: 10.1002/chem.201302736. [DOI] [PubMed] [Google Scholar]
- 119.Lee H., Lytton-Jean A. K. R., Chen Y. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nature Nanotechnology. 2012;7(6):389–393. doi: 10.1038/nnano.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee D.-J., Kessel E., Edinger D., et al. Dual antitumoral potency of EG5 siRNA nanoplexes armed with cytotoxic bifunctional glutamyl-methotrexate targeting ligand. Biomaterials. 2016;77:98–110. doi: 10.1016/j.biomaterials.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 121.O'Shannessy D. J., Yu G., Smale R., et al. Folate receptor alpha expression in lung cancer: diagnostic and prognostic significance. Oncotarget . 2012;3(4):414–425. doi: 10.18632/oncotarget.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Parker N., Turk M. J., Westrick E., Lewis J. D., Low P. S., Leamon C. P. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Analytical Biochemistry. 2005;338(2):284–293. doi: 10.1016/j.ab.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 123.Li S.-D., Chono S., Huang L. Efficient gene silencing in metastatic tumor by siRNA formulated in surface-modified nanoparticles. Journal of Controlled Release. 2008;126(1):77–84. doi: 10.1016/j.jconrel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li J., Yang Y., Huang L. Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. Journal of Controlled Release. 2012;158(1):108–114. doi: 10.1016/j.jconrel.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang Y., Hu Y., Wang Y., Li J., Liu F., Huang L. Nanoparticle delivery of pooled siRNA for effective treatment of non-small cell lung caner. Molecular Pharmaceutics. 2012;9(8):2280–2289. doi: 10.1021/mp300152v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen W.-H., Lecaros R. L. G., Tseng Y.-C., Huang L., Hsu Y.-C. Nanoparticle delivery of HIF1α siRNA combined with photodynamic therapy as a potential treatment strategy for head-and-neck cancer. Cancer Letters. 2015;359(1):65–74. doi: 10.1016/j.canlet.2014.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Guo J., Ogier J. R., Desgranges S., Darcy R., ODriscoll C. Anisamide-targeted cyclodextrin nanoparticles for siRNA delivery to prostate tumours in mice. Biomaterials. 2012;33(31):7775–7784. doi: 10.1016/j.biomaterials.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 128.Gregoriadis G., Senior J. Targeting of small unilamellar liposomes to the galactose receptor in vivo. Biochemical Society Transactions. 1984;12(2):337–339. doi: 10.1042/bst0120337. [DOI] [PubMed] [Google Scholar]
- 129.Park I. K., Park Y. H., Shin B. A., et al. Galactosylated chitosan-graft-dextran as hepatocyte-targeting DNA carrier. Journal of Controlled Release. 2000;69:97–108. doi: 10.1016/s0168-3659(00)00298-4. [DOI] [PubMed] [Google Scholar]
- 130.Han L., Tang C., Yin C. Dual-targeting and pH/redox-responsive multi-layered nanocomplexes for smart co-delivery of doxorubicin and siRNA. Biomaterials. 2015;60:42–52. doi: 10.1016/j.biomaterials.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 131.Han L., Tang C., Yin C. Oral delivery of shRNA and siRNA via multifunctional polymeric nanoparticles for synergistic cancer therapy. Biomaterials. 2014;35(15):4589–4600. doi: 10.1016/j.biomaterials.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 132.Lee J. H., Engler J. A., Collawn J. F., Moore B. A. Receptor mediated uptake of peptides that bind the human transferrin receptor. European Journal of Biochemistry. 2001;268(7):2004–2012. doi: 10.1046/j.1432-1327.2001.02073.x. [DOI] [PubMed] [Google Scholar]
- 133.Oh S., Kim B. J., Singh N. P., Lai H., Sasaki T. Synthesis and anti-cancer activity of covalent conjugates of artemisinin and a transferrin-receptor targeting peptide. Cancer Letters. 2009;274(1):33–39. doi: 10.1016/j.canlet.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 134.Gao L.-Y., Liu X.-Y., Chen C.-J., et al. Core-Shell type lipid/rPAA-Chol polymer hybrid nanoparticles for invivo siRNA delivery. Biomaterials. 2014;35(6):2066–2078. doi: 10.1016/j.biomaterials.2013.11.046. [DOI] [PubMed] [Google Scholar]
- 135.Zhu L., Perche F., Wang T., Torchilin V. P. Matrix metalloproteinase 2-sensitive multifunctional polymeric micelles for tumor-specific co-delivery of siRNA and hydrophobic drugs. Biomaterials. 2014;35(13):4213–4222. doi: 10.1016/j.biomaterials.2014.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Malhotra M., Tomaro-Duchesneau C., Saha S., Prakash S. Systemic siRNA delivery via peptide-tagged polymeric nanoparticles, targeting PLK1 gene in a mouse xenograft model of colorectal cancer. International Journal of Biomaterials. 2013;2013 doi: 10.1155/2013/252531.252531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hong S., Zhang X., Chen J., Zhou J., Zheng Y., Xu C. Targeted gene silencing using a follicle-stimulating hormone peptide-conjugated nanoparticle system improves its specificity and efficacy in ovarian clear cell carcinoma in vitro. Journal of Ovarian Research. 2013;6(1, article no. 80) doi: 10.1186/1757-2215-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang X.-Y., Chen J., Zheng Y.-F., et al. Follicle-stimulating hormone peptide can facilitate paclitaxel nanoparticles to target ovarian carcinoma in vivo. Cancer Research. 2009;69(16):6506–6514. doi: 10.1158/0008-5472.CAN-08-4721. [DOI] [PubMed] [Google Scholar]
- 139.Shah V., Taratula O., Garbuzenko O. B., Taratula O. R., Rodriguez-Rodriguez L., Minko T. Targeted nanomedicine for suppression of CD44 and simultaneous cell death induction in ovarian cancer: An optimal delivery of siRNA and anticancer drug. Clinical Cancer Research. 2013;19(22):6193–6204. doi: 10.1158/1078-0432.CCR-13-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sioud M., Mobergslien A. Efficient siRNA targeted delivery into cancer cells by gastrin-releasing peptides. Bioconjugate Chemistry. 2012;23(5):1040–1049. doi: 10.1021/bc300050j. [DOI] [PubMed] [Google Scholar]
- 141.Gao Y., Wang Z.-Y., Zhang J., et al. RVG-peptide-linked trimethylated chitosan for delivery of siRNA to the brain. Biomacromolecules. 2014;15(3):1010–1018. doi: 10.1021/bm401906p. [DOI] [PubMed] [Google Scholar]
- 142.Gooding M., Malhotra M., McCarthy D. J., et al. Synthesis and characterization of rabies virus glycoprotein-tagged amphiphilic cyclodextrins for siRNA delivery in human glioblastoma cells: In vitro analysis. European Journal of Pharmaceutical Sciences. 2015;71:80–92. doi: 10.1016/j.ejps.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 143.Yang L., Sajja H. K., Cao Z., et al. uPAR-targeted optical imaging contrasts as theranostic agents for tumor margin detection. Theranostics. 2014;4(1):106–118. doi: 10.7150/thno.7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.van Waarde A., Rybczynska A. A., Ramakrishnan N. K., Ishiwata K., Elsinga P. H., Dierckx R. A. J. O. Potential applications for sigma receptor ligands in cancer diagnosis and therapy. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2015;1848(10):2703–2714. doi: 10.1016/j.bbamem.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 145.Pirollo K. F., Zon G., Rait A., et al. Tumor-targeting nanoimmunoliposome complex for short interfering RNA delivery. Human Gene Therapy. 2006;17(1):117–124. doi: 10.1089/hum.2006.17.117. [DOI] [PubMed] [Google Scholar]
- 146.Chen Y., Wang W., Lian G., et al. Development of an MRI-visible nonviral vector for siRNA delivery targeting gastric cancer. International Journal of Nanomedicine. 2012;7:359–368. doi: 10.2147/IJN.S24083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Alshaer W., Hillaireau H., Vergnaud J., Ismail S., Fattal E. Functionalizing Liposomes with anti-CD44 Aptamer for Selective Targeting of Cancer Cells. Bioconjugate Chemistry. 2015;26(7):1307–1313. doi: 10.1021/bc5004313. [DOI] [PubMed] [Google Scholar]
- 148.Sánchez-Martín D., Cuesta Á. M., Fogal V., Ruoslahti E., Álvarez-Vallina L. The multicompartmental p32/gClqR as a new target for antibody-based tumor targeting strategies. The Journal of Biological Chemistry. 2011;286(7):5197–5203. doi: 10.1074/jbc.M110.161927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pu K., Yuan L., Chen L., et al. Identification of VEGFR2-binding peptides using high throughput bacterial display methods and functional assessment. Current Cancer Drug Targets. 2015;15(2):158–170. doi: 10.2174/156800961502150320112339. [DOI] [PubMed] [Google Scholar]
- 150.Kim B., Yang J., Hwang M., et al. Aptamer-modified magnetic nanoprobe for molecular MR imaging of VEGFR2 on angiogenic vasculature. Nanoscale Research Letters. 2013;8(1):1–10. doi: 10.1186/1556-276X-8-399. [DOI] [PMC free article] [PubMed] [Google Scholar]