

Abstract
RB loss occurs commonly in neoplasia but its contributions to advanced cancer have not been assessed directly. Here we show that RB loss in multiple murine models of cancer produces a prometastatic phenotype. Gene expression analyses showed that regulation of the cell motility receptor RHAMM by the RB/E2F pathway was critical for epithelial–mesenchymal transition, motility, and invasion by cancer cells. Genetic modulation or pharmacologic inhibition of RHAMM activity was sufficient and necessary for metastatic phenotypes induced by RB loss in prostate cancer. Mechanistic studies in this setting established that RHAMM stabilized F-actin polymerization by controlling ROCK signaling. Collectively, our findings show how RB loss drives metastatic capacity and highlight RHAMM as a candidate therapeutic target for treating advanced prostate cancer.
Introduction
The retinoblastoma tumor suppressor protein (RB) is a central regulator of cell-cycle progression and proliferation. RB physically associates with E2F transcription factors to repress E2F-mediated transcriptional program (1). Upon mitogenic signaling, D-type cyclins complex with cyclin-dependent kinases 4 and 6 (CDK4/6), triggering hyperphosphorylation of the RB protein and resulting in functional inactivation of the RB pathway and acceleration of E2F-mediated gene transcription. Both RB and E2F play a central role in essential biological programs, and mutation or genetic ablation of the RB1 gene leads to dysfunction of RB-E2F pathway in multiple cancers (2) and results in increased proliferation. Recently, these events have been implicated in other vital cellular processes, including angiogenesis, epithelial-to-mesenchymal transition, migration, invasion, and metastasis (3).
Recent bioinformatics and cancer genomic studies have shown that homozygous deletion of the RB locus is a central feature of metastatic castrate-resistant prostate cancer (mCRPC). Loss of RB function facilitates the development of a castrate-resistant phenotype (CRPC) via E2F-mediated upregulation of the androgen receptor (AR). Despite this, castrate resistance and the formation of metastatic lesions remain distinct biological processes. Thus, loss of RB likely facilitates both disease states. In addition, RB loss is also a reliable genomic alteration in neuroendocrine prostate carcinoma, particularly small-cell prostate cancer, with clinical studies associating the loss of p16, a CDK4/6 inhibitor, with distant organ metastases, implying that loss of RB-mediated transcriptional repression may result in a prometastatic phenotype (4). While RB activation has been shown to function as a key suppressor of multiple stages of metastatic progression within the context of breast cancer (5), the direct impact of RB loss on the metastatic potential of prostate cancer is unknown.
We sought to delineate the impact of RB function on migration, invasion, and metastatic formation using a panel of human isogenic cancer models. In this study, we show that loss of RB function alters cytoskeletal organization, induces EMT, and increases migration, invasion, and metastases. Our data nominates the receptor for hyaluronan acid–mediated motility (RHAMM; ref. 6) as an RB/E2F–regulated target gene with sufficient function to trigger the metastatic program and serve as a biomarker for advanced cancers. Accordingly, overexpression of RHAMM recapitulates the RB loss phenotype; conversely, inhibition of RHAMM either through genetic or pharmacologic approaches, reverses the process. Activation of the RB pathway via CDK4/6 inhibition restricts metastatic tumor burden by transcriptionally repressing RHAMM. Finally, RHAMM function is mediated via F-actin and Rho kinase activation. In summary, our studies identify loss of RB activity as a key regulator of cancer metastasis in mCRPC.
Materials and Methods
Cell culture
LNCaP, LAPC4, C4-2, PC3, PC3-ML, RWPE1, H1299, LN18, and Saos2 cells were maintained in improved minimum essential medium (IMEM) supplemented with 5% FBS (heat-inactivated FBS). LAPC4 cells were maintained in Iscove’s modified Dulbecco’s medium supplemented with 10% ΔFBS (Atlanta Biologicals). Cell lines used were not cultured longer than 6 months after receipt from the original source of ATCC. They were routinely verified to be mycoplasma-free using the Universal Mycoplasma Detection Kit (ATCC). Cell lines were authenticated using STR profiling (DDC Medical).
Genetic manipulation of RB and RHAMM
RB knockdown was carried out as described previously (4, 7). RHAMM knockdown was generated through lentiviral infection using RHAMM shRNA I (Santa Cruz Biotechnology), or RHAMM shRNA II (OriGene) in LNCaP, PC3, and PC3-ML cells. Similarly, RHAMM overexpression was carried out via lentiviral infection using RHAMM cDNA (GeneCopoeia). Knockdown and overexpression were verified using qRT-PCR and immunobloting analysis, as described previously (8, 9). RHAMM shRNA sequences are presented in the Supplementary Table S1.
RNA analysis
Total RNA was isolated from (i) RB-proficient and RB-deficient H1299, LNCaP, PC3, and PC3-ML cells treated with a CDK4/6 inhibitor, PD 0332991 at 500 nmol/L; (ii) RHAMM-overexpressing and RHAMM knockdown LNCaP, PC3, and PC3-ML cells; (iii) in vivo tumors, metastases, and whole blood from mice inoculated with control, RB-deficient, and RHAMM knockdown PC3-ML cells (Invitrogen). Five micrograms of total RNA was reverse transcribed with random hexamers or VILO kit (Invitrogen). Approximately 50 or 100 ng of cDNA was applied for PCR or semiquantitative or real-time PCR. Real-time PCR was performed with an ABI Step-One apparatus using Power SYBR Green Master Mix and using the target mRNA primers for pRB, RHAMM, cyclin E, E-cadherin, N-cadherin, vimentin, as described in refs. 10 and 11. Signals were normalized with an internal control GAPDH and quantitated using ΔΔCt method. Primers were presented in the Supplementary Table S1.
Microarray profiling of RB target genes
DNA-free total RNA was isolated from RB-proficient H1299 cells (non–small cell lung cancer) and PC3-ML treated with PD 0332991. Quantificiation was performed with a Nanodrop ND-100 spectrophotometer, and RNA quality assessment was analyzed with an Agilent 2200 TapeStation (Agilent Technologies). Fragmented biotin-labeled cDNA (from 100 ng of RNA) was synthesized using the GeneChip WT Plus Kit (Affymetrix). Human Transcriptome Array 2.0 (Affymetrix), was hybridized with 5 μg fragmented and biotin-labeled cDNA in 200 μL of hybridization cocktail. Target denaturation was performed at 99°C for 5 minutes, 45°C for 5 minutes, followed by hybridization with rotation at 60 rpm for 16 hours at 45°C. Arrays were then washed and stained using Gene chip Fluidic Station 450, using Affymetrix GeneChip hybridization Wash & Stain Kit. Chips were scanned on an Affymetrix Gene Chip Scanner 3000, using Command Console Software. Quality control of the experiment was performed by Expression Console Software v 1.4.1. Chp file was generated by sst-rma normalization from Affymetrix cel file using the Expression Console Software. Experimental conditions were compared with control using the transcriptome array console software. Differentially expressed gene lists were subjected to pathway analysis using Ingenuity Pathway Analysis (IPA) software and Gene Set Enrichment Analysis (GSEA). The microarray data were deposited at Gene Expression Omnibus (GEO): GSE87879 (H1299) and GSE87886 (PC3-ML).
Immunoblot analysis
For protein analysis, RB-proficient and -deficient cells (LNCaP, PC3, LAPC4, C4-2, PC3-ML), RHAMM-overexpressing cells (PC3, LNCaP, PC3-ML), and RHAMM knockdown cells (PC3, PC3-ML, LNCaP) were utilized. Briefly, the cells were harvested by trypsinization, and cell lysis was carried out in RIPA buffer [150 mmol/L NaCl, 1% NP40, 0.5% deoxycholate, 0.1% SDS, 50 mmol/L Tris (pH 8.0)] supplemented with protease inhibitors, phosphatase inhibitors, and phenylmethylsulfonyl fluoride. After brief sonication, lysates were clarified, protein concentrations were determined using Bio-Rad Protein Assay Reagent, and an equal amount of protein was subjected to SDS-PAGE and transferred onto Immobilin-P PVDF transfer membranes (Millipore). The membranes were immunoblotted for RB (BD Biosciences), phospho-RB (phospho-serine 780), RHAMM, GAPDH, F-actin (phalloidin, Invitrogen Inc), cofilin, phospho-cofilin (S3) (Cell Signaling Technology), ROCK II, F-actin, lamin B, E-cadherin, vimentin, E2F1, E2F2, and RNRII (Santa Cruz Biotechnology and Abcam) by standard techniques and visualized using Enhanced Western Lightning Chemiluminescence (Perkin-Elmer Life Sciences). The signals were normalized with the internal control lamin B or GAPDH.
In silico analysis of RHAMM in patient-derived prostate specimens
Three publicly available human prostate cancer microarray datasets were used to evaluate transcriptional relationships between the RHAMM transcript (HMMR) and other molecular and clinical variables. Initially, a dataset provided by Memorial Sloan Kettering Cancer Center (New York, NY; ref. 12; GSE21034) was used to demonstrate correlation between transcripts of interest by computing Pearson correlation coefficient for HMMR, E2F1, CCNA1, and MKI67 with respect to the RB1 tumor suppressor transcript within both tumor and normal sample subsets. Differential expression between tumor and normal for these transcripts was also evaluated using a two-sided t test.
Molecular modeling of cancer metastases through RB–RHAMM–Rho kinase pathway
Control, RB-deficient, and RHAMM-overexpressing PC3 cells were treated with 10 μmol/L Y27632, a Rho kinase inhibitor (Sigma-Aldrich), or vehicle and analyzed by confocal microscopy. In addition, cell migration and invasion assays were performed. Total cell extracts were probed for ROCK II, phospho-cofilin, and total cofilin. RHAMM protein was immunoprecipitated and blotted for F-actin. In addition, cell lysates were generated from lung metastases and probed for RHAMM and F-actin.
Correlation of RHAMM with recurrence and metastasis in clinical samples
Differential expression of the RHAMM transcript was evaluated in metastatic human prostate cancer samples versus primary tumor samples, using a two-sided t test, within the normalized data of three independent microarray datasets (12–14). Two of these datasets (GSE21034 and GSE25136) include clinical time to recurrence and were used to evaluate the association of RHAMM with recurrence-free survival. Kaplan–Meier curves and log-rank P values were used to evaluate survival differences associated with low and high expression of the HMMR transcript.
Statistical analysis
Statistical analyses were performed using GraphPad Prism (version 6.0) software (GraphPad Software, Inc., La Jolla, CA). All of the data was analyzed for statistical significance using Student’s t-test/one-way ANOVA. For all experiments, P < 0.05 was considered statistically significant.
For details, please refer to the Supplementary information for additional material and methods.
Results
RB loss promotes cancer metastasis
To explore the impact of loss of RB on the metastatic potential of prostate cancer, RB knockdown was verified in isogenic prostate cancer cells (Fig. 1A). To investigate a metastatic phenotype independent of hormone sensitivity, an AR-negative cell line (PC3) was utilized, in addition to the AR-positive LNCaP cell line. Strikingly, RB loss altered cell morphology, resulting in cell elongation and filopodia development, as noted by F-actin staining, a typical promigratory and invasive phenotype (Fig. 1B) with quantification in Supplementary Fig. S1A (15). Prior studies have also shown that invasive phenotypes exhibit altered cell surface markers, particularly downregulation of E-cadherin and upregulation of vimentin (16). In the current study, RB loss similarly decreased expression of E-cadherin (Fig. 1C, left) and increased expression of vimentin (Fig. 1C, right) compared with RB-proficient cells with quantification in Supplementary Fig. S1A. Collectively these data suggest that RB loss alters cell morphology and deregulates factors known to promote a metastatic phenotype.
Figure 1.
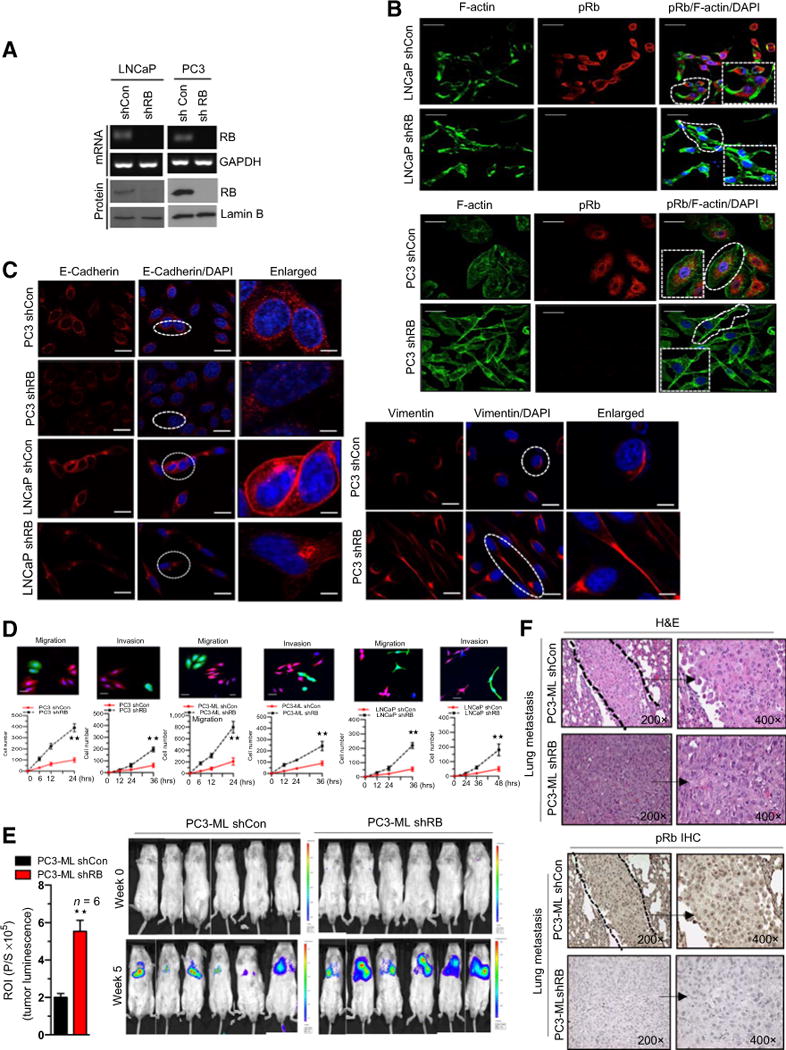
RB loss promotes cancer metastasis. A, RT-PCR analysis of RB, GAPDH mRNA, RB, and lamin B immunoblot in shCon and shRB LNCaP and PC3 cells. B, Confocal microscopic images of F-actin, pRb, and DAPI immunofluorescence in shCon and shRB LNCaP and PC3 cells. C, Confocal microscopic images of E-cadherin in shCon and shRB PC3 and LNCaP cells (left) and confocal microscopic images of vimentin in shCon and shRB PC3 cells (right). D, Graphic representation of quantitative migration kinetics with noncoated Boyden chamber and cell invasion kinetics with Matrigel-coated Boyden chamber in RB-proficient and -deficient PC3, PC3-ML, and LNCaP cells. E, Graphic representation of tumor luminescence from shCon and shRB PC3-ML tumor metastases in male SCIDs, with representative images (left and right). F, hematoxylin and eosin (H&E) and pRb staining of RB-proficient and -deficient PC3-ML tumor metastases at ×200 and ×400 (right). Each group contained a minimum of 6 animals. Each data point is a mean ± SD from three or more independent experiments. **, P < 0.05 was considered as statistically significant. Scale bar, 50 μm.
Given that all stages of prostate cancer depend on AR function (17), even in the advanced metastatic setting, we investigated the increased metastatic potential of RB loss in AR-positive cell lines. In the presence of androgens, RB loss promoted increased migratory and invasive capacity across multiple tumor models (Fig. 1D; Supplementary Fig. S1B–S1D); previous publications have demonstrated that this is not due to alterations in cellular proliferation (4). In parallel, we sought to generalize our findings beyond prostate cancer; using isogenic non–small cell lung cancer and glioblastoma cell lines, RB knockdown augmented the migratory and invasive capacity of these cells (Fig. 1D; Supplementary Fig. S1E). Utilizing an in vivo metastatic tail vein injection model, RB-deficient prostate cancer cells had increased lung metastases as compared with RB-proficient cells, as noted with tumor luminescence (Fig. 1E) and IHC (Fig. 1F). Collectively, these data suggest that RB loss alters migratory capacity and increases measured circulating tumor cells thereby promoting a metastatic phenotype.
Transcriptome array predicts RHAMM as an E2F target gene and RHAMM expression inversely correlates with RB status
The canonical function of RB is through formation of an inhibitory complex with E2F transcription factors. To investigate whether RB/E2F target genes are involved in cancer metastases, microarray analysis of RB-proficient PC3-ML and H1299 cells treated with a CDK4/6 inhibitor was performed. As expected, the top hit was E2F targets (Fig. 2A; Supplementary Fig. S2A). Given our hypothesis that RB functions to inhibit metastatic formation, we interrogated the E2F Targets implicated in epithelial-to-mesenchymal transformation. HMMR, the gene product of RHAMM, was among the top hits of downregulated genes upon RB activation in IPA analysis (Supplementary Fig. S2B). Several cancer models have implicated RHAMM in augmenting cell migration and invasion (18).
Figure 2.
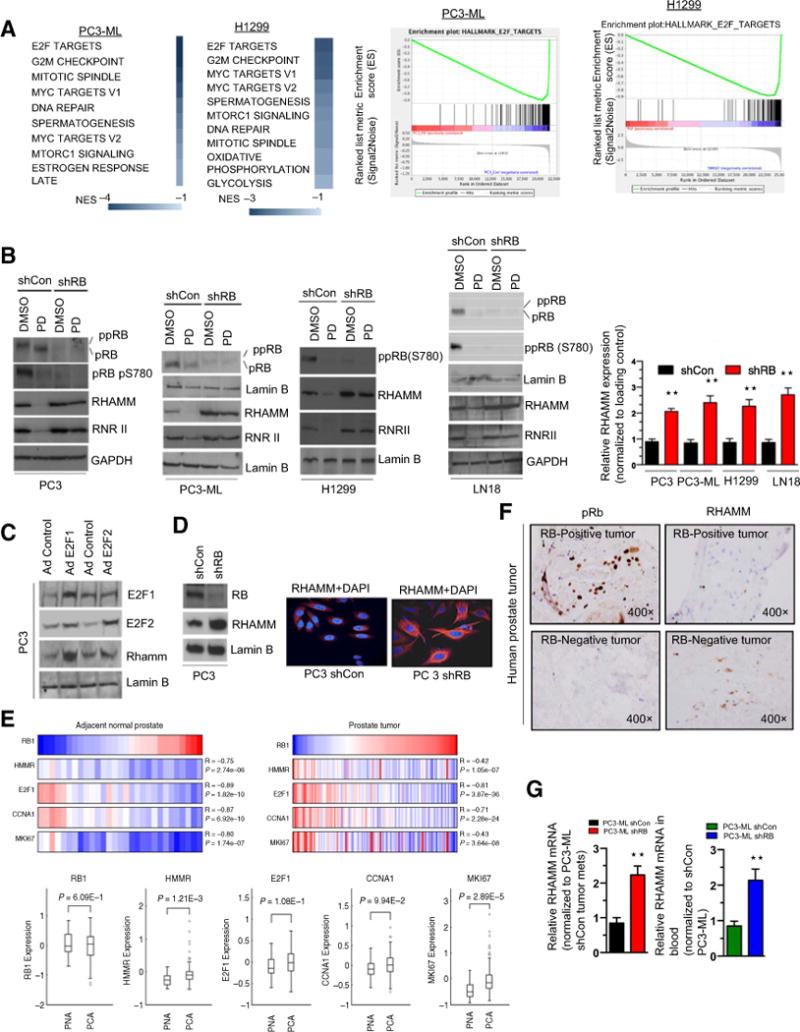
Transcriptome array predicts RHAMM as an E2F target gene and RHAMM expression inversely correlates with RB status. A, KEGG pathway analysis of differentially expressed transcripts from microarray analysis of RB-proficient parental PC3-ML and H1299 cells treated with either DMSO or PD 0332991 (left). GSEA analysis of the E2F target hallmarks in PC3-ML and H1299 cells (right). B, Western blotting analysis of RB, phospho RB pS780, RHAMM, RNRII, and lamin B in shCon and shRB PC3-ML, PC3, H1299, LN18 cells (left) and quantitation of RHAMM protein analysis in response to DMSO or PD 0332991 (right). C, Immunoblotting analysis of E2F1, E2F2, RHAMM, and lamin B in control and ectopically expressed adenovirus harboring E2F1 or E2F2 cDNA in PC3 cells. D, Immunoblotting analysis of RB, RHAMM, and lamin B in shCon and shRB PC3 cells (left) with immunofluorescence confocal microscopic images of RHAMM in shCon and shRB PC3 cells (right). E, In silico analysis and heatmap of HMMR, E2F1, CCNA1, and MKI67 with respect to RB1 transcript status in normal prostate (top left) and prostate tumor clinical specimens (top right) and quantitation (box plot) of HMMR, E2F1, CCNA1, and MKI67 with respect to RB1 transcript in normal and prostate tumors specimens (bottom). F, IHC analysis of pRb and RHAMM in RB-proficient and RB-deficient human prostate clinical specimens; representative samples are shown (×400). G, qRT-PCR analysis of RHAMM mRNA from lung metastases and whole blood of shCon and shRB PC3-ML tumor–bearing animals. Each data point is a mean ± SD from three or more independent experiments. **, P < 0.05 was considered as statistically significant. Scale bar, 50 μm.
We next sought to confirm that RHAMM was controlled by RB/E2F. Activation of RB-proficient cells via CDK 4/6 inhibition decreased RHAMM expression in multiple cancer models, while RB knockdown conversely increased RHAMM expression (Supplementary Fig. S2C–S2E), indicating that RHAMM expression is an RB-mediated event. This was confirmed at the protein level, whereby introduction of the CDK4/6 inhibitor resulted in hypophosphorylation of RB at S780, with concomitant diminished expression of RHAMM and RNR II, a known RB target (Fig. 2B; Supplementary Fig. S2E, right). As RB suppression is regulated via its interaction with E2F, we also utilized ectopic expression of E2F1 and E2F2 and noted an induction of the expression of RHAMM (Fig. 2C). Furthermore, immunofluorescence staining demonstrated higher RHAMM levels in setting of RB loss (Fig. 2D; Supplementary Fig. S2D), confirming the protein analysis.
In silico analysis demonstrated an inverse correlation between expression of RB1 transcript and RHAMM (HMMR) transcript in both normal prostate and prostate tumors (Fig. 2E). Furthermore, as previously demonstrated (19), RHAMM expression is highly elevated in prostate cancer compared with normal prostate tissue. E2F1, CCNA1, and MKI67 were utilized as controls and demonstrated similar trends (Fig. 2E). In addition, immunohistochemical analysis of primary prostate tumors suggested an inverse correlation between RB and RHAMM protein levels (Fig. 2F). Concomitantly, using human-specific primers, higher levels of HMMR mRNA were noted in both metastatic samples and whole blood from mice containing RB-deficient lung metastases than in RB-proficient controls (Fig. 2G). Collectively, these findings support RHAMM as an RB/E2F–regulated gene.
RB/E2F complex regulates RHAMM transcription
RB/E2F complexes regulate transcription primarily by binding to promoter regions and altering chromatin structure (20). An in silico analysis of RB/E2F putative–binding sites within 1 Kb of the RHAMM promoter revealed three potential binding sites (Fig. 3A). Binding affinities on the RHAMM promoter were further confirmed through an in silico docking model (Fig. 3B; Supplementary Table S2). Further chromatin immunoprecipitation indicated that upon CDK4/6 inhibition, RB binds robustly on the RB/E2F site II (Fig. 3C, top; Supplementary Fig. Fig. S3A) with consonant deacetylation of histone 4 (Fig. 3C, bottom; Supplementary Fig. S3B), indicating that RB binding results in a closed chromatin state that inhibits transcription. Conversely, no binding was detected in the context of RB loss. Furthermore, RB failed to associate with putative/predicted RB/E2F site I and RB/E2F site III.
Figure 3.
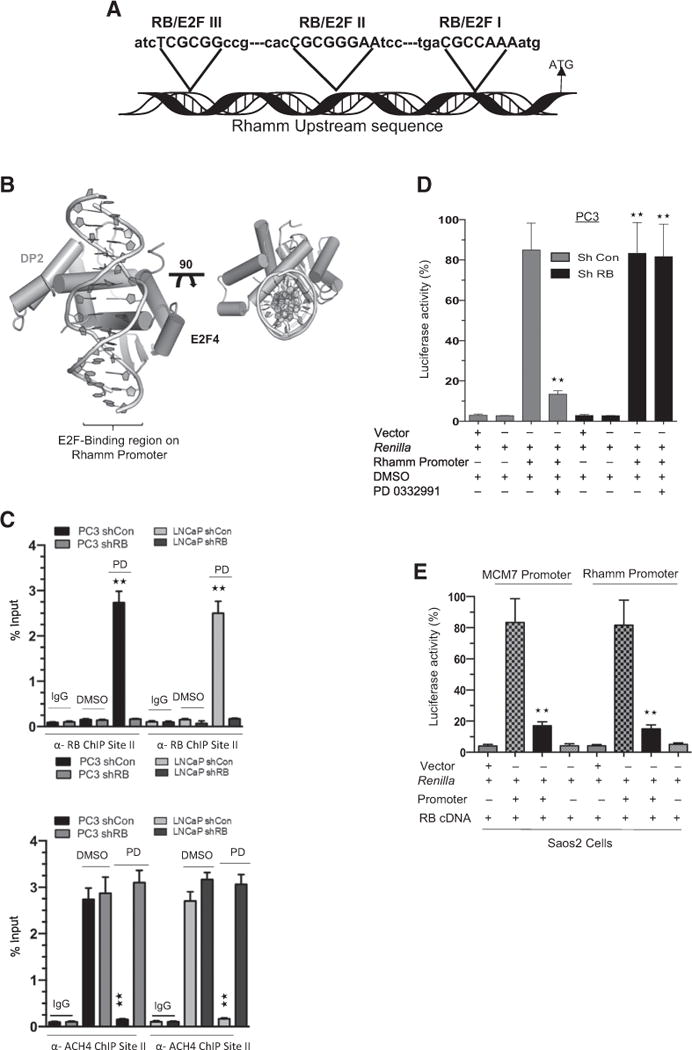
RB/E2F complex transcriptionally regulates RHAMM. A, Schematic illustration shows the location of three putative RB/E2F–binding sites on the RHAMM promoter. B, Docking model shows the RB/E2F binding region on the RHAMM promoter. C, Anti-RB (top) and anti-ACH4 (bottom) chromatin immunoprecipitation (ChIP) analysis at the RB/E2F–binding site II on the RHAMM promoter. shCon and shRB PC3 cells were treated with either DMSO or PD 0332991. D, Transcription analysis of RHAMM promoter via luciferase assay in shCon and shRB PC3 cells in response to PD 0332991 or DMSO. E, RB-specific transcription activation of RHAMM promoter and known RB target promoter MCM7 in RB-deficient Saos2 cells in response to ectopic expression of RB-cDNA or vector control. Each data point is a mean ± SD from three or more independent experiments. **, P < 0.05 was considered as statistically significant.
Using a luciferase reporter construct (Supplementary Fig. S3C), RHAMM promoter activity was repressed via CDK4/6 inhibition in the RB-proficient cells, whereas RB-deficient cells failed to alter promoter activity (Fig. 3D; Supplementary Fig. S3D). To demonstrate RB specificity on transcriptional control of RHAMM, we utilized RB-deficient Saos2 cells. Ectopic expression of RB cDNA repressed the promoter activities of RHAMM and MCM7 (a bona fide RB target; Fig. 3E; ref. 21). Collectively, these results demonstrate that RHAMM gene transcription is mediated via RB/E2F transcriptional control.
RHAMM overexpression mimics the RB loss metastatic phenotype and is a clinically meaningful marker of metastatic disease
Given the novel role of RB in regulating RHAMM transcription, we hypothesized that overexpression of RHAMM would recapitulate the metastatic phenotype. Stable overexpression was confirmed via protein analysis (Supplementary Fig. S4A). RHAMM overexpression altered cellular morphology (Fig. 4A), resulting in increased F-actin–rich filopodia. As filopodia interact with their environment via cell adhesion molecules such as cadherins and vimentin (22), we investigated the influence of RHAMM overexpression on these cell surface molecules. There was evidence of epithelial-to-mesenchymal transition with decreased levels of E-cadherin with increased N-cadherin and vimentin at both the transcript and protein levels (Fig. 4B and C; Supplementary Fig. S4A). RHAMM overexpression did not alter cellular proliferation (Fig. 4D), but increased migratory and invasive capacity compared with control cells (Fig. 4E; Supplementary Fig. S4B). In addition, RHAMM overexpression augmented migration and invasion in RWPE 1 cells (Supplementary Fig. S4C) as well as H1299 cells (Supplementary Fig. S4D).
Figure 4.
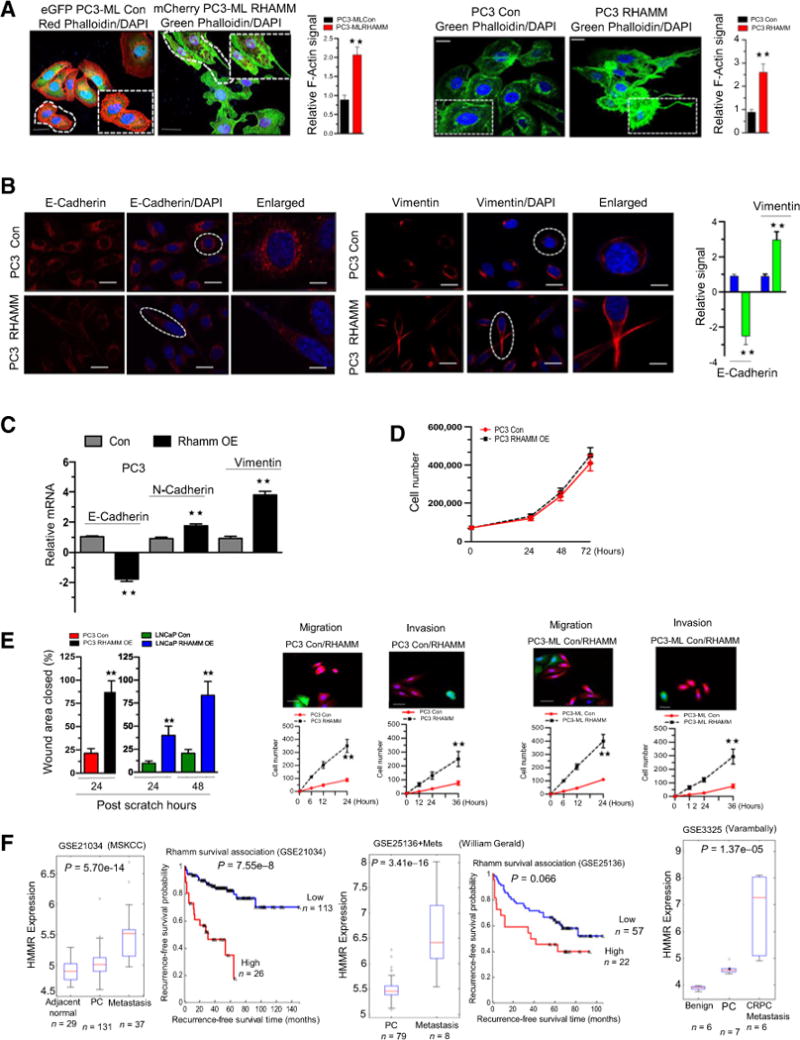
RHAMM overexpression mimics the RB loss metastatic phenotype and is a clinically meaningful marker of metastatic disease. A, Confocal microscopic images of F-actin staining (phalloidin) in control and RHAMM-overexpressing PC3-ML and PC3 cells with quantification (left and right). B, Immunofluorescence of confocal microscopic images of E-cadherin (left) and vimentin (middle) in control and RHAMM-overexpressing PC3 cells with quantification (right). C, qRT-PCR analysis of E-cadherin, N-cadherin, and vimentin mRNA. D, Short-term growth assay in control and RHAMM-overexpressing PC3 cells. E, Graphic representation of scratch assay in control and RHAMM-overexpressing PC3 and LNCaP cells (left), quantitative cell migration, and invasion kinetics in control and RHAMM-overexpressing PC3 and PC3-ML (right). F, Analysis of differential expression of HMMR transcript in human prostate metastatic samples versus primary tumor samples (GSE21034, ref. 12; GSE25136, ref. 13; GSE3225, ref. 14) and Kaplan–Meier survival curve (P values are presented in figure). For each data point, there is a mean ± SD from three or more independent experiments. **, P < 0.05 was considered as statistically significant except in silico analysis. Scale bar, 50 μm.
Using in silico analysis, we compared RHAMM transcript in metastatic versus localized prostate cancer, and found that RHAMM transcript was overexpressed in metastatic disease compared with primary prostate tumors. Elevated RHAMM transcript expression was associated with poor outcome in two independent datasets (P = 7.55 × 10−8, and P = 0.066; Fig. 4F; refs. 12, 19, 23). Thus, RHAMM overexpression mimics the RB-loss metastatic phenotype and is a clinically meaningful marker of metastatic disease.
Inhibition of RHAMM activity reverses the RB loss prometastatic phenotype
Given that inhibition of RHAMM has been shown to decrease migration and invasion in the context of breast cancer (24), we employed stable shRNA against RHAMM and knockdown as verified by RHAMM mRNA and protein (Supplementary Fig. S5A and S5B, left). Genetic knockdown of RHAMM was carried out using two different RHAMM shRNA constructs and resulted in diminished migration and invasion (Fig. 5A); however, it did not significantly alter overall proliferation (Supplementary Fig. S5A and S5B, right). This suggested that RHAMM specifically affected cell motility. Genetic loss of RHAMM reduced metastatic lung tumor burden in vivo (Fig. 5B). Low levels of RHAMM expression were confirmed in both metastatic lung lesions, whole blood samples (Fig. 5C), and by IHC (Fig. 5D). These findings implicate the significance of RHAMM in metastatic formation.
Figure 5.
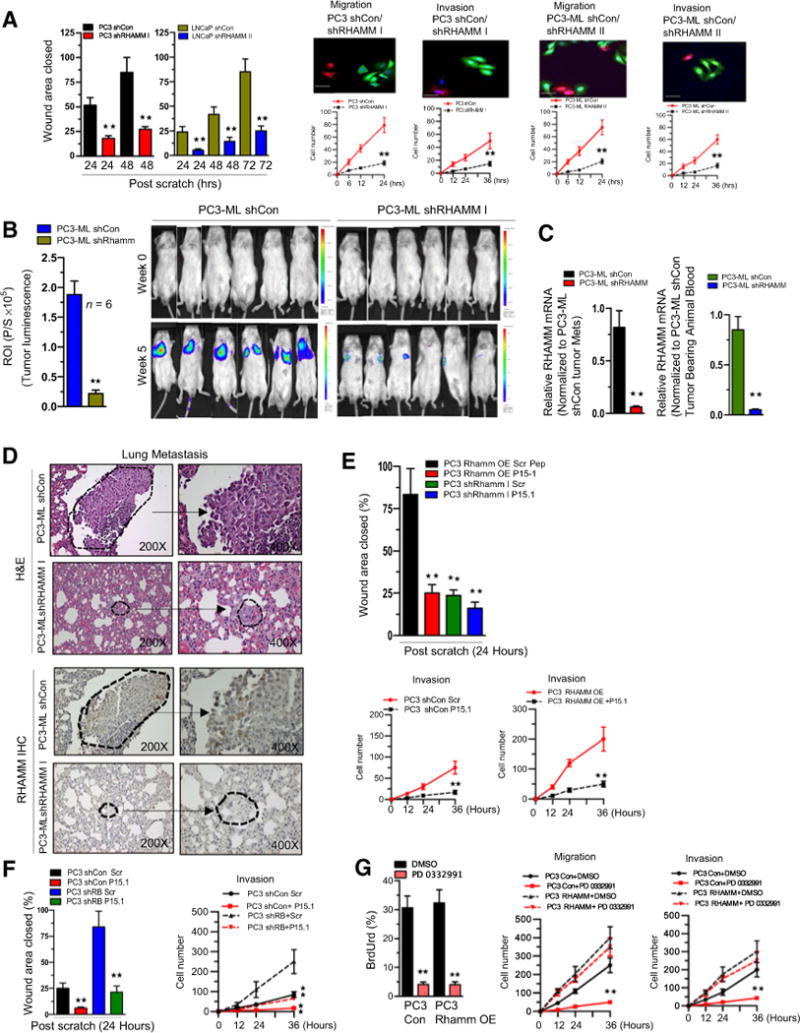
Inhibition of RHAMM activity reverses the RB loss prometastatic phenotype. A, Graphic representation of scratch assay (left), confocal microscopic images and quantitative cell migration and invasion kinetics in RHAMM-proficient and -deficient PC3 and PC3-ML (right). B, Graphic representation of tumor luminescence in shCon and shRHAMM PC3 cells tumor lung metastases (left) and representative images (right). Each group contained at least 6 animals per treatment condition. C, qRT-PCR analysis of RHAMM mRNA from lung metastases and whole blood of shCon and shRHAMM PC3-ML tumor–bearing animals. D, hematoxylin and eosin (H&E) and RHAMM protein IHC staining of lung metastases at ×200 and ×400 (middle). E, Graphic representation of scratch assay in control, shRHAMM I, and RHAMM-overexpressing PC3 cells in response to RHAMM peptide mimetic (top) and quantitative cell invasion kinetics in control and RHAMM-overexpressing PC3 cells in response to RHAMM peptide mimetic (bottom). F, Graphic representation of scratch assay (left) and quantitative invasion kinetics (right) in shCon and shRB PC3 cells in response to RHAMM peptide mimetic. G, Bromodeoxyuridine (BrdUrd) incorporation (left), quantitative cell migration and invasion kinetics (right) in RHAMM-overexpressing PC3 or control cells in response to DMSO or PD 0332991. For each data point, there is a mean ± SD from three or more independent experiments. **, P < 0.05 was considered as statistically significant. Scale bar, 50 μm.
Pharmacologic targeting of RHAMM was accomplished using a previously described RHAMM peptide mimetic (18). Introducing the RHAMM peptide mimetic decreased migration and invasion in both control cells (Supplementary Fig. S5C), RHAMM-overexpressing cells (Fig. 5E), and in the setting of RB knockdown (Fig. 5F; Supplementary Fig. S5D). Intriguingly, RB activation via CDK4/6 inhibition in RHAMM-overexpressing and control cells decreased cell-cycle progression (Fig. 5G, left), but did not alter overall invasion or migration rates (Fig. 5G, right). Thus, the prometastatic nature conferred via RB knockdown is mediated via RHAMM activity and inhibition of this pathway is capable of abrogating the RB loss–driven prometastatic phenotype.
Reprogramming of RB/E2F pathway via CDK4/6 inhibition restricts metastases
Given that loss of RB promotes a prometastatic phenotype, we next sought to determine whether RB activation through inhibiting CDK4/6 activity would affect metastatic progression. As previously shown with PD 0332991, a selective CDK4/6 inhibitor, hypophosphorylation of RB at serine 780 inhibits E2F-mediated transcriptional activity (Fig. 6A; ref. 25). Activation of RB decreased cellular migratory and invasive capacity in vitro (Fig. 6B).
Figure 6.
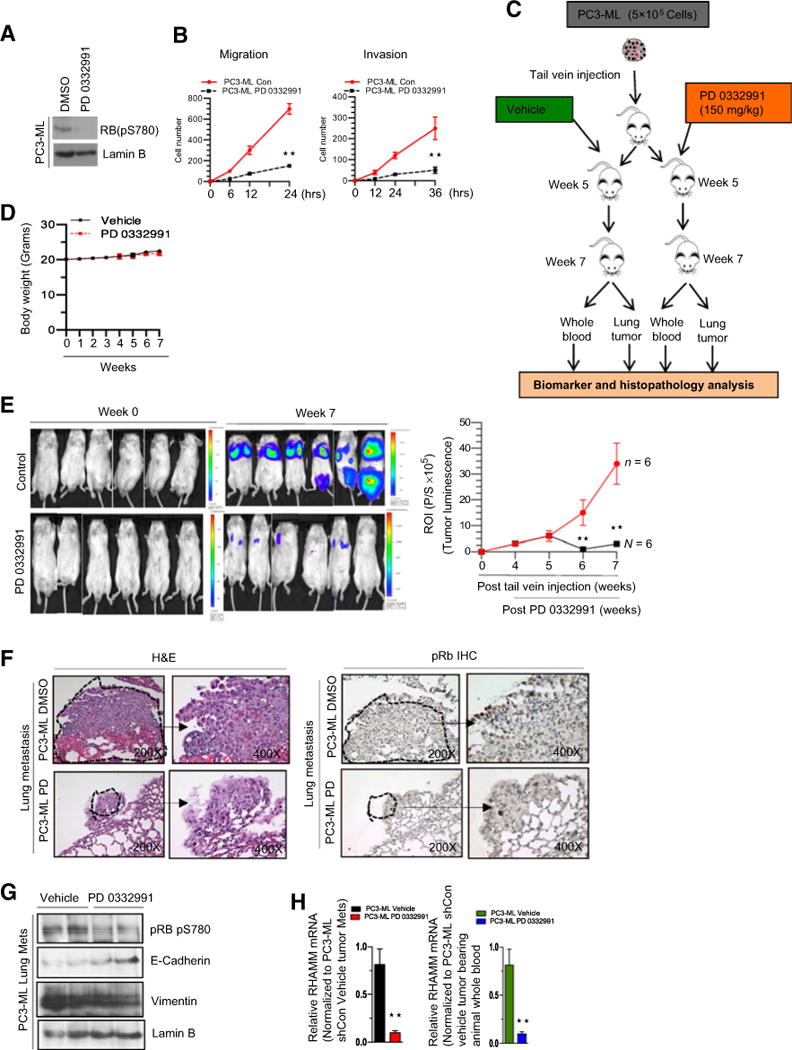
Reprogramming of RB/E2F pathway via CDK4/6 inhibition restricts metastases. A, Western blotting analysis of phospho RB pS780 and lamin B. B, Quantitative cell migration and invasion kinetics in PC3-ML cells treated with DMSO or PD 0332991. C, Schematic representation of experimental design showing experimental metastasis and timing of drug treatments and endpoint analysis. D, Graphic representation of weekly body weights in response to PD 0332991 treatment. E, Representative images with indicated time points (left) and graphic representation of tumor luminescence (right). F, Hematoxylin and eosin (H&E) and pRb IHC staining (×200 and ×400) of lung metastases in response to vehicle or PD 0332991. Arrow, enlarged region. G, Western blotting analysis of phospho RB pS780, E-cadherin, vimentin, and lamin B in PC3-ML–induced lung metastases in response to DMSO or PD 0332991. H, qRT-PCT analysis of RHAMM mRNA from lung tissue and whole blood of PC3-ML tumor–bearing animals treated with DMSO or PD 0332991. Each experimental group used 6 or more animals. Each data point is a mean ± SD from three or more independent experiments. **, P < 0.05 was considered as statistically significant.
To further investigate this concept in vivo, we performed tail vein injections of luciferase-tagged RB-proficient PC3-ML cells in a mouse model. After injection, mice were randomized to either vehicle or PD 0332991 as shown in the schematic illustration (Fig. 6C). There was no significant difference in body weight between the treatment groups (Fig. 6D). At time of sacrifice, vehicle-treated animals had higher lung tumor luminescence compared with the CDK4/6 inhibitor–treated cohort (Fig. 6E). Furthermore, Rb IHC and hematoxylin and eosin analysis was performed on the metastatic lung tumors revealing that animals treated with the CDK4/6 inhibitor had lower tumor burden as compared with vehicle-treated controls (Fig. 6F).
Tumor protein analysis confirmed that treatment with the CDK 4/6 inhibitor resulted in activated RB with hypophosphorylation at serine 780 as well as in vivo E-cadherin elevation and decreased vimentin compared with the vehicle control (Fig. 6G). Further qRT-PCR analysis of RHAMM mRNA in lung tumors and RNA from whole blood demonstrated that RB repressed RHAMM transcript (Fig. 6H). Together, these results establish a new paradigm that RB regulates metastatic potential, ultimately promoting the concept that RB-mediated target gene regulation controls metastasis.
RHAMM signaling via Rho-associated protein kinase impacts the prometastatic phenotype
RHAMM is known to function through multiple downstream pathways (26). Thus, to further elucidate the specific mechanism whereby RB loss mediates enhanced migration and invasion through RHAMM activity, we performed coimmunoprecipitation assays to determine RHAMM protein–protein interactions. As described previously (27), RHAMM directly interacts with F-actin filaments (Fig. 7A), resulting in the stabilization of actin polymerization. F-actin stabilization is mediated through phosphorylated cofilin via ROCKII function (28). As such, RB-deficient and RHAMM-overexpressing cells (Fig. 7B) induced elevated levels of phosphorylated cofilin, as demonstrated via protein analysis and immunofluorescence assays (Fig. 7C and D). We challenged this pathway pharmacologically by introducing Y27632, a ROCK II inhibitor (29). Inhibition of ROCK II function diminished phosphorylated cofilin (Fig. 7E) and decreased cellular migration and invasion (Fig. 7F) in both the RB knockdown and RHAMM overexpression setting. Furthermore, immunoblot analysis of lung metastases (from the previously described tail vein injections) revealed that RB activation via CDK4/6 inhibition significantly decreased levels of RHAMM and F-actin compared with vehicle-treated controls (Fig. 7G). Collectively, these findings demonstrate that RB loss results in a derepression of RHAMM function, driving F-actin stabilization via the ROCK signaling pathway and enhancing migration, invasion, and metastatic growth. Conversely, in the RB-proficient setting, activation of RB restricts metastatic tumor burden by transcriptionally repressing RHAMM, thus inhibiting the prometastatic signaling cascade (Fig. 7H).
Figure 7.
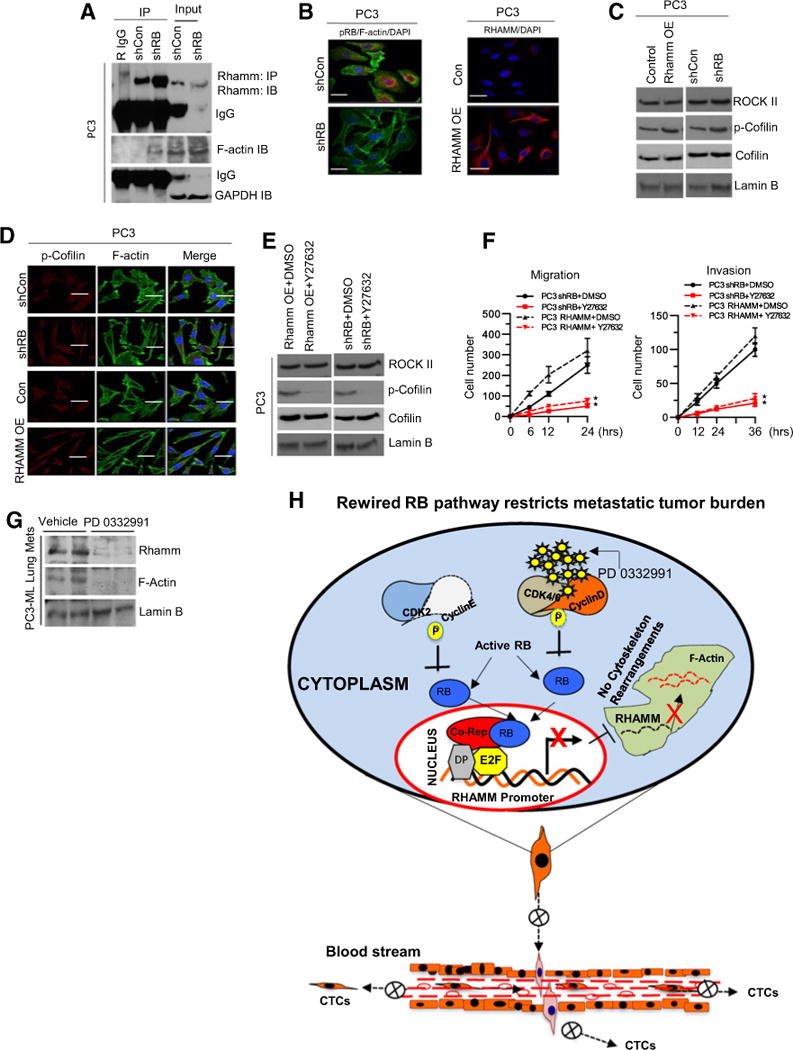
RHAMM signaling via Rho-associated protein kinase impacts the prometastatic phenotype. A, Coimmunoprecipitation of RHAMM and immunoblotting of RHAMM, F-actin, and loading control GAPDH in control and RB-deficient PC3 cells. B, Immunofluorescence confocal images of pRB and F-actin in RB-proficient and -deficient PC3 cells (left) and confocal images of RHAMM in control and RHAMM-overexpressing PC3 cells (right). C, Western blot analysis of ROCK II, p-Cofilin, cofilin, and lamin B in control, RHAMM-overexpressing, and RB-deficient PC3 cells. D, Immunoflourescence confocal images of localized F-actin, pRb, p-Cofilin, and RHAMM in control, RHAMM-overexpressing, and RB-deficient PC3 cells. E, Immunoblotting analysis of ROCK II, p-Cofilin, cofilin, and lamin B in RHAMM-overexpressing and RB-deficient PC3 cells treated with DMSO or Rho kinase inhibitor (Y27632). F, Quantitative cell migration and invasion kinetics in RB-deficient and RHAMM-overexpressing PC3 cells in response to DMSO or Y27632. G, Immunoblotting analysis of RHAMM, F-actin, and lamin B in PC3-ML–induced lung metastases treated with control or PD 0332991. H, Schematic illustration of the proposed working model. Each data point is a mean ± SD from three or more independent experiments. **, P < 0.05 was considered as statistically significant. Scale bar, 50 μm.
Discussion
To our knowledge, the current study is the first to provide a clinically relevant function for RB in mediating the formation of metastatic prostate cancer. Our data strongly support the hypothesis that RB perturbation results in a lethal metastatic phenotype mediated via RHAMM deregulation. This conclusion is supported by several key observations: (i) RB loss of function is overrepresented in metastatic prostate cancer and many other cancers; (ii) RB depletion is sufficient to induce a promigratory, invasive, and EMT phenotype; (iii) reprogramming the RB pathway restricts the metastatic potential of prostate cancer and circulating tumor cell markers in vivo; (iii) RB/E2F complex regulates expression of RHAMM, a key driver of metastatic potential; (iv) the RB loss–driven metastatic phenotype can be rescued through either genetic or pharmacologic impingement of RHAMM activity in vivo; and (v) upon RB loss, RHAMM signals via activated cofilin and stabilization of F-actin and alters cytoskeleton structure. On the basis of these findings, we propose a new paradigm for RB function in protecting against metastatic disease. These data describe an important role of RB/E2F signaling outside of canonical cell-cycle control in cancer biology.
Functionally inactive RB is a critical mediator of the transition to castrate resistance as RB binds to regions responsible for controlling AR gene expression, resulting in repression of transcription at these loci (30). Consequently, RB loss or aberrant E2F activation facilitates bypass of hormonal therapy. In addition, RB status informs the therapeutic response of prostate cancer, with RB loss causing sensitivity to radiotherapy (7) and chemotherapy (4, 31), while functionally active RB is necessary for utilization of CDK4/6 inhibitors (Fig. 6) (32). As well, RB loss is nearly universal in human prostatic small-cell carcinoma (33), and recent mouse models demonstrate the formation of neuroendocrine prostate tumors within the context of RB loss (34).
The RB/E2F pathway appears to facilitate multiple steps of the metastatic process. RB inactivation has been demonstrated to result in the downregulation of the epithelial marker E-cadherin and development of the EMT phenotype (Fig. 1; Supplementary Fig. S1) with highly invasive characteristics in breast cancer (35). Conversely, activation of the RB pathway in preclinical models inhibits metastatic tumor burden and circulating tumor markers (Fig. 6). Gene expression studies have observed a lack of RB expression in nearly half of all metastatic hepatocellular carcinoma cases (36). E2Fs have been shown to increase the invasive capacity of cells in multiple cancer models (37, 38), and E2F expression levels are increased in metastatic tumors compared with primary tumors (39).
Despite these findings, prior research had not established the underlying mechanism driving the functional relevance of alterations in the RB/E2F pathway to metastasis. In the current study, we demonstrate that RB/E2F forms a repressive complex on the RHAMM promoter, and loss of RB allows for the upregulation of RHAMM expression and function (Fig. 2, 3). RHAMM is a glycosaminoglycan and extracellular matrix–binding protein that has been implicated in cell growth, differentiation, and motility (23, 40). Overexpression of RHAMM is associated with development of metastases (Fig. 4; ref. 41) and RHAMM is highly overexpressed in prostate cancer metastases compared with localized prostate cancer or benign prostate gland (Fig. 2; ref. 42). Furthermore, high RHAMM expression is associated with biochemical failure in Gleason score 7 disease (19). It is also one of the three-protein signatures in a multi-biomarker predictive model that best predicts biochemical failure at 3 and 5 years (19). Interestingly, overexpression of RHAMM is not cancer specific; multiple studies have shown elevated RHAMM levels in many types of solid tumors including head and neck (43), breast (44), and colon cancer (45). In all of these cancers, high RHAMM expression is associated with poor clinical outcome.
In order for metastasis to occur, a series of coordinated processes, including local invasion, intravasation, and extravasation must occur (46). Tumor cell motility is mediated by members of the Rho family of GTPase through their action on actin assembly, actomyosin contractility, and microtubules (47). Activities of these proteins facilitate the formation of protrusions at the leading edge of cells to direct migration and regulate the disassembly of focal adhesions at the rear of the cell, and they are critical to invasion and progression of prostate cancer (48, 49). The RHO effectors, ROCK I and ROCK II, function to increase phosphorylation of MLC through inactivation of MLC phosphatase (50) and induce activation of LIM-kinase 1 (LIMK). LIMK phosphorylates cofilin on serine 3 inhibit actin-depolymerizing activity (51). The current study demonstrates that loss of RB regulation results in elevated RHAMM levels, leading to actin polymerization via increased cofilin phosphorylation (Fig. 7). This process is reversed with pharmacologic inhibition of ROCK II function inhibiting migration and invasion.
Collectively, we demonstrate a novel role for RB in mediating metastatic formation through transcriptional regulation of RHAMM. Loss of RB results in a proinvasive, migratory, and EMT phenotype that can be directly reversed via genetic or pharmacologic RHAMM blockade. Given the increasing importance of RB status in clinical decision making and increased utilization of RB as a biomarker (NCT02059213, NCT02218606), we propose that RHAMM activity and its downstream effectors, such as ROCK II, cofilin, and F-actin, may be potential therapeutic targets.
Supplementary Material
Acknowledgments
The authors thank members of the K. Knudsen laboratory for input and commentary.
Grant Support
R.B. Den was supported by a Young Investigator Award from the Prostate Cancer Foundation, a Physician Research Training Award from the Department of Defense Grant (PC101841), and grant #IRG 08-060-04 from the American Cancer Society. K.E. Knudsen was supported by PA Cure Award, NIH grant R01 CA099996, and a Movember-PCF Challenge Award.
S.G. Zhao reports receiving a commercial research grant from the Prostate Cancer Foundation and has provided expert testimony for GenomeDx Biosciences. F.Y. Feng is a founder and president of PFS Genomics and is a consultant/advisory board member for Medivation/Astellas, GenomeDx, and Celgene.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential: No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: C. Thangavel, J.L. Dean, K.J. Pienta, A.P. Dicker, R.B. Den
Development of methodology: C. Thangavel, E. Boopathi, Y. Liu, S. Ciment, J.L. Dean, M.A. Price, J.B. McCarthy, E.A. Turely, A.P. Dicker, R.B. Den
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): C. Thangavel, E. Boopathi, Y. Liu, A. Haber, J.L. Dean, A.E. Snook, J.R. Hernandez, R. Birbe, F.Y. Feng, K.E. Knudsen, R.B. Den
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C. Thangavel, E. Boopathi, Y. Liu, A. Ertel, A. Bhardwaj, S. Addya, P. Cotzia, J.L. Dean, C. McNair, S.G. Zhao, R. Birbe, K.E. Knudsen, R.B. Den
Writing, review, and/or revision of the manuscript: C. Thangavel, A. Haber, A. Bhardwaj, N. Williams, P. Cotzia, J.L. Dean, S.G. Zhao, R. Birbe, J.B. McCarthy, E.A. Turely, F.Y. Feng, K.E. Knudsen, R.B. Den
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): C. Thangavel, S. Ciment, P. Cotzia, J.L. Dean, A.P. Dicker, R.B. Den Study supervision: R.B. Den
References
- 1.Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–97. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manning AL, Dyson NJ. RB: mitotic implications of a tumour suppressor. Nat Rev Cancer. 2012;12:220–6. doi: 10.1038/nrc3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knudsen ES, McClendon AK, Franco J, Ertel A, Fortina P, Witkiewicz AK. RB loss contributes to aggressive tumor phenotypes in MYC-driven triple negative breast cancer. Cell Cycle. 2015;14:109–22. doi: 10.4161/15384101.2014.967118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma A, Comstock CE, Knudsen ES, Cao KH, Hess-Wilson JK, Morey LM, et al. Retinoblastoma tumor suppressor status is a critical determinant of therapeutic response in prostate cancer cells. Cancer Res. 2007;67:6192–203. doi: 10.1158/0008-5472.CAN-06-4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim KJ, Godarova A, Seedle K, Kim MH, Ince TA, Wells SI, et al. Rb suppresses collective invasion, circulation and metastasis of breast cancer cells in CD44-dependent manner. PLoS One. 2013;8:e80590. doi: 10.1371/journal.pone.0080590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maxwell CA, McCarthy J, Turley E. Cell-surface and mitotic-spindle RHAMM: moonlighting or dual oncogenic functions? J Cell Sci. 2008;121(Pt 7):925–32. doi: 10.1242/jcs.022038. [DOI] [PubMed] [Google Scholar]
- 7.Thangavel C, Boopathi E, Ciment S, Liu Y, R ON, Sharma A, et al. The retinoblastoma tumor suppressor modulates DNA repair and radioresponsiveness. Clin Cancer Res. 2014;20:5468–82. doi: 10.1158/1078-0432.CCR-14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kouvidi K, Berdiaki A, Nikitovic D, Katonis P, Afratis N, Hascall VC, et al. Role of receptor for hyaluronic acid-mediated motility (RHAMM) in low molecular weight hyaluronan (LMWHA)-mediated fibrosarcoma cell adhesion. J Biol Chem. 2011;286:38509–20. doi: 10.1074/jbc.M111.275875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meier C, Spitschak A, Abshagen K, Gupta S, Mor JM, Wolkenhauer O, et al. Association of RHAMM with E2F1 promotes tumour cell extravasation by transcriptional up-regulation of fibronectin. J Pathol. 2014;234:351–64. doi: 10.1002/path.4400. [DOI] [PubMed] [Google Scholar]
- 10.Augello MA, Burd CJ, Birbe R, McNair C, Ertel A, Magee MS, et al. Convergence of oncogenic and hormone receptor pathways promotes metastatic phenotypes. J Clin Invest. 2013;123:493–508. doi: 10.1172/JCI64750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu D, Liu J, Lin B, Liu S, Hou R, Hao Y, et al. Lewis y regulate cell cycle related factors in ovarian carcinoma cell RMG-I in vitro via ERK and Akt signaling pathways. Int J Mol Sci. 2012;13:828–39. doi: 10.3390/ijms13010828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun Y, Goodison S. Optimizing molecular signatures for predicting prostate cancer recurrence. Prostate. 2009;69:1119–27. doi: 10.1002/pros.20961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8:393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9:446–54. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- 16.Majid S, Dar AA, Saini S, Shahryari V, Arora S, Zaman MS, et al. miRNA-34b inhibits prostate cancer through demethylation, active chromatin modifications, and AKT pathways. Clin Cancer Res. 2013;19:73–84. doi: 10.1158/1078-0432.CCR-12-2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Augello MA, Den RB, Knudsen KE. AR function in promoting metastatic prostate cancer. Cancer Metastasis Rev. 2014;33:399–411. doi: 10.1007/s10555-013-9471-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tolg C, Hamilton SR, Zalinska E, McCulloch L, Amin R, Akentieva N, et al. A RHAMM mimetic peptide blocks hyaluronan signaling and reduces inflammation and fibrogenesis in excisional skin wounds. Am J Pathol. 2012;181:1250–70. doi: 10.1016/j.ajpath.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizzardi AE, Vogel RI, Koopmeiners JS, Forster CL, Marston LO, Rosener NK, et al. Elevated hyaluronan and hyaluronan-mediated motility receptor are associated with biochemical failure in patients with intermediate-grade prostate tumors. Cancer. 2014;120:1800–9. doi: 10.1002/cncr.28646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brehm A, Kouzarides T. Retinoblastoma protein meets chromatin. Trends Biochem Sci. 1999;24:142–5. doi: 10.1016/s0968-0004(99)01368-7. [DOI] [PubMed] [Google Scholar]
- 21.Thangavel C, Boopathi E, Ertel A, Lim M, Addya S, Fortina P, et al. Regulation of miR106b cluster through the RB pathway: mechanism and functional targets. Cell Cycle. 2013;12:98–111. doi: 10.4161/cc.23029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- 23.Hamilton SR, Fard SF, Paiwand FF, Tolg C, Veiseh M, Wang C, et al. The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J Biol Chem. 2007;282:16667–80. doi: 10.1074/jbc.M702078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci U S A. 2014;111:E89–98. doi: 10.1073/pnas.1319190110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rocca A, Farolfi A, Bravaccini S, Schirone A, Amadori D. Palbociclib (PD 0332991): targeting the cell cycle machinery in breast cancer. Expert Opin Pharmacother. 2014;15:407–20. doi: 10.1517/14656566.2014.870555. [DOI] [PubMed] [Google Scholar]
- 26.Park D, Kim Y, Kim H, Kim K, Lee YS, Choe J, et al. Hyaluronic acid promotes angiogenesis by inducing RHAMM-TGFbeta receptor interaction via CD44-PKCdelta. Mol Cells. 2012;33:563–74. doi: 10.1007/s10059-012-2294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Assmann V, Jenkinson D, Marshall JF, Hart IR. The intracellular hyaluronan receptor RHAMM/IHABP interacts with microtubules and actin filaments. J Cell Sci. 1999;112(Pt 22):3943–54. doi: 10.1242/jcs.112.22.3943. [DOI] [PubMed] [Google Scholar]
- 28.Pritchard CA, Hayes L, Wojnowski L, Zimmer A, Marais RM, Norman JC. B-Raf acts via the ROCKII/LIMK/cofilin pathway to maintain actin stress fibers in fibroblasts. Mol Cell Biol. 2004;24:5937–52. doi: 10.1128/MCB.24.13.5937-5952.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rath N, Olson MF. Rho-associated kinases in tumorigenesis: re-considering ROCK inhibition for cancer therapy. EMBO Rep. 2012;13:900–8. doi: 10.1038/embor.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, Thangavel C, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010;120:4478–92. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Leeuw R, Berman-Booty LD, Schiewer MJ, Ciment SJ, Den RB, Dicker AP, et al. Novel actions of next-generation taxanes benefit advanced stages of prostate cancer. Clin Cancer Res. 2015;21:795–807. doi: 10.1158/1078-0432.CCR-14-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Comstock CE, Augello MA, Goodwin JF, de Leeuw R, Schiewer MJ, Ostrander WF, Jr, et al. Targeting cell cycle and hormone receptor pathways in cancer. Oncogene. 2013;32:5481–91. doi: 10.1038/onc.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20:890–903. doi: 10.1158/1078-0432.CCR-13-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parisi T, Bronson RT, Lees JA. Inactivation of the retinoblastoma gene yields a mouse model of malignant colorectal cancer. Oncogene. 2015;34:5890–9. doi: 10.1038/onc.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arima Y, Inoue Y, Shibata T, Hayashi H, Nagano O, Saya H, et al. Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Res. 2008;68:5104–12. doi: 10.1158/0008-5472.CAN-07-5680. [DOI] [PubMed] [Google Scholar]
- 36.Hui AM, Li X, Makuuchi M, Takayama T, Kubota K. Over-expression and lack of retinoblastoma protein are associated with tumor progression and metastasis in hepatocellular carcinoma. Int J Cancer. 1999;84:604–8. doi: 10.1002/(sici)1097-0215(19991222)84:6<604::aid-ijc11>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 37.Yoon SO, Shin S, Mercurio AM. Ras stimulation of E2F activity and a consequent E2F regulation of integrin alpha6beta4 promote the invasion of breast carcinoma cells. Cancer Res. 2006;66:6288–95. doi: 10.1158/0008-5472.CAN-06-0826. [DOI] [PubMed] [Google Scholar]
- 38.Zhang SY, Liu SC, Johnson DG, Klein-Szanto AJ. E2F-1 gene transfer enhances invasiveness of human head and neck carcinoma cell lines. Cancer Res. 2000;60:5972–6. [PubMed] [Google Scholar]
- 39.Thomassen M, Tan Q, Kruse TA. Gene expression meta-analysis identifies metastatic pathways and transcription factors in breast cancer. BMC Cancer. 2008;8:394. doi: 10.1186/1471-2407-8-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tolg C, McCarthy JB, Yazdani A, Turley EA. Hyaluronan and RHAMM in wound repair and the “cancerization” of stromal tissues. BioMed Res Int. 2014;2014:103923. doi: 10.1155/2014/103923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hall CL, Turley EA. Hyaluronan: RHAMM mediated cell locomotion and signaling in tumorigenesis. J Neurooncol. 1995;26:221–9. doi: 10.1007/BF01052625. [DOI] [PubMed] [Google Scholar]
- 42.Gust KM, Hofer MD, Perner SR, Kim R, Chinnaiyan AM, Varambally S, et al. RHAMM (CD168) is overexpressed at the protein level and may constitute an immunogenic antigen in advanced prostate cancer disease. Neoplasia. 2009;11:956–63. doi: 10.1593/neo.09694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmitt A, Barth TF, Beyer E, Borchert F, Rojewski M, Chen J, et al. The tumor antigens RHAMM and G250/CAIX are expressed in head and neck squamous cell carcinomas and elicit specific CD8+ T cell responses. Int J Oncol. 2009;34:629–39. doi: 10.3892/ijo_00000188. [DOI] [PubMed] [Google Scholar]
- 44.Wang C, Thor AD, Moore DHII, Zhao Y, Kerschmann R, Stern R, et al. The overexpression of RHAMM, a hyaluronan-binding protein that regulates ras signaling, correlates with overexpression of mitogen-activated protein kinase and is a significant parameter in breast cancer progression. Clin Cancer Res. 1998;4:567–76. [PubMed] [Google Scholar]
- 45.Zlobec I, Terracciano L, Tornillo L, Gunthert U, Vuong T, Jass JR, et al. Role of RHAMM within the hierarchy of well-established prognostic factors in colorectal cancer. Gut. 2008;57:1413–9. doi: 10.1136/gut.2007.141192. [DOI] [PubMed] [Google Scholar]
- 46.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 47.Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–9. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 48.Gingell JC, Desai KM. Peyronie’s disease. Br J Urol. 1989;63:223–6. doi: 10.1111/j.1464-410x.1989.tb05179.x. [DOI] [PubMed] [Google Scholar]
- 49.Zhou C, Ling MT, Kin-Wah Lee T, Man K, Wang X, Wong YC. FTY720, a fungus metabolite, inhibits invasion ability of androgen-independent prostate cancer cells through inactivation of RhoA-GTPase. Cancer Lett. 2006;233:36–47. doi: 10.1016/j.canlet.2005.02.039. [DOI] [PubMed] [Google Scholar]
- 50.Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, et al. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol. 1999;147:1023–38. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–12. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.