

Abstract
There is great potential for engineering cellular therapeutics by repurposing biological systems. Here, we report utilization of the granzyme-perforin pathway of cytotoxic lymphocytes as a cell-to-cell protein delivery module. We designed and constructed granzyme B-derived chaperone molecules fused to a fluorescent protein payload and expressed these constructs in natural killer (NK) cells. Using confocal microscopy and flow cytometry, we investigated the co-localization of the chaperones with lytic granules and the chaperone-mediated transfer of the fluorescent protein payload from NK to target cells in co-culture experiments. A synthetic chaperone consisting of the granzyme B ER signal peptide and a domain encompassing putative N-linked glycosylation sites in granzyme B is insufficient for payload transfer to target cells, whereas full-length granzyme B is sufficient for payload delivery. Combining our functional data with an analysis of the crystal structure of granzyme B suggests that the necessary motifs for granzyme B loading into lytic granules are dispersed throughout the primary amino acid sequence and are only functional when contiguous in the tertiary structure. These results illustrate that by using granzyme B as a molecular chaperone the granzyme-perforin pathway can be exploited as a programmable molecular delivery system for cell-based therapies.
Keywords: synthetic immunology, molecular delivery, engineered cells
Graphical Abstract
Introduction
With their ability to sense and integrate a wide range of signals, actively move to specific tissue compartments, and actuate context-dependent responses, engineered cell-based therapeutics are emerging as the next major class of medical intervention.1 Chimeric antigen receptor T cells (CAR Ts) are highly effective in treating hematological malignancies,2, 3, 4 and many mesenchymal stem cell therapies5 are at various stages of development for use in cardiac,6, 7 neurological,8 and malignant9, 10, 11 disease. These advances are a result of recombining the diverse functionality of biological systems to generate new functional biological molecules and pathways.12, 13
For any useful cellular device, three primary components are required: input modules, control logic, and output modules. Significant progress has been made with the former two, including a variety of transcriptional control circuits14 as well as highly specific and configurable chimeric antigen receptors15 and synthetic notch receptors.16, 17 Output modules for cellular devices have been less developed and investigated. Most rely on passive, nonspecific secretion, activated by a combination of sensory input modules, modulated by control circuits.18 This approach has potential for a variety of metabolic derangements,19 but also has a significant limitation in that transcriptionally activated secretion creates a temporal delay between activation and actuation. For many applications, an ideal output module would have the deliverable payload pre-positioned for rapid actuation and delivery. Furthermore, secretion only achieves regional specificity at best. In many cases cell-specific delivery would be extremely valuable, both as a research tool, as well as in therapeutic contexts.
To date the most investigated and best understood cell-to-cell delivery systems are bacterial in origin, specifically bacterial type III and type IV secretion systems.20, 21, 22 Other synthetic biology approaches have also been developed using, for example, invasin triggered by environmental signals or quorum sensing.23, 24 These designs achieve cell-specific delivery, yet they retain two major weaknesses. First, they are for the most part transcriptionally controlled, resulting in a lag between activation and delivery, and second, they are bacterial in origin, likely raising prohibitive immunological obstacles to human therapeutic applications.
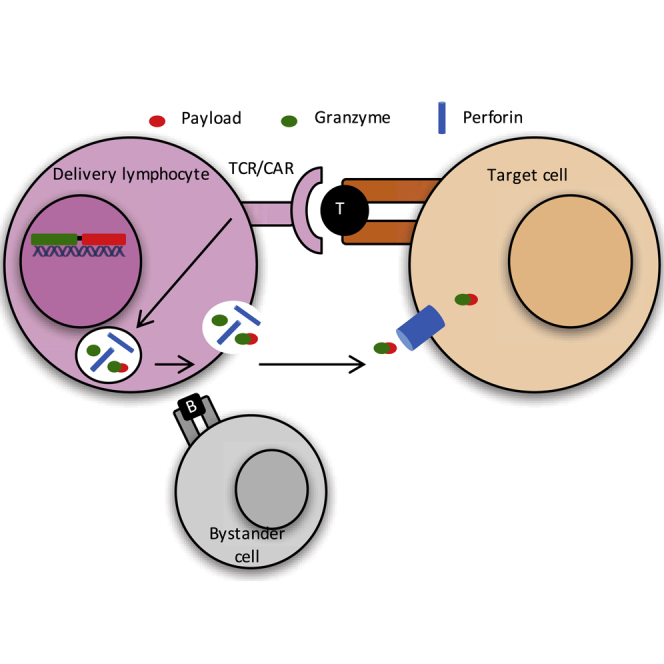
We set out to develop a cell-to-cell delivery module in which payload delivery could be rapidly initiated by surface receptors, with this activation resulting in the transfer of the payload from the delivery cell to a target cell in a cell-specific manner. Further, we desired that delivery originate from a pre-positioned source, such that the time between initiation of target recognition and completion of payload delivery is a short, tightly coupled process. The granzyme-perforin pathway, in the context of a cytotoxic lymphocyte chassis, is a unique and potentially broadly applicable cell-to-cell delivery system that fulfilled these criteria. This pathway is one of the main ways in which cytotoxic lymphocytes kill target cells.25 Its main constituent components are the serine protease granzymes and the pore forming protein perforin, both of which are stored in membrane-bound secretory lysosomes, or lytic granules, in the cytosol of cytotoxic lymphocytes.26 Upon target cell recognition, the cytotoxic lymphocyte forms a tight apposition with the target cell, forming the immunological synapse. Surface receptor signaling results in the endocytic release of granzymes and perforin from the lytic granules into the synapse between the two cells.27 Perforin then inserts in the target cell membrane and oligomerizes to form transient pores, through which granzyme diffuses into the target cell.28, 29, 30 Finally, granzyme cleaves caspases and BH3-interacting domain death agonist (BID) to initiate target cell apoptosis. Importantly, surrounding bystander cells typically do not receive appreciable quantities of granzymes.31, 32, 33 In summary, surface receptor-mediated target cell recognition results in specific, cytoplasm-to-cytoplasm intercellular transfer of granzymes to that same target cell. More broadly, cytotoxic lymphocytes have exceptional utility as a cellular chassis because they are targetable, expandable, and amenable to genetic manipulation.34
This combination of the granzyme-perforin pathway and cytotoxic lymphocyte chassis fulfill all of the requirements for a cellular device output module listed above: native and engineered targeting modules (T cell receptors or chimeric antigen receptors) that naturally interface with a signaling cascade that initiates release of pre-positioned molecules, that are delivered in a cell-specific manner, as defined by the original surface receptor recognition of the target cell.
Granzyme B (GZB) is a well-studied effector molecule that transits the granzyme-perforin pathway. Here, we engineer granzyme B-derived chaperones and trace chaperone mediated trafficking of a functional fluorescent protein payload through this pathway from effector to target cells. This demonstrates the implementation of a programmable cell-to-cell transfer module that can potentially be used to deliver different ectopic protein payloads to targeted tissues or cells.
Results
Design of Granzyme B-Derived Molecular Chaperones
Granzyme B is synthesized as a pre-pro-protein, with an 18 amino acid N-terminal endoplasmic reticulum (ER) signal peptide, followed by an inhibitory dipeptide, followed by the rest of the protein.26 Upon initiation of translation, the ER signal peptide directs the nascent protein to the ER, where it is co-translationally inserted into the ER. As the protein is synthesized in the ER, an N-glycan is added, which targets the protein to the Golgi network once synthesis is complete. In the Golgi, the N-glycan is further phosphorylated. This phosphosugar moiety on granzyme B then binds to the mannose-6-phosphate receptor, which targets the protein to lytic granules, where it is sequestered until target cell recognition, resulting in granzyme B release into the immune synapse.27 Importantly, recent work has shown that following release into the immune synapse, the trafficking of granzyme B to the target cell membrane and entry into the target cell via perforin pores is likely a result of passive diffusion only.28, 29, 33
We used this information to guide our design of chaperones for granzyme-perforin-mediated delivery. Since the steps in this process that are downstream of lytic granule exocytosis appear to be passive, we hypothesized that a chaperone that successfully directed a payload to be loaded into lytic granules would also be sufficient for payload delivery to the target cell. In designing such a chaperone, we adopted two strategies: rational and empirical (schematics for all constructs used in this paper are found in Figure 1).
Figure 1.

Design of Payload Delivery Module Chaperones
(A) Granzyme B as a model protein that transits the granzyme-perforin pathway. The full-length coding sequence is shown in green, with the ER signal peptide in light green (GZBSS). The two putative N-linked glycosylation motifs are shown in blue (NLG), with the encompassing putative sort motif (GZBSM) in yellow. The boundaries of the first and second halves of granzyme B used in this study (GZB1 and GZB2) are shown above the full-length coding sequence. (B) Schematic of the constructs used in this study. crmCherry (MCH, red), an RFP protein was used as a model payload, and a flexible glycine serine linker (GSL, purple) was used to join GZB, GZB1, and GZB2 to MCH.
For our rational design, we sought to develop a set of minimal granzyme B domains that would shuttle a protein payload to lytic granules. We took this set to be an ER localization domain and a lytic granule localization domain. For the former, we used the granzyme B ER signal peptide (GZBSS). For the latter, we used a 53 amino acid motif surrounding two computationally predicted N-linked glycosylation sites (GZBSM). Our final rational design consisted of GZBSS at the N terminus, followed by the model payload, followed by GZBSM. For a model payload, we selected crmCherry (hereafter mCherry or MCH), a derivative of the mCherry red fluorescent protein that is known to be stable in the acidic environment of lysosomes.35
The behavior of chimeric proteins consisting of domains derived from multiple proteins that have been rearranged is unpredictable. Therefore, we also selected full-length granzyme B as an empirical chaperone. Our rationale for this choice was that if there were unknown domains within granzyme B other than the region surrounding the N-linked glycosylation sites that were necessary for lytic granule loading or if the necessary domains are adjacent to the N-linked glycosylation sites only in the tertiary structure of granzyme B, then they would be captured in the full-length protein. As a first step toward delineating the location of these critical motifs within full-length granzyme B, we also investigated the first and second halves of granzyme B (GZB1 and GZB2, respectively) as independent chaperones. If either of these halves were functional as a chaperone, not only would it indicate that a minimal motif was present in them, from a practical standpoint this would be useful as it would yield a smaller chaperone, potentially much smaller if the process were repeated and the minimal motif further constrained. We placed the GZBSS ER signal peptide at the N terminus of both GZB1 and GZB2. To keep these granzyme B domains in as native a form as possible, we fused the MCH payload to the C terminus of GZB, with the two proteins connected by a flexible glycine serine linker.
As controls, we also generated two additional constructs: MCH alone and GZBSS followed by MCH.
Screening Chaperone Designs by Assessing Lytic Granule Co-localization Using Confocal Microscopy
Since our hypothesis was that lytic granule loading of a payload would be sufficient for payload delivery, we first investigated the subcellular localization of the chaperones using confocal microscopy. Using electroporation, we expressed the candidates in the natural killer cell line YT-Indy, which retains a functional granzyme-perforin pathway and has well-characterized target cell lines.36 Following enrichment for mCherry+ cells via cell sorting, we stained the cells for the lysosomal and lytic granule marker Lamp1 and then acquired images of the cells using confocal microscopy. As expected, the Lamp1 distribution was punctate in nature, but the MCH distribution was highly variable (Figure 2).
Figure 2.

Subcellular Distribution of Candidate Chaperone-mCherry Fusion Proteins
YT-Indys expressing the candidate fusion proteins (labeled at left) were stained for the lytic granule marker Lamp1 and imaged using confocal microscopy. Shown are merged red (mCherry) and green (Lamp1) channels for three representative cells for each sample. White scale bar, 5 μm.
Due to the range of phenotypes observed in the images, we sought to evaluate the degree of payload (MCH) co-localization with Lamp1 in an unbiased manner. To do this, we developed a semi-automated image filtration and analysis pipeline. The algorithm is illustrated in Figure 3 and seeks to eliminate both local background and bleed as well as pixel noise. This is achieved using both local and global image information to filter each pixel. This filtering is critical to enable quantitation of co-localization, as it eliminates the background noise from the regions of the image in which there are no cells, as well as regions that are adjacent to granules that have moderate signal intensity, both of which could give a spurious contribution to any quantitative metric of co-localization. The efficacy of this method can be observed by examining the progression of the columns from top to bottom of Figure 3: note that pixel intensities of the punctate structures remain relatively intact, whereas the binarized images (showing the extent of the background signal) change from containing large homogeneous structures to puncta that closely resemble those in the pixel intensity images. To our knowledge, this combination of local background subtraction with global pixel noise filtration has not been reported previously. We feel that it is particularly useful for studies involving small, subcellular structures, and in the hopes that it will be of use to the community, we have included the source code that implements the algorithm in the Supplemental Information.
Figure 3.

Automated Image Noise Filtering Pipeline
All panels are derived from a single channel of a single image. The first column consists of pixel intensity traces of a single horizontal line of pixels from the corresponding two-dimensional image in the second column (this line is shown in blue throughout the figure). The third column shows binarized versions of the middle column: all pixels with intensity greater than 0 are set to 1. The first row is the raw image data. The second row is the local background of the image. The third row is the background subtracted image, literally the second row subtracted from the first. The section of the images enclosed by the orange boxes in the third row have been enlarged and are displayed in the fourth row, to better show the pixel noise (small fluctuations near zero in the first column). The horizontal purple line is the threshold that will be applied to filter pixel noise. The fifth row shows the final resulting image after the threshold has been applied. The bottom row also shows the final processed image but zoomed back out to allow for comparison to rows 1–3. As in row 3, the orange boxes correspond to the region enlarged in rows 4 and 5.
Using this approach, we quantified the co-localization between MCH and Lamp1 in these filtered images using the Manders M1 coefficient and Pearson correlation coefficient (PCC) (Figure 4). Both metrics indicated that MCH alone had a low degree of co-localization with lytic granules, which would be expected as the lytic granules are small dense granules, and unfused MCH is distributed throughout the cytosol. These quantitative metrics were less useful for differentiating between the remaining fusion proteins. This motivated us to qualitatively assess the morphology of the distributions of MCH and Lamp1 and use this as a guide to selecting candidate chaperones. GZBSS-MCH had a perinuclear and membrane distribution, consistent with it entering the secretory pathway. GZB-MCH displayed a punctate cytosolic distribution consistent with granule loading. GZBSS-MCH-GZBSM exhibits a heterogeneous distribution that is partially punctate and granular (similar to GZB-MCH) and partially of moderate intensity, diffuse, and cytosolic (similar to unfused MCH). GZB1-MCH has an interesting membranous phenotype, which we hypothesize might be a result of this fusion protein entering the secretory pathway. GZB2-MCH has a highly punctuate distribution, but one that is considerably divergent from that of the Lamp1 distribution. The marked difference in the morphology of the subcellular localization of the two halves of granzyme B is interesting, and we discuss this further below. In summary, our assessment of the subcellular distribution of the various chaperone mCherry fusion proteins led us to conclude that GZB-MCH and GZBSS-MCH-GZBSM had the most potential as candidate chaperones.
Figure 4.

Quantitative Assessment of Candidate Chaperone Co-localization with Lytic Granules
Two co-localization metrics were calculated: Manders M1 (quantifying the fraction of pixel intensity of mCherry-positive pixels that also contain Lamp1 signal) and Pearson correlation coefficient (PCC, quantifying the degree to which red and green pixel intensities are correlated). These were calculated using both channels from each image. Each circle is the score of a single image, and each image contained between two and five cells. Overlaid are box and whisker plots.
Transfer of Fusion Proteins from Effector to Target Cells
We next characterized the capacity of two candidate chaperones (GZBSS-MCH and GZBSS-MCH-GZBSM) to facilitate transfer of the payload through the granzyme perforin pathway to target cells. To do this, we conducted a series of co-culture experiments, again using mCherry as a fluorescent reporter payload that was easily traceable. Effectors expressing a variety of mCherry fusion proteins were co-cultured with fluorescently labeled target cells, and then analyzed for evidence of mCherry in the target cell populations. We used the B cell lymphoblastoid cell line 721.221 (hereafter 721)37 as target cells, as they are a well known YT-Indy target.
We started by testing GZB-MCH, GZBSS-MCH, and MCH alone. GZB-MCH was the chaperone our microscopy images had suggested was most likely to load payloads into lytic granules, while MCH alone was clearly not loaded into lytic granules and thus a good control. GZBSS-MCH was included to confirm that it was not being loaded into lytic granules and hence would not be transferred to target cells. YT-Indy cells were transfected with plasmids coding for these chaperones and then FACS enriched for RFP+ cells. The various effector cell types were separately co-cultured with 721 target cells that had been labeled with a fluorescent dye, to distinguish between the effector and target cells. This mixed cell population was then analyzed via flow cytometry (Figure 5). Target 721 cells that were co-cultured with YT-Indys expressing GZB-MCH show an increase in mCherry signal, as compared to 721 s alone, 721 s co-cultured with unmodified YT-Indys, and 721 s co-cultured with YT-Indys expressing either MCH or GZBSS-MCH. Notably, this increase is most prominent in the dead cell fraction (DAPI+) of the target cells. Since the majority of these cells are dying due to YT-Indy attack, DAPI positivity can be viewed as a proxy for YT-Indy targeting. Therefore, the increase in MCH signal in dead (DAPI+) targets co-cultured with YT-Indys expressing GZB-MCH, but not MCH or GZBSS-MCH, suggests that GZB-MCH is transferred to target cells specifically via chaperone-mediated trafficking through the granzyme-perforin pathway.
Figure 5.

Transfer of Granzyme B mCherry Fusion Protein to Target Cells
YT-Indys expressing various mCherry fusion proteins were co-cultured with CFSE-labeled target 721 cells, and the mixed population was analyzed via flow cytometry. (A) Gating strategy for isolating target cells. Debris was eliminated (top panel), and then FITC+ targets selected (bottom panel). Not shown is an intermediate hierarchical gating step in which doublets are excluded using forward and side scatter width versus height gates. (B) Target 721 cell mCherry fluorescence. Each column is labeled by the effector that was present in the co-culture, but only target cells are plotted, using the gating from (A). Each column contains the same data showing target cell populations from all co-cultures, but only a single target population is highlighted in blue, which corresponds to the effector partner that was present in the co-culture partner.
We then investigated if the rationally designed chaperone (GZBSS-MCH-GZBSM) would perform similarly to GZB-MCH. We conducted the same type of experiment as above, comparing YT-Indys expressing GZBSS-MCH, GZBSS-MCH-GZBSM, and GZB-MCH. We selected GZBSS as the comparator so that all constructs would have the same N-terminal ER signal peptide and potential for secretion. This would allow us to differentiate between non-specific mCherry signal in target cells and mCherry signal in target cells resulting from chaperone-mediated transfer. The data (Figure 6) are consistent with our initial co-culture experiments and indicate that GZB transfers MCH to target cells, but GZBSS or GZBSS in combination with GZBSM does not. The fact that GZBSS-MCH-GZBSM did not transfer MCH to target cells is interesting. Given the mixed phenotype observed in YT-Indys expressing this construct in our microscopy images, we thought there was a possibility that it might also traffic to target cells. The fact that it does not provides useful information concerning the nature and location of the granzyme B motifs responsible for its trafficking through the granzyme-perforin pathway, which we discuss below.
Figure 6.

Comparison of MCH Payload Transfer to Target Cells by the Two Granzyme B-Derived Chaperones
YT-Indys expressing GZBSS-MCH (red), GZBSS-MCH-GZBSM (green), or GZB-MCH (blue) fusion proteins were co-cultured with CFSE-labeled target 721 cells, and the mixed population was analyzed via flow cytometry. The same gating strategy from Figure 5 was used, and only target cells are plotted. Live and dead cells were selected using a DAPI versus MCH dot plot. Bar plots show mean fluorescent intensity of the RFP channel (MCH MFI) of 721 target cells, with error bars denoting the standard deviation of duplicate samples. p values were calculated using Tukey’s HSD test applied to the results of a single factor ANOVA that was conducted for each target cell population separately.
Finally, to confirm at the protein level that granzyme B transfers the MCH payload to target cells, we repeated the above experiments, with the additional, post-co-culture, step of collecting live and dead (DAPI− and DAPI+, respectively) 721 target cells from each co-culture via fluorescence-activated cell sorting (FACS). Data from this sort is shown in Figure 7A, and it is consistent with the flow cytometry data from our other experiments. Whole-cell lysates from the sorted target cell populations were then size-separated by gel electrophoresis and probed for mCherry via western blot (Figure 7B). A prominent 60-kDa band consistent with GZB-MCH is observed in lysates of 721s co-cultured with YT-Indys expressing GZB-MCH. There is also a background band of approximately 30 kDa, consistent with unfused mCherry in the lysate of 721s co-cultured with YT-Indys expressing GZBSS-MCH. This is not unexpected, as the ER signal peptide in GZBSS-MCH directs mCherry to the secretory pathway,38 resulting in extracellular mCherry, some of which is likely taken up by the target 721s. However, if non-specific uptake were the main mechanism of MCH transfer from effector to target cell for all samples, then we would not expect to see any difference between 721s co-cultured with YT-Indys expressing GZB-MCH compared to 721s co-cultured with YT-Indys expressing GZBSS-MCH. Instead, of the DAPI+ 721 samples, only the sample from targets that were co-cultured with YT-Indys expressing GZB-MCH has detectable amounts of mCherry protein, and this band is detected at approximately 60 kDa, the expected size of GZB-MCH. Conversely, there is no detectable analogous 30-kDa band corresponding to MCH in the lysates from DAPI+ targets co-cultured with YT-Indys expressing GZBSS-MCH. That the putative GZB-MCH band in the DAPI+ 721 sample co-cultured with YT-Indys expressing GZB-MCH is even detectable is noteworthy given the actual amount of protein loaded is quite small, as demonstrated by the lack of a vinculin-loading control band. This is despite equal cell numbers for all lanes being sorted and lysed and is because the DAPI+ dead cells are apoptotic and rapidly degrading, which results in a loss of protein.
Figure 7.

Western Blot Confirmation of GZB-MCH Fusion Protein Transfer to Target Cells
(A) FACS sort data. YT-Indys expressing either GZB-MCH or GZBSS-MCH were co-cultured with CFSE-labeled target 721 cells, stained with DAPI, and FACS sorted. Target cells were first selected (upper left panel) and then divided into live and dead (upper right), which were sorted separately and analyzed in (B). The bottom panel shows the mCherry fluorescence of targets (bottom left), live targets (bottom middle), and dead targets (bottom right), for 721s co-cultured alone (blue), with YT-Indys expressing GZBSS-MCH (orange), or co-cultured with YT-Indys expressing GZB-MCH (red). (B) Western blot of sorted target cell populations from (A). Equal cell-equivalent amounts of whole-cell lysates of sorted target 721 populations were separated by gel electrophoresis, transferred to blots, and probed for mCherry and vinculin (as a loading control). Note that there is a faint, non-specific background band at roughly 60 kDa in all live lanes. This band is not specific for MCH, and we have observed it across a variety of cell lines from various lineages. It is not visible in the dead lanes simply due to the low amount of protein in these lanes, as discussed in the main text. Expected protein sizes: MCH = 30 kDa; GZB-MCH = 60 kDa; vinculin = 130 kDa. Numbers displayed are sizes in kDa of the protein ladder.
Taken together, these results and analysis suggest that while there is some background, non-specific 721 uptake of MCH from the co-culture media, YT-Indys expressing GZB-MCH specifically transfer the fusion protein to targeted 721s, while YT-Indys expressing GZBSS-MCH and GZBSS-MCH-GZBSM do not specifically transfer MCH to targeted 721s. Thus, granzyme B appears to be a suitable chaperone protein for delivery of protein payloads via the granzyme-perforin pathway.
Discussion
Cellular therapeutics that repurpose and recombine biological function in a cellular chassis are transforming medicine.1 These efforts will rely heavily on the development of modules and systems that perform specific sensory, computational, and effector functions.12, 13 We report here our efforts to develop a cell-to-cell delivery module for cellular therapeutics by repurposing the granzyme-perforin pathway of cytotoxic lymphocytes. Our results support the use of granzyme B as a molecular chaperone for inserting protein payloads into this pathway and facilitating payload delivery to target cells.
We hypothesized that lytic granule loading of a payload would be sufficient for transfer to a target cell, and that loading could be achieved by fusing a chaperone to the payload. We designed two candidate chaperones derived from granzyme B, fused them to mCherry, and investigated their subcellular localization in the natural killer cell line YT-Indy.
All constructs containing an N-terminal ER signal peptide exhibited a high degree of co-localization with Lamp1, as opposed to MCH alone. This result is best understood by considering the biological distribution of Lamp1 and the cellular compartments in which the co-localization occurs. The primary route of newly synthesized Lamp1 follows the secretory pathway to exosomes at the cell membrane and is then recaptured in early endosomes and eventually fuses with nascent lysosomes.38, 39 In the case of GZBSS-MCH, the ER signal peptide would direct the protein to the secretory pathway, so GZBSS-MCH is expected to be found co-localized with Lamp1 in a perinuclear distribution in the ER and Golgi and in punctate granules at the cell membrane, but not in cytoplasmic lytic granules. This is what we observe in cells expressing GZBSS-MCH, in contrast to those expressing GZB-MCH, in which the observed co-localization is primarily in cytoplasmic puncta, consistent with lytic granules. These observations are supported by our co-culture experiments, which indicated that GZB-MCH was transferred from effector to target cell, but not GZBSS-MCH. This interpretation is predicated on the assumption that entry into the target cell via perforin pores is passive, which, while historically controversial, is supported by most recent experimental and theoretical data.26, 28, 29, 33, 40
We postulated that combining an ER signal peptide with a putative N-linked glycosylation motif would be sufficient for payload delivery, but our co-culture results clearly refuted this. That GZBSS-MCH-GZBSM did not transfer to target cells has several interesting implications surrounding the intracellular trafficking of granzyme B. The first is that the putative N-linked glycosylation sites we computationally identified and their flanking amino acids are insufficient for granule loading. While we cannot rigorously exclude the possibility, we do not believe that these results are simply due to faulty glycosylation of GZBSM, since this process occurs co-translationally and only depends on local sequence.41 This suggests then that GZBSM is not being phosphorylated, likely because the binding domain for the GlcNAc-1-phosphotransferase, which adds a phosphate group to the mannose of the N-linked glycan in the Golgi,38 is not faithfully recapitulated in GZBSS-MCH-GZBSM. This could either be due to a lack of actual amino acids that are present elsewhere in the full-length granzyme B protein or that the phosphotransferase-binding domain is conformation dependent, as has been suggested elsewhere in the literature,42 or both. Much work has been invested into characterizing this domain, but its exact nature remains elusive. Our results suggest that whatever the exact composition, its constituent residues are likely distributed throughout the primary amino acid sequence and hence were not captured in GZBSM, which is why it failed to facilitate transfer of the MCH payload to target cells. This conclusion is consistent with the location of the asparagine residues within the context of GZBSM and the full-length granzyme B protein. As shown in Figure 8, both of the N-linked glycosylation sites we computationally identified are located immediately adjacent to residues that lie external to GZBSM. In particular, N104 is located at a junction in which residues on one side of N104 are located within GZBSM, while those on the other side are located in the other half of the granzyme B protein (Figures 8A and 8C). Also of note is that the other N-linked glycosylation site (N71) is surrounded by a triangular pattern of lysines (Figure 8D), a pattern which some experimental data suggests is the phosphotransferase binding site.43, 44
Figure 8.

Spatial Context of Putative N-linked Glycosylation Sites in Granzyme B
(A) Schematic of primary amino acid structure of granzyme B. The coloring corresponds to the crystal structures below. Numbers are amino acid residues. (B and C) Three-dimensional crystal structure of granzyme B, colored as in (A), highlighting the potential importance of surface-exposed residues that are immediately adjacent (B, ADJ-N71, purple ; C, ADJ-N104, red) to the N-linked glycosylation sites (blue), but are not contained within the GZBSM (yellow). Note that these regions are quite far from the GZBSM in primary amino acid space, as shown in (A), and in (B) and (C) by the labeling of representative amino acids in these regions. (D) Lysines have been colored in white, to show their inverted triangular pattern surrounding the N71 putative glycosylation site. (E) Location of putative N-linked glycosylation sites (blue) throughout the protein. The residues have been colored from red through white, to cyan, according to their position in the primary amino acid sequence.
The conclusion that the domains necessary for lytic granule (LG) loading lie throughout the granzyme protein is further supported by our microscopy data, which showed that neither half of granzyme B co-localized with Lamp1 in a similar fashion to full-length granzyme B. This data is also consistent with the arginine residue structural analysis presented above. Beyond corroboration, the morophological appearances of GZB1-MCH and GZB2-MCH are interesting in their own right. In particular, the membranous distribution of GZB1-MCH suggests it is entering the secretory pathway, similar to GZBSS-MCH, and further suggests that the absence of key domains found in GZB2 are preventing GZB1-MCH from being sorted to LGs. Interpreting the highly punctate distribution of GZB2-MCH that is nevertheless distinct from that of Lamp1 is more challenging. It is possible that the sorting domains found in GZB2—that presumably make up part of the overall complement of domains in full-length granzyme B responsible for its localization to LGS—are degenerate or otherwise used by a variety of sorting pathways in the Golgi. This degeneracy could result in GZB2-MCH being sorted into another, unknown, granular compartment distinct from LGs or other granules containing Lamp1. However, this is speculative and would require experimental investigation.
In summary, our results argue that granzyme B trafficking to lytic granules requires residues or domains beyond those immediately flanking the putative N-linked glycosylation motifs. In particular, this data implies that the GlcNAc-1-phosphotransferase binding domain is not a contiguous amino acid sequence, but rather a conformation-dependent motif composed of residues located throughout the length granzyme B. By extension, this analysis and data suggest that barring fine-scale delineation of the exact residues and three-dimensional motifs necessary for LG loading, using full-length granzyme B will likely be necessary for delivery of a payload to a target cell, at least in the near term. Both the flow cytometry and western blot data from our co-culture experiments demonstrated that it is also sufficient: YT-Indys expressing GZB-MCH transfer it to 721 target cells. Importantly, these same data also indicated a background level of accumulation of mCherry signal in target cells co-cultured with YT-Indy cells expressing the comparator constructs (GZBSS-MCH and MCH alone). While this might initially appear to undermine the utility of this system, we in fact believe the opposite: it demonstrates the need for specific, cell-to-cell delivery, the activation of which is controlled by surface receptor interactions. In the case of comparator effector populations expressing GZBSS-MCH or MCH, the mCherry signal is the same in both live and dead target cells, indicating a non-specific effect. If these effector cells were used to deliver the payload, the specificity of delivery would be at best localized. However, in the case of the GZB-MCH-expressing effector population, there is a significant increase in the RFP signal in dead target cells, indicating that YT-Indy-targeted 721s specifically received the most GZB-MCH. Furthermore, our observations consistently have been that MCH is much brighter than GZBSS-MCH, which is in turn brighter than GZB-MCH. If the transfer were non-specific and occurred at roughly equal rates for all mCherry fusion proteins, then we would expect to see 721s co-cultured with YT-Indys expressing MCH alone to display the greatest increase in RFP signal, followed by those co-cultured with GZBSS-MCH and finally those with GZB-MCH. Instead, we see the opposite: with the greatest increase in RFP signal in cells co-cultured with YT-Indys expressing GZB-MCH, despite this fusion protein having the dimmest fluorescent intensity. Together, this data suggests there is a basal level of background accumulation of the mCherry payload in all cases, but substantial, target-specific transfer of the payload in the case of YT-Indys expressing GZB-MCH.
The successful transfer of GZB-MCH highlights two unique and highly desirable features of the granzyme-perforin pathway: modularity and prepositioning. The first is important in that all that is required to deliver a payload is to fuse it to the chaperone. In principle, no further modifications are required, regardless of the payload. This modularity suggests that the system might be widely applicable as a means of cellular delivery, either in cytotoxic lymphocytes, or in the long term, in other, orthogonal, highly engineered cellular chassis. The second advantage is that, as opposed to producing a payload in response to target cell recognition using transcriptional control, a pre-synthesized payload loaded into a lytic granule can be released on the timescale during which the immune synapse remains intact, and hence cell-to-cell specificity is maintained.
A critical consideration in using this system is the design and construction of functional fusion proteins that traffic the pathway and remain functional in the target cell. Several issues bear discussion in this regard. Future payloads may not be stable in the harsh environment of the lytic granule, which is acidic and contains many proteases. This might limit the range of applications such a system could be used for, although it is possible that the payload could be engineered to increase its ability to survive the lytic granule, for example by removing a protease cleavage site. The size of the payload is also important, since the internal lumen of the perforin pore has been observed to be 10–20 nm,28 which sets an upper limit on the payload size. However, the diameter of granzyme B is only 5 nm,45 leaving an appreciable window for a variety of payloads. The diffusivity of the fusion protein is important to consider, as this dictates the propensity for the granzyme fusion protein to both spread throughout the synapse, translocate through the pore, and escape the synapse.33 While empirical measurement of the fusion protein diffusivity would be a substantial undertaking, we can at least make estimates regarding the change in diffusivity using the Stokes-Einstein relationship, in which the diffusivity D, is proportional to the inverse of the radius r of the molecule. (The exact relationship is D = KBT/(6πηr), where KBT is the thermal energy and η is the solvent viscosity. Note that aside from r, all other terms are constants between different proteins). The radius of granzyme B and mCherry have both been experimentally determined to be approximately 2.5 nm. Thus, the radius of the fusion protein could be expected to be approximately the average of its long and short dimensions (that is 2.5 and 5 nm), or 3.25 nm. Thus, the ratio of the radius of the fusion protein to wild-type granzyme is approximately 1.3. Therefore, using the Stokes-Einstein relationship, we would predict that the diffusivity of the fusion protein would be approximately 77% that of the wild-type granzyme. Of course, this analysis only considers the size of the protein and ignores any residue-specific protein-protein interactions, intraprotein effects, effects on solubility, and many other factors. We have conducted a computational study of some of these factors and have found that the rate of perforin pore formation is by far the most important factor in determining the efficacy of granzyme B internalization.33 Finally, the effect of each fusion partner on the other’s function is an important consideration. We have attempted to mitigate this by joining the two by a flexible glycine-serine linker, this has proven successful in maintaining the function of a variety of payloads fused to granzyme B (data not shown). However, in the absence of reliable computational prediction of protein structure and function, the function of both granzyme and the payload of each new fusion would have to be empirically validated, although we note that as more fusions are generated, design rules may become apparent.
As currently implemented, this approach relies upon using a cytotoxicy lymphocyte chassis as the delivery cell. Therefore, any eventual application would have to consider the native cytotoxic effector mechanisms of the lymphocyte chassis. Unmodified cytotoxic lymphocytes are appropriate vehicles to deliver payloads to target cells with the intent of killing them, as would be the case with tumor cells. However, for other applications—for example delivery of pro-survival factors in degenerative diseases, deficient enzymes in metabolic diseases, reprogramming cells, or delivery of gene-editing tools in research contexts—the granzyme-perforin delivery functionality would have to be decoupled from the delivery cell cytotoxicity. This may be possible either through attenuation or knockout of the native effector mechanisms in a cytotoxic lymphocyte or by reconstituting the pathway in an independent, non-cytolytic cell chassis. While both of these suggestions would be challenging undertakings, there have been some remarkable successes in similar endeavors, most notably the reconstitution of basic T cell receptor signaling.46 The granzyme chaperone itself should be readily catalytically inactivated, as has been previously reported.47 Alternatively, other classes of chaperone could be considered. Promising candidates include other lytic granule constituents, such as other granzymes, and the cathepsins, as well as derivatives thereof. However, we feel strongly that even if all of these approaches were found to be intractable, this system warrants further investigation as a potential therapeutic modality in the context of CAR T cell therapy for cancer.
We have repurposed the granzyme-perforin pathway as a cell-to-cell delivery module for cellular therapeutics. By facilitating targeted transfer of arbitrary payloads with single-cell precision, this system is an important addition to the part set of synthetic immunology.
Materials and Methods
Computational Identification of N-Linked Glycosylation Motifs
The granzyme B coding sequence was downloaded from NCBI RefSeq gene (NCBI: NG_028340.1). We then used NetNGlyc 1.048 to predict putative N-linked glycosylation sites, of which there were two, 33 residues apart. Since the NX(S/T) consensus sequence is necessary but insufficient for glycosylation and the glycosylation occurs co-translationally,49 it follows that local sequence context surrounding the consensus site is critical. Therefore, we extracted a 53-amino acid domain from granzyme B, extending from 10 amino acids N-terminal of the first putative glycosylation site, to 10 amino acids C-terminal of the second site. Intriguingly, this domain was also present in human granzyme H.
Plasmids
A custom mammalian expression vector was used in this work (Figure S1). This pdL vector was constructed in house, based on a pcDNA3.1(+) (Thermo Fisher Scientific) backbone. Specifically, the mammalian and bacterial selectable markers and all origins of replication are derived from pcDNA3.1(+), corresponding to bases 1670 (CGATTTCGGCCTATTGGTTA…) to 5396 (…TAAACAAATAGGGGTTCCGC). A custom expression cassette was cloned into this backbone. This cassette consisted of eukaryotic and prokaryotic promoters and ribosomal binding sequences, followed by the open reading frame, followed by eukaryotic and prokaryotic transcriptional termination sites. For the mammalian promoter, we used the CAG promoter for its ability to drive high levels of expression in a variety of tissues. The sequence was amplified from pEMS1157.50 This was followed by a hybrid T7 prokaryotic promoter, taken from pCMVTnT (Promega). This was followed by consensus Shine-Dalgarno and Kozak sequences. Following this is the open reading frame, which varies by plasmid. Following the end of the coding sequence, there is a BGH poly(A) sequence, and then a T7 terminator (with both sequences taken from pcDNA3.1(+)). Restriction enzyme cleavage sites flank all components to facilitate subcloning.
The full plasmid sequence for the base pdL vector is in the Supplemental Information, along with the full coding sequence for all plasmids used. All plasmids were constructed through a combination of PCR, synthesis, and restriction/ligation cloning. All PCR amplicons and coding sequences were sequence verified.
Cell Culture
YT-Indy and 721.221 cells were a gift from Judy Lieberman (Harvard University). YT-Indy cells were cultured in RPMI 1640 media, supplemented with 20% heat inactivated fetal bovine serum, 1× GlutaMAX, 1 mM sodium pyruvate, 10 mM HEPES, 0.1 mM beta-mercaptoethanol. 721 cells were cultured in DMEM, supplemented with 10% heat inactivated fetal bovine serum and 1× GlutaMAX. All cell culture reagents were purchased from Thermo Fisher Scientific.
Transfection
YT-Indy cells were electroporated using the Neon system (Thermo Fisher Scientific), using the 100 μL tip. 6 × 106 cells were washed once in PBS, and resuspended in Buffer R along with 20 μg plasmid DNA, in a final volume of 110 μL. The extra volume ensures no bubbles are generated in aspirating the cell mixture into the electroporation tip. Critically, the plasmid DNA must be of a concentration of at least 1 μg/μL, and it must be prepared using an endotoxin-free method. The quality of the plasmid prep greatly influences the electroporation efficiency as well as the post-electroporation viability. The apparatus was prepared as in the manufacturer’s manual, using the E2 electrolytic buffer. The electroporation conditions were 3 × 10 ms pulses at 1250 V. The electroporated cells were then immediately added to 5 mL media spread across two wells of a 6-well plate.
Flow Cytometry
Cells were harvested and resuspended in PBS supplemented with 10% complete media and 1 μg/mL of DAPI (Sigma) as a viability stain. If cells were to be sorted, they were passed through a 35-μm nylon filter (BD Falcon). Cells were kept on ice and then analyzed on a BD Fortessa II or sorted on either a BD Aria III or Fusion. For sorting, cells were sorted into complete media. In all flow cytometry experiments, two initial gating steps were used. Debris was excluded by excluding cells at the bottom left corner of a propidium iodide (PI) versus FSC-A (forward scatter area) gate. Doublets were excluded using a hierarchical gating scheme: all cells with a wider pulse width signal were excluded first in FSC-W versus FSC-H (forward scatter width versus height) and then SSC-W versus SSC-H (side scatter width versus height). All flow cytometry data was analyzed in FlowJo.
Microscopy
Transfected cells were first FACS sorted for moderate intensity RFP+ cells. 2.5 × 105 cells resuspended in 50 μL complete media (RPMI-1640 supplemented with 10% fetal calf serum, 2 mM glutamine, 1 mM pyruvate, 2 μM 2-mercaptoethanol, 50 U/mL penicillin, and 50 μg/mL streptomycin) were adhered to 0.01% poly-L lysine (Sigma) coated, pre-cleaned 12-mm coverslips (#1.0, Fisherbrand) for 15 min at 37°C. Cells were fixed with 2% paraformaldehyde (Electron Microscopy) for 15 min, washed with PBS, and then permeabilized with 0.1% Triton X-100 (Sigma) for 1 min. Samples were washed with PBS and then blocked in 10% goat serum in PBS (blocking buffer) (Jackson Immunoresearch Labs) for 1 hr. Subsequently, cells were stained with polyclonal mouse anti-Lamp1 primary antibody (Abcam cat. #24170) at 1:250 dilution in blocking buffer for 1 hr. Samples were washed with PBS and then stained with Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (Thermo Fisher Scientific cat. #A-11008) at 1:1,000 dilution in PBS for 45 min. After washing with PBS, coverslips were then mounted on glass microscope slides using Prolong Diamond (Thermo Fisher Scientific) overnight. All steps were completed at room temperature unless otherwise noted.
The following day, samples were imaged using a spinning disk confocal system (3i Intelligent Imaging Innovations) based on an inverted Zeiss Axiovert 200M microscope equipped with 100 NA 1.45 Oil Plan Fluor objective and a QuantEM 512SC Photometrics camera. Ten images were acquired for each sample, with each image containing two to five cells in the field of view. All exposure parameters were kept constant across all samples.
Image Analysis
Image filtering was done using a custom script written in MATLAB. The green channel (Lamp1) was filtered as follows: The localized background of the image was calculated for each pixel as the median intensity of a 25 × 25 pixel square centered on that pixel. This background pixel intensity was subtracted from the original pixel intensity. Any pixels with negative intensity after this step were set to zero. This step aids in distinguishing small punctate structures from one another. Next, a pixel noise threshold was calculated as follows. First the median absolute deviation (MAD) of all nonzero pixels from the raw image was calculated. From this, the standard deviation of the pixel intensity was approximated as 1.4 times the MAD, which is a reasonable estimate of the pixel noise. Finally, the noise threshold was taken as 6 times this value (that is 6 × 14 × MAD). Any pixels in the background subtracted image whose intensity were below this value were set to zero. The red channel (mCherry) was filtered in the same way, except localized background was not subtracted. The MATLAB script implementing this algorithm is in the Supplemental Information.
Colocalization analysis was also conducted in MATLAB. For paired red and green channel images, with pixel intensities and respectively, PCC was calculated as
| (Equation 1) |
where and are the mean pixel intensities. The Manders M1 coefficient was calculated as
| (Equation 2) |
These co-localization scores were calculated separately for each sample of each image and then plotted using RStudio.
Cell Labeling
Cells were fluorescently labeled with carboxyfluorescein succinimidyl ester (CFSE; eBioscience) following the manufacturer’s protocol, except that only one PBS wash prior to labeling was done and only one media wash after labeling was done.
Co-culture Experiments
YT-Indys were transfected with mCherry fusion proteins, and 48 hr later FACS sorted for viable RFP+ cells. The following day 4 × 105 YT-Indy effector cells were combined with 1 × 105 CFSE-labeled target 721 cells at a 4:1 effector:target (E:T) ratio in a final volume of 500 μL YT-Indy media in 5 mL polystyrene round-bottom tubes (BD Falcon). The cell suspension was gently pelleted by spinning it at 200 × g for 15 s. The tubes were then incubated at 37°C for 90 min, and then prepared for flow cytometry or FACS sorting as above.
Statistical Analysis
mCherry median fluorescent intensity was tabulated for each target cell population using flow cytometry data from above. For each target cell population, a single factor analysis of variance was conducted to determine if the MCH median fluorescent intensity (MFI) means were the same for all effector cell populations using the model MFI ∼EffectorPopulation. We then used these results as input for a Tukey’s honest significant difference (HSD) test of the difference between sample means within each target cell population. Statistical tests were conducted in R, using the aov and TukeyHSD commands respectively.
Western Blotting
3 × 104 cells were sorted into PBS in microcentrifuge tubes. Cells were kept on ice thereafter. Cells were then pelleted, resuspended in 10 μL PBS, and lysed directly by adding 10 μL 2× Laemmli sample buffer. Samples were incubated at 95°C for 10 min and then stored at −20°C.
For blotting, samples were boiled again at 95°C for 10 min and then loaded onto pre-cast 4%–12% Bis-Tris polyacrylamide gels (Thermo Fisher Scientific). Proteins were size separated by gel electrophoresis by running the gel at 150 V for 75 min. Proteins were transferred to a nitrocellulose membrane using a standard wet transfer at 300 mA for 2 hr.
The blot was cut horizontally at 100 kDa and then was blocked in tris-buffered saline with Tween 20 (TBS-T) with 5% skim milk powder at room temperature for 1 hr and then incubated with primary antibody in sealed pouches at 4°C overnight. The primary antibodies used were rabbit anti-mCherry (Biovision cat. #5993-100) and rabbit anti-vinculin (Abcam cat. #EPR8185) as a loading control. The dilutions were 1:500 (mCherry) with 5% skim milk powder, 1:10,000 (vinculin) with 2% skim milk powder, both in TBS-T. Blots were then washed with TBS-T and incubated with horseradish-peroxidase-conjugated goat anti-rabbit secondary antibody (Santa Cruz Biotechnology cat. #sc-2004) for 1 hr. The dilution was 1:5,000 in TBS-T, with 5% skim milk powder (anti-mCherry) and 2% skim milk powder (anti-Vinculin). Finally, the blots were washed with TBS-T and then developed using Bio-Rad Clarity Western ECL (enhanced chemiluminescence) substrate reagent, following the manufacturer’s protocol. Blots were imaged using a Bio-Rad Chemidoc MP Imaging System, with exposure times ranging from 1 to 100 s.
Crystal Structure Analysis
To visualize the location of the various motifs of granzyme B in the three dimensional protein, we downloaded the granzyme B crystal structure from the Protein Data Bank (PDB: 1FQ3) and rendered the base crystal structure and custom annotations using PyMOL (Schroedinger). Surface-exposed residues adjacent to the N-linked glycosylation sites were determined by first selecting all residues that were within 15Å of the glycosylated residue. We then selected the subset of these residues that were surface exposed, using a custom PyMOL script written by Jason Vertrees.
Author Contributions
D.J.W. and R.A.H. designed the research and the chaperones. D.J.W. and L.D. generated the plasmids and performed the western blots. D.J.W. and L.A. acquired the microscopy images. D.J.W. conducted the co-culture experiments, developed the image-analysis methods, and conducted statistical and crystal structure analyses. D.J.W. and R.A.H. analyzed the data and wrote the paper.
Conflicts of Interest
R.A.H. and D.J.W. are listed as inventors on a patent application (WO2015157864A1) that covers some of the work presented in this manuscript.
Acknowledgments
We thank Daniel Coombs and Josh Scurll, as well as Dave Savage for helpful discussion. We thank Mike Gold for microscope use. This work was supported by BioCanRx (Biotherapeutics for Cancer Treatment funded by the Networks of Centres of Excellence), Genome BC, the BC Cancer Foundation, and the Canadian Cancer Society (grant 703329). D.J.W. is supported by a VCH-CIHR-UBC MD/PhD Studentship Award and a CIHR Frederick Banting and Charles Best Canada Graduate Scholarship Doctoral Research Award.
Footnotes
Supplemental Information includes one figure, a base pdL plasmid sequence, coding sequence inserts, and MATLAB source code for image filtering algorithm and can be found with this article online at https://doi.org/10.1016/j.omtm.2017.10.003.
Supplemental Information
References
- 1.Fischbach M.A., Bluestone J.A., Lim W.A. Cell-based therapeutics: the next pillar of medicine. Sci. Transl. Med. 2013;5:179ps7. doi: 10.1126/scitranslmed.3005568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davila M.L., Riviere I., Wang X., Bartido S., Park J., Curran K., Chung S.S., Stefanski J., Borquez-Ojeda O., Olszewska M. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’souza N., Rossignoli F., Golinelli G., Grisendi G., Spano C., Candini O., Osturu S., Catani F., Paolucci P., Horwitz E.M., Dominici M. Mesenchymal stem/stromal cells as a delivery platform in cell and gene therapies. BMC Med. 2015;13:186. doi: 10.1186/s12916-015-0426-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suresh S.C., Selvaraju V., Thirunavukkarasu M., Goldman J.W., Husain A., Alexander Palesty J., Sanchez J.A., McFadden D.W., Maulik N. Thioredoxin-1 (Trx1) engineered mesenchymal stem cell therapy increased pro-angiogenic factors, reduced fibrosis and improved heart function in the infarcted rat myocardium. Int. J. Cardiol. 2015;201:517–528. doi: 10.1016/j.ijcard.2015.08.117. [DOI] [PubMed] [Google Scholar]
- 7.Cheng Z., Ou L., Zhou X., Li F., Jia X., Zhang Y., Liu X., Li Y., Ward C.A., Melo L.G., Kong D. Targeted migration of mesenchymal stem cells modified with CXCR4 gene to infarcted myocardium improves cardiac performance. Mol. Ther. 2008;16:571–579. doi: 10.1038/sj.mt.6300374. [DOI] [PubMed] [Google Scholar]
- 8.van Velthoven C.T., Braccioli L., Willemen H.L., Kavelaars A., Heijnen C.J. Therapeutic potential of genetically modified mesenchymal stem cells after neonatal hypoxic-ischemic brain damage. Mol. Ther. 2014;22:645–654. doi: 10.1038/mt.2013.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sage E.K., Kolluri K.K., McNulty K., Da Silva Lourenco S., Kalber T.L., Ordidge K.L., Davies D., Gary Lee Y.C., Giangreco A., Janes S.M. Systemic but not topical TRAIL-expressing mesenchymal stem cells reduce tumour growth in malignant mesothelioma. Thorax. 2014;69:638–647. doi: 10.1136/thoraxjnl-2013-204110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niess H., Bao Q., Conrad C., Zischek C., Notohamiprodjo M., Schwab F., Schwarz B., Huss R., Jauch K.W., Nelson P.J., Bruns C.J. Selective targeting of genetically engineered mesenchymal stem cells to tumor stroma microenvironments using tissue-specific suicide gene expression suppresses growth of hepatocellular carcinoma. Ann. Surg. 2011;254:767–774. doi: 10.1097/SLA.0b013e3182368c4f. discussion 774–775. [DOI] [PubMed] [Google Scholar]
- 11.Sage E.K., Thakrar R.M., Janes S.M. Genetically modified mesenchymal stromal cells in cancer therapy. Cytotherapy. 2016;18:1435–1445. doi: 10.1016/j.jcyt.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim W.A. Designing customized cell signalling circuits. Nat. Rev. Mol. Cell Biol. 2010;11:393–403. doi: 10.1038/nrm2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodsworth D.J., Holt R.A. Cell-based therapeutics: making a faustian pact with biology. Trends Mol. Med. 2017;23:104–115. doi: 10.1016/j.molmed.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Stanton B.C., Siciliano V., Ghodasara A., Wroblewska L., Clancy K., Trefzer A.C., Chesnut J.D., Weiss R., Voigt C.A. Systematic transfer of prokaryotic sensors and circuits to mammalian cells. ACS Synth. Biol. 2014;3:880–891. doi: 10.1021/sb5002856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadelain M., Brentjens R., Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roybal K.T., Rupp L.J., Morsut L., Walker W.J., McNally K.A., Park J.S., Lim W.A. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. 2016;164:770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morsut L., Roybal K.T., Xiong X., Gordley R.M., Coyle S.M., Thomson M., Lim W.A. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016;164:780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kemmer C., Gitzinger M., Daoud-El Baba M., Djonov V., Stelling J., Fussenegger M. Self-sufficient control of urate homeostasis in mice by a synthetic circuit. Nat. Biotechnol. 2010;28:355–360. doi: 10.1038/nbt.1617. [DOI] [PubMed] [Google Scholar]
- 19.Ye L., Chang Y.H., Xiong Q., Zhang P., Zhang L., Somasundaram P., Lepley M., Swingen C., Su L., Wendel J.S. Cardiac repair in a porcine model of acute myocardial infarction with human induced pluripotent stem cell-derived cardiovascular cells. Cell Stem Cell. 2014;15:750–761. doi: 10.1016/j.stem.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green E.R., Mecsas J. Bacterial secretion systems: an overview. Microbiol. Spectr. 2016;4 doi: 10.1128/microbiolspec.VMBF-0012-2015. 10.1128/microbiolspec.VMBF-0012-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costa T.R.D., Felisberto-Rodrigues C., Meir A., Prevost M.S., Redzej A., Trokter M., Waksman G. Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 2015;13:343–359. doi: 10.1038/nrmicro3456. [DOI] [PubMed] [Google Scholar]
- 22.Reeves A.Z., Spears W.E., Du J., Tan K.Y., Wagers A.J., Lesser C.F. Engineering Escherichia coli into a protein delivery system for mammalian cells. ACS Synth. Biol. 2015;4:644–654. doi: 10.1021/acssynbio.5b00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson J.C., Clarke E.J., Arkin A.P., Voigt C.A. Environmentally controlled invasion of cancer cells by engineered bacteria. J. Mol. Biol. 2006;355:619–627. doi: 10.1016/j.jmb.2005.10.076. [DOI] [PubMed] [Google Scholar]
- 24.Anderson J.C., Voigt C.A., Arkin A.P. Environmental signal integration by a modular AND gate. Mol. Syst. Biol. 2007;3:133. doi: 10.1038/msb4100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barry M., Bleackley R.C. Cytotoxic T lymphocytes: all roads lead to death. Nat. Rev. Immunol. 2002;2:401–409. doi: 10.1038/nri819. [DOI] [PubMed] [Google Scholar]
- 26.Voskoboinik I., Whisstock J.C., Trapani J.A. Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 2015;15:388–400. doi: 10.1038/nri3839. [DOI] [PubMed] [Google Scholar]
- 27.de Saint Basile G., Ménasché G., Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat. Rev. Immunol. 2010;10:568–579. doi: 10.1038/nri2803. [DOI] [PubMed] [Google Scholar]
- 28.Law R.H.P., Lukoyanova N., Voskoboinik I., Caradoc-Davies T.T., Baran K., Dunstone M.A., D’Angelo M.E., Orlova E.V., Coulibaly F., Verschoor S. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. 2010;468:447–451. doi: 10.1038/nature09518. [DOI] [PubMed] [Google Scholar]
- 29.Lopez J.A., Susanto O., Jenkins M.R., Lukoyanova N., Sutton V.R., Law R.H., Johnston A., Bird C.H., Bird P.I., Whisstock J.C. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood. 2013;121:2659–2668. doi: 10.1182/blood-2012-07-446146. [DOI] [PubMed] [Google Scholar]
- 30.Thiery J., Keefe D., Boulant S., Boucrot E., Walch M., Martinvalet D., Goping I.S., Bleackley R.C., Kirchhausen T., Lieberman J. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat. Immunol. 2011;12:770–777. doi: 10.1038/ni.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stinchcombe J.C., Majorovits E., Bossi G., Fuller S., Griffiths G.M. Centrosome polarization delivers secretory granules to the immunological synapse. Nature. 2006;443:462–465. doi: 10.1038/nature05071. [DOI] [PubMed] [Google Scholar]
- 32.Cartwright A.N.R., Griggs J., Davis D.M. The immune synapse clears and excludes molecules above a size threshold. Nat. Commun. 2014;5:5479. doi: 10.1038/ncomms6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woodsworth D.J., Dunsing V., Coombs D. Design parameters for granzyme-mediated cytotoxic lymphocyte target-cell killing and specificity. Biophys. J. 2015;109:477–488. doi: 10.1016/j.bpj.2015.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinrichs C.S., Rosenberg S.A. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol. Rev. 2014;257:56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang L., Pike D., Sleat D.E., Nanda V., Lobel P. Potential pitfalls and solutions for use of fluorescent fusion proteins to study the lysosome. PLoS One. 2014;9:e88893. doi: 10.1371/journal.pone.0088893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montel A.H., Morse P.A., Brahmi Z. Upregulation of B7 molecules by the Epstein-Barr virus enhances susceptibility to lysis by a human NK-like cell line. Cell. Immunol. 1995;160:104–114. doi: 10.1016/0008-8749(95)80015-b. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu Y., DeMars R. Production of human cells expressing individual transferred HLA-A,-B,-C genes using an HLA-A,-B,-C null human cell line. J. Immunol. 1989;142:3320–3328. [PubMed] [Google Scholar]
- 38.Braulke T., Bonifacino J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta. 2009;1793:605–614. doi: 10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 39.Saftig P., Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- 40.Lopez J.A., Jenkins M.R., Rudd-Schmidt J.A., Brennan A.J., Danne J.C., Mannering S.I., Trapani J.A., Voskoboinik I. Rapid and unidirectional perforin pore delivery at the cytotoxic immune synapse. J. Immunol. 2013;191:2328–2334. doi: 10.4049/jimmunol.1301205. [DOI] [PubMed] [Google Scholar]
- 41.Breitling J., Aebi M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013;5:a013359. doi: 10.1101/cshperspect.a013359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Meel E., Lee W.S., Liu L., Qian Y., Doray B., Kornfeld S. Multiple domains of GlcNAc-1-phosphotransferase mediate recognition of lysosomal enzymes. J. Biol. Chem. 2016;291:8295–8307. doi: 10.1074/jbc.M116.714568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warner J.B., Thalhauser C., Tao K., Sahagian G.G. Role of N-linked oligosaccharide flexibility in mannose phosphorylation of lysosomal enzyme cathepsin L. J. Biol. Chem. 2002;277:41897–41905. doi: 10.1074/jbc.M203097200. [DOI] [PubMed] [Google Scholar]
- 44.Qian Y., Lee I., Lee W.S., Qian M., Kudo M., Canfield W.M., Lobel P., Kornfeld S. Functions of the alpha, beta, and gamma subunits of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. J. Biol. Chem. 2010;285:3360–3370. doi: 10.1074/jbc.M109.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waugh S.M., Harris J.L., Fletterick R., Craik C.S. The structure of the pro-apoptotic protease granzyme B reveals the molecular determinants of its specificity. Nat. Struct. Biol. 2000;7:762–765. doi: 10.1038/78992. [DOI] [PubMed] [Google Scholar]
- 46.James J.R., Vale R.D. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature. 2012;487:64–69. doi: 10.1038/nature11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurschus F.C., Kleinschmidt M., Fellows E., Dornmair K., Rudolph R., Lilie H., Jenne D.E. Killing of target cells by redirected granzyme B in the absence of perforin. FEBS Lett. 2004;562:87–92. doi: 10.1016/S0014-5793(04)00187-5. [DOI] [PubMed] [Google Scholar]
- 48.Gupta R., Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002;2002:310–322. [PubMed] [Google Scholar]
- 49.Blázquez M., Shennan K.I.J. Basic mechanisms of secretion: sorting into the regulated secretory pathway. Biochem. Cell Biol. 2000;78:181–191. [PubMed] [Google Scholar]
- 50.Portales-Casamar E., Swanson D.J., Liu L., de Leeuw C.N., Banks K.G., Ho Sui S.J., Fulton D.L., Ali J., Amirabbasi M., Arenillas D.J. A regulatory toolbox of MiniPromoters to drive selective expression in the brain. Proc. Natl. Acad. Sci. USA. 2010;107:16589–16594. doi: 10.1073/pnas.1009158107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.