

Abstract
Increasing CO2 concentration in the atmosphere is believed to have a profound impact on the global climate. To reverse the impact would necessitate not only curbing the reliance on fossil fuels but also developing effective strategies capture and utilize CO2 from the atmosphere. Among several available strategies, CO2 reduction via the electrochemical or photochemical approach is particularly attractive since the required energy input can be potentially supplied from renewable sources such as solar energy. In this Review, an overview on these two different but inherently connected approaches is provided and recent progress on the development, engineering, and understanding of CO2 reduction electrocatalysts and photocatalysts is summarized. First, the basic principles that govern electrocatalytic or photocatalytic CO2 reduction and their important performance metrics are discussed. Then, a detailed discussion on different CO2 reduction electrocatalysts and photocatalysts as well as their generally designing strategies is provided. At the end of this Review, perspectives on the opportunities and possible directions for future development of this field are presented.
Keywords: CO2 reduction, electrocatalysis, nanotechnology, photocatalysis
1. Introduction
Energy shortage and environmental pollution are two major global challenges facing the human society. Current world energy consumption is highly dependent upon fossil fuels. Concerns are growing that the increasing human activities would not only accelerate the consumption of fossil fuels but also result in escalated greenhouse gas emission and breaks the carbon balance in the natural world.1, 2 Since late 19th century, CO2 concentration in the atmosphere has increased from 280 to 400 ppm (Figure 1 ).3 This has resulted in the continuous rise of the global average temperature. How to effectively reduce the atmospheric CO2 level and further utilize it has become an important research topic worldwide. Strategies are now being actively sought to mitigate CO2 emission via improving the combustion efficiency of fossil fuels or exploring clean and renewable energy sources (e.g., wind, tide, and solar energy).4, 5 Alternatively, great efforts are also being actively undertaken to develop carbon capture and storage (CCS) techniques that fix atmospheric CO2 and store it underground in a supercritical state.6 However, the CCS technique itself is energy intensive and nonrenewable.
Figure 1.
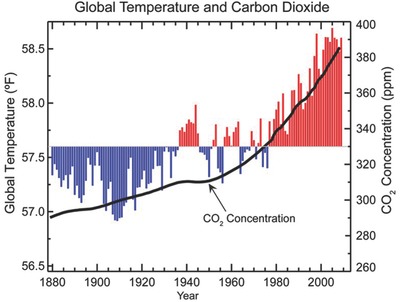
Atmospheric CO2 concentration and corresponding global average temperature since the late 19th century. Red bars indicate temperatures above and blue bars indicate temperatures below the 1901–2000 average temperature. Adopted from the website of National Ocean and Atmospheric Administration (NOAA).3 Copyright 2017, NOAA.
In nature, the photosynthesis of green plants plays an indispensable role in maintaining the carbon/oxygen cycle which is vital for the maintenance of life on earth. It is consisted of two sequential steps known as the light and dark reactions (Figure 2 a).7 In the light reaction, chlorophyll adsorbs sunlight, converts it to the chemical energy stored in adenosine triphosphate (ATP), and meanwhile oxidizes water to O2. In the dark reaction, CO2 is fixed and reduced stepwise to form carbohydrates using energy stored in ATP. The natural photosynthesis essentially provides the energy needed for most lives on this planet, and is the basis for the survival of mankind.
Figure 2.
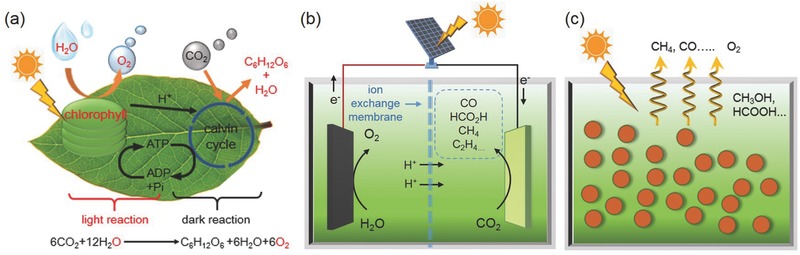
Analogy among a) natural photosynthesis, b) electrochemical synthesis on electrocatalysts powered by a photovoltaic cell, and c) photochemical synthesis on powdery photocatalysts.
For more than three decades, researchers have been ambitiously attempting to mimic what Mother Nature does and pursuing artificial photosynthesis that aims at the spontaneous transformation of atmospheric CO2 and water to chemical fuels using sunlight as the sole energy input.8, 9 Although still at very low efficiency currently, artificial photosynthesis is believed to have the great potential to make a substantial contribution to our future energy supply. It is now generally approached from two directions. As schematically illustrated in Figure 2b, the first route uses photovoltaic (PV) cells to generate a sufficient photovoltage which is then supplied to the cathode for the CO2 reduction and the anode for the water oxidation. Proper electrocatalysts are employed on the two electrodes so as to expedite the reaction rate and improve the reaction selectivity. The advantage of this route is the flexibility in the design of PV and electrocatalyst pairs. Components can be individually optimized and then combined together to enable the best overall performance. The second route is the direct photocatalytic approach where light‐absorbing semiconductor particles (photocatalysts) decorated with suitable electrocatalysts (commonly referred as cocatalysts in photocatalysis) are dispersed in aqueous solution and achieve light harvesting, charge separation, and interfacial charge transfer to drive corresponding reactions all within particles (Figure 2c). The merit of the second route is its wireless configuration that renders the device design much more straightforward and compact. At this moment, it is too early to judge which route would eventually dominate in the future. Their future success strongly relies on the development of high‐performance CO2 reduction electrocatalysts or photocatalysts.
Since CO2 is a thermodynamically stable molecule, its multistep reduction via the electrochemical or photochemical approach is significantly more challenging than the splitting of water, and confronted with many fundamental technical hurdles.10 The history of electrocatalytic CO2 reduction can be traced back to 19th century. In 1870, Royer first reported the reduction of CO2 to formic acid on Zn electrodes.11 Between 1970s and 1980s, a series of seminal works published by Japanese scientists marked the advent of a new phase in electrocatalytic CO2 reduction research. Ito and Murata examined the electrocatalytic performances of several metals such as In, Cd, Sn, Zn, and Pb for reducing CO2 to formic acid.12 Hori et al. discovered that polycrystalline Cu electrodes in aqueous media could generate short‐chain hydrocarbons with a promising activity.13 Studies on photocatalytic CO2 reduction commenced in 1970s. In 1978, Halmannn first observed that CO2 was reduced to CH3OH and CO on a p‐type GaP electrode under light illumination.14 In the year later, Inoue et al. reported that formic acid, formaldehyde, and methyl alcohol were produced from the photocatalytic reduction of CO2 in the aqueous suspensions of semiconductors such as TiO2, ZnO, CdS, GaP, and SiC.15 Following these pioneering works on electrocatalytic or photocatalytic CO2 reduction, increasing attention has been attracted to this field, and many exciting progresses have been made in recent years.
In this paper, we present an overview of the recent progress on electrocatalytic or photocatalytic CO2 reduction. Several previous high‐quality review articles on similar topics are available.16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26 Given the recent heightened research activities and increasingly deepened understanding of these two processes, we believe that an up‐to‐date account on their status and existing challenges is necessary so as to provide readers with a current snapshot of this rapidly evolving area. Even though the two approaches involve dissimilar experimental techniques, their nature is essentially identical—that is how to activate the chemically inert CO2 molecule and promote its conversion under external energy stimuli. In addition, the surface charge transfer step in photocatalysis is in fact an electrochemical process and can be enhanced via the incorporation of proper cocatalysts. These are the reasons why we think electrocatalytic and photocatalytic CO2 reduction are inherently connected and decide to discuss them together here. This review starts with a brief description about the basic principles and important performance merits of electrocatalytic and photocatalytic CO2 reduction. It is then followed by detailed discussions on different catalysts for electrocatalytic and photocatalytic CO2 reduction and their several designing strategies. At the end, we present our brief perspectives on the possible future development of this field.
2. Fundamentals of Electrocatalytic and Photocatalytic CO2 Reduction
CO2 is one of the most stable molecules due to the strong C=O double bond with bonding energy of 750 kJ mol−1—considerably larger than that of C—C (336 kJ mol−1), C—O (327 kJ mol−1), or C—H bond (411 kJ mol−1). CO2 reduction via either the electrocatalytic or the photocatalytic approach is a thermodynamically uphill reaction and demands significant energy input to break the C=O bond. To make it even more complicated, CO2 reduction may proceed via several different reaction pathways with the transfer of 2, 4, 6, 8, 12 or even more electrons and yielding diverse reduction products including carbon monoxide (CO), formic acid (HCOOH), methane (CH4), ethylene (C2H4), and many others depending on the nature of the electrocatalysts or photocatalysts as well as the actual experimental conditions.27, 28 As a result, electrocatalytic or photocatalytic CO2 reduction is generally suffered from very limited efficiency and poor selectivity.
2.1. Fundamentals of Electrocatalytic CO2 Reduction
Table 1 summarizes the equilibrium potentials (vs the standard hydrogen electrode, SHE) of CO2 reduction to different products in pH 7.0 aqueous solution.29 Even though some reactions (i.e., reduction to CH4, methanol or C2H4) are thermodynamically more favorable than the two‐electron hydrogen evolution reaction (HER), the kinetics of the CO2 reduction is substantially more sluggish. This is because after its chemical absorption on the working electrode, the first electron transfer to form CO2 •− anion radical does not initiate until at—1.90 V in order to reorganize the originally linear molecule into a bent anion radical (Figure 3 ).30, 31 The formation of this intermediate state imposes a significant overpotential to the reaction and is frequently identified as the rate determining step.32, 33 Once CO2 •− is formed, it may be subsequently reduced via the protonation of its oxygen atom, resulting in the formation of •COOH. This intermediate is then reduced to CO and released from the electrode surface. Alternatively, CO2 •− may also be reduced via the protonation of its carbon atom to form HCOO• instead of •COOH at high overpotentials, which is further reduced to formate (HCOO−). As a result, most CO2 reduction electrocatalysts yield CO or formate as the primary reduction products. Only on very few electrocatalysts (e.g., Cu), CO can be further reduced to hydrocarbons. The reaction mechanism of these electrocatalysts is not clearly understood, but it is believed to proceed stepwise via the H addition, the scission of C—O bond, and the coupling of C—C bond.34, 35, 36
Table 1.
Standard electrochemical potentials for CO2 reduction
| Reduction potentials of CO2 | E° [V] vs SHE at pH 7 |
|---|---|
| CO2 + e− → CO2 − | −1.9 |
| CO2 + 2H+ + 2e− → HCOOH | −0.61 |
| CO2 + 2H+ + 2e− → CO + H2O | −0.52 |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | −0.34 |
| CO2 + 4H+ + 4e− → HCHO + H2O | −0.51 |
| CO2 + 6H+ + 6e− → CH3OH + H2O | −0.38 |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 |
| 2H+ + 2e– → H2 | −0.42 |
Figure 3.
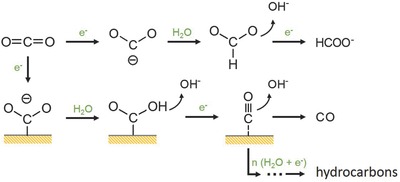
Possible reaction pathways for electrocatalytic CO2RR on metal electrodes in aqueous solutions. Adapted with permission.45 Copyright 1994, Elsevier.
There are several important performance metrics in the evaluation of CO2 reduction electrocatalysts as listed in the following:
Onset potential: onset potential refers to the working potential where the electrocatalytic current starts to take off from the background. It is not a well‐defined parameter since sometimes it is difficult to determine the exact current onset point especially when the capacitive contribution is significant. To avoid the confusion, onset potential now is frequently defined as the potential where the electrocatalytic current of a certain product reaches a given level (e.g., 0.1 mA cm−2). Such a level also varies from study to study. Care therefore needs to be taken when comparing the onset potential across the literature.
Tafel slope: Tafel slope (b) can be experimentally derived by plotting the overpotential (η) with respect to the logarithm of the current density (log j) and fitting the linear region of the curve with the Tafel equation (η = b log j + a). It indicates the overpotential increment necessary to raise the current density of a certain product by tenfold. A smaller Tafel slope corresponds to a steep rise of the current density with the increasing overpotential and is a highly desirable characteristic of electrocatalysts. For multielectron transfer process such CO2 reduction reaction, the Tafel slope may also provide valuable insights into the rate determining step and possible reaction pathway. For example, when the formation of CO2 •– anion radical is rate determining, the ideal Tafel slope should be per decade.37
Turnover frequency (TOF): TOF is defined as the rate of electrochemical conversions per electrocatalytic site at certain overpotential. It reflects the intrinsic activity of an electrocatalyst and allows the comparison among different materials regardless of their actual geometric parameters or areal loading. Unfortunately, except for a few special cases where material surface activity sites can be clearly quantified (e.g., Pt and Pd),38 it is highly challenging to calculate the TOF value of most electrocatalysts due to the structural ambiguity of the active sites and the difficulty in precisely counting them. Many studies often assume all the added catalysts effectively participate in the reaction. Thus derived TOF values are underestimated (sometime by orders of magnitude), but they may still provide some insights into the intrinsic activity.
Faradaic efficiency: Faradaic efficiency of a certain product is defined as the ratio of charges transferred to this product relative to the total charges passed through the circuit. It reflects the selectivity of electrocatalysts. Since CO2 reduction kinetics is slow, it is usually accompanied by the considerable cogeneration of H2 from HER. High Faradaic efficiency (>80%) toward the desirable products is one of the many requirements for good CO2 reduction electrocatalysts.
Stability: Besides its activity and selectivity, any electrocatalyst should have sufficient long‐term stability in order to be considered for practical applications. It is usually evaluated via the cyclic voltammetry (CV) cycling, galvanostatic or potentiostatic polarizations. Evaluating the stability of electrocatalysts and understanding their possible degradation mechanism is a critical step toward the continuous optimization of electrocatalysts.
2.2. Fundamentals of Photocatalytic CO2 Reduction
A typical process of photocatalytic CO2 reduction on a semiconductor photocatalyst is schematically illustrated in Figure 4 .20 It consists of five sequential steps—light absorption, charge separation, CO2 adsorption, surface redox reaction, and product desorption. The first step is the absorption of photons to generate electron and hole pairs. Illumination of a photocatalyst with the incident light excites electrons from the valance band (VB) to the conduction band (CB), leaving an equal number of holes in VB. In order for these photogenerated electrons or holes to be energetically favorable to reduce CO2 or oxidize water, photocatalysts should possess suitable band structure. Their CB edge must be more negative than the redox potential of CO2 reduction (summarized in Table 1), and the VB edge should be more positive than the redox potential of water oxidation (0.817 V vs SHE in pH 7.0 aqueous solution). The band gap has to be sufficiently large since we need to additionally take into consideration of the large overpotentials associated with these two electrochemical reactions. On the other hand, the band gap of photocatalysts cannot be too large as this would limit their effective utilization of the solar spectrum. For example, one of the most well studied semiconductors—TiO2 has a band gap of 3.2 eV. It only absorbs photons of light in the ultraviolet domain (<400 nm), which accounts for less than 5% of the entire solar spectrum.39 Given these two criteria, the ideal band gap is estimated to be 1.8–2.0 eV. However, most photocatalyst materials at present have band gaps off the ideal range. Strategies such as doping and solid solution are being actively sought to carefully engineer photocatalyst band structures as will be discussed later.
Figure 4.
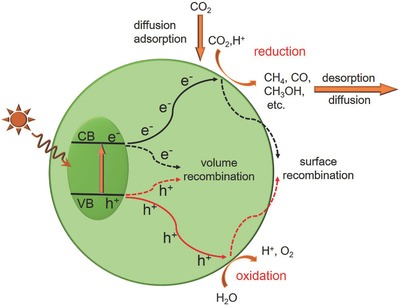
Schematic showing the five fundamental steps in photocatalytic CO2 reduction. Adopted with permission.20 Copyright 2014, Springer.
The second step is the spatial separation of photogenerated electrons and holes. This process is in direct competition with the charge recombination. Whether the charge separation is dominant over the recombination or vice versa depends on the relative time scale between the lifetime of photogenerated carriers and the recombination rate and is determined by a complex interplay among material crystallinity, dimension, surface properties, and many other structural factors. Pronounced recombination would result in the significant loss of free charge carriers and the release of harvested energy as heat. To enhance the overall photocatalytic efficiency therefore requires improving the separation efficiency of photogenerated carriers and suppressing their possible recombination. It may be achieved via the proper modification of material structures, such as selective surface treatments.
The third step is the CO2 adsorption. It is the prerequisite for the electron transfer from the photocatalyst to the CO2 molecule. In general, photocatalysts with high surface areas can provide more active sites for the CO2 adsorption. Another possible route to improve the CO2 adsorption is the alkali modification of the photocatalyst surface, as has been demonstrated for TiO2.40, 41 Due to the Lewis acidity of CO2 molecules, the reaction between CO2 and the alkaline photocatalyst surface would lead to the formation of intermediates such as bidentate carbonate, favoring the activation and subsequent reduction of CO2 molecules.
The forth step is the surface redox reaction. After migrating to the surface, photogenerated electrons and holes can separately drive different half reactions: electrons for reducing CO2 to CO, CH4, HCOOH, CH3OH or other hydrocarbons, and holes for oxidizing water to molecular O2. This step is a purely electrochemical process. The introduction of cocatalysts for CO2 reduction or water oxidation would dramatically enhance the interfacial charge transfer rate, and hence the overall solar‐to‐fuel conversion efficiency (SFE). Optimal electrocatalysts identified from electrochemical studies are also good candidates as cocatalysts, given that a suitable interface is established between the semiconductor photocatalyst and cocatalyst to enable the rapid charge transfer across it. This step also clearly reflects the interesting connection between electrocatalysis and photocatalysis. After the photocatalytic reaction is complete, the last step is the product desorption. If the product cannot be timely released from the catalyst surface, the reaction then is terminated and the catalyst becomes “poisoned.”
For photocatalytic CO2 reduction, the reaction can be carried out in liquid or gas phase medium. Most current studies refer the gas phase medium, where the suspended powdery photocatalyst directly reacts with surrounding CO2 and water vapor under light irradiation. The activity of photocatalysts are usually quantified using the production rate of a certain product (in terms of µmol h−1, or µmol h−1 g−1 when normalized with respect to the mass of photocatalyst) under given conditions including temperature (usually room temperature), pressure (usually 1 atm), and light intensity (usually 1 sun or 100 mW cm−2). TOF values of photocatalysts are similarly calculated based on the specific surface area of catalyst powders. Selectivity of photocatalysts is assessed by comparing the relative production rates of different gaseous or liquid products (including H2). In addition, photocatalysis has the following two important performance metrics that are frequently cited in literature:
Apparent quantum efficiency (AQE) or external quantum efficiency (EQE): AQE or EQE is defined as the number ratio of electrons transferred toward a certain product relative to incident photons at a given wavelength. They can be expressed as the product of the efficiencies of light absorption, charge separation, and surface redox reaction. Photocatalysts therefore have to be efficient at all the three steps in order to have great AQE or EQE values.
SFE: SFE is defined as the ratio of converted chemical energy relative to the incident solar energy. It can also be understood as the integral of AQE or EQE over the entire solar spectrum. By comparison, AQE or EQE reflects the energy conversion efficiency of photocatalysts at a particular wavelength, while SFE reflects the overall energy conversion efficiency of photocatalysts. The ideal limiting SFE at a single absorber particle is η = 14.4% based on a light absorber with a 2.0 eV band gap.42 It is suggested that a >10% SFE is required for photocatalysis to be an economically viable resource.43, 44
One critical issue that needs special attention for the CO2 reduction research is the possible carbon contamination. Studies suggest that organic substances including solvents, reactants, and surfactants used for the catalyst preparation may leave carbonaceous residues in the final product, and, during electrocatalysis or photocatalysis (particularly the latter), may decompose to small molecules such as CO and CH4, causing the overestimation of catalytic activities.16 It is therefore necessary to confirm that the measured products are indeed from the CO2 reduction rather than the decomposition of carbonaceous residues. Isotopic 13CO2 labeling is an effective technique to verify the origin of reduction products and has been widely employed in many studies. Additionally, the possible carbon contamination may also be ruled out by carrying out control experiments in an inert gas environment (N2 or Ar) under otherwise identical conditions. Compared to isotopic 13CO2 labeling, control experiments in N2 or Ar are generally more cost and time effective.
3. Electrocatalytic Materials for CO2 Reduction
In this part, we aim to review different electrocatalysts that have been developed for CO2 reduction reaction (CO2RR) in recent years. They can be generally categorized into metals, metal chalcogenides and carbonaceous materials (Table 2 ). In what follows, we will review the current development status of these materials.
Table 2.
Summary of CO2 reduction electrocatalysts from recent literature
| Electrocatalyst | Electrolyte | Selectivity and activity | Stability | Reference |
|---|---|---|---|---|
| Cu NCs with 44 nm edge length | 0.1 m KHCO3 | J tot = ≈5.7 mA cm−2, F.E. CO2RR 80%, ethylene 41%, methane 20% @ −1.1 V vs RHE | – | 53 |
| Cu mesopore electrode (width/depth) | 0.1 m KHCO3 | J tot = 14.3 mA cm−2, F.E. C2H4 38% (30 nm/40 cm) C2H6 46%(30 nm/70 nm) @ −1.7 V vs NHE; onset potential −0.96 V vs NHE | – | 226 |
| 3D porous hollow fiber Cu electrode | 0.3 m KHCO3 | J tot = ≈10 mA cm−2, F.E. CO 75% @ −0.4 V vs RHE | 24 h @ −0.4 V vs RHE | 227 |
| Cu NPs 13.1 nm | 0.1 m KHCO3 | J tot = 20 mA cm−2, H2 0.078, CO 0.016, CH4 0.0018, C2H4 0.0006 (Vol. % cm−2) @ −1.1 V vs RHE | – | 54 |
| Cu NPs | 0.1 m NaHCO3 | J tot = ≈9 mA cm−2, F.E. CH4 80%, H2 13% @ −1.25 V vs RHE | 1 h @ −1.25 V vs RHE | 55 |
| OD Cu films | 0.5 m NaHCO3 | J tot = 2.7 mA cm−2, F.E. CO ≈40%, HCO2H 33% @ −0.5 V vs RHE | 7 h @ −0.5 V vs RHE | 48 |
| Plasma‐activated Cu | 0.1 m KHCO3 | F.E. C2H4 60% @ −0.9 V vs RHE; onset E: −0.5 V vs RHE | – | 59 |
| OD Au NPs | 0.5 m NaHCO3 | J tot = 6 mA cm−2, F.E. CO 98% @ −0.4 V vs RHE | 8 h @ −0.4 V vs RHE | 65 |
| Au25 cluster | 0.1 m KHCO3 | J tot = ≈14.3 mA cm−2, F.E. CO 99.6% @ −0.89 V vs RHE | – | 228 |
| Au NPs 8 nm | 0.5 m KHCO3 | F.E. CO 90% @ −0.67 V vs RHE | – | 63 |
| Au NWs | 0.5 m KHCO3 | J tot = 1.84 A g−1, F.E. CO 94% @ −0.35 V vs RHE | 12 h @ −0.35 V vs RHE | 67 |
| Au/carbon nanotubes (CNT) | 0.5 m NaHCO3 | J tot = 15 A g−1, F.E. CO 94% @ −0.5 V vs RHE | 12 h @ −0.5 V vs RHE | 66 |
| Nanoporous Ag | 0.5 m KHCO3 | J tot = 18 mA cm−2, F.E. CO ≈92% @ −0.6 V vs RHE | 2 h @ −0.6 V vs RHE | 74 |
| Mesostructured Ag | 0.1 m KHCO3 | F.E. CO > 80% @ −0.7 V vs RHE | – | 75 |
| Oxide‐derived Ag | 0.1 m KHCO3 | J tot = 1.15 mA cm−2, F.E. CO ≈89% @ −0.8 V vs RHE | – | 73 |
| Anodized polycrystalline Ag | 0.1 m aqueous KHCO3 | J tot = 1.15 mA cm−2, F.E. CO ≈89% @ −0.8 V vs RHE | – | 106 |
| Graphene confined Sn quantum sheets | 0.1 m NaHCO3 | J tot = 21.1 mA cm−2, F.E. HCOOH 89% @ −1.8 V vs SHE | 18 h @ −1.8 V vs RHE | 79 |
| Sn/SnOx on Ti foil | 0.5 m NaHCO3 | F.E. CO ≈40.6% HCOOH ≈56.8% @ −0.7 V vs RHE | – | 65 |
| ≈5 nm Sn NPs/graphene | 0.1 m NaHCO3 | J tot = 10.2 mA cm−2 , F.E. HCOOH 93.6% @ −1.8 V vs SHE | 18 h @ −1.8 V vs SHE | 105 |
| Au3Cu | 0.1 m KHCO3 | J tot = 3 mA cm−2, F.E. CO 64.7% HCOOH 3.11% @ −0.73 V vs RHE | – | 92 |
| Cu‐In | 0.1 m KHCO3 | J tot = 0.53 mA cm−2, F.E. CO 90% @ −0.5 V vs RHE | 7 h @ −0.6 V vs RHE | 93 |
| Pd Icosahedra/C | 0.1 m KHCO3 | F.E. CO 91.1% @ −0.8 V vs RHE | 10 h @ −0.9 V vs RHE | 229 |
| Mo‐Bi bimetallic chalcogenide | 1‐butyl‐3‐methylimidazolium tetrafluoroborate (BMIM‐BF4) in MeCN | J tot = 12.1 mA cm−2, F.E. MeOH 71.2% @ −0.7 V vs SHE | – | 104 |
| Vertically aligned Mo0.95Nb0.05S2 | 50 vol % 1‐ethyl‐3‐methylimidazolium tetrafluoroborate (EMIM‐BF4) and water | J tot = 237 mA cm−2, F.E. CO 82% @ −0.8 V vs RHE | – | 103 |
| Bulk MoS2 | 96 mol% water and 4 mol% EMIM‐BF4 | J tot = ≈65 mA cm−2, F.E. CO ≈98% @ −0.764 V vs RHE | – | 101 |
| WSe2 | 50 mol% water and 50 mol% EMIM‐BF4 | J tot = 330 mA cm−2, F.E. CO ≈85% @ −0.764 V vs RHE | – | 102 |
| Boron‐doped diamond (BDD) | MeOH containing tetrabutylammonium perchlorate (TBAP) | J tot = ≈0.1 mA cm−2, F.E. HCHO ≈74% @ −1.7 V vs Ag/Ag+ | 20 h @ −1.7 V vs Ag/Ag+ | 108 |
| Polyethylenimine‐Nitrogen doped carbon nanotubes (PEI‐NCNT) | 0.1 m KHCO3 | J tot = −9.5 mA cm−2, F.E. HCOOH 87% @ −1.8 V vs SCE | 24 h @ −1.8 V vs SCE | 105 |
| polyacrylonitrile (PAN)‐based CNFs | EMIM‐BF4 | J tot = 3.86 mA cm−2, F.E. CO 98% @ −0.573 V vs SHE | 9 h @ −0.573 V vs SHE | 107 |
| N‐doped nanodiamond/Si rod array | 0.5 m NaHCO3 | J tot = −0.69–0.89 mA cm−2, F.E. CH3COO− 91.2–91.8% @ −0.8–−1.0 V vs RHE | – | 109 |
| N‐doped graphene quantum dots (QDs) | 1 m KOH | J tot = 100 mA cm−2, total F.E. for CO2RR products 90% C2H4 46% C2H5OH 21% CO 23% @ −0.86 V vs RHE | – | 110 |
3.1. Metals
Elemental metals are among the earliest investigated CO2RR electrocatalysts. In a series of seminal works published between 1980s and 1990s, Hori et al. first reported that CO, CH4, formate, and other hydrocarbons were detected from the electrocatalytic CO2RR on various metal electrodes in aqueous KHCO3 electrolyte solution.13, 27, 45 Based on the reduction products, these metals are divided to three groups. The first group includes Sn, Pb, Bi, In, etc. They hardly adsorb the CO2 •− intermediate. Desorbed CO2 •− tends to be protonated at the carbon atom and ultimately transforms to formate or formic acid as the major reduction product. The second group includes Au, Ag, Zn, Pd, Ga, etc. They can bind the CO2 •− intermediate, catalyze the cleavage of C—O bond in CO2, and allow resultant CO to easily desorb from the electrode as the major reduction product. The third group includes Pt, Ti, Ni, Fe, etc. They have low HER overpotentials and strong CO adsorption properties, giving rise to H2 as the major production. In addition to the three groups, Cu is the only elemental metal capable of producing C1–C3 hydrocarbons at significant rates. It is suggested that the adsorption of CO on Cu is suitable for its further reduction to hydrocarbons or alcohols at high overpotentials through COH or CHO intermediates.36
Theoretical simulation has been proven a powerful tool to understand the electrocatalytic CO2RR activity and selectivity on different metals. Nørskov and co‐workers used density functional theory (DFT) calculations to describe trends in catalytic activity for CO2 reduction to CO as a function of the adsorption energies of the two reaction intermediates—COOH and CO.46 They revealed that on Au and Ag, the reaction rate was limited by CO2 activation, and resultant CO desorbed easily from their surfaces; whereas on Pd, Ni, Pt, and Rh, CO2 activation and conversion to adsorbed CO was facile, and the reaction rate was mainly limited by the desorption of CO due to its strong affinity. Cu had intermediate bonding strength with both CO and COOH in comparison with others. Unfortunately, all the metals were suggested to be well off the ideal activity since the CO and COOH adsorption energies were essentially linearly correlated, making it impractical to adjust one parameter without affecting the other. Studt and co‐workers compared the CO2 reduction to formic acid and its competing reactions on 27 different metal surfaces and found that Pb (211) surface was one of the most promising monometallic surfaces for the reduction of CO2 to formic acid with a virtually zero overpotential and very high selectivity, while Cd, Tl, and Sn surfaces were selective toward formic acid production but required relatively high overpotentials (0.2–0.4 V).47
3.1.1. Copper
As introduced above, Cu is the only known elemental metal that can reduce CO2 beyond CO or formic acid. In addition to the two‐electron reduction products, methane, ethane, ethylene, ethyne, methanol, and ethanol are all measured as the possible reduction products. Recent work by Jaramillo and co‐workers reported the identification of 16 different reduction products on metallic Cu surface (Figure 5 a).36 Aside from common products, some aldehydes, ketones, carboxylic acids, esters, and hydrocarbons such as paraffins and olefins containing up to six carbon atoms could also be generated as minor products on Cu electrocatalysts. It was proposed that these C≥2 products might be formed via enol‐like surface intermediates. Nevertheless, deep reduction of CO2 is kinetically challenging: these higher value chemicals generally cannot be produced at significant rates until at very negative potentials (←0.8 V vs reversible hydrogen electrode or RHE) and their Faradaic efficiency is usually <30%.34, 36, 48
Figure 5.
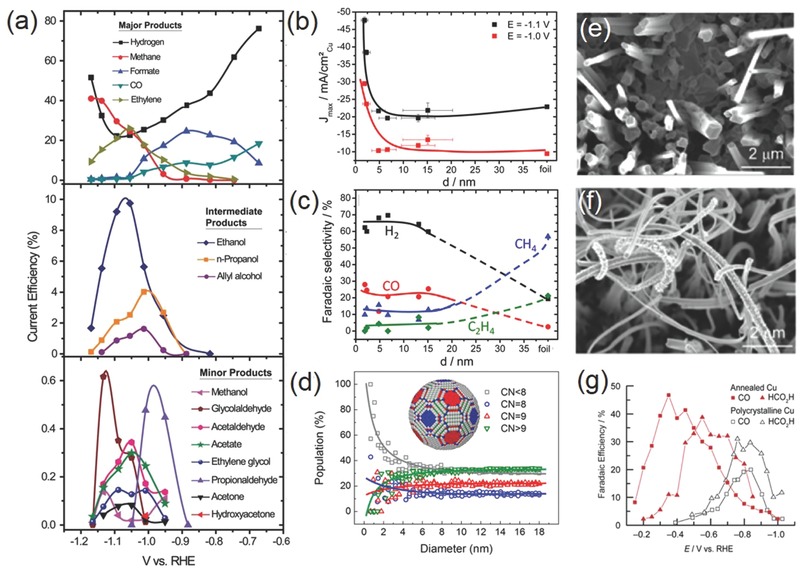
a) Faradaic efficiency as a function of potential for major (top), intermediate range (middle), and minor (bottom) products on a metallic Cu surface. Reproduced with permission.36 Copyright 2012, The Royal Society of Chemistry. Particle size dependence of b) current density and c) Faradaic efficiency for different CO2RR products on Cu NPs; d) population of surface atoms with certain coordination number (CN) as a function of particle diameter. Reproduced with permission.54 Copyright 2014, American Chemical Society. Scanning electron microscope (SEM) images of e) an annealed Cu electrode and f) the same electrode after CO2RR; g) Faradaic efficiency for CO and HCOOH as a function of potential on polycrystalline Cu and annealed Cu. Reproduced with permission.48 Copyright 2012, American Chemical Society.
To promote the electrocatalytic performance of Cu, different strategies have been exploited. The morphology of Cu catalysts has a profound influence on the catalytic activity and product selectivity. On single crystal Cu electrodes, the selectivity toward hydrocarbons (mainly methane and ethylene) is strongly dependent on the electrode surface. Hori et al. first showed that ethylene formation was favored on Cu (100) surface, whereas methane was the main hydrocarbon product on Cu (111) terraces.49 Similar results were also observed by Koper and co‐workers.50, 51 It is generally accepted that CO is a key intermediate in the formation of both methane and ethylene. Even though the exact reaction mechanism remains elusive, experimental evidence was shown by Koper and co‐workers that ethylene was formed on Cu (100) surface presumably via electron‐mediated dimerization reaction of two adsorbed CO molecules.52 Dimerization of CO on Cu (111) surface was unfavorable compared to the protonation of CO to CHO, which subsequently led to the formation of methane. However, a different insight was provided by Novskov and co‐workers suggesting that the formation of ethylene instead proceeded through the chemical dimerization of CHO intermediate based on DFT calculations.35 In spite of the debatable reaction pathway, this unique morphological dependence of hydrocarbon selectivity can be capitalized in the design and engineering of practical Cu‐based CO2RR electrocatalysts. For example, Nilsson and co‐workers reported a simple in‐situ synthesis of nanocube‐covered Cu surface having predominant (100) exposure for efficient and selective ethylene production.53 Its ethylene selectivity was measured to increase by more than two orders of magnitude compared to polycrystalline Cu with nearly complete suppression of the methane signal.
Particle size is also an important structural parameter for Cu‐based CO2RR electrocatalysts. Strasser and co‐workers discovered that Cu nanoparticles (NPs) exhibited dramatically enhanced total current density and higher selectivity toward CO and H2 as their particle size was decreased, particularly for those under 5 nm, while hydrocarbon selectivity was increasingly suppressed (Figure 5b–d).54 This experimental observation was rationalized by DFT calculations, which showed that smaller Cu NPs could provide more undercoordinated atoms as strong binding sites to key intermediates such as H and COOH, thus accelerating HER and the reduction of CO2 to CO while decreasing further recombination reaction to hydrocarbons. However, conflicting results were also disclosed by Alivisatos and co‐workers showing that Cu NPs (≈7 nm, grew to ≈25 nm during electrochemical experiments) exhibited an enhanced methanation current density four times greater than that of Cu foil, and an average Faradaic efficiency of 80% during extended electrolysis.55 The marked difference in reaction selectivity might be caused by the different synthetic approaches and measurement conditions employed.
Furthermore, surface roughening of Cu electrode materials is an effective route to promote their CO2RR performance. This is not only owing to the enlarged surface area but also due to the generation of a significant number of active surface sites such as edges, steps, and defects, which are suggested to have lower energy barriers for the formation of key CO2 reduction intermediates (e.g., CO and CHO).56 Indeed, evidence from thermal desorption spectroscopy demonstrated that CO bind considerably strongly onto Cu step edges, kinks, or defects than terrace sites.57 Surface roughening of Cu can be achieved in several different ways such as thermal annealing or electropolishing. Li and Kanan roughened Cu foils by first annealing them in air and then electrochemically reducing formed microthick Cu2O films to Cu nanoparticles (Figure 5e–g).48 Comparing to polycrystalline Cu, they observed that roughened Cu electrodes were capable of efficient CO2 reduction to CO and HCOOH at much lower overpotentials with greater current density and stability. In a follow‐up study, the same research group revealed that the roughening process generated a high density of grain boundaries which could support surface active sites normally unstable on individual nanoparticles.58 Cuenya and co‐workers employed facile and tunable plasma treatments to roughen Cu surfaces, and found that the optimal sample demonstrated a lower overpotential (−0.5 V vs RHE) and record selectivity (60% at −0.9 V) toward ethylene.59 Besides larger surface area and the increasing number of low‐coordinated sites, the authors suggested that the stable oxide layer formed during plasma treatment played a key role for enhancing the reaction activity and ethylene selectivity.
Aside from aforementioned electrode structural parameters, electrolyte cations or anions that were once thought to be idle may directly or indirectly impact the electrocatalytic process. It was not until very recently that their roles in CO2RR received careful investigations. Based on their experimental observation, Bell and co‐workers proposed that the pK a for cation hydrolysis decreased from Li+ to Cs+ in their bicarbonate electrolyte, and larger cations such as K+, Rb+, and Cs+ had pK a values sufficiently low that they could act as buffering agents and lower the local pH near the cathode, leading to increased ethylene selectivity and lowered Faradaic efficiency for H2 on electropolished Cu foils.60 The CO2RR performance of Cu can also be tuned with the addition of halide anions to the electrolyte. Strasser and co‐workers demonstrated that adding Cl− and Br− led to an increased CO selectivity compared with the halide‐free electrolyte.61 By contrast, in the presence of I– the CO selectivity declined and methane formation was enhanced up to six times. It was suggested that adsorbed I– anions on the cathode could favor the protonation of CO intermediate to CHO—a key step toward methane formation.
3.1.2. Gold
Among various bulk metals, Au generally exhibits the highest activity and selectivity for reducing CO2 to CO,45, 62 and has attracted much attention over the last five years. Current research efforts are mainly centered on the development of nanostructured Au materials with further improved CO2RR performance and the understanding of its possible reaction mechanism at the atomic level.
Like Cu NPs, the reaction activity and selectivity of Au NPs strongly depends on their particle size. Smaller NPs are not necessarily more desirable for electrocatalytic CO2RR. Sun and co‐workers prepared monodispersed Au NPs having diameters of 4, 6, 8, and 10 nm and found that the 8 nm Au NPs exhibited the optimal activity and Faradaic efficiency for CO (90% at −0.67 V vs RHE) (Figure 6 a–c).63 DFT calculations disclosed that edge sites on the Au NP surface facilitated CO formation by stabilizing key intermediates such as COOH while the corner sites were active for HER owing to their increased affinity toward H. The highest selectivity observed with 8 nm Au NPs was attributed to their optimal ratio of edge sites over corner sites. Similar size dependence was also reported by Cuenya and co‐workers for Au NPs of 1–8 nm.64 As the NP size decreased, the authors observed a dramatic increase in current density and a significant decline in CO selectivity. It was rationalized by the increasing low‐coordinated sites that were suggested to favor HER over CO2 reduction.
Figure 6.
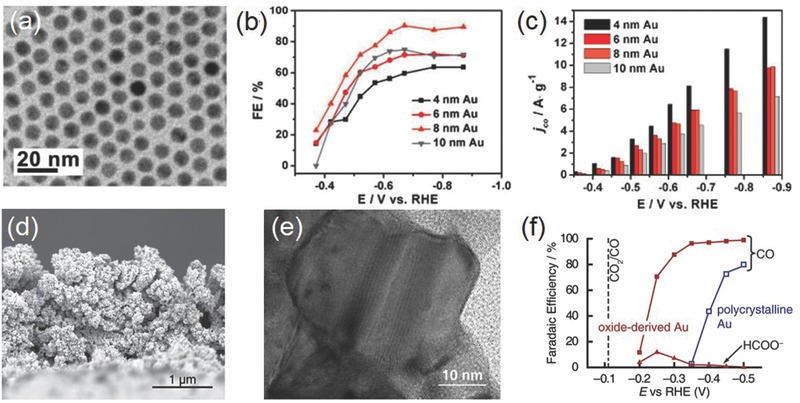
a) Transmission electron microscopy (TEM) image of 8 nm Au NPs; b) potential‐dependent Faradaic efficiency for CO on Au NPs with different sizes; c) current densities for CO formation at various potentials. Reproduced with permission.63 Copyright 2013, American Chemical Society. d) Cross‐sectional SEM image and e) high‐magnification TEM image of oxide‐derived Au NPs; f) Faradaic efficiency for CO and formate on oxide‐derived Au NPs in 0.5 m NaHCO3. Reproduced with permission.65 Copyright 2012, American Chemical Society.
The CO2RR activity of Au can be dramatically boosted by properly engineering its surface defects. Kanan and co‐workers prepared oxide‐derived Au by simply applying a periodic square‐wave potential routine on Au electrodes and then electrochemically reducing thick Au oxide films (Figure 6d–f).65 It was suggested that unique metastable structures were resulted from the reduction of the oxide film. This oxide‐derived Au exhibited selective CO2 reduction to CO in aqueous solution at overpotentials as low as 140 mV, high CO Faradaic efficiency (>80% at potentials more cathodic than −0.3 V), and stability for at least 8 h. Its exceptional activity was attributed to the surface metastable structures stabilizing the CO2 •− intermediate and accelerating the reduction process, although no detail was given on the nature of these metastable sites. The same research group later reported that grain boundaries were beneficial toward the CO2RR activity of Au.66 They uncovered that there was a linear correlation between the grain boundary surface density and specific activity for CO2 reduction on vapor deposited Au NPs on carbon nanotubes.
In addition to NPs, Au‐based materials with other nanoscale morphology have also been actively pursued for CO2RR. There is an enriched library of Au nanostructures with the precisely controlled geometry that provides the necessary material basis for electrochemical studies. For example, Sun and co‐workers prepared ultrathin Au nanowires by seed‐mediated growth method.67 The product was featured with dominant surface edge sites, and capable of highly efficient CO2 reduction to CO with low onset potential of −0.2 V versus RHE, high Faradaic efficiency of 94%, and mass activity of 1.84 A g−1 Au at −0.35 V. Stable Au concave rhombic dodecahedra were prepared by Nam and co‐workers by adding 4‐aminothiophenol during seed‐mediated growth to bind and stabilize various high‐index crystal planes such as (331), (221) and (553).68 Electrochemical measurement showed that concave rhombic dodecahedra exhibited improved CO selectivity (>80% between −0.8 and −0.4 V) and mass activity compared to Au films, cubes, or rhombic dodecahedra.
3.1.3. Silver
Ag is the second noble metal that can enable the highly selective reduction of CO2 to CO, but it is relatively less active than Au due to its intrinsically weaker binding toward reaction intermediates.45, 62, 69 For bulk Ag metal, Hori and co‐workers found that the electrocatalytic activity of CO2 reduction to CO was substantially faster on atomically stepped Ag (110) than that on flat Ag (100) or Ag (111).70 Such a dependence now is understood as Ag (110) binds COOH more strongly than other facets.69 In addition to CO and H2 as the major products, Jaramillo and co‐workers identified four minor reduction products including formate, methane, methanol, and ethanol.71 They found that H2 was dominant at low and high overpotentials, while CO overtook H2 in the medium overpotential region. Those minor products, on the other hand, only appeared at very negative potentials (←1.2 V vs RHE) since they involved the further reduction of CO intermediate that was only weakly adsorbed on Ag.
Nanostructured Ag is considerably more attractive than bulk Ag metal for CO2RR. Studies on Ag NPs by Masel and co‐workers showed an increasing CO2RR current density as their size decreased from 200 to 5 nm.72 However, the current density significantly dropped if the size further decreased to 1 nm. The authors interpreted their results as a consequence of variations in the binding energy of intermediates when the particle size decreased. Nevertheless, a different view was held by Jung and co‐workers proposing that the highest CO2RR activity for 5 nm Ag NPs was due to the optimal ratio of edge sites that were calculated to be the most CO2RR active—like in the case of Au NPs.69 Porous Ag also represents a popular choice of materials. Smith and co‐workers reported that the oxide‐derived Ag electrode from the anodization of Ag foil in alkaline solutions had a highly porous structure (Figure 7 a,b).73 It attained ≈80% Faradaic efficiency for reducing CO2 to CO at a moderate overpotential of 0.49 V, much enhanced than untreated polycrystalline Ag (≈4%) under identical conditions. This improvement was likely correlated with the nanostructured surface populated with highly active sites for stabilizing COOH intermediate as well as a high local pH arising from porosity‐induced transport limitation. A nanoporous silver was synthesized by Jiao and co‐workers from two‐step dealloying of an Ag‐Al precursor.74 Electrochemical measurements demonstrated that it was capable of reducing CO2 to CO with ≈92% selectivity at a rate >3000 times higher than bulk Ag under moderate overpotentials (<0.5 V). These authors likewise attributed this superior activity to the curved surface having a high density of step sites with possibly higher‐index facets that stabilized CO2 •− intermediate, therefore lowering the thermodynamic barrier for its reduction. More recently, Surendranath and co‐workers developed mesostructured Ag inverse opal electrodes and showed that with the increasing electrode mesostructural roughness, its specific activity for CO2 reduction to CO systematically rose by threefold and that for catalyzed HER systematically declined by tenfold (Figure 7c,d).75 They suggested that the mesostructured‐induced transport limitation was the primary cause for the possibility of tuning both catalyst selectivity and efficiency.
Figure 7.
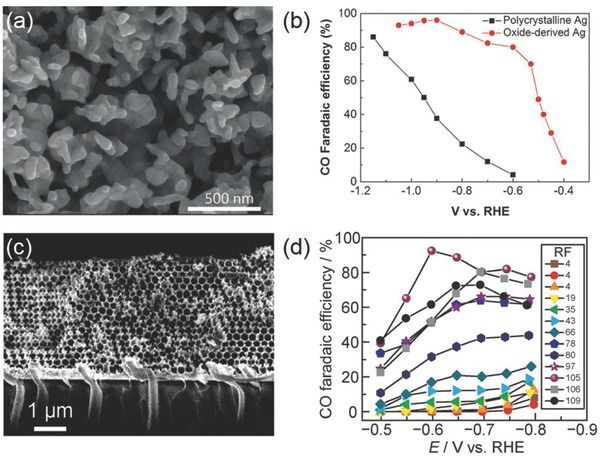
a) SEM image of oxide‐derived Ag; b) Faradaic efficiency for CO on polycrystalline Ag and oxide‐derived Ag. Reproduced with permission.73 c) Cross‐sectional SEM image of an Ag‐IO film; d) potential‐dependent Faradaic efficiency for CO on Ag films with varying roughness factors. Reproduced with permission.75
3.1.4. Tin
According to Hori et al., bulk Sn metal electrode requires large overpotential (>0.86 V) in order to generate moderate current density (5.0 mA cm−2) for CO2 reduction to formic acid with the Faradaic efficiency of 88.4%.45 It is worth noting that in contrast to Au and Ag, the surface of Sn (particularly nanostructured Sn) rapidly gets oxidized upon exposure to air. This surface oxide layer may not be fully reduced even during CO2RR electrocatalysis, and therefore would greatly influence the electrochemical process. In order to elucidate the possible effect of the surface oxide layer, Kanan and co‐workers compared the activity of an Sn electrode with native SnOx layer and an electrode etched to expose metallic Sn0 surface (Figure 8 a–c).65 The latter exhibited higher overall current densities but almost exclusively H2 over the entire potentials range examined. It was proposed that SnOx directly participated in CO2RR pathway by stabilizing CO2 •− intermediate, otherwise HER dominated because the electron transfer to CO2 was prohibitively slow on metallic Sn. Zhou and co‐workers further explored the dependence of CO2RR selectivity on the surface SnOx thickness and uncovered that the electrode with an initial SnOx thickness of ≈3.5 nm delivered the maximum Faradaic efficiency of 64% for formate while CO production reached its highest Faradaic efficiency of 35% with an initial SnOx layer thickness of 7.0 nm.76 Further thickening the oxide layer resulted in a heightened HER rate. Even though the surface oxide layer cannot be not fully reduced during CO2RR, their partial reduction may result in the formation of a high density of grain boundaries that are usually beneficial to CO2RR as demonstrated by Spurgeon and co‐workers.77
Figure 8.
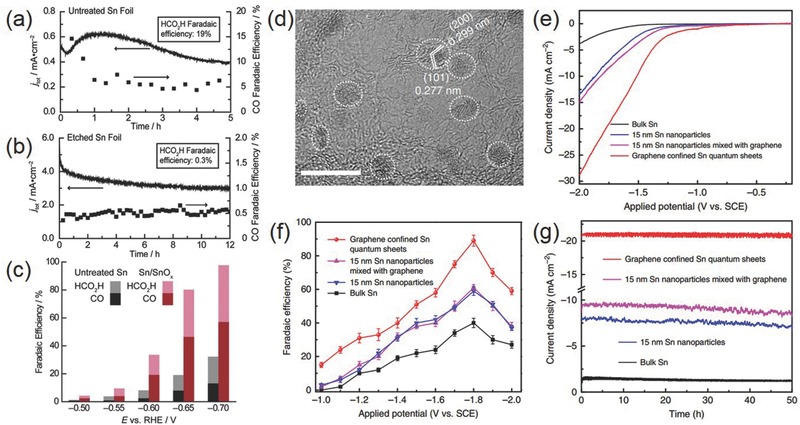
a,b) Change of the total current density and CO Faradaic efficiency with time on a) untreated Sn and b) etched Sn at −0.7 V versus RHE in 0.5 m NaHCO3; c) their potential‐dependent Faradaic efficiency for CO and formic acid. Reproduced with permission.65 Copyright 2012, American Chemistry Society. d) High‐magnification TEM image of Sn quantum sheets confined in graphene; e) polarization curves, f) potential‐dependent Faradaic efficiency for formate, and g) chronoamperometry stability at −1.8 V versus SCE on Sn quantum sheets confined in graphene as well as several control samples in 0.1 m NaHCO3 aqueous solution. Reproduced with permission.79 Copyright 2016, Nature Publishing Group.
Hybridizing Sn or SnOx nanoparticles with conductive carbon materials is proven a successful strategy to promote the CO2RR performance. Meyer and co‐workers prepared ≈5 nm SnO2 NPs uniformly deposited on graphene.78 CO2 reduction to formate was found to take place at overpotentials as low as ≈340 mV and with Faradaic efficiency of >93% at −1.8 V versus SHE in 0.1 m NaHCO3 aqueous solutions. This notable activity was believed to arise from the compromise between the adsorption strength of CO2 •− and its subsequent kinetic activation on the nanoscale Sn surface as well as the electronic interactions between the graphene support and metal NPs. Xie and co‐workers recently showed that metallic Sn quantum sheets confined in graphene displayed a large current density of 21.1 mA cm−2, Faradaic efficiency of ≈90% and great stability for over 50 h at −1.8 V versus saturated calomel electrode (SCE) for selective CO2 reduction to formate (Figure 8d–g).79 It was suggested as the collective result of the highly conductive graphene support facilitating the rate‐determining electron transfer step from CO2 to CO2 •− and the graphene confined Sn quantum sheets stabilizing the CO2 •− radical with its low‐coordinated sites.
3.1.5. Other Metals
There are many other metals investigated for CO2RR electrocatalysis. Pd can reduce CO2 to CO but its Faradaic efficiency is generally much lower than that on Au or Ag due to the competing HER.45, 80 One possible remedy to this poor selectivity is to control its particle size below 5 nm for optimal edge and corner site density.81 Occasionally, formate is identified as the main reduction product on Pd as probably mediated by the surface PdHx.82 There are also a handful of reports about Zn‐, In‐, or Bi‐based materials, mostly prepared from electrodeposition, for selectively reducing CO2 to CO.83, 84, 85, 86, 87, 88, 89 Even though Co is commonly regarded as HER‐active, partially oxidized Co ultrathin layers surprisingly reduce CO2 to formate at a very high Faradaic efficiency.90, 91 Besides pure metals, alloying of two different metals is explored so as to tune the binding strength of key intermediates through geometric and electronic effects, and therefore optimize the CO2RR reaction activity and selectivity. Thus far, most research attention has been focused on Cu‐based alloys, such as Cu–Au, Cu–Pd, Cu–In and Cu–Pt and Cu–Sn, and some showed very exciting results.92, 93, 94, 95, 96 It can be reasonably expected that the alloying strategy would soon be expanded to other combination for better possibilities.
3.2. Metal Chalcogenides
Layered transition metal dichalcogenides (TMD) such as MoS2, MoSe2, and WS2 have been widely investigated as the HER electrocatalysts.97 However, it is not until recent years that their potential in CO2RR starts to be unveiled. Nørskov and co‐workers used DFT calculations to explore the binding properties of CO2RR intermediates (COOH, CHO, and CO) on MoS2 and MoSe2 edges.98, 99 COOH and CHO were found to prefer bridging S or Se atoms, while CO was selectively adsorbed on the metal atom. These authors argued that in this way, the active edges may break the scaling relations observed between intermediates on transition metals, making them potentially more attractive for CO2RR than even the best transition metals. Because TMD materials are excellent HER electrocatalysts, CO2RR of TMD materials usually have to be carried out in mixture solution of ionic liquid (e.g., EMIM‐BF4) and water in order to suppress HER. Some ionic liquids are reported to form a stable complex with the CO2 •− intermediate so that they can lower the activation energy barrier for effective CO2 reduction.100
In 2014, Salehi‐Khojin and co‐workers first experimentally demonstrated bulk MoS2 as a highly efficient electrocatalyst for selectively reducing CO2 to CO with a small overpotential of 54 mV in a mixture of 96 mol% water and 4 mol% EMIM‐BF4.101 An impressive cathodic current density of ≈65 mA cm−2 and a CO Faradaic efficient of 98% were delivered at η = 0.65 V, much improved than Au and Ag NPs. In a follow‐up study, the same research group compared the CO2RR performance of four different TMD materials (MoS2, MoSe2, WS2, and WSe2) with similar sizes and identified that WSe2 nanoflakes were the most active.102 At η = 54 mV, it exhibited an exceptional current density of ≈19 mA cm−2, CO Faradaic efficiency of 24%, and TOF of 0.28 s−1 in 50:50 vol% ionic liquid/water electrolyte; at η = 0.65 V, the recorded current density for WSe2 reached 330 mA cm−2 with a Faradaic efficiency of ≈85%—an unprecedented activity surpassing any other known CO2RR electrocatalysts (Figure 9 ). DFT calculations revealed that unlike transition metal surfaces, COOH formation on TMD edges was exergonic, and CO was also stabilized, indicating the formation of CO from CO2 was kinetically favorable. The authors also built a proof‐of‐concept artificial leaf by coupling Si photovoltaic cells with oxygen evolution reaction (OER) and CO2RR electrocatalysts in a wireless configuration and achieved concurrent production of CO and oxygen under light illumination. Moreover, proper doping of TMD materials may further push their performance to the limit. 5% Nb doped vertically aligned MoS2 was found to exhibit the smallest onset overpotential of 31 mV, and one order of magnitude higher CO formation TOF than pristine MoS2 within an overpotential range of 50–150 mV.103 The presence of Nb was suggested to facilitate the rapid release of CO from the TMD edge. Despite these exciting progresses on this family of materials, it should be noted that their excellent CO2RR activities are yet to be confirmed by other research groups.
Figure 9.
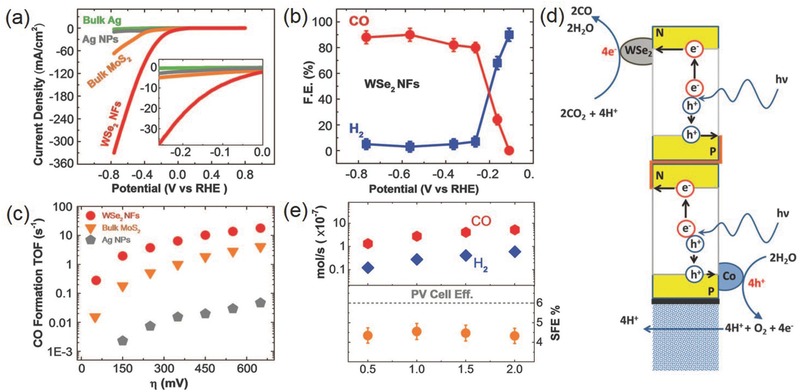
a) CV curves of WSe2 NFs, bulk MoS2, Ag NPs, and bulk Ag in CO2‐saturated EMIM‐BF4/H2O solution; b) potential‐dependent Faradaic efficiency for CO and H2 on WSe2 NFs; c) CO formation TOF of WSe2 NFs, bulk MoS2, and Ag NPs; d) schematic showing an artificial leaf with WSe2 cocatalyst for reducing CO2 to CO under light illumination. e) Product formation rates under different light illumination intensities using the WSe2/IL cocatalyst system. Reproduced with permission.102 Copyright 2016, American Association for the Advancement of Science.
CO2 may also be reduced to liquid products on metal chalcogenides. Han and co‐workers recently showed that Mo‐Bi sulfide could reduce CO2 to methanol in 0.5 m BMIM‐BF4 in acetonitrile.104 Its Faradaic efficiency reached a record value of 71.2% with a current density of 12.1 mA cm−2 at −0.7 V versus SHE. The synergy between Mo and Bi was speculated as the main origin of its high activity, where Bi favored the transformation of CO2 to CO, and Mo promoted the further hydrogenation of CO to methanol.
3.3. Carbonaceous Materials
Carbonaceous materials are also applied for CO2RR electrocatalysis by virtue of their good electrical conductivity, low cost, chemical stability and usually large surface area. However, pristine carbonaceous materials are generally inert and have negligible activity for CO2RR because they can hardly activate CO2 molecule or adsorb CO2 •− intermediate.105, 106 It is a different scenario when carbonaceous materials are properly doped with heteroatoms (e.g., N, B, P, and S). Doping introduces structural defects or induces charge/spin densities on the adjacent carbon atoms, therefore significantly altering the interaction between carbonaceous materials and CO2 or reaction intermediates.
Up to now, various forms of heteroatom functionalized carbonaceous materials including carbon nanotubes, nanofibers, graphene sheets, graphene quantum dots and nanodiamonds have been pursued and investigated in CO2 reduction. Salehi‐Khojin and co‐workers prepared N‐doped carbon nanofibers (CNFs) from pyrolyzing electrospun polyacrylonitrile precursor.107 It exhibited a small onset overpotential of 170 mV for selectively reducing CO2 to CO in EMIM‐BF4 and delivered more than an order of magnitude higher current density than bulk Ag under similar experimental conditions. The electrocatalytically active sites were proposed to be highly positive carbon atoms adjacent to electronegative N dopants. Liquid C1 products are identified from the CO2RR on some carbonaceous materials. For example, Einaga and co‐workers discovered the sp3‐bonded carbon in B‐doped diamond (BDD) electrode selectively reduced CO2 to formaldehyde with Faradaic efficiency up to 74% in methanol or seawater, even though the current density was considerably low (<0.3 mA cm−2).108 Meyer and co‐workers achieved Faradaic efficiency for formate production of 87% with current densities of 9.5 mA cm−2 at −1.8 V versus SCE on polyethylenimine‐enhanced N‐doped carbon nanotubes in 0.1 m KHCO3 solution.105 In addition, C2–C3 products are also obtained. Quan and co‐workers observed that N‐doped nanodiamond (NDD) demonstrated Faradaic efficiency of ≈77% toward acetate production and ≈15% toward formate formation at a potential range of −0.8–−1 V versus RHE.109 It was the first example of CO2RR electrocatalysts other than Cu that were able to convert CO2 to a C2 product with such high selectivity. Using N‐doped graphene quantum dots, Ajayan and co‐workers reported that CO2 could be reduced into multicarbon hydrocarbons and oxygenates with total Faradaic efficiency of CO2 reduction of up to 90% and selectivity for ethylene and ethanol reaching 45%.110 The C2 and C3 product distribution and production rate was comparable to those obtained with Cu‐based electrocatalysts.
3.4. General Material Designing Strategies
Based on the above discussion, a few general material designing strategies can be identified across various CO2RR electrocatalysts and are summarized as follows. (1) Nanostructuring: Nanostructuring so far is the most common and popular approach to promote the performance of catalyst materials.111, 112 When the size of catalyst particles is reduced down to the nanoscale, their surface areas are dramatically enhanced, which more than often leads to heightened activities. However, one thing worth noting here is that unlike most other catalytic reactions, smaller particle size is not always beneficial to CO2RR as we have seen for Cu, Au, and Ag metals. This is because too small particles have an increasing number of low‐coordinated sites that favor HER over CO2RR. Apart from the particle size, we now are able to precisely tune the morphology of catalyst materials at the atomic scale with the assistance of nanotechnology, and many delicate nanostructures such as nanocubes, nanosheets, nanowires and so on can be readily prepared by carefully controlling the reaction conditions. This brings us the possibility to explore the morphology dependence of their catalytic activity and selectivity and to gain deeper insight into their structure–property correlation. Moreover, nanostructuring may also introduce structural defects such as vacancies and grain boundaries with local chemical environment and electronic structure distinct from the bulk. They may serve as the CO2RR active sites with unexpected performance. (2) Doping or alloying: Doping or alloying with foreign atoms is an effective way to adjust the binding energy of CO2 and key reduction intermediates on catalyst surfaces, and thus has the great potential to modulate the CO2RR activity and selectivity. The proper choice of dopant types, doping level or alloying composition can be guided by DFT‐based computations. Cu is the only elemental metal capable of producing C1–C3 hydrocarbons at significant rates due to its suitable binding energy toward the CO intermediate. Stronger CO binding (on catalysts such as Fe, Ni, Co, and Pt) encourages HER, whereas weaker CO binding (on catalysts such as Sn, Pb and Hg) favors formate formation.113, 114 By combing metals having strong CO binding property with metals having weak CO binding property, one may arrive at an intermediate CO‐catalyst interaction that promotes the formation of higher‐value reduction products. (3) Hybridization with carbon: Electrocatalysis relies on the conduction of electricity to drive catalytic reactions. A high surface area and conductive carbon support (such as graphene, carbon nanotubes, carbon fibers, and so on) would not only greatly benefit the electron transport to and from electrocatalysts but also improve their dispersion. The synergy between the support and the electrocatalyst may also afford an unexpected gain in activity and selectivity. However, care should be taken when designing such hybrid electrocatalysts since many carbon materials may contain metal impurities (such as Fe, Co, and Mo that are often used as the catalyst seed for the growth of carbon nanotubes) that promote the HER side reaction.
4. Photocatalytic Materials for CO2 Reduction
In addition to electrical energy, light can also provide the necessary energy stimulus in order to break the C=O bond in CO2. Since the first report about the photocatalytic application of TiO2 by Fujishima and Honda in 1972,115 there have been increasing research activities on semiconductor photocatalytic materials, initially for water splitting and more recently for CO2 reduction. A number of strategies have been developed and implemented by engineering their structures at different scales so as to promote their efficiency in light absorption, charge separation and interfacial charge transfer. In this part, we aim to review different photocatalysts (Table 3 ) and their design strategies and to highlight our current understanding of these complex systems.
Table 3.
Summary of CO2 reduction photocatalysts from recent literature
| Photocatalyst | Cocatalyst | Light source | Experimental condition | Main products and highest yield | Reference |
|---|---|---|---|---|---|
| Anatase TiO2 (0.1 g) | – | 300 W Xe lamp | CO2 and H2O vapor | CH4 1.35 µmol h−1 g−1 | 199 |
| TiO2 single crystals (0.02 g) | Pt | 400 W Xe lamp | CO2 and H2O vapor | CH4 1361 µmol h−1 g−1 (QE (CH4) = 2.41%). | 211 |
| Commercial P25 (1.25 cm2) | Au–Cu nanoalloys | Sun simulated light (1000 W Xe lamp) | CO2 and H2O vapor | CH4 2300 µmol h−1 g−1 | 212 |
| Rutile TiO2 modified anatase TiO2 nanorods (0.1 g) | – | 300 W Hg lamp | CO2 and H2O vapor | CH4 2.5 µmol h−1 g−1 | 230 |
| Degussa P25 (0.05 g) | Cu2+, Cu+, and Cu+/Cu0 | 150W solar simulator | CO2 and H2O vapor | CO 25 µmol g−1, CH4 25 µmol g−1 | 231 |
| Defective TiO2 (anatase, rutile, and brookite) (0.1 g) | – | A 150 W solar simulator | CO2 and H2O vapor | CH4 17 µmol g−1 | 120 |
| TiO2 (0.1 g) | Ag | 8 W Hg lamp | CO2 bubbled solution | CH4 9 µmol g−1, CH3OH 1.8 µmol g−1 | 232 |
| Anatase TiO2 nanosheets exposed with 95% of {100} facets (0.04 g) | – | 300 W Xe lamp | CO2 and H2O vapor | CH4 5.8 ppm g−1 h−1 | 233 |
| Codoped TiO2 (0.1 g) | – | 300 W Xe lamp (λ > 420 nm) | CO2 and H2O vapor | CO 1.9, CH4 0.09 µmol h−1 g−1 | 174 |
| TiO2 (0.4 g) | Ag | 500 W Xe lamp (λ > 420 nm) | CO2 saturated H2O | CH3OH 400 µmol g−1 (3 h) | 234 |
| Defective single‐unit‐cell BiVO4 layers (0.2 g) | – | 300 W Xe lamp (AM1.5) | CO2 saturated water | Methanol 398.3 µmol h−1 g−1, | 131 |
| BiVO4 and CuGaS2 (0.05 g) | CoOx/Pt | 300 W Xe lamp (λ > 420 nm) | CO2 saturated K2SO3 solution | CO 6 µmol h−1 g−1 | 220 |
| ZnAl2O4‐modified ZnGaNO (0.1 g) | Pt | 300 W Xe‐lamp (λ > 420 nm) | CO2 and H2O vapor | CH4 9.2 µmol h−1 g−1 | 148 |
| Ni doped anatase TiO2 (0.5 g) | – | 18 W cm−2 Hg lamp | CO2 saturated water | CO 14 µmol g−1 | 235 |
| AgBr/TiO2 (0.5 g) | – | 150 W Xe lamp (λ > 420 nm) | CO2 saturated KOH solution | CH4 128.56, CH3OH 77.87, C2H5OH 13.28, CO 32.14 mol g−1, | 236 |
| Titanate nanosheet‐assembled Yolk@Shell Microspheres (0.1 g) | – | 150 W Xe lamp (λ > 420 nm) | CO2 saturated water (NaHCO3+HCl) | CH3OH 2.1 µmol h−1 g−1 | 185 |
| Graphene‐Ti0.91O2 hollow spheres (0.01 g) | – | 300 W Xe‐lamp | CO2 and H2O vapor | CO 9, CH4 1 µmol h−1 g−1 | 184 |
| Nifion coated TiO2 particles (unspecified) | Pd (1 wt%) | 300 W Xe‐lamp | CO2 saturated Na2CO3 | CH4 6, C2H6 5 µmol h−1 | 237 |
| Degussa P25 (unspecified) | Pt‐Cu2O | 200 W Xe lamp (λ = 320–780 nm). | CO2 and H2O vapor | CH4 33, CO 8.3, H2 25 µmol h−1 g−1 | 238 |
| Sandwich‐like graphene‐TiO2 hybrid sheets (0.1 g) | – | 300 W Xe‐lamp | CO2 and H2O vapor | C2H6 16.8, CH4 8 µmol h−1 g−1 | 239 |
| Porous silica supported Cu/TiO2 catalysts (0.1 g) | – | Xe‐lamp 2.4 mW cm−2 | CO2 and H2O vapor | CO 45, CH4 13.2 µmol h−1 g−1 (QE (CO2) = 1.41%) | 240 |
| SrTiO3/TiO2 coaxial nanotube arrays (0.005 g) | Au–Cu Alloy NPs | 300 W Xe lamp | CO2 bubbled N2H4·H2O solution | CO 165 ppm cm−1 h−1 | 241 |
| TiO2/ZnO powder (0.1 g) | – | 300 W Xe lamp (60 mW m−2) | CO2 and H2O vapor | 55 µmol h−1 g−1 | 242 |
| In doped anatase TiO2 | – | 400 W Hg lamp | CO2, He, and H2O vapor | CH4 243.75, CO 81.25 µmol h−1 g−1 | 243 |
| Anatase TiO2 single crystals with {101} facets (0.1 g) | RuO2 | 300 W Xe lamp | CO2 and H2O vapor | CH4 1.8, H2 80, O2 15 µmol h−1 g−1 | 244 |
| Graphene–WO3 nanobelt (0.1 g) | – | 300 W Xe lamp (λ > 400 nm) | CO2 and H2O vapor | CH4 0.1, O2 3.5 µmol h−1 | 245 |
| WO3 (0.1 g) | – | 300 W Xe lamp | CO2 and H2O vapor | CH4 16 µmol g−1 | 126 |
| Nb3O8‐nanosheets (unspecified) | amorphous Cu clusters | Hg‐Xe lamp (240–300 nm) | 0.5 m KHCO3 aqueous solution (PH = 12) | CO 0.07 µmol h−1 | 246 |
| Defective single‐unit‐cell BiVO4 (0.2 g) | – | 300 W Xe lamp | CO2 and H2 vapor | CH3OH 398.3 µmol g−1 h−1 (AQE = 5.95%, 350 nm) | 131 |
| Single unit cell Bi2WO6 (0.2 g) | – | 300 W Xe lamp (100 mW cm−2) | CO2 and H2O vapor | Methanol 502 µmol g−1 h−1 | 183 |
| NaTaO3 (0.07 g) | Pt or Ru | 300 W Xe lamp (λ > 200 nm) | CO2, H2O, and H2 vapor | Pt/NaTaO3 (CO 139.1 µmol g−1 h−1) | 247 |
| Single‐crystalline Zn2GeO4 nanobelts | RuO2/Pt | 300 W Xe lamp | CO2 and H2O vapor | CH4 6 µmol h−1 g−1 | 134 |
| Porous Ga2O3 (0.05 g) | – | 300 W Xe lamp | CO2 and H2O vapor | CH4 (170 ppm) CO quantum yield 3.993% | 248 |
| Mesoporous ZnGa2O4 (0.1 g) | RuO2 | 300 W Xe lamp | CO2 and H2O vapor | 249 | |
| In2O3 nanobelts coated with carbon layer (0.2 g) | Pt | 300 W Xe lamp | CO2 saturated H2O (10% triethanolamine (TEOA)) | CO, 126.6 CH4 µmol h−1 | 250 |
| Cu2O/reduced graphene oxide (RGO) (0.5 g) | – | 300 W Xe lamp | CO2 and H2O vapor | CO 50 ppm h−1 g−1 | 251 |
| Nitrogen doped ZnO (0.01 g) | Cu | 8 W fluorescent tube | CO2 and H2O vapor molar ratio of 6.7 (CO2:H2O) | CO (0.73 µmol h−1 g−1), CH3OH, CH4, H2 | 252 |
| CeO2 (0.1 g) | Pt | 300 W Xe lamp | CO2 and H2O vapor | CH4 1.12 µmol h−1 g−1. | 253 |
| Ni/SiO2·Al2O3 (1.5 cm2) | – | Solar simulator | CO2, N2, H2 vapor | CH4 (highest selectivity 99.9%), CO, C2H6, | 254 |
| Co3O4 with exposed {112} facets ([Ru(bpy)3]Cl2 as a photosensitizer) (0.01 g) | – | 300 W Xe lamp (λ > 420 nm) | CO2 saturated acetonitrile/TEOA/H2O (3:1:1) solution | CO 1297, H2 502 µmol g−1 h−1 | 255 |
| Graphene oxide (GO)‐CdS nanorods (0.01 g) | – | 300 W Xe lamp (λ > 420 nm) (150 mW cm−2) | CO2 and H2O vapor | CH4 2.51 µmol h−1 g−1 | 256 |
| Cu2S nanorod (unspecified) | Pt | 450 W Xe lamp | 1 m Na2CO3 | CO 3.02, CH4 0.13 µmol h−1 g−1. | 257 |
| Bi2S3 (0.01 g) | – | 250 W Hg lamp | CO2 saturated methanol | HCOOH 700 µmol g−1 (4 h) | 139 |
| GaN nanowire arrays (3.5 cm2) | Rh/Cr2O3 | 300 W Xe lamp | CO2 and H2O vapor | CH4 ≈3.5 µmol g−1 h−1 in 24 h. | 144 |
| MgAl layered double oxide (LDO) grafted TiO2 (0.1 g) | – | 450 W Xe lamp (λ > 400 nm) | CO2 and H2O vapor (reaction temperature at 150 °C) | CO 1 µmol h−1 g−1 | 258 |
| Mg doped InGaN/GaN nanowire (3 cm2) | Pt | 300 W Xe lamp (AM1.5 G filter) | CO2 and H2 vapor (1:4) | CH3OH 500 µmol g−1 h−1 | 145 |
| ZnCu–M(III) (M = Al, Ga) LDH (0.1 g) | – | 500 W Xe lamp | CO2 and H2 vapor | CH3OH 0.49, CO 0.62 µmol h−1 g−1 | 155 |
| Ni/Mg/Zn‐Ga/Al/In‐LDH (0.1 g) | – | 200 W Hg‐Xe lamp | CO2 and H2 vapor | CO 3.21, O2 17 µmol h−1 g−1 | 158 |
| MgAl‐LDH (unspecified) | Pd | 500 W Xe lamp | CO2 saturated water | CH4 3.7 µmol | 259 |
| Defect rich Zn‐Al LDH nanosheet (0.1 g) | – | 300 W Xe lamp | CO2 and H2 vapor | CO 8 µmol h−1 g−1 | 159 |
| C3N4 (0.008 g) | C3N4 | 400 W Hg lamp | Solution of 4:1 v/v solvent (MeCN, N,N′‐dimethylacetamide (DMA), MeOH, or water):TEOA | HCOOH, TON(>1000), AQY (5.7%, 400 nm) | 172 |
| Graphene–g‐C3N4 hybrid (unspecified) | – | 15 W daylight bulb (8.5 mW cm−2) | CO2 and H2O vapor | CH4 5.87 µmol g−1 | 260 |
4.1. Compositions of Semiconductor Photocatalysts
4.1.1. Metal Oxide
Metal oxides are a very common type of photocatalyst materials for CO2 reduction. A large number of them consist of transition metal cations (e.g., Ti4+, Zr4+, Nb5+, Ta5+, W6+, and Mo6+) with the d 0 configuration. Their conduction bands are composed of vacant metal d orbitals and usually more negative than 0 V, while their valence bands are composed of O 2p orbitals and usually more positive than 3 eV.116 The band structure of these metal oxides can generally enable the simultaneous CO2 reduction and water oxidation, but their wide band gap more than often restricts the utilization of solar spectrum only within the UV region.117, 118 TiO2 is the most representative and well‐studied d 0 metal oxide semiconductor photocatalyst with the advantages of low cost, low toxicity, and chemical stability.119 Among its three polymorphs that naturally exist, the anatase form of TiO2 receives wide attention and is shown to be highly active in photocatalytic CO2 reduction.16 Comparatively, rutile is less active due to its fast charge recombination, and brookite, on the other hand, is rarely investigated for photocatalysis probably due to the past difficulty in obtaining phase‐pure brookite. However, increasing evidence now suggests that pure brookite has a high activity for photocatalytic CO2 reduction, and oxygen‐deficient brookite is even more appealing than anatase due to its enhanced interaction with and charge transfer to the CO2 molecule (Figure 10 a).120, 121 The photocatalytic activity of TiO2 also depends on exposed crystal facets. Even though thermodynamically less stable than other low‐index facets such as {101} and {001}, the {010} facet of anatase TiO2 is shown to be more active (Figure 10b), presumably benefited from its more favorable surface atomic structure having 100% five‐coordinated Ti atoms and slightly more negative conduction band.122, 123, 124 DFT calculations also predict that the interaction of CO2 with {010} was stronger than its interactions with {101} or {001}.125 Apart from TiO2, other transition metal oxides such as WO3 and ZrO2 as well as oxysalts such as titanates (ATiO3, A = Na, Sr, Ca, or Pb), tantalates (ATaO3, A = Li, Na, or K), niobates (ANbO3, A = Na, K), tungstate (Bi2WO6), and vanadates (BiVO4, InVO4, Na2V6O16·xH2O) have also been explored in photocatalytic CO2 reduction.126, 127, 128, 129, 130, 131, 132 Some of them show considerable visible light activity. Furthermore, many main group metal oxides with metal cations (e.g., In3+, Ga3+, Ge4+, Sn4+, and Sb5+) in the d 10 configuration are also photocatalytically active. Their conduction bands consist of hybridized sp orbitals with large dispersion and are able to provide photogenerated electrons with high reducing power.116, 118 Unfortunately, their higher conduction bands translate to further broadened band gaps. For example, two popular examples—Zn2Ga2O4 and ZnGeO4 have band gaps of 4.5 and 4.4 eV, respectively, making them only responsive to the deep UV light.133, 134
Figure 10.
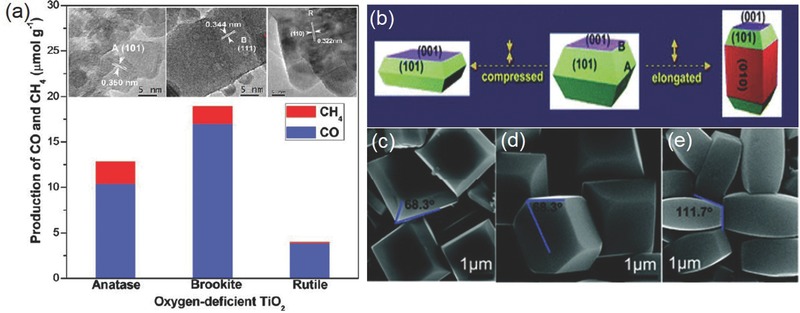
a) Production rates of CO and CH4 on three TiO2 nanocrystal polymorphs (anatase, rutile, and brookite). Reproduced with permission.120 Copyright 2012, American Chemical Society. b) Schematic of anatase TiO2 with different percentages of {101}, {001}, and {010} facets and c–e) SEM images of corresponding synthetic products. Reproduced with permission.122
4.1.2. Metal Sulfide
Metal sulfides represent another large group of photocatalyst materials for CO2 reduction. Compared to their oxide counterparts, metal sulfides possess higher valence bands mainly of the S 3p character and have narrower band gaps. It is, however, a general concern that photogenerated holes on their valance band may not be energetic enough to oxidize water and would instead result in their irreversible photocorrosion. As a result, hole scavengers are frequently added in order to extend their stability. CdS is a well‐known visible light photocatalyst having a band gap (2.4 eV) that matches well with the solar spectrum.116 In 1988, Eggins et al. first reported the photocatalytic performance of CdS for CO2 reduction under visible light, yielding formaldehyde, methanol, formate, acetate, and glyoxylate as the main products.135 Wang and Wang recently coupled CdS with Co‐ZIF‐9 as the cocatalyst and achieved the conversion from CO2 to CO with a high apparent quantum yield of 1.93% under monochromatic irradiation of 420 nm.136 ZnS also attracts considerable attention for photocatalytic CO2 reduction. Its conduction band has very high energy and extremely low redox potential (−1.8–−2.0 V vs SHE) and can enable the one‐electron reduction of CO2 to CO2 •− as confirmed by electron paramagnetic resonance spin‐trapping experiments.137 Macyk and co‐workers demonstrated that ZnS nanoparticles functionalized with Ru cocatalyst photocatalyzed CO2 reduction to formic acid and CO with traces of methane.137 The photocatalytic activity and selectivity were found to depend on nanoparticle size and solvent polarity. The same research group also showed that when coupled with the oxidation of acetylacetone by photogenerated holes from its valence band, photocatalytic CO2 reduction on ZnS would lead to the formation of carboxylic acids.138 In addition to above compounds, other sulfides and solid solutions were also investigated.139, 140, 141, 142
4.1.3. Metal Nitride
Metal nitrides or oxynitrides also have narrow band gaps due to their high valence bands mainly composed of N 2p orbitals. Despite their desirable visible light absorbance, little photocatalytic activity, however, was generally observed for d 0 transition metal nitrides and oxynitrides such as Ta3N5, TaON, MTaO2N (M = Ca, Sr, Ba and La) probably due to their low conduction band edge. It is suggested that d 10 metal with broadly dispersed conduction bands are more promising toward photocatalytic CO2 reduction.116, 143 Gallium‐based nitrides are the most studied. Mi and co‐workers reported that GaN nanowires reduced CO2 to CH4 and CO at a high conversion rate using sunlight as the only energy input.144 Decoration of the nanowire surface with Rh/Cr2O3 or Pt cocatalyst nanoparticles enhanced the reaction rate and selectivity toward CH4, reaching ≈3.5 and ≈14.8 µmol gcat −1 h−1 for CH4, respectively. In their follow‐up study, these authors employed multiband InGaN/GaN nanowires to realize the rapid transformation of CO2 to methanol.145 The photocatalytic activity was further boosted with the incorporation of Mg‐dopant, which was believed to promote CO2 adsorption and reduce surface potential barrier based on DFT calculations. The optimal conversion rate was measured to be 0.5 mmol gcat −1 h−1 with high stability over 10 h under visible light illumination (>400 nm). It is worth noting that compared to sulfides, GaN is known for its improved resistance to photocorrosion so that no hole scavenger is necessary. Other nitrides and oxynitrides investigated for photocatalytic CO2 reduction include ZnGeNO and ZnGaNO.146, 147, 148
4.1.4. Layered Double Hydroxide (LDH)
LDH with the general formula of [M2+ 1− xM3+ x(OH)2][An − x / n·mH2O] (where M2+, M3+, and An− are divalent cation, trivalent cation, and interlayer anion, respectively) is a class of layered materials comprising positively charged metal hydroxide layers and charge‐balancing anions between the layers.149 Many LDH compounds (such as Ti‐based LDHs) are known to be excellent photocatalysts for water splitting.150, 151, 152 Their potential in photocatalytic CO2 reduction was first uncovered by Izumi's group in 2011.153 Up to now, LDHs containing Zn2+, Cu2+, Mg2+, and Ni2+ combined with Al3+, Ga3+, Cr3+, and In3+ are shown to be active for reducing CO2 to CO or methanol as main products.154 Inclusion of Cu ions within the host layers or replacing the interlayer anions with cuprous anions generally improves the selectivity for methanol over CO production.155 For example, a methanol selectivity as high as 68% was reported using ZnCuGa‐CO3 LDH.156 Tanak and co‐workers compared the photocatalytic activity of several different LDH materials (composed of divalent Ni/Mg/Zn and trivalent Al/Ga/In) in aqueous solution and concluded that Ni‐Al LDH had the highest activity (110.9 µmol in 8 h) with a CO selectivity of 88.4%.157, 158 Zhang and co‐workers recently showed that reducing the thickness of ZnAl LDH nanosheets dramatically enhanced their photocatalytic activity relative to their bulk counterpart, giving rise to a remarkable CO formation rate up to 7.6 µmol g−1 h−1.159
4.1.5. Metal‐Organic Framework (MOF)
MOFs are a family of porous materials with crystalline and open structures consisting of metal ions or clusters coordinated with organic ligands.160 Since both the organic ligands and metal ions can be systematically varied, MOFs possess extraordinary chemical and functional versatility. Most uniquely, they may contain photosensitizers and catalytic centers in a single solid, and thereby represent promising alternatives to conventional semiconductors for photocatalysis. For example, many Ti‐based MOFs combine the photocatalytic activity of titanium oxide clusters with the light adsorbing properties of organic linkers and are photocatalytically active under UV–vis light. Li and co‐workers first reported Ti‐containing MIL‐125‐NH2 with the 2‐aminoterephthalate linker as the photocatalyst for CO2 reduction to formate under visible light irradiation.161 It was followed by Uribe‐Romo and co‐workers, who prepared a series of Ti‐based MOFs isoreticular to MIL‐125‐NH2, where the amine functionality was decorated with alkyl chains of varying length and connectivity.162 The authors observed that by successively increasing the alkyl substitution, resulted MOFs displayed a gradually decreased bandgap from 2.56 to 2.29 eV and increased photocatalytic reaction rates and quantum yield for reducing CO2 to formate. In particular, MIL‐125‐NHCyp (Cyp = cyclopentyl) exhibited the largest AQE of 1.8%, as attributed to its long‐lived excited‐state and narrow bandgap compared to the parent MIL‐125‐NH2.
4.1.6. Metal‐Free Material
Carbonaceous materials attract growing interest for the solar fuel production in recent years. One of the good examples is graphitic carbon nitride (g‐C3N4). It has a layered structure analogous to graphite and ideally built from tri‐s‐triazine units.163 In 2009, Domen and co‐workers first reported its photocatalytic activity for hydrogen production under visible light.164 Great efforts have been invested on C3N4 to explore its potential for photocatalytic CO2 reduction since 2013.165, 166, 167, 168 Various reduction products such as CO, CH4, C2H6, HCOOH, and CH3OH are measured.169, 170, 171 Peng and co‐workers were among the first to study the photocatalytic performance of C3N4, and observed that porous C3N4 derived from urea yielded methanol and ethanol while nonporous C3N4 derived from melamine only converted CO2 to ethanol.165 Their AQE were 0.18% and 0.08% respectively. By hybridizing C3N4 with Ru complexes, Maeda and co‐workers achieved the selective conversion of CO2 to HCOOH.172 Their best photocatalyst had a TON of greater than 1000 (20 h) under visible light irradiation with an AQE of 5.7% at 400 nm, both of which were the highest values ever reported under similar conditions. Moreover, exfoliation of bulk C3N4 powder to atomic layer thick nanosheets represents an effective route to further boost its photocatalytic performance thanks to the enlarged surface area and enhanced charge separation property of nanosheets.173 Ye and co‐workers observed that the hybrid of exfoliated C3N4 nanosheets with a Zr‐based MOF (UiO‐66) exhibited better charge separation efficiency and prolonged lifetime of photogenerated carriers.171 A much higher photocatalytic activity for CO2 reduction to CO (9.9 µmol g−1 h−1) was accordingly measured under visible light.
4.2. Photocatalytic Materials Designing Strategies
4.2.1. Band Structure Engineering
As mentioned above, the band structure of a semiconductor photocatalyst determines its capability to absorb light and energize surface redox reactions. Unfortunately, most photocatalyst materials have band structures far from ideal: some (such as TiO2) with band gaps too wide to effectively utilize the solar spectrum, and others (such as transition metal sulfides) with suitable band gaps but improper CB or VB edges for driving CO2 reduction or water oxidation. Band structure engineering therefore is frequently sought as a possible strategy to enhance the visible light activity of photocatalysts. It is commonly accomplished via ion doping: cation doping generally modifies the CB, and anion doping modifies the VB. Much knowledge has been accumulated on this approach.
Taking TiO2 for example, studies were shown that the cation doping of TiO2 by V, Cr, Mn, Fe, and Ni afforded an obvious redshift of its absorption band, and the extent of the shift depended on the amount and the type of the doping.39 It is suggested that the doping results in the formation of some localized states below the CB edge of Ti 3d orbitals and consequently obvious band gap narrowing.116 Ye and co‐workers reported that the codoping of mesoporous TiO2 altered both its CB and VB structure and increased its visible light absorption.174 The main CO2 reduction products were CO and CH4, and their selectivity could be tuned by adjusting the Co doping level. An optimal activity of 90 µmol g−1 h−1 for CH4 and 1.94 mmol g−1 h−1 for CO was achieved at the Co/Ti molar ratio of 2.5%.
Compared to cation doping, anion doping is even more desirable. This is because for most oxides (e.g., TiO2, ZrO2, Nb2O5, and In2O3), there is a large room for raising their VB edges without compromising their capability to oxidize water. Partially replacing O in the lattice with other nonmetal elements such as B, C, N, S, and P is proven effective (Figure 11 a).175 Additional electronic states above the valence band edge introduced by the nonmetal doping were found responsible for the visible‐light response as corroborated by DFT calculations (Figure 11b) and X‐ray photoelectron spectroscopy characterizations.175, 176 Among various nonmetal doping systems, N‐doping is the most widely studied. Asahi et al. first reported that N‐doping of TiO2 could lead to a significant band‐gap narrowing.177 The resultant yellowish film exhibited a much improved optical absorption at a wavelength of less than 500 nm. Most remarkably, Cheng and co‐workers demonstrated that red anatase TiO2 with a band gap as small as 1.94 V could be prepared by high‐concentration B/N codoping.178 Its light absorption edge was accordingly extended up to ≈700 nm covering the full visible light spectrum. The band structure of such a B/N codoped TiO2 was close to the ideal state for efficient photocatalysis. Several types of N‐doped TiO2 were previously investigated for photocatalytic CO2 reduction. N‐doped mesoporous TiO2 was developed by Li et al.179 With the introduction of 0.2 wt% Pt as the cocatalyst, this photocatalyst enabled a CH4 production rate of 2.9 µmol gcat −1 h−1. Grimes and co‐workers demonstrated that N‐doped TiO2 nanotube arrays loaded with both Cu and Pt nanoparticles had a hydrocarbon production rate of 111 ppm cm−2 h−1 or 160 µL g−1 h−1 under outdoor global AM 1.5 sunlight.180
Figure 11.
a) Diffuse reflectance spectra of pure TiO2, C‐doped TiO2, S‐doped TiO2, and N‐doped TiO2 showing the prominent effect of anion doping. Reproduced with permission.175 Copyright 2008, American Chemical Society. b) Calculated density of state (DOS) of pure TiO2 and N‐doped TiO2 with different concentrations of O vacancies. Reproduced with permission.176 Copyright 2009, American Chemical Society.
4.2.2. Nanostructure Design
Rapid spatial separation of photogenerated electrons and holes hold the second decisive key to high‐performance photocatalysis. The average distance that photogenerated carriers migrate from the bulk to the surface is known as the diffusion length. If the diffusion length could be significantly shortened to the nanoscale, the probability of charge separation would be dramatically enhanced and that of charge recombination would be suppressed.181 As a result, proper nanostructure design of photocatalysts is beneficial to their photocatalytic performance. Nanostructured materials have large specific surface areas and short diffusion length. Their photogenerated carriers more likely reach the surface and participate in the surface electrochemical reaction before the recombination takes place.
A myriad of nanostructured materials at different dimensions are developed for photocatalytic CO2 reduction. 1D nanostructures in the form of nanorods, nanowires, nanotubes, and nanobelts have attracted great interest. These materials are usually of single‐crystalline phase that can enable the rapid transport of photogenerated charges. For example, Ye and co‐workers prepared ultrathin W18O49 nanowires with diameter below 1 nm (Figure 12 a).182 They showed strong light absorption from the visible to the near‐infrared region, and was able to reduce CO2 to CH4 in the absence of any cocatalyst at an impressive formation rate of 666 ppm g−1 h−1. Zou and co‐workers prepared single‐crystalline Zn2GeO4 nanobelts that was featured with thickness as small as ≈7 nm and aspect ratio up to 10 000 (Figure 12b,c).134 Their high crystallinity and ultralong/ultrathin geometry configuration facilitated the migration and separation of photogenerated carriers and consequently resulted in an improved photocatalytic activity for reducing CO2 to CH4.
Figure 12.
a) TEM image of W18O49 nanowires for selectively reducing CO2 to CH4. Reproduced with permission.182 b,c) SEM and TEM images of Zn2GeO4 nanoribbons. Reproduced with permission.134 Copyright 2010, American Chemical Society. d) Schematic of the photocatalytic CO2 reduction to methanol on the single‐unit‐cell Bi2WO6 layers; e) TEM image of Bi2WO6 layers; f) methanol formation rate on Bi2WO6 layers and bulk Bi2WO6, g) stability of methanol formation on Bi2WO6 layers. Reproduced with permission.183
In recent years, the great potentials of 2D nanostructures such as nanosheets or nanoflakes start to be gradually unveiled. These 2D materials often have high specific surface areas that can provide abundant active sites for the CO2 adsorption and photocatalytic reaction. Xie and co‐workers developed a series of 2D atomic thick semiconductor materials for photocatalytic CO2 reduction. They demonstrated that Bi2WO6 atomic layers could be prepared through a lamellar Bi‐oleate intermediate (Figure 12d,e).183 The reduced thickness afforded dramatically improved carrier separation efficiency, as evidenced by the increased carrier lifetime from 14.7 to 83.2 ns based on time‐resolved fluorescence spectroscopy measurements. As a result, the product exhibited a high methanol formation rate of 75 µmol g–1 h–1—125 times higher than that of bulk Bi2WO6 and satisfactory stability over 2 d (Figure 12f,g). In their latest report, the same research group followed a similar approach and measured an even better photocatalytic performance for o‐BiVO4 atomic layers, reaching a remarkable methanol formation rate up to 398.3 µmol g–1 h–1 and an AQE of 5.96% at 350 nm.131
In addition to above 1D and 2D materials, more complex and hierarchical structures have also been developed and demonstrated for photocatalytic CO2 reduction. Zou and co‐workers prepared unique hollow spheres composed of single‐layered titanium oxide nanosheets and graphene via layer‐by‐layer assembly.184 They displayed enhanced photocatalytic activity, with a CO production rate of 8.91 µmol g−1 h−1 and a CH4 production rate of 1.14 µmol g−1 h−1, markedly improved over individual titanium oxide nanosheets or P25 TiO2. This enhancement was interpreted as the consequence of the ultrathin nature of titanium oxide nanosheets and their intimate contact with graphene, facilitating the rapid charge separation. Their hollow structure was also believed to induce the multiscattering of the incident light and benefit the light adsorption. Yu and co‐workers prepared hierarchical amine‐functionalized titanate yolk@shell microspheres via one‐pot hydrothermal method.185 The final product had multilevel porous structures composed of basic building blocks including aggregated nanoparticles as the yolk and self‐assembled 2D nanosheets as the shell. Thanks to the hierarchical structure and amine functionality, the yolk@shell microspheres showed enhanced visible light absorption and CO2 adsorption properties, and could selectively reduce CO2 to methanol at an impressive rate of ≈2 µmol g−1 h−1.
4.2.3. Heterostructure Design
Another strategy to facilitate the spatial separation of photogenerated electrons and holes is via the heterostructure design. When two semiconductor materials are coupled together, they may form three possible types of heterostructures namely straddling gap (type I), staggered gap (type II), and broken gap (type III), depending on their relative band positions.186 Among them, the type II of heterosturcture is the most desirable since it promotes the spatial charge separation by transferring electrons to one material with the lower CB and holes to another material with the higher VB, as is shown in Figure 13 a.187 Coupling semiconductors with staggered gaps is an approach frequently adopted in photocatalysis. TiO2 is a common component of many heterostructures. Dong and co‐workers described a hierarchical assembly of ultrathin ZnIn2S4 nanosheets on TiO2 electrospun nanofibers (Figure 13b–d).188 These two components had low lattice mismatch and suitable band alignment to form type II heterostructure. The fast separation of photogenerated carriers was attested by the reduced decay lifetime and quenched ZnIn2S4 photoluminescence signal. When further functionalized with plasmonic Au or Ag nanoparticles, the optimal photocatalyst exhibited ≈16‐fold improvement for the photocatalytic reduction of CO2 to CH4 compared to pure ZnIn2S4. N‐type TiO2 can also be coupled with p‐type semiconductors such as CuO or Cu2O to form type II p−n hereojunctions.189, 190, 191, 192 Schaak and co‐workers synthesized a CuO‐TiO2− xNx p–n junction though the reactive template method.193 It photocatalytically reduced CO2 to methane under simulated solar irradiation at a rate of 41.3 ppm g−1 h−1—much enhanced over pure CuO or TiO2− xNx. Apart from TiO2, silver halides are also frequently investigated for forming heterostructures.194 Chai and co‐workers developed AgX/pCN (X = Cl and Br) heterojunction photocatalysts by depositing AgX (X = Cl and Br) on protonated graphitic carbon nitride.195 The optimal photocatalyst achieved a total CH4 evolution rate of 10.92 µmol g−1 h−1, which were 34.1 and 4.2 times higher than individual AgBr and pCN respectively due to the improved separation efficiency of photogenerated electron/hole pairs at the heterojunction interface.
Figure 13.
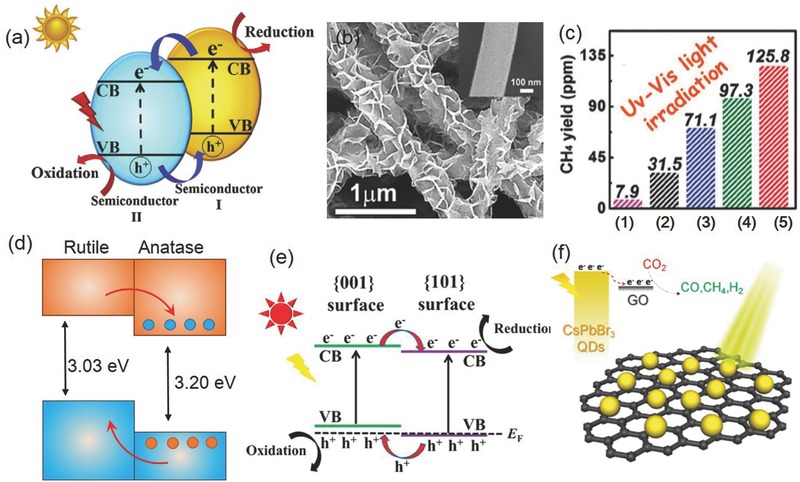
a) Schematic of the conventional type‐II heterojunction photocatalyst. Reproduced with permission.40 Copyright 2017, Elsevier. b) SEM image of ZnIn2S4/TiO2; c) comparison of CH4 yield from photocatalytic CO2 reduction on 1) ZnIn2S4, 2) TiO2, 3) ZnIn2S4/TiO2, 4) Au/ZnIn2S4/TiO2, and 5) Ag/ZnIn2S4/TiO2 after UV–vis irradiation for 4 h. Reproduced with permission.188 d) Proposed VB and CB alignment for the anatase/rutile interface. Reproduced with permission.197 Copyright 2013, Nature Publishing Group. e) Schematic {001}/{101} surface heterojunction. Reproduced with permission.199 Copyright 2014, American Chemical Society. f) Schematic of CO2 photoreduction over the CsPbBr3 QD/GO photocatalyst. Reproduced with permission.202 Copyright 2017, American Chemical Society.
In addition to heterostructures formed from materials with distinctive compositions, this concept may also be extended to some single‐component systems (sometimes termed homojunction), where different phases or surface facets coexist, and have varying energetics to allow for the spatial separation of photogenerated carriers.196 A good example is the anatase and rutile polymorphs of TiO2. When brought together, they form a type II, staggered band alignment with ≈0.4 eV difference in band position.197 Such a difference is significant enough to promote the migration of electrons from rutile to anatase, and holes from anatase to rutile, rendering the mixed‐phase photocatalyst generally superior to individual polymorphs. Another example is the homojunction formed by α/β phase Ga2O3 showing over three folds higher activity for photocatalytic water splitting than individual α‐ or β‐Ga2O3.198 The influence of different surface facets is also documented in literature. Jaroniec and co‐workers showed that the {101} and {001} facets of anatase TiO2 exhibited slightly different band edge positions based on the DFT calculations (Figure 13e).199 The coexistence of these two facets in single TiO2 particle would create a surface heterojunction, which facilitated the migration of the photogenerated electrons and holes to the {101} and {001} facets, respectively (Figure 13f). The ratio of different crystal facets substantially influenced the photocatalytic activity of TiO2, and the optimal {101}/{001} ratio was experimentally identified to be 45:55 for the photocatalytic conversion of CO2 to CH4.
Furthermore, even though carbon nanomaterials (e.g., graphene or carbon nanotubes) are typically not light responsive, they are often employed to form hybrid materials with light‐absorbing semiconductor photocatalysts, which can also be regarded as a special type of heterostructures. These carbon nanomaterials have large surface areas for supporting photocatalysts, may enable fast extraction of photogenerated carriers from the semiconductor, and hence improved photocatalytic activity. There are many successful demonstrations along this line.200, 201 For example, in a most recent report, Kuang and co‐workers showed that supporting CsPbBr3 quantum dots on graphene oxide considerably improved their photocatalytic activity, reaching an average CO formation rate of 4.9 µmol g−1 h−1 and CH4 formation rate of 2.1 µmol g−1 h−1 under AM 1.5G simulated illumination.202
4.2.4. Defect Engineering
Defects play a crucial role in catalysis. It may change the interaction between the catalyst surface and the target molecule, consequently lower the reaction activation energy or even alter the reaction pathway. Engineering the type and density of defects on catalyst surfaces is an important means to tune their activities. Oxygen vacancies are among the most common defects in oxide or hydroxide surfaces and are suggested to greatly affect the photocatalytic CO2 reduction.203, 204, 205 Among many experimental supports was the low‐temperature scanning tunneling microscopy visualizing that CO2 molecules were preferably absorbed at the oxygen vacancies (Figure 14 a).206 Quantum mechanical modeling also indicated that the electron transfer from the CB of stoichiometric anatase TiO2 to CO2 was not energetically favorable, but defects on anatase TiO2 surface could promote the electron transfer to CO2.204
Figure 14.
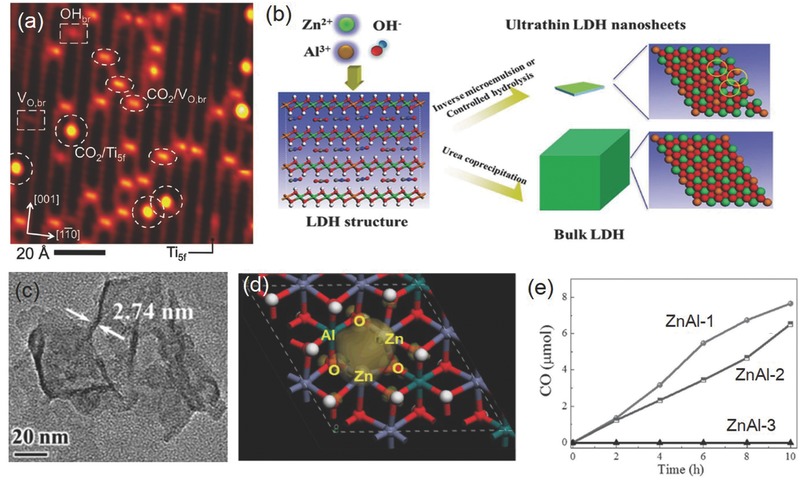
a) Scanning tunneling microscope (STM) image of CO2 molecules adsorbed on TiO2 (110) plane. Reproduced with permission.206 Copyright 2011, American Chemical Society. b) Schematic showing the formation of coordinatively unsaturated ZnAl‐LDH nanosheets; c) TEM image of coordinatively unsaturated ZnAl‐LDH nanosheets; d) charge density distribution for the valence band maximum of Vo‐doped ZnAl‐LDH; e) time‐dependent CO yields on different ZnAl‐LDH samples. Reproduced with permission.159
Many semiconductor photocatalysts containing rich structural defects have been investigated for CO2 reduction with improved performances. Wang and co‐workers showed that the CO2 photoreduction over defective CeO2 selectively yielded CO at a decent formation rate of ≈4 µmol g–1 h–1, and eliminating these defects by postsynthetic annealing fully deactivated the photocatalyst.207 The authors proposed that oxygen vacancies together with local strain promoted the CO2 capture and activation on the CeO2 surface, therefore lowering its reaction barrier. Self‐doped SrTiO3− δ powders were prepared by Ye and co‐workers and analyzed to contain Ti3+ ions and oxygen vacancies by forming Ruddlesden–Popper crystallographic shears.205 These defects not only induced an in‐gap band to enhance the visible light absorption of the photocatalyst but also improved the chemical adsorption of CO2 on the surface based on temperature programmed desorption experiments. A better photocatalytic activity for converting CO2 to CH4 was therefore resulted. Zhang and co‐workers recently reported that abundant oxygen vacancies and coordinatively unsaturated Zn+ centers were created when the thickness of ZnAl LDH nanosheets were reduced to two repeat stacking layers (Figure 14b–d).159 Thus formed Zn+‐Vo complexes served as the active sites for the efficient adsorption of CO2 and H2O molecules, significantly improving the photocatalytic activity for CO2 reduction to CO.
4.2.5. Cocatalyst Loading
Even when photogenerated carriers are rapidly separated and migrate to the surface, they may not readily participate in the surface redox reactions since both CO2 reaction and water oxidation involve multistep proton‐coupled electron transfer and are notorious for their highly sluggish nature. A common and often necessary strategy to improve the photocatalytic performance is to introduce auxiliary cocatalysts to the surface of the semiconductor photocatalyst.208 These cocatalysts markedly change the energetics of the charge transfer process at the surface and increase the catalytic turnover rates, making the production rate of solar fuels a dominant process over the charge recombination or reverse reactions.209 In addition, the timely consumption the photogenerated charges on the cocatalyst would also slow down the photocorrosion of semiconductor photocatalysts and improve their stability. As reviewed in Section 3, many different CO2RR electrocatalysts have been developed in the last five years. In principle, they are all potential cocatalyst materials for photocatalytic CO2 reduction, provided a favorable interface is established between the photocatalyst and cocatalyst to facilitate the charge transfer from the former to the latter.
Noble metals including Pt, Au, Pd, and Ag represent the most widely used cocatalyst materials for photocatalytic CO2 reduction. They are usually deposited onto the photocatalyst surface via either chemical reduction or photochemical reduction of corresponding precursors. These noble metals can often serve as the electron sink to concentrate photogenerated electrons from photocatalysts, and consequently reduce the possibility of electron‐hole recombination.209 Within their presence, the selectivity of CO2 photoreduction is generally shifted in favor of CH4 over CO or other hydrocarbon products. Using P25 TiO2 as the model photocatalyst, Wang and co‐workers demonstrated that the ability of noble metal cocatalysts to promote the CH4 formation rate increased in the order Ag < Rh < Au < Pd < Pt.210 The most effective cocatalyst was identified to be Pt, presumably due to its efficient extraction of photogenerated electrons from TiO2. It was also recognized that the size of cocatalyst particles affected the photocatalytic activity and selectivity. CH4 formation was more favored on smaller Pt nanoparticles. TiO2 loaded with ≈3 nm (mean size) Pt nanoparticles was measured to produce CH4 about four times faster than TiO2 loaded with ≈5 nm Pt nanoparticles. Additional introduction of MgO in the photocatalyst further doubled the CH4 formation rate and boosted its selectivity to >99%. Biswas and co‐workers also studied the size effect of Pt cocatalyst on the photocatalytic performance of TiO2.211 The optimal particle size was measured to be ≈1 nm. With such a cocatalyst particle size, a peak CH4 formation rate of 1361 µmol g–1 h–1 was observed, and the corresponding quantum yield was calculated to be 2.41% (Figure 15 a). By contrast, larger or smaller cocatalyst nanoparticles led to markedly reduced photocatalytic activity. The difference was rationalized based on the relative energy band alignment of TiO2 and Pt as its size varied: too small Pt particle size would lift its energy level above the bottom of the TiO2 CB due to the quantum confinement, retarding the electron transfer from photocatalyst to Pt cocatalyst, whilst for bigger Pt nanoparticles, their properties approached bulk Pt and could act as recombination centers by capturing both photogenerated electrons and holes.
Figure 15.
a) Schematic of photocatalytic CO2 reduction on nanostructured TiO2 films deposited with Pt cocatalyst particles of varying sizes. Different alignments between TiO2 band structure and Pt work function is suggested to be responsible for the observed different photocatalytic activities. Reproduced with permission.211 Copyright 2012, American Chemical Society. b) High‐resolution TEM image of an Au−Cu nanoparticle deposited on the TiO2 surface as the cocatalyst for selectively reducing CO2 to CH4. Reproduced with permission.212 Copyright 2014. American Chemical Society. c) Schematic showing the M. thermoacetica–CdS hybrid system for the photosynthetic conversion of CO2 to acetic acid. Reproduced with permission.213 Copyright 2016, American Association for the Advancement of Science.
Aside from monometallic cocatalysts, bimetallic cocatalysts are also considered for photocatalytic CO2 reduction. As previously mentioned, alloying of different metals offers the probability to tailor their electrocatalytic activity and selectivity. Garcia and co‐workers showed that Au–Cu nanoalloy cocatalyst greatly promoted the photocatalytic activity of commercial P25 TiO2 (Figure 15b).212 The optimal Au/Cu ratio was determined to be 1: 2. Under this condition, the photocatalyst exhibited an excellent CH4 formation rates up to 2.2 mmol g−1 h−1 and minimal concomitant H2 generation.
Very recently, it was showed bacterial or enzymatic materials might also be used as the cocatalyst to promote the performance of photocatalysts. The proof of concept was first demonstrated by Yang and co‐workers by integrating the non‐photosynthetic CO2‐reducing bacterium M. thermoacetica with CdS nanoparticles (Figure 15c).213 The hybrid system selectively (≈90%) photoreduced CO2 to acetic acid. The optimal production rate was reported to be ≈520 × 10−6 m h−1 with a quantum yield of ≈52% under 485 nm light illumination. This study represented an important leap in the pursuit of efficient artificial photosynthesis and uncovered the great potential of inorganic‐biological hybrid systems.
4.2.6. Z‐Scheme System Construction
Suitable band structure, efficient charge separation, and rapid surface charge transfer are key characteristics of successful photocatalysts. Yet it is highly challenging (if not impossible) for a single‐component photocatalyst to simultaneously meet all these criteria. In nature, photosynthesis takes place in two individual but well concerted steps in photosystems I and II that harvest 700 and 680 nm photons, respectively.214, 215 The sites for oxygen evolution and CO2 fixation are also spatially separated. Such a natural two‐step process inspires the design of Z‐scheme systems for artificial photosynthesis.216 Instead of using a single‐component photocatalyst to fulfill all the functions, one may combine two different light‐absorbing semiconductor materials through a redox mediator, which electrically connects the two parts by mediating the electron transfer from the CB of one semiconductor with relatively lower energy to the VB of another semiconductor (Figure 16 a). In this configuration, the cathodic and anodic half reactions can be decoupled, and the selection criteria for photocatalyst materials can be considerably relaxed. Z‐scheme can more efficiently utilize visible light, capitalize the strongly reducing electrons from one catalyst and the strongly oxidizing holes from another, and achieve overall photocatalytic activities that are not attainable with single‐component systems. Many successful Z‐scheme designs are now available. Arai and co‐workers constructed a Z‐scheme system by combining InP/Ru complex polymer (for CO2 reduction to formate) with TiO2 (for water oxidation).217 The selectivity for formate reached >70% and the overall conversion efficiency from solar energy to chemical energy was 0.03–0.04%, which approached that of natural photosynthesis in plants. The conversion efficiency was further increased to 0.14% in a follow‐up work by the same research group when TiO2 in the Z‐scheme was replaced by SrTiO3 since its higher conduction band facilitated the electron transfer to InP.218 Sulfide‐based photocatalysts are generally susceptible to photocorrosion. Z‐scheme is an effective strategy to alleviate their photocorrosion by rapidly filling the photogenerated holes in the VB of sulfide photocatalysts. Kudo and co‐workers studied the combination of several different p‐type sulfide photocatalysts (such as ZnS, AgGaS2, AgInS2, CuGaS2, CuInS2, and many others) and n‐type oxide photocatalysts (such as TiO2 and BiVO4) in the Z‐scheme configuration for photocatalytic water splitting or CO2 reduction, using RGO as the solid state electron mediator (Figure 16b).219, 220 In particular, the combination of CuGaS2 and BiVO4 was demonstrated to reduce CO2 to CO with a good short‐term stability even though it was accompanied by a significant cogeneration of H2. The low CO selectivity could probably be improved by introducing proper cocatalysts. Z‐scheme may also be formed between a molecular photosensitizer and a semiconductor photocatalyst.221 Ishitani and co‐workers constructed a hybrid photocatalyst by combining Ru(II) binuclear complex and Ag‐loaded TaON together with organic hole scavenger.222 HCOOH was detected as the main reduction product. Its maximum TON for HCOOH was measured to be 750 under visible‐light irradiation for 24 h, and its optimal external quantum efficiency was 0.48% at 400 nm.
Figure 16.
a) Schematic of Z‐scheme photocatalytic mechanism. Reproduced with permission.216 b) Schematic of the Z‐scheme system for water splitting and CO2 reduction by coupling Pt‐loaded metal sulfide and CoOx/BiVO4 using RGO as the solid state electron mediator. Reproduced with permission.220 Copyright 2014, American Chemical Society.
5. Conclusion and Perspectives
This review summarizes recent advances in CO2 reduction to useful chemical fuels via the electrochemical or photochemical approach. Even though some significant advances have been achieved in the past decades for both electrocatalytic and photocatalytic CO2 reduction, their reaction activity and selectivity are still rather low. At this moment, there is no detailed technoeconomical analysis available to estimate the target production cost of chemical fuels from CO2 reduction in order to compete with other fuel production technologies. For water splitting, US Department of Energy has specified the solar‐to‐hydrogen commercialization target is $2.00–$4.00 per kg of H2 and the minimum solar energy conversion efficiency should be 5% and preferably >10% to compete with gasoline.223 We therefore envisage that a comparable solar‐to‐fuel efficiency is necessary for photocatalytic or PV+electrocatalytic CO2 reduction to become a commercial reality. Even though the solar to fuel (STF) efficiency of most current CO2 reduction systems is still well below the minimum target, two recent breakthroughs are worth highlighting here. By integrating a photovoltaic cell with biocompatible Earth‐abundant inorganic catalysts and bacterium Ralstonia eutropha, Nocera and co‐workers achieved an artificial photosynthetic process for carbon fixation into biomass and liquid fuels at an efficiency of ≈10%.224 Gratzel and co‐workers demonstrated atomic layer deposition of SnO2 on CuO nanowires as a bifunctional electrocatalyst for both CO2RR and OER, and when coupled with a GaInP/GaInAs/Ge photovoltaic cell, enabled an solar‐to‐CO conversion efficiency of 13.4%.225 The efficiencies reported in the above two studies already exceed that of natural photosynthetic systems and are suggestive of the great potential of the CO2 reduction technology.
To further enhance the CO2 reduction performance, improvements can be possibly made from the following two directions. Seeking new material compositions and structures would continue to be at the heart of electrocatalytic and photocatalytic CO2 reduction research. Priority should be given to exploration guided by theoretical computation. Ample examples have been shown that theoretical computation is a powerful tool in predicting new catalysts by comparing the energy barriers and overpotentials of reaction intermediates on certain crystal surfaces of each catalyst. In the future, the subjects of computation may be extended from currently predominant metallic catalysts (due to their well‐defined surface geometry) to more complicated systems including pure compounds (oxides, sulfides, and so on) or even hybrids. For photocatalytic CO2 reduction, the exploration of new materials and structures can also be greatly accelerated by borrowing knowledge from photocatalytic water splitting. Photocatalytic CO2 reduction and photocatalytic water splitting only differs in their surface reaction step. If strategies (such as incorporation of proper cocatalysts) can be undertaken to significantly shift the cathodic reaction selectivity away from HER to CO2RR, essentially all existing photocatalysts for water splitting can be transformed to those for CO2 reduction.
Increasing understanding of the reaction pathway and mechanism would help us toward the design of better CO2 reduction catalysts. A clear picture of how CO2 is adsorbed, transformed and desorbed is still missing at this moment. Of particular interest is to understand the rate determining step in CO2 reduction and the surface binding of its reaction intermediates so that we can accordingly strengthen or weaken the intermediate binding and expedite the overall reaction rate. Many advanced electronic and spectroscopic tools such as aberration‐corrected transmission electron microscopy (TEM), X‐ray absorption spectroscopy, electron spin resonance, and time‐resolved fluorescence spectroscopy are now available to us with unprecedented capabilities. They may offer detailed information about the catalyst structure at different levels and insights about the catalytic process. We will also see more and more in situ characterization techniques such as in situ Raman and attenuated total reflection infrared spectroscopy brought in to shed new light on the binding configuration and environment of reaction intermediates.
CO2 reduction is of both fundamental and practical significance. In spite of serious challenges, there is no reason to doubt its great potential and impact. More efforts are called for in the fundamental understanding, materials development, and catalyst engineering of CO2 reduction to make it a viable technology for a carbon neutral future.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
J.W. and Y.H. contributed equally to this work. The authors acknowledge supports from the Ministry of Science and Technology of China (2017YFA0204800), the National Natural Science Foundation of China (51472173 and 51522208), the Natural Science Foundation of Jiangsu Province (BK20140302 and SBK2015010320), the Priority Academic Program Development of Jiangsu Higher Education Institutions, Collaborative Innovation Center of Suzhou Nano Science and Technology, and the “111” project.
Biographies
Jinghua Wu received his Bachelor of Science (BS) degree from Shandong University, China in 2006 and his PhD degree from Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences (CAS) in 2012. He then joined Professor Takayoshi Sasaki's group as a postdoctoral researcher in the National Institute for Materials Science (NIMS), Japan. Now he works in Professor Yanguang Li's group as a postdoctoral researcher in the Institute of Functional Nano and Soft Materials (FUNSOM) at Soochow University. His research interest focuses on developing low‐dimensional materials for photocatalytic CO2 reduction.

Yang Huang received her BS in Chemistry from Xiamen University, China in 2010 and obtained her PhD in Chemistry from Fujian Institute of Research on the Structure, CAS in 2015. Now she works as a postdoctoral researcher under the supervision of Prof. Yanguang Li in FUNSOM at Soochow University. Her current research focuses on developing nanostructured electrocatalysts for CO2 reduction and HER.

Yanguang Li is a Professor in FUNSOM at Soochow University, China. He received his BS in Chemistry from Fudan University, China in 2005 and obtained his PhD in Chemistry from Ohio State University in 2010. He then moved to the Stanford University and completed a postdoctoral training before taking the current faculty position in 2013. His research focuses on nanostructured functional materials for energy applications, particularly in the realm of electrocatalysis, photocatalysis, and advanced batteries.

Wu J., Huang Y., Ye W., Li Y., Adv. Sci. 2017, 4, 1700194 https://doi.org/10.1002/advs.392
References
- 1. Vitousek P. M., Mooney H. A., Lubchenco J., Melillo J. M., Science 1997, 277, 494. [Google Scholar]
- 2. Wang W.‐H., Himeda Y., Muckerman J. T., Manbeck G. F., Fujita E., Chem. Rev. 2015, 115, 12936. [DOI] [PubMed] [Google Scholar]
- 3. National Ocean and Atmospheric Administration (NOAA) , National Centers for Environmental Information, Global Climate Change Indicators, https://www.ncdc.noaa.gov/monitoring‐references/faq/indicators.php (accessed: June 2017). [Google Scholar]
- 4. Hoffert M. I., Caldeira K., Benford G., Criswell D. R., Green C., Herzog H., Jain A. K., Kheshgi H. S., Lackner K. S., Lewis J. S., Science 2002, 298, 981. [DOI] [PubMed] [Google Scholar]
- 5. Tee S. Y., Win K. Y., Teo W. S., Koh L. D., Liu S., Teng C. P., Han M. Y., Adv. Sci. 2017, 4, 1600337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nielsen C. J., Herrmann H., Weller C., Chem. Rev. 2012, 41, 6684. [DOI] [PubMed] [Google Scholar]
- 7. Barber J., Chem. Soc. Rev. 2009, 38, 185.19088973 [Google Scholar]
- 8. Concepcion J. J., House R. L., Papanikolas J. M., Meyer T. J., Proc. Natl. Acad. Sci. USA 2012, 109, 15560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Armaroli N., Balzani V., Angew. Chem., Int. Ed. 2007, 46, 52. [DOI] [PubMed] [Google Scholar]
- 10. Hu B., Guild C., Suib S. L., J. CO2 Util. 2013, 1, 18. [Google Scholar]
- 11. Royer M. E., Compt. Rend. 1870, 70, 731. [Google Scholar]
- 12. Ito K., Murata T., Bull. Nagoya Ind. Technol. 1975, 271, 369. [Google Scholar]
- 13. Hori Y., Kikuchi K., Murata A., Suzuki S., Chem. Lett. 1986, 15, 897. [Google Scholar]
- 14. Halmann M., Nature 1978, 275, 115. [Google Scholar]
- 15. Inoue T., Fujishima A., Konishi S., Honda K., Nature 1979, 277, 637. [Google Scholar]
- 16. Li K., Peng B., Peng T., ACS Catal. 2016, 6, 7485. [Google Scholar]
- 17. Habisreutinger S. N., Schmidt‐Mende L., Stolarczyk J. K., Angew. Chem., Int. Ed. 2013, 52, 7372. [DOI] [PubMed] [Google Scholar]
- 18. Tu W., Zhou Y., Zou Z., Adv. Mater. 2014, 26, 4607. [DOI] [PubMed] [Google Scholar]
- 19. Low J., Yu J., Ho W., J. Phys. Chem. Lett. 2015, 6, 4244. [DOI] [PubMed] [Google Scholar]
- 20. Li X., Wen J., Low J., Fang Y., Yu J., Sci. China Mater. 2014, 57, 70. [Google Scholar]
- 21. Mao J., Li K., Peng T., Catal. Sci. Technol. 2013, 3, 2481. [Google Scholar]
- 22. Zhu D. D., Liu J. L., Qiao S. Z., Adv. Mater. 2016, 28, 3423. [DOI] [PubMed] [Google Scholar]
- 23. Qiao J., Liu Y., Hong F., Zhang J., Chem. Soc. Rev. 2014, 43, 631. [DOI] [PubMed] [Google Scholar]
- 24. Yuan L., Xu Y.‐J., Appl. Surf. Sci. 2015, 342, 154. [Google Scholar]
- 25. Liu J., Guo C., Vasileff A., Qiao S., Small Methods 2017, 1, 1600006. [Google Scholar]
- 26. Zhu Y. P., Guo C., Zheng Y., Qiao S.‐Z., Acc. Chem. Res. 2017, 50, 915. [DOI] [PubMed] [Google Scholar]
- 27. Hori Y., Murata A., Takahashi R., J. Chem. Soc., Faraday Trans. 1 1989, 85, 2309. [Google Scholar]
- 28. Lim R. J., Xie M., Sk M. A., Lee J.‐M., Fisher A., Wang X., Lim K. H., Catal. Today 2014, 233, 169. [Google Scholar]
- 29. Bard A. J., Parsons R., Jordan J., Standard Potentials in Aqueous Solution, CRC Press, New York: 1985. [Google Scholar]
- 30. Leitner W., Angew. Chem., Int. Ed. 1995, 34, 2207. [Google Scholar]
- 31. Benson E. E., Kubiak C. P., Sathrum A. J., Smieja J. M., Chem. Soc. Rev. 2009, 38, 89. [DOI] [PubMed] [Google Scholar]
- 32. Chaplin R. P. S., Wragg A. A., J. Appl. Electrochem. 2003, 33, 1107. [Google Scholar]
- 33. Vassiliev Y. B., Bagotsky V. S., Osetrova N. V., Khazova O. A., Mayorova N. A., J. Electroanal. Chem. Interfacial Electrochem. 1985, 189, 271. [Google Scholar]
- 34. Gattrell M., Gupta N., Co A., J. Electroanal. Chem. 2006, 594, 1. [Google Scholar]
- 35. Peterson A. A., Abild‐Pedersen F., Studt F., Rossmeisl J., Norskov J. K., Energy Environ. Sci. 2010, 3, 1311. [Google Scholar]
- 36. Kuhl K. P., Cave E. R., Abram D. N., Jaramillo T. F., Energy Environ. Sci. 2012, 5, 7050. [Google Scholar]
- 37. Gileadi E., Electrode Kinetics for Chemists, Engineers, and Materials Scientists, Wiley‐VCH, Weinheim, Germany: 1993. [Google Scholar]
- 38. Garsany Y., Baturina O. A., Swider‐Lyons K. E., Kocha S. S., Anal. Chem. 2010, 82, 6321. [DOI] [PubMed] [Google Scholar]
- 39. Chen X., Mao S. S., Chem. Rev. 2007, 107, 2891. [DOI] [PubMed] [Google Scholar]
- 40. Low J., Cheng B., Yu J., Appl. Surf. Sci. 2017, 392, 658. [Google Scholar]
- 41. Huang Q., Yu J., Cao S., Cui C., Cheng B., Appl. Surf. Sci. 2015, 358, 350. [Google Scholar]
- 42. Bolton J. R., Strickler S. J., Connolly J. S., Nature 1985, 316, 495. [Google Scholar]
- 43. Jacobsson T. J., Fjällström V., Sahlberg M., Edoff M., Edvinsson T., Energy Environ. Sci. 2013, 6, 3676. [Google Scholar]
- 44. Cox C. R., Lee J. Z., Nocera D. G., Buonassisi T., Proc. Natl. Acad. Sci. USA 2014, 111, 14057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hori Y., Wakebe H., Tsukamoto T., Koga O., Electrochim. Acta 1994, 39, 1833. [Google Scholar]
- 46. Hansen H. A., Varley J. B., Peterson A. A., Norskov J. K., J. Phys. Chem. Lett. 2013, 4, 388. [DOI] [PubMed] [Google Scholar]
- 47. Yoo J. S., Christensen R., Vegge T., Nørskov J. K., Studt F., ChemSusChem 2016, 9, 358. [DOI] [PubMed] [Google Scholar]
- 48. Li C. W., Kanan M. W., J. Am. Chem. Soc. 2012, 134, 7231. [DOI] [PubMed] [Google Scholar]
- 49. Hori Y., Takahashi I., Koga O., Hoshi N., J. Mol. Catal. A: Chem. 2003, 199, 39. [Google Scholar]
- 50. Schouten K. J. P., Pérez Gallent E., Koper M. T., ACS Catal. 2013, 3, 1292. [Google Scholar]
- 51. Schouten K. J. P., Qin Z., Gallent E. P., Koper M. T., J. Am. Chem. Soc. 2012, 134, 9864. [DOI] [PubMed] [Google Scholar]
- 52. Schouten K., Kwon Y., Van der Ham C., Qin Z., Koper M., Chem. Sci. 2011, 2, 1902. [Google Scholar]
- 53. Roberts F. S., Kuhl K. P., Nilsson A., Angew. Chem., Int. Ed. 2015, 54, 5179. [DOI] [PubMed] [Google Scholar]
- 54. Reske R., Mistry H., Behafarid F., Roldan Cuenya B., Strasser P., J. Am. Chem. Soc. 2014, 136, 6978. [DOI] [PubMed] [Google Scholar]
- 55. Manthiram K., Beberwyck B. J., Alivisatos A. P., J. Am. Chem. Soc. 2014, 136, 13319. [DOI] [PubMed] [Google Scholar]
- 56. Tang W., Peterson A. A., Varela A. S., Jovanov Z. P., Bech L., Durand W. J., Dahl S., Nørskov J. K., Chorkendorff I., Phys. Chem. Chem. Phys. 2012, 14, 76. [DOI] [PubMed] [Google Scholar]
- 57. Vollmer S., Witte G., Wöll C., Catal. Lett. 2001, 77, 97. [Google Scholar]
- 58. Li C. W., Ciston J., Kanan M. W., Nature 2014, 508, 504. [DOI] [PubMed] [Google Scholar]
- 59. Mistry H., Varela A. S., Bonifacio C. S., Zegkinoglou I., Sinev I., Choi Y.‐W., Kisslinger K., Stach E. A., Yang J. C., Strasser P., Cuenya B. R., Nat. Commun. 2016, 7, 12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Singh M. R., Kwon Y., Lum Y., Ager J. W. 3rd, Bell A. T., J. Am. Chem. Soc. 2016, 138, 13006. [DOI] [PubMed] [Google Scholar]
- 61. Varela A. S., Ju W., Reier T., Strasser P., ACS Catal. 2016, 6, 2136. [Google Scholar]
- 62. Hori Y., Murata A., Kikuchi K., Suzuki S., J. Chem. Soc., Chem. Commun. 1987, 10, 728. [Google Scholar]
- 63. Zhu W., Michalsky R., Metin O., Lv H., Guo S., Wright C. J., Sun X., Peterson A. A., Sun S., J. Am. Chem. Soc. 2013, 135, 16833. [DOI] [PubMed] [Google Scholar]
- 64. Mistry H., Reske R., Zeng Z., Zhao Z. J., Greeley J., Strasser P., Cuenya B. R., J. Am. Chem. Soc. 2014, 136, 16473. [DOI] [PubMed] [Google Scholar]
- 65. Chen Y., Li C. W., Kanan M. W., J. Am. Chem. Soc. 2012, 134, 19969. [DOI] [PubMed] [Google Scholar]
- 66. Feng X., Jiang K., Fan S., Kanan M. W., J. Am. Chem. Soc. 2015, 137, 4606. [DOI] [PubMed] [Google Scholar]
- 67. Zhu W., Zhang Y. J., Zhang H., Lv H., Li Q., Michalsky R., Peterson A. A., Sun S., J. Am. Chem. Soc. 2014, 136, 16132. [DOI] [PubMed] [Google Scholar]
- 68. Lee H.‐E., Yang K. D., Yoon S. M., Ahn H.‐Y., Lee Y. Y., Chang H., Jeong D. H., Lee Y.‐S., Kim M. Y., Nam K. T., ACS Nano 2015, 9, 8384. [DOI] [PubMed] [Google Scholar]
- 69. Back S., Yeom M. S., Jung Y., ACS Catal. 2015, 5, 5089. [Google Scholar]
- 70. Hoshi N., Kato M., Hori Y., J. Electroanal. Chem. 1997, 440, 283. [Google Scholar]
- 71. Hatsukade T., Kuhl K. P., Cave E. R., Abram D. N., Jaramillo T. F., Phys. Chem. Chem. Phys. 2014, 16, 13814. [DOI] [PubMed] [Google Scholar]
- 72. Salehi‐Khojin A., Jhong H.‐R. M., Rosen B. A., Zhu W., Ma S., Kenis P. J. A., Masel R. I., J. Phys. Chem. C 2013, 117, 1627. [Google Scholar]
- 73. Ma M., Trzesniewski B. J., Xie J., Smith W. A., Angew. Chem., Int. Ed. 2016, 55, 9748. [DOI] [PubMed] [Google Scholar]
- 74. Lu Q., Rosen J., Zhou Y., Hutchings G. S., Kimmel Y. C., Chen J. G., Jiao F., Nat. Commun. 2014, 5, 3242. [DOI] [PubMed] [Google Scholar]
- 75. Yoon Y., Hall A. S., Surendranath Y., Angew. Chem., Int. Ed. 2016, 55, 15282. [DOI] [PubMed] [Google Scholar]
- 76. Wu J., Risalvato F. G., Ma S., Zhou X.‐D., J. Mater. Chem. A 2014, 2, 1647. [Google Scholar]
- 77. Kumar B., Atla V., Brian J. P., Kumari S., Nguyen T. Q., Sunkara M., Spurgeon J. M., Angew. Chem., Int. Ed. 2017, 56, 3645. [DOI] [PubMed] [Google Scholar]
- 78. Zhang S., Kang P., Meyer T. J., J. Am. Chem. Soc. 2014, 136, 1734. [DOI] [PubMed] [Google Scholar]
- 79. Lei F., Liu W., Sun Y., Xu J., Liu K., Liang L., Yao T., Pan B., Wei S., Xie Y., Nat. Commun. 2016, 7, 12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ohkawa K., Hashimoto K., Fujishima A., Noguchi Y., Nakayama S., J. Electroanal. Chem. 1993, 345, 445. [Google Scholar]
- 81. Gao D., Zhou H., Wang J., Miao S., Yang F., Wang G., Wang J., Bao X., J. Am. Chem. Soc. 2015, 137, 4288. [DOI] [PubMed] [Google Scholar]
- 82. Min X., Kanan M. W., J. Am. Chem. Soc. 2015, 137, 4701. [DOI] [PubMed] [Google Scholar]
- 83. Won da H., Shin H., Koh J., Chung J., Lee H. S., Kim H., Woo S. I., Angew. Chem., Int. Ed. 2016, 55, 9297. [DOI] [PubMed] [Google Scholar]
- 84. Rosen J., Hutchings G. S., Lu Q., Forest R. V., Moore A., Jiao F., ACS Catal. 2015, 5, 4586. [Google Scholar]
- 85. Quan F., Zhong D., Song H., Jia F., Zhang L., J. Mater. Chem. A 2015, 3, 16409. [Google Scholar]
- 86. DiMeglio J. L., Rosenthal J., J. Am. Chem. Soc. 2013, 135, 8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Medina‐Ramos J., DiMeglio J. L., Rosenthal J., J. Am. Chem. Soc. 2014, 136, 8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ding C., Li A., Lu S.‐M., Zhang H., Li C., ACS Catal. 2016, 6, 6438. [Google Scholar]
- 89. Detweiler Z. M., White J. L., Bernasek S. L., Bocarsly A. B., Langmuir 2014, 30, 7593. [DOI] [PubMed] [Google Scholar]
- 90. Gao S., Jiao X., Sun Z., Zhang W., Sun Y., Wang C., Hu Q., Zu X., Yang F., Yang S., Liang L., Wu J., Xie Y., Angew. Chem., Int. Ed. 2016, 55, 698. [DOI] [PubMed] [Google Scholar]
- 91. Gao S., Lin Y., Jiao X., Sun Y., Luo Q., Zhang W., Li D., Yang J., Xie Y., Nature 2016, 529, 68. [DOI] [PubMed] [Google Scholar]
- 92. Kim D., Resasco J., Yu Y., Asiri A. M., Yang P., Nat. Commun. 2014, 5, 4948. [DOI] [PubMed] [Google Scholar]
- 93. Rasul S., Anjum D. H., Jedidi A., Minenkov Y., Cavallo L., Takanabe K., Angew. Chem., Int. Ed. 2015, 54, 2146. [DOI] [PubMed] [Google Scholar]
- 94. Ma S., Sadakiyo M., Heima M., Luo R., Haasch R. T., Gold J. I., Yamauchi M., Kenis P. J., J. Am. Chem. Soc. 2017, 139, 47. [DOI] [PubMed] [Google Scholar]
- 95. Guo X., Zhang Y., Deng C., Li X., Xue Y., Yan Y.‐M., Sun K., Chem. Commun. 2015, 51, 1345. [DOI] [PubMed] [Google Scholar]
- 96. Li Q., Fu J., Zhu W., Chen Z., Shen B., Wu L., Xi Z., Wang T., Lu G., Zhu J.‐J., J. Am. Chem. Soc. 2017, 139, 4290. [DOI] [PubMed] [Google Scholar]
- 97. Zeng M., Li Y., J. Mater. Chem. A 2015, 3, 14942. [Google Scholar]
- 98. Hong X., Chan K., Tsai C., Nørskov J. K., ACS Catal. 2016, 6, 4428. [Google Scholar]
- 99. Chan K., Tsai C., Hansen H. A., Nørskov J. K., ChemCatChem 2014, 6, 1899. [Google Scholar]
- 100. Rosen B. A., Salehi‐Khojin A., Thorson M. R., Zhu W., Whipple D. T., Kenis P. J., Masel R. I., Science 2011, 334, 643. [DOI] [PubMed] [Google Scholar]
- 101. Asadi M., Kumar B., Behranginia A., Rosen B. A., Baskin A., Repnin N., Pisasale D., Phillips P., Zhu W., Haasch R., Klie R. F., Kral P., Abiade J., Salehi‐Khojin A., Nat. Commun. 2014, 5, 4470. [DOI] [PubMed] [Google Scholar]
- 102. Asadi M., Kim K., Liu C., Addepalli A. V., Abbasi P., Yasaei P., Phillips P., Behranginia A., Cerrato J. M., Haasch R., Zapol P., Kumar B., Klie R. F., Abiade J., Curtiss L. A., Salehi‐Khojin A., Science 2016, 353, 467. [DOI] [PubMed] [Google Scholar]
- 103. Abbasi P., Asadi M., Liu C., Sharifi‐Asl S., Sayahpour B., Behranginia A., Zapol P., Shahbazian‐Yassar R., Curtiss L. A., Salehi‐Khojin A., ACS Nano 2017, 11, 453. [DOI] [PubMed] [Google Scholar]
- 104. Sun X., Zhu Q., Kang X., Liu H., Qian Q., Zhang Z., Han B., Angew. Chem., Int. Ed. 2016, 55, 6771. [DOI] [PubMed] [Google Scholar]
- 105. Zhang S., Kang P., Ubnoske S., Brennaman M. K., Song N., House R. L., Glass J. T., Meyer T. J., J. Am. Chem. Soc. 2014, 136, 7845. [DOI] [PubMed] [Google Scholar]
- 106. Wu J., Yadav R. M., Liu M., Sharma P. P., Tiwary C. S., Ma L., Zou X., Zhou X. D., Yakobson B. I., Lou J., Ajayan P. M., ACS Nano 2015, 9, 5364. [DOI] [PubMed] [Google Scholar]
- 107. Kumar B., Asadi M., Pisasale D., Sinha‐Ray S., Rosen B. A., Haasch R., Abiade J., Yarin A. L., Salehi‐Khojin A., Nat. Commun. 2013, 4, 2819. [Google Scholar]
- 108. Nakata K., Ozaki T., Terashima C., Fujishima A., Einaga Y., Angew. Chem., Int. Ed. 2014, 53, 871. [DOI] [PubMed] [Google Scholar]
- 109. Liu Y., Chen S., Quan X., Yu H., J. Am. Chem. Soc. 2015, 137, 11631. [DOI] [PubMed] [Google Scholar]
- 110. Wu J., Ma S., Sun J., Gold J. I., Tiwary C., Kim B., Zhu L., Chopra N., Odeh I. N., Vajtai R., Yu A. Z., Luo R., Lou J., Ding G., Kenis P. J., Ajayan P. M., Nat. Commun. 2016, 7, 13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mistry H., Varela A. S., Kühl S., Strasser P., Cuenya B. R., Nat. Rev. Mater. 2016, 1, 16009. [Google Scholar]
- 112. Cao S., Tao F. F., Tang Y., Li Y., Yu J., Chem. Soc. Rev. 2016, 45, 4747. [DOI] [PubMed] [Google Scholar]
- 113. He J., Dettelbach K. E., Salvatore D. A., Li T., Berlinguette C. P., Angew. Chem., Int. Ed. 2017, 129, 6164. [DOI] [PubMed] [Google Scholar]
- 114. Kuhl K. P., Hatsukade T., Cave E. R., Abram D. N., Kibsgaard J., Jaramillo T. F., J. Am. Chem. Soc 2014, 136, 14107. [DOI] [PubMed] [Google Scholar]
- 115. Fujishima A., Honda K., Nature 1972, 238, 37. [DOI] [PubMed] [Google Scholar]
- 116. Chen X., Shen S., Guo L., Mao S. S., Chem. Rev. 2010, 110, 6503. [DOI] [PubMed] [Google Scholar]
- 117. Kudo A., Miseki Y., Chem. Soc. Rev. 2009, 38, 253. [DOI] [PubMed] [Google Scholar]
- 118. Inoue Y., Energy Environ. Sci. 2009, 2, 364. [Google Scholar]
- 119. Sohn Y., Huang W., Taghipour F., Appl. Surf. Sci. 2017, 396, 1696. [Google Scholar]
- 120. Liu L., Zhao H., Andino J. M., Li Y., ACS Catal. 2012, 2, 1817. [Google Scholar]
- 121. Rodriguez M. M., Peng X., Liu L., Li Y., Andino J. M., J. Phys. Chem. C 2012, 116, 19755. [Google Scholar]
- 122. Pan J., Liu G., Lu G. Q. M., Cheng H. M., Angew. Chem., Int. Ed. 2011, 50, 2133. [DOI] [PubMed] [Google Scholar]
- 123. Pan J., Wu X., Wang L., Liu G., Lu G. Q., Cheng H.‐M., Chem. Commun. 2011, 47, 8361. [DOI] [PubMed] [Google Scholar]
- 124. Liu G., Jimmy C. Y., Lu G. Q. M., Cheng H.‐M., Chem. Commun. 2011, 47, 6763. [DOI] [PubMed] [Google Scholar]
- 125. Indrakanti V. P., Kubicki J. D., Schobert H. H., Energy Fuels 2008, 22, 2611. [Google Scholar]
- 126. Chen X., Zhou Y., Liu Q., Li Z., Liu J., Zou Z., ACS Appl. Mater. Interfaces 2012, 4, 3372. [DOI] [PubMed] [Google Scholar]
- 127. Lo C.‐C., Hung C.‐H., Yuan C.‐S., Wu J.‐F., Sol. Energy Mater. Sol. Cells 2007, 91, 1765. [Google Scholar]
- 128. Teramura K., Okuoka S.‐I., Tsuneoka H., Shishido T., Tanaka T., Appl. Catal., B 2010, 96, 565. [Google Scholar]
- 129. Zhou H., Guo J., Li P., Fan T., Zhang D., Ye J., Sci. Rep. 2013, 3, 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zhou Y., Tian Z., Zhao Z., Liu Q., Kou J., Chen X., Gao J., Yan S., Zou Z., ACS Appl. Mater. Interfaces 2011, 3, 3594. [DOI] [PubMed] [Google Scholar]
- 131. Gao S., Gu B., Jiao X., Sun Y., Zu X., Yang F., Zhu W., Wang C., Feng Z., Ye B., Xie Y., J. Am. Chem. Soc. 2017, 139, 3438. [DOI] [PubMed] [Google Scholar]
- 132. Feng S., Chen X., Zhou Y., Tu W., Li P., Li H., Zou Z., Nanoscale 2014, 6, 1896. [DOI] [PubMed] [Google Scholar]
- 133. Yan S. C., Ouyang S. X., Gao J., Yang M., Feng J. Y., Fan X. X., Wan L. J., Li Z. S., Ye J. H., Zhou Y., Angew. Chem., Int. Ed. 2010, 122, 6544. [Google Scholar]
- 134. Liu Q., Zhou Y., Kou J., Chen X., Tian Z., Gao J., Yan S., Zou Z., J. Am. Chem. Soc 2010, 132, 14385. [DOI] [PubMed] [Google Scholar]
- 135. Eggins B. R., Irvine J. T., Murphy E. P., Grimshaw J., J. Chem. Soc., Chem. Commun. 1988, 16, 1123. [Google Scholar]
- 136. Wang S., Wang X., Appl. Catal., B 2015, 162, 494. [Google Scholar]
- 137. Baran T., Dibenedetto A., Aresta M., Kruczala K., Macyk W., Chempluschem 2014, 79, 708. [Google Scholar]
- 138. Baran T., Wojtyla S., Dibenedetto A., Aresta M., Macyk W., Appl. Catal., B 2015, 178, 170. [Google Scholar]
- 139. Chen J., Qin S., Song G., Xiang T., Xin F., Yin X., Dalton Trans. 2013, 42, 15133. [DOI] [PubMed] [Google Scholar]
- 140. Liu J.‐Y., Garg B., Ling Y.‐C., Green Chem. 2011, 13, 2029. [Google Scholar]
- 141. Li X., Liu H., Luo D., Li J., Huang Y., Li H., Fang Y., Xu Y., Zhu L., Chem. Eng. J. 2012, 180, 151. [Google Scholar]
- 142. Peng H., Lu J., Wu C., Yang Z., Chen H., Song W., Li P., Yin H., Appl. Surf. Sci. 2015, 353, 1003. [Google Scholar]
- 143. Sato J., Kobayashi H., Inoue Y., J. Phys. Chem. C 2003, 107, 7970. [Google Scholar]
- 144. AlOtaibi B., Fan S., Wang D., Ye J., Mi Z., ACS Catal. 2015, 5, 5342. [Google Scholar]
- 145. AlOtaibi B., Kong X., Vanka S., Woo S. Y., Pofelski A., Oudjedi F., Fan S., Kibria M. G., Botton G. A., Ji W., Guo H., Mi Z., ACS Energy Lett. 2016, 1, 246. [Google Scholar]
- 146. Zhang N., Ouyang S., Kako T., Ye J., Chem. Commun. 2012, 48, 1269. [DOI] [PubMed] [Google Scholar]
- 147. Liu Q., Zhou Y., Tian Z., Chen X., Gao J., Zou Z., J. Mater. Chem. 2012, 22, 2033. [Google Scholar]
- 148. Yan S., Yu H., Wang N., Li Z., Zou Z., Chem. Commun. 2012, 48, 1048. [DOI] [PubMed] [Google Scholar]
- 149. Wang Q., O'Hare D., Chem. Rev. 2012, 112, 4124. [DOI] [PubMed] [Google Scholar]
- 150. Gunjakar J. L., Kim T. W., Kim H. N., Kim I. Y., Hwang S.‐J., J. Am. Chem. Soc. 2011, 133, 14998. [DOI] [PubMed] [Google Scholar]
- 151. Gomes Silva C., Bouizi Y., Fornes V., Garcia H., J. Am. Chem. Soc. 2009, 131, 13833. [DOI] [PubMed] [Google Scholar]
- 152. Lee Y., Choi J. H., Jeon H. J., Choi K. M., Lee J. W., Kang J. K., Energy Environ. Sci. 2011, 4, 914. [Google Scholar]
- 153. Ahmed N., Shibata Y., Taniguchi T., Izumi Y., J. Catal. 2011, 279, 123. [Google Scholar]
- 154. Izumi Y., Coord. Chem. Rev. 2013, 257, 171. [Google Scholar]
- 155. Ahmed N., Morikawa M., Izumi Y., Catal. Today 2012, 185, 263. [Google Scholar]
- 156. Morikawa M., Ahmed N., Yoshida Y., Izumi Y., Appl. Catal., B 2014, 144, 561. [Google Scholar]
- 157. Iguchi S., Teramura K., Hosokawa S., Tanaka T., Phys. Chem. Chem. Phys. 2015, 17, 17995. [DOI] [PubMed] [Google Scholar]
- 158. Teramura K., Iguchi S., Mizuno Y., Shishido T., Tanaka T., Angew. Chem., Int. Ed. 2012, 124, 8132. [DOI] [PubMed] [Google Scholar]
- 159. Zhao Y., Chen G., Bian T., Zhou C., Waterhouse G. I., Wu L. Z., Tung C. H., Smith L. J., O'Hare D., Zhang T., Adv. Mater. 2015, 27, 7824. [DOI] [PubMed] [Google Scholar]
- 160. Luo X.‐L., Yin Z., Zeng M.‐H., Kurmoo M., Inorg. Chem. Front. 2016, 3, 1208. [Google Scholar]
- 161. Fu Y., Sun D., Chen Y., Huang R., Ding Z., Fu X., Li Z., Angew. Chem., Int. Ed. 2012, 124, 3420. [DOI] [PubMed] [Google Scholar]
- 162. Logan M. W., Ayad S., Adamson J. D., Dilbeck T., Hanson K., Uribe‐Romo F. J., J. Mater. Chem. A 2017, 5, 11854. [Google Scholar]
- 163. Wang Y., Wang X., Antonietti M., Angew. Chem., Int. Ed. 2012, 51, 68. [DOI] [PubMed] [Google Scholar]
- 164. Wang X., Maeda K., Thomas A., Takanabe K., Xin G., Carlsson J. M., Domen K., Antonietti M., Nat. Mater. 2009, 8, 76. [DOI] [PubMed] [Google Scholar]
- 165. Mao J., Peng T., Zhang X., Li K., Ye L., Zan L., Catal. Sci. Technol. 2013, 3, 1253. [Google Scholar]
- 166. Ong W.‐J., Tan L.‐L., Ng Y. H., Yong S.‐T., Chai S.‐P., Chem. Rev. 2016, 116, 7159. [DOI] [PubMed] [Google Scholar]
- 167. Ong W.‐J., Front. Mater. 2017, 4, 11. [Google Scholar]
- 168. Ye S., Wang R., Wu M.‐Z., Yuan Y.‐P., Appl. Surf. Sci. 2015, 358, 15. [Google Scholar]
- 169. Kuriki R., Sekizawa K., Ishitani O., Maeda K., Angew. Chem., Int. Ed. 2015, 54, 2406. [DOI] [PubMed] [Google Scholar]
- 170. Ong W.‐J., Tan L.‐L., Chai S.‐P., Yong S.‐T., Mohamed A. R., Nano Energy 2015, 13, 757. [Google Scholar]
- 171. Shi L., Wang T., Zhang H., Chang K., Ye J., Adv. Funct. Mater. 2015, 25, 5360. [Google Scholar]
- 172. Kuriki R., Sekizawa K., Ishitani O., Maeda K., Angew. Chem. Int. Ed. 2015, 54, 2406. [DOI] [PubMed] [Google Scholar]
- 173. Yang S., Gong Y., Zhang J., Zhan L., Ma L., Fang Z., Vajtai R., Wang X., Ajayan P. M., Adv. Mater. 2013, 25, 2452. [DOI] [PubMed] [Google Scholar]
- 174. Wang T., Meng X., Liu G., Chang K., Li P., Kang Q., Liu L., Li M., Ouyang S., Ye J., J. Mater. Chem. A 2015, 3, 9491. [Google Scholar]
- 175. Chen X., Burda C., J. Am. Chem. Soc. 2008, 130, 5018. [DOI] [PubMed] [Google Scholar]
- 176. Wang J., Tafen D. N., Lewis J. P., Hong Z., Manivannan A., Zhi M., Li M., Wu N., J. Am. Chem. Soc. 2009, 131, 12290. [DOI] [PubMed] [Google Scholar]
- 177. Asahi R., Morikawa T., Ohwaki T., Aoki K., Taga Y., Science 2001, 293, 269. [DOI] [PubMed] [Google Scholar]
- 178. Liu G., Yin L.‐C., Wang J., Niu P., Zhen C., Xie Y., Cheng H.‐M., Energy Environ. Sci. 2012, 5, 9603. [Google Scholar]
- 179. Li X., Zhuang Z., Li W., Pan H., Appl. Catal. A 2012, 429, 31. [Google Scholar]
- 180. Varghese O. K., Paulose M., LaTempa T. J., Grimes C. A., Nano Lett. 2009, 9, 731. [DOI] [PubMed] [Google Scholar]
- 181. Hochbaum A. I., Yang P., Chem. Rev. 2010, 110, 527. [DOI] [PubMed] [Google Scholar]
- 182. Xi G., Ouyang S., Li P., Ye J., Ma Q., Su N., Bai H., Wang C., Angew. Chem., Int. Ed. 2012, 51, 2395. [DOI] [PubMed] [Google Scholar]
- 183. Liang L., Lei F., Gao S., Sun Y., Jiao X., Wu J., Qamar S., Xie Y., Angew. Chem., Int. Ed. 2015, 54, 13971. [DOI] [PubMed] [Google Scholar]
- 184. Tu W., Zhou Y., Liu Q., Tian Z., Gao J., Chen X., Zhang H., Liu J., Zou Z., Adv. Funct. Mater. 2012, 22, 1215. [Google Scholar]
- 185. Liu S., Xia J., Yu J., ACS Appl. Mater. Interfaces 2015, 7, 8166. [DOI] [PubMed] [Google Scholar]
- 186. Yuan Y.‐P., Ruan L.‐W., Barber J., Loo S. C. J., Xue C., Energy Environ. Sci. 2014, 7, 3934. [Google Scholar]
- 187. Wang H., Zhang L., Chen Z., Hu J., Li S., Wang Z., Liu J., Wang X., Chem. Soc. Rev. 2014, 43, 5234. [DOI] [PubMed] [Google Scholar]
- 188. Zhang Z., Huang Y., Liu K., Guo L., Yuan Q., Dong B., Adv. Mater. 2015, 27, 5906. [DOI] [PubMed] [Google Scholar]
- 189. Cheng W.‐Y., Yu T.‐H., Chao K.‐J., Lu S.‐Y., Int. J. Hydrogen Energy 2013, 38, 9665. [Google Scholar]
- 190. Yang L., Luo S., Li Y., Xiao Y., Kang Q., Cai Q., Environ. Sci. Technol. 2010, 44, 7641. [DOI] [PubMed] [Google Scholar]
- 191. Shifu C., Sujuan Z., Wei L., Wei Z., J. Nanosci. Nanotechnol. 2009, 9, 4397. [DOI] [PubMed] [Google Scholar]
- 192. Roy S. C., Varghese O. K., Paulose M., Grimes C. A., ACS Nano 2010, 4, 1259. [DOI] [PubMed] [Google Scholar]
- 193. In S. I., Vaughn D. D., Schaak R. E., Angew. Chem., Int. Ed. 2012, 51, 3915. [DOI] [PubMed] [Google Scholar]
- 194. Li H., Gan S., Wang H., Han D., Niu L., Adv. Mater. 2015, 27, 6906. [DOI] [PubMed] [Google Scholar]
- 195. Ong W.‐J., Putri L. K., Tan L.‐L., Chai S.‐P., Yong S.‐T., Appl. Catal., B 2016, 180, 530. [Google Scholar]
- 196. Li H., Zhou Y., Tu W., Ye J., Zou Z., Adv. Funct. Mater. 2015, 25, 998. [Google Scholar]
- 197. Scanlon D. O., Dunnill C. W., Buckeridge J., Shevlin S. A., Logsdail A. J., Woodley S. M., Catlow C. R. A., Powell M. J., Palgrave R. G., Parkin I. P., Nat. Mater. 2013, 12, 798. [DOI] [PubMed] [Google Scholar]
- 198. Wang X., Xu Q., Li M., Shen S., Wang X., Wang Y., Feng Z., Shi J., Han H., Li C., Angew. Chem., Int. Ed. 2012, 51, 13089. [DOI] [PubMed] [Google Scholar]
- 199. Yu J., Low J., Xiao W., Zhou P., Jaroniec M., J. Am. Chem. Soc. 2014, 136, 8839. [DOI] [PubMed] [Google Scholar]
- 200. Xiang Q., Cheng B., Yu J., Angew. Chem., Int. Ed. 2015, 54, 11350. [DOI] [PubMed] [Google Scholar]
- 201. Gusain R., Kumar P., Sharma O. P., Jain S. L., Khatri O. P., Appl. Catal., B 2016, 181, 352. [Google Scholar]
- 202. Xu Y.‐F., Yang M.‐Z., Chen B.‐X., Wang X.‐D., Chen H.‐Y., Kuang D.‐B., Su C.‐Y., J. Am. Chem. Soc 2017, 139, 5660. [DOI] [PubMed] [Google Scholar]
- 203. Indrakanti V. P., Kubicki J. D., Schobert H. H., Energy Environ. Sci. 2009, 2, 745. [Google Scholar]
- 204. Indrakanti V. P., Schobert H. H., Kubicki J. D., Energy Fuels 2009, 23, 5247. [Google Scholar]
- 205. Xie K., Umezawa N., Zhang N., Reunchan P., Zhang Y., Ye J., Energy Environ. Sci. 2011, 4, 4211. [Google Scholar]
- 206. Acharya D., Camillone N. III, Sutter P., J. Phys. Chem. C 2011, 115, 12095. [Google Scholar]
- 207. Jiang D., Wang W., Gao E., Sun S., Zhang L., Chem. Commun. 2014, 50, 2005. [DOI] [PubMed] [Google Scholar]
- 208. Wenderich K., Mul G., Chem. Rev. 2016, 116, 14587. [DOI] [PubMed] [Google Scholar]
- 209. Yang J., Wang D., Han H., Li C., Acc. Chem. Res. 2013, 46, 1900. [DOI] [PubMed] [Google Scholar]
- 210. Xie S., Wang Y., Zhang Q., Deng W., Wang Y., ACS Catal. 2014, 4, 3644. [Google Scholar]
- 211. Wang W.‐N., An W.‐J., Ramalingam B., Mukherjee S., Niedzwiedzki D. M., Gangopadhyay S., Biswas P., J. Am. Chem. Soc. 2012, 134, 11276. [DOI] [PubMed] [Google Scholar]
- 212. Neaţu Ş., Maciá‐Agulló J. A., Concepción P., Garcia H., J. Am. Chem. Soc. 2014, 136, 15969. [DOI] [PubMed] [Google Scholar]
- 213. Sakimoto K. K., Wong A. B., Yang P., Science 2016, 351, 74. [DOI] [PubMed] [Google Scholar]
- 214. Dau H., Zaharieva I., Acc. Chem. Res. 2009, 42, 1861. [DOI] [PubMed] [Google Scholar]
- 215. Berardi S., Drouet S., Francàs L., Gimbert‐Suriñach C., Guttentag M., Richmond C., Stoll T., Llobet A., Chem. Soc. Rev. 2014, 43, 7501. [DOI] [PubMed] [Google Scholar]
- 216. Zhou P., Yu J., Jaroniec M., Adv. Mater. 2014, 26, 4920. [DOI] [PubMed] [Google Scholar]
- 217. Sato S., Arai T., Morikawa T., Uemura K., Suzuki T. M., Tanaka H., Kajino T., J. Am. Chem. Soc. 2011, 133, 15240. [DOI] [PubMed] [Google Scholar]
- 218. Arai T., Sato S., Kajino T., Morikawa T., Energy Environ. Sci. 2013, 6, 1274. [Google Scholar]
- 219. Iwashina K., Iwase A., Ng Y. H., Amal R., Kudo A., J. Am. Chem. Soc. 2015, 137, 604. [DOI] [PubMed] [Google Scholar]
- 220. Iwase A., Yoshino S., Takayama T., Ng Y. H., Amal R., Kudo A., J. Am. Chem. Soc. 2016, 138, 10260. [DOI] [PubMed] [Google Scholar]
- 221. Kumagai H., Sahara G., Maeda K., Higashi M., Abe R., Ishitani O., Chem. Sci. 2017, 8, 4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Nakada A., Nakashima T., Sekizawa K., Maeda K., Ishitani O., Chem. Sci. 2016, 7, 4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Pinaud B. A., Benck J. D., Seitz L. C., Forman A. J., Chen Z., Deutsch T. G., James B. D., Baum K. N., Baum G. N., Ardo S., Energy Environ. Sci. 2013, 6, 1983. [Google Scholar]
- 224. Liu C., Colón B. C., Ziesack M., Silver P. A., Nocera D. G., Science 2016, 352, 1210. [DOI] [PubMed] [Google Scholar]
- 225. Schreier M., Héroguel F., Steier L., Ahmad S., Luterbacher J. S., Mayer M. T., Luo J., Grätzel M., Nat. Energy 2017, 2, 17078. [Google Scholar]
- 226. Yang K. D., Ko W. R., Lee J. H., Kim S. J., Lee H., Lee M. H., Nam K. T., Angew. Chem., Int. Ed. 2017, 56, 796. [DOI] [PubMed] [Google Scholar]
- 227. Kas R., Hummadi K. K., Kortlever R., de Wit P., Milbrat A., Luiten‐Olieman M. W., Benes N. E., Koper M. T., Mul G., Nat. Commun. 2016, 7, 10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Kauffman D. R., Alfonso D., Matranga C., Qian H., Jin R., J. Am. Chem. Soc. 2012, 134, 10237. [DOI] [PubMed] [Google Scholar]
- 229. Huang H., Jia H., Liu Z., Gao P., Zhao J., Luo Z., Yang J., Zeng J., Angew. Chem., Int. Ed. 2017, 56, 3594. [DOI] [PubMed] [Google Scholar]
- 230. Wang P.‐Q., Bai Y., Liu J.‐Y., Fan Z., Hu Y.‐Q., Catal. Commun. 2012, 29, 185. [Google Scholar]
- 231. Liu L., Gao F., Zhao H., Li Y., Appl. Catal., B 2013, 134, 349. [Google Scholar]
- 232. Koci K., Mateju K., Obalova L., Krejcikova S., Lacny Z., Placha D., Capek L., Hospodkova A., Solcova O., Appl. Catal., B 2010, 96, 239. [Google Scholar]
- 233. Yang J.‐S., Liao W.‐P., Wu J.‐J., ACS Appl. Mater. Interfaces 2013, 5, 7425. [DOI] [PubMed] [Google Scholar]
- 234. Liu E., Kang L., Wu F., Sun T., Hu X., Yang Y., Liu H., Fan J., Plasmonics 2014, 9, 61. [Google Scholar]
- 235. Kwak B. S., Vignesh K., Park N.‐K., Ryu H.‐J., Baek J.‐I., Kang M., Fuel 2015, 143, 570. [Google Scholar]
- 236. Asi M. A., He C., Su M., Xia D., Lin L., Deng H., Xiong Y., Qiu R., Li X.‐Z., Catal. Today 2011, 175, 256. [Google Scholar]
- 237. Kim W., Seok T., Choi W., Energy Environ. Sci. 2012, 5, 6066. [Google Scholar]
- 238. Zhai Q., Xie S., Fan W., Zhang Q., Wang Y., Deng W., Wang Y., Angew. Chem., Int. Ed. 2013, 52, 5776. [DOI] [PubMed] [Google Scholar]
- 239. Tu W., Zhou Y., Liu Q., Yan S., Bao S., Wang X., Xiao M., Zou Z., Adv. Funct. Mater. 2013, 23, 1743. [Google Scholar]
- 240. Li Y., Wang W.‐N., Zhan Z., Woo M.‐H., Wu C.‐Y., Biswas P., Appl. Catal., B 2010, 100, 386. [Google Scholar]
- 241. Kang Q., Wang T., Li P., Liu L., Chang K., Li M., Ye J., Angew. Chem., Int. Ed. 2015, 54, 841. [DOI] [PubMed] [Google Scholar]
- 242. Xi G., Ouyang S., Ye J., Chem. Eur. J. 2011, 17, 9057. [DOI] [PubMed] [Google Scholar]
- 243. Tahir M., Amin N. S., Appl. Catal., B 2015, 162, 98. [Google Scholar]
- 244. Jiao W., Wang L., Liu G., Lu G. Q., Cheng H.‐M., ACS Catal. 2012, 2, 1854. [Google Scholar]
- 245. Wang P.‐Q., Bai Y., Luo P.‐Y., Liu J.‐Y., Catal. Commun. 2013, 38, 82. [Google Scholar]
- 246. Yin G., Nishikawa M., Nosaka Y., Srinivasan N., Atarashi D., Sakai E., Miyauchi M., ACS Nano 2015, 9, 2111. [DOI] [PubMed] [Google Scholar]
- 247. Li M., Li P., Chang K., Wang T., Liu L., Kang Q., Ouyang S., Ye J., Chem. Commun. 2015, 51, 7645. [DOI] [PubMed] [Google Scholar]
- 248. Park H.‐A., Choi J. H., Choi K. M., Lee D. K., Kang J. K., J. Mater. Chem. 2012, 22, 5304. [Google Scholar]
- 249. Yan S. C., Ouyang S. X., Gao J., Yang M., Feng J. Y., Fan X. X., Wan L. J., Li Z. S., Ye J. H., Zhou Y., Zou Z. G., Angew. Chem., Int. Ed. 2010, 49, 6400. [DOI] [PubMed] [Google Scholar]
- 250. Pan Y.‐X., You Y., Xin S., Li Y., Fu G., Cui Z., Men Y.‐L., Cao F.‐F., Yu S.‐H., Goodenough J. B., J. Am. Chem. Soc. 2017, 139, 4123. [DOI] [PubMed] [Google Scholar]
- 251. An X., Li K., Tang J., Chemsuschem 2014, 7, 1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Núñez J., Víctor A., Jana P., Coronado J. M., Serrano D. P., Catal. Today 2013, 209, 21. [Google Scholar]
- 253. Li P., Zhou Y., Zhao Z., Xu Q., Wang X., Xiao M., Zou Z., J. Am. Chem. Soc. 2015, 137, 9547. [DOI] [PubMed] [Google Scholar]
- 254. Sastre F., Puga A. V., Liu L., Corma A., García H., J. Am. Chem. Soc. 2014, 136, 6798. [DOI] [PubMed] [Google Scholar]
- 255. Gao C., Meng Q., Zhao K., Yin H., Wang D., Guo J., Zhao S., Chang L., He M., Li Q., Adv. Mater. 2016, 28, 6485. [DOI] [PubMed] [Google Scholar]
- 256. Yu J., Jin J., Cheng B., Jaroniec M., J. Mater. Chem. A 2014, 2, 3407. [Google Scholar]
- 257. Manzi A., Simon T., Sonnleitner C., Döblinger M., Wyrwich R., Stern O., Stolarczyk J. K., Feldmann J., J. Am. Chem. Soc. 2015, 137, 14007. [DOI] [PubMed] [Google Scholar]
- 258. Zhao C., Liu L., Rao G., Zhao H., Wang L., Xu J., Li Y., Catal. Sci. Technol. 2015, 5, 3288. [Google Scholar]
- 259. Hong J., Zhang W., Wang Y., Zhou T., Xu R., Chemcatchem 2014, 6, 2315. [Google Scholar]
- 260. Ong W.‐J., Tan L.‐L., Chai S.‐P., Yong S.‐T., Chem. Commun. 2015, 51, 858. [DOI] [PubMed] [Google Scholar]