

Abstract
Protein folding is a fundamental life process with many implications throughout biology and medicine. Consequently, there have been enormous efforts to understand how proteins fold. Almost all of this effort has focused on water-soluble proteins, however, leaving membrane proteins largely wandering in the wilderness. The neglect has not occurred because membrane proteins are unimportant, but rather because they present many theoretical and technical complications. Indeed, quantitative membrane protein folding studies are generally restricted to a handful of well-behaved proteins. Single-molecule methods may greatly alter this picture, however, because the ability to work at or near infinite dilution removes aggregation problems, one of the main technical challenges of membrane protein folding studies.
Graphical abstract
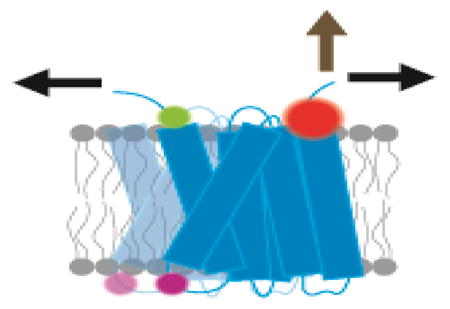
Unlike soluble proteins that fold in a homogeneous solvent, membrane proteins fold in a complex membrane that presents a variety of environments from an apolar core to a polar and charged interfacial region and finally bulk water. The complexity of the bilayer greatly adds to the challenge of studying membrane protein folding because the forces that drive folding will change continuously in different regions. Moreover, membranes themselves vary greatly in composition and properties. Thus, membrane protein folding is not just one tough problem, but a continuum of tough problems and it is easy to despair of ever getting our minds around it. Nevertheless, an understanding of life will not be complete without an understanding of this central process.
Single-molecule methods: freedom from aggregation?
Anyone who works with purified membrane proteins has likely experienced the problem of aggregation. Unfolded membrane proteins appear to be particularly susceptible to aggregation, a problem that becomes especially acute when studying proteins in detergents because detergents tend to destabilize membrane protein structure. Thus, bulk unfolding studies of most membrane proteins are plagued by aggregation, which often precludes detailed physical studies of the folding process. Single-molecule techniques may therefore be particularly useful for membrane protein folding studies because it is possible to use very low concentrations, or in the case of tethered membrane proteins, effectively infinite dilution.
Membrane protein classes
There are two main classes of membrane proteins: those that span the membrane via helical secondary structure and those that span as β-barrels. The β-barrel class resides in the outer membranes of bacteria, mitochondria, and chloroplasts, while the helical class is generally found everywhere else. Helix bundle membrane proteins are by far the most abundant class. The in vivo folding of the two classes of proteins is entirely different [1] and involves separate cellular machinery to catalyze the processes [2–7]. Here we will focus on techniques for studying the helical class, although some of the methods will be applicable to both.
Folding in two stages
Helical membrane proteins can be inserted into the membrane in a variety of ways [2,7–10], but for the vast majority of helix bundle membrane proteins, insertion occurs via the Sec translocon. Membrane protein folding in vivo can be conceptually divided into two primary stages [11,12]. In the first step, the transmembrane segments are cotranslationally inserted in a particular orientation via a ribosome-Sec translocon complex driven by sequence features of each transmembrane segment [7,13–20]. Once the initial insertion and topology are established, the transmembrane helices can fold into a final structure in the second step. Oligomerization and cofactor addition, which may also be considered a third stage [21–23], complete the folding process. While early models of membrane protein folding envisioned simple assembly of the inserted helices in the second stage of folding, we now know that in some cases helices can completely flip in the membrane after insertion [24–31]. Moreover, about 10% of helices in membrane protein structures do not traverse the membrane, and presumably become oriented only during second-stage folding.
Once inserted, the protein will remain ensconced in a membrane environment and exist in equilibrium between the second-stage unfolded and folded states. Second-stage folding is analogous to the soluble protein folding problem which envisions a conformational search in an energy landscape with a major valley in the landscape directing the search toward the native conformation [32]. It is the nature of the energy landscape and the search process that we seek to understand in our studies of membrane protein folding.
Measuring membrane protein assembly with single-molecule fluorescence methods
Studies of membrane protein oligomerization are a convenient way to learn about the energetics of interactions in the membrane [33–50], and fluorescence has been an effective tool for measuring association of membrane proteins [51–62]. The sensitivity of fluorescence allows for oligomerization studies at the single-molecule level.
To study G-protein coupled receptor (GPCR) oligomerization, Mathiasen et al. reconstituted donor and acceptor labeled GPCRs (β2-adrenergic receptor, cannabinoid receptor type 1, or opsin) in proteoliposomes that also bore a fluorescently labeled lipid (Figure 1A) [63]. The vesicles were tethered to a biotinylated surface by a neutravidin-mediated linkage to biotinylated lipid, and then analyzed as single fluorescent spots by confocal microscopy. By visualizing individual proteoliposomes, it was possible to distinguish between empty, donor-only, acceptor-only and un-reconstituted aggregates from proteoliposomes containing both donor and acceptor labeled protein. In bulk measurements, it would be impossible to distinguish these different possibilities and all of them would contribute to a spurious average FRET signal. By measuring FRET and receptor density in the donor-plus-acceptor proteoliposomes, it was possible to see that FRET exceeded what would be expected by random collisions of molecules, indicating a favorable free energy of association for the β2-adrenergic receptor (β2AR). It was also possible to extract free energies of association by plotting the observed FRET efficiencies of each proteoliposome against both the acceptor-labeled receptor concentration and total labeled receptor concentration in the proteoliposomes, and then fitting for the association constant and the FRET efficiency of a bound dimer. This fit takes into account FRET from random collisions and the ratio of donor- to acceptor-labeled receptors. The calculated dissociation free energy for β2AR was ~19.5 kJ/mol (~4.66 kcal/mol) using a mole fraction standard state. Interestingly, the dissociation free energy was different in the agonist bound and antagonist bound receptor suggesting that there could be interplay between receptor concentration and receptor signaling. In addition to the ability to distinguish particles that simply contribute noise, this approach only requires a small amount of material. A limitation of the technique, however, is that dilution of the receptors is constrained to the bilayer area of a single liposome so that only weak interactions are measurable.
Figure 1. Single-molecule fluorescence measurements of membrane protein oligomerization.

(A) Measuring membrane protein oligomerization in isolated lipid vesicles immobilized on a surface via DNA-biotin-avidin tethers. Expected fluorescence signals from a line across the confocal plane are plotted from aggregated protein (i), empty vesicles (ii), homo-oligomers (iii), monomers (iv), and hetero-oligomers (v). (B) Large multilamellar vesicles containing fluorescently labeled protein (i) are converted to small unilammelar vesicles (ii) by extrusion, preserving the oligomeric equilibrium distribution in the large vesicles. Photobleaching events are counted in extruded vesicles, from which the fraction of dimeric complexes can be calculated as a function of protein concentrations (iii).
The Robertson group was able to expand the range of accessible dissociation constants to very strong complexes, by developing a technique that enables much greater dilutions [64] (Figure 1B). In their technique the protein is first reconstituted in large multilamellar vesicles where the protein to lipid ratio can be very low (as low as 7.5 × 10−9 protein/lipid). A monomer-dimer equilibrium is established and then assessed by extrusion into small unilamellar vesicles. The presence of monomers and dimers in the individual vesicles can then be measured by simply counting photobleaching events. This is a clever technique that should allow the energetics of protein-protein interfaces in the membrane to be studied in detail. This method can measure strong transmembrane interactions, and allowed equilibrium measurements of dimerization of the E. coli ClC Cl−/H+ antiporter, one of the most stable dimers studied so far with an association free energy of ~11.4 kcal/mol (mole fraction standard state). Perhaps by incorporating the steric trapping approach [65], it would be possible to push the limits even further.
Observing folding by single-molecule fluorescence
Although single-molecule fluorescence methods have been extensively applied to soluble proteins, the approach has not been translated extensively to membrane protein folding in spite of the potential advantages. In one example, Hartmann et al. were able to observe the folding of the small membrane protein mistic in detergent using single-molecule FRET [66]. Mistic was labeled with fluorescence donors and acceptors and driven to unfold with urea. (Figure 2) A small confocal volume was illuminated and FRET measurements were performed as single molecules floated in an out of the confocal volume. They observed population peaks at high FRET (folded) and low FRET (unfolded), and the unfolded fraction increased at higher urea concentrations as expected. However, there was also a significant fraction at intermediate FRET values. They were able to show that the intermediate FRET values were due to rapid folding/unfolding transitions while passing through the confocal volume, allowing them to measure rates. The beauty of this approach is that it can be performed at extremely low concentrations (~5 pM), greatly reducing the potential for aggregation problems.
Figure 2. Single-molecule FRET measurements of membrane protein folding.

(A) Example trajectory of a membrane protein undergoing folding and unfolding transitions through the confocal excitation volume. (B) Mistic in a micelle environment unfolded with urea (triangles). (C) Histogram of FRET efficiencies from recorded single-molecule bursts. Low FRET efficiencies represent the fraction of unfolded molecules (green), while high FRET efficiencies represent folded molecules (red). The mid-range FRET efficiencies arise from molecules undergoing fast folding and unfolding transitions during single bursts, reflecting the unfolding and folding rates, ku and kf.
Force spectroscopy using an atomic force microscope (AFM)
Optical tweezer and atomic force microscopy methods for applying force to single molecules were first employed to study protein folding in 1997 [67]. Magnetic tweezers were added to the repertoire in the 2000’s [68] and force spectroscopy is now an important tool in the arsenal for studying protein folding. The first efforts to employ forced unfolding of membrane proteins occurred in 2000 [69]. In this work, bacteriorhodopsin in the natural two-dimensional crystalline array called the purple membrane was adsorbed onto a flat surface, and the AFM tip was adsorbed to a single protein after many cycles of pressing the tip into the purple membrane and retracting from the surface. The stylus was then lifted off the surface of the membrane with increasing force (Figure 3). Monitoring the extension of the polypeptide chain as the force increased produced a force extension curve that revealed specific intermediates corresponding to sequential segments of the polypeptide chain being pulled out of the membrane (Figure 3B).
Figure 3.

Unfolding of a membrane protein using atomic force spectroscopy. (A) Schematic of forced unfolding of a membrane protein. The AFM stylus is shown in gray and the black arrows show the direction of tip movement. (B) Illustration of representative force-distance curves from unfolding, refolding, and a second cycle of unfolding. Force peaks matching the unfolding segments in the first pull are highlighted in blue. Misfolded segments whose force peaks do not match are highlighted in red.
Many attempts either failed to adsorb to the protein or attached to a non-terminal cytoplasmic loop of bacteriorhodopsin. To compare a collection of equivalent pulling events, only force spectra that extend to the full length of bacteriorhodopsin before the final rupture peak were analyzed. The unfolded regions of the polypeptide could be fit to a worm-like chain model as a function of force so the length of the protein being stretched could be measured and then mapped onto the structure. In this manner it was possible to identify which parts of the protein are unfolded at each force peak.
Since this pioneering work, the Müller group has employed this forced unfolding approach in many interesting applications. Figure 4 summarizes the main unfolding segments identified in the membrane proteins examined to date. As this is a single-molecule technique, every unfolding trajectory can be slightly different since there is no population averaging. Moreover, the unfolding intermediates and trajectories can change with pulling speed [70]. Nevertheless, the dominant folding domains can be identified. For the most part, transmembrane helices are extracted as pairs. Removal of helical hairpins certainly makes topological and energetic sense as it preserves the original topology of the remaining segments. However, extraction of single helices is not infrequent, particularly at higher pulling forces [70]. Unraveling of single helices must lead to some sort of distorted structure in which parts of the transmembrane helices are extended in the membrane or where a loop is pulled into the membrane, possibly in a helical structure. The latter situation would at least allow satisfaction of backbone hydrogen bonds, but would require pulling hydrophilic regions into the bilayer. No doubt the choice between removal of hairpins or smaller segments represents a complex balancing of forces.
Figure 4.

Membrane protein unfolding domains from AFM-based single-molecule force spectroscopy of (A) bacteriorhodopsin pulled from the C-terminus [69], (B) halorhodopsin pulled from the C-terminus [112] and (C) N-terminus [112], (D) LacY pulled from the C-terminus [87], (E) DtpA pulled from the N-terminus [86], (F) and C-terminus [86], (G) β2AR pulled from the N-terminus [88], (H) bovine rhodopsin pulled from the N-terminus [84], and (I) NhaA pulled from the C-terminus [72] and (J) N-terminus [72]. Unfolding domains are highlighted by color and the start of each segment is labeled with the contour length of the corresponding force peak. Black arrows show position of the applied pulling force.
Refolding of AFM extracted proteins
Remarkably, extraction/unfolding of helical membrane proteins can be at least partially reversible [71–73]. To study refolding, extension is stopped before pulling the final segment, so that the polypeptide chain is still anchored to the membrane. The AFM tip is then lowered back to the bilayer surface so that the extracted portions can reinsert. In some cases, repeated unfolding shows the same set of force peaks as seen in the initial unfolding process, indicating complete refolding. In other cases, only some of the force peaks are restored indicating partial refolding. Refolding of bacteriorhodopsin displayed “snap-in” force peaks for two pairs of refolding helices [71]. Integrating these snap-in peaks provides a measure of the work performed by the refolding protein against the AFM cantilever. However, this attempt to quantify the free energy of refolding helices is complicated by the fact that the speed of the cantilever back towards the surface may be faster than the time for refolding, which can suppress refolding peaks [72]. It is also unclear whether the helices are undergoing a first-stage insertion or making tertiary contacts as well. Indeed, sometimes the unfolding rupture forces were lower after refolding, suggesting that complete reformation of tertiary structure does not always occur. The ability of a membrane protein partially extracted from the bilayer to refold is quite remarkable and only possible because the protein is tethered and cannot aggregate with other proteins.
Energetics extraction and folding using an AFM
Though atomic force spectroscopy observes membrane protein unfolding under non-equilibrium conditions, the energetics of membrane protein folding can be extrapolated back to zero force. Early experiments unfolded bR at different pulling speeds, correlating the force of unfolding domains with pulling speed [70]. Using Monte Carlo simulations, as originally used for the unfolding of spectrin domains, the data can be fit for two parameters: the distance to the transition state and the unfolding rate at zero force [74]. Observation of force peaks during refolding of bR also permits the calculation of the upper limit on insertion and refolding free energy of transmembrane helices back into the membrane [71]. The Müller lab later employed the equilibrium free energy derivation by Hummer and Szabo [75,76] based on Jarzynski’s theorem relating irreversible work to equilibrium free energy differences to determine the unfolding free energies (ΔGu) of several membrane proteins that exhibit irreversible unfolding [77]. The free energy of unfolding cannot be properly calculated for the first pair of helices due to non-specific interactions between the AFM tip and the membrane surface, so the free energy contribution of the first unfolding segment was estimated by multiplying the average free energy per residue by the length of the first helical pair. The ΔGu of bacteriorhodopsin and halorhodopsin, which share a common fold, were found to be 290.5 and 205.7 kcal/mol, respectively, while that of NhaA, a sodium antiporter consisting of 12 transmembrane helices, was found to be 485.0 kcal/mol. The unfolding free energy of bR is much greater than measured by other unfolding studies in detergent (~20 kcal/mol) [78] and under native conditions (~11.2 – 12.3 kcal/mol) [79], and in fact is much closer to the ~200 kcal/mol expected from the water-octanol partitioning free energies [80]. This discrepancy suggests that the majority of the free energy difference measured by atomic force spectroscopy is from transfer of the protein into an aqueous environment (first stage folding) rather than tertiary structure formation (second stage folding).
Detection of ligand binding in force spectroscopy using an AFM
One of the most surprising discoveries of membrane protein force spectroscopy is the ability to detect stabilization by ligands from increased unfolding forces of individual structural segments. For example in the case of NhaA, Na+ binding increased the occurrence and unfolding force of a segment in helix V, which bears key Na+ binding residues, implying that Na+ binding stabilizes a ligand binding site around helix V [81,82]. Stabilization of β2AR by agonists is primarily confined to the structural core of the protein, while the neutral antagonist alprenolol only stabilized the first helix [83]. Not all ligands have stabilizing effects confined to specific structural segments. The inverse agonist carazalol stabilized segments throughout β2AR [83] and in the case of bovine rhodopsin, titrating in Zn2+ globally raised the unfolding force of structural segments across the protein unfolded by force spectroscopy [84].
Studies of inhibitors have revealed the specific interactions that can stabilize an alternate conformation of a membrane protein. The NhaA inhibitor 2-aminoperimidine mimics the interaction of the natural substrate at helix V, but also forms stabilizing interactions with helix IX, evidenced by the higher unfolding forces measured for segments around helix IX [81]. Later force spectroscopy experiments demonstrated that the inhibitor helps lock the antiporter in an alternate conformation by kinetically trapping helix IX in a narrow energy well [85]. The dipeptide and tripeptide permease A (DtpA) is also locked in an inactive conformation by an inhibitor that stabilizes helix II [86].
In LacY, substrate binding altered unfolding barriers for all segments of the protein [87]. These results are quite surprising because by the time the final segment of LacY is being extracted from the membrane almost the entire structure of LacY is already gone, including most of the sugar binding pocket. How then could a sugar remain bound and alter the energetics of extraction of the final domain? One possibility is that the protein is extracted at such a high rate that the structure has not had time to readjust and the sugar has not had time to diffuse away.
Effects of lipid composition on extraction unfolding using an AFM
One of the advantages of force spectroscopy is the ability to study folding in true membranes, providing an opportunity to study the effects of changes in lipid composition. For example, force spectroscopy showed that addition of the cholesterol analog cholesteryl hemisuccinate broadly stabilized β2AR, increasing the magnitude of unfolding force peaks [88]. LacY is a particularly interesting case as remarkable work in the Dowhan group has shown that an entire domain of the protein comprising 6 transmembrane helices, can completely flip in a bilayer upon depletion of PE. [28–31] Consistent with these results, two different patterns of force peaks were observed with the most obvious changes occurring around helices VII and VIII [89], exactly where the inversion occurs. The probability of these patterns changed with lipid composition as expected. Surprisingly, the unfolding segments in the N-terminal domain are essentially the same for both topologies, even though they are being pulled out in opposite orientations. These results suggest there is some sort of intrinsic domain stability that is being probed by force spectroscopy.
Detection of chaperone assisted refolding using an AFM
Force spectroscopy of LacY has recently been used to observe chaperone folding of a complex membrane, in a purified system for the first time to our knowledge [73]. YidC is a translocon-associated protein that can assist the insertion and assembly of many membrane proteins and can catalyze the insertion of single TM proteins [2,90,91]. YidC is known to be required for the folding of LacY during normal biogenesis [92,93], although the mechanism for assisting folding remains unknown. To test whether YidC can assist the folding of mechanically unfolded LacY, Serdiuk et al. partially pulled LacY out of the bilayer and then lowered the AFM tip back down to the membrane surface and held the protein at zero force for a time. In the absence of YidC only a fraction of the molecules partially refolded and none completely refolded. In the presence of YidC, however, the degree of misfolding was dramatically reduced and some of the LacY molecules were found to completely refold as assessed by recovery of the normal force spectrum. Thus, YidC clearly assists refolding of mechanically unfolded and extracted LacY. A caveat to these studies, however, is that folding from a partially extracted LacY protein is different from the in vivo process because it convolutes first- and second-stage folding. Indeed, YidC is known to assist the insertion of TM helices although this may be a different activity than is needed for second stage folding of LacY.
A quantum leap in AFM capabilities
Forced unfolding studies of membrane proteins have recently been dramatically advanced by the Perkins lab [94] through the development of new AFM technology that enables microsecond time resolution and enhanced force sensitivity. Using their improved instrument, they were able to resolve many more bacteriorhodopsin unfolding transitions than were observable previously, including reversible unfolding/folding transitions involving only a few amino acids. The new technique demonstrates that unfolding is a much more rich and complex process than previously imagined, which will certainly take a while to fully explore and appreciate.
Single-molecule tweezers for second-stage folding
Work on forced unfolding and extraction of membrane proteins has yielded many surprises. It is certainly not obvious that one would be able to pull an extremely hydrophobic membrane protein out of a bilayer and then reinsert it. Similarly, it is unexpected that large portions of a folded protein can be extracted and the structure of the remaining protein remains sufficiently intact to preserve substrate binding pockets. The location of unfolding domains is also not easily predictable.
While these are fascinating insights into membrane protein structure, force spectroscopy using AFM so far has been limited to pulling the protein vertically out of the bilayer. This is not a physiologically relevant folding process as it combines both the first and second folding stages in a complex way. Indeed the overall energetics of the extraction process appear to be dominated by removal from the bilayer. For example, as noted above, the estimated ΔG for unfolding of bacteriorhodpsin measured in AFM experiments is ~290.5 kcal/mol [77], while the measured free energy for second stage folding within a bicelle is only about 11.2 – 12.3 kcal/mol [79].
In contrast to force spectroscopy using an AFM, optical and magnetic tweezers offer a method to pull membrane proteins along the plane of the membrane rather than orthogonal to it. To accomplish this goal, we have used magnetic tweezers to perform forced unfolding experiments on membrane proteins [95,96]. In our approach, which follows methods pioneered by the Marqusee and Bustamante labs [97–99], we attach the N- and C-termini of a membrane protein to DNA handles. One DNA handle is tethered to a surface and the other to a magnetic bead, allowing us to pull on the protein with a magnet. The protein is ensconced in a bilayer-like disc called a bicelle. Although bicelles are not true bilayers, they are a reasonable surrogate that is particularly convenient because they readily form in lipid-amphiphile mixtures [100,101]. (Figure 5A)
Figure 5.

Forced unfolding of GlpG using magnetic tweezers. (A) Schematic of the experimental set up for unfolding a membrane protein in a bicelle using magnetic tweezers. (B) Force-extension curves for repeated unfolding and refolding of GlpG. (C) The energy landscape for second-stage folding of GlpG in bicelles determined from magnetic tweezer experiments [95].
We tested this approach on GlpG, a six transmembrane segment rhomboid protease that has been studied by SDS denaturation [102–104] and also by steric trapping [103], making it a good point of comparison for protein folding studies. Attachment of the DNA handles at the N and C termini of the transmembrane domain of GlpG places the mechanical unfolding force along the surface of the membrane.
Figure 5B shows typical results of unfolding GlpG. Initially we see stretching of the DNA and then at ~25 pN we see a jump in extension, or rip, corresponding to the unfolding of GlpG. When the force is subsequently lowered we don’t see any refolding events. However, if we maintain a low force for a time, we can see that complete refolding occurs because a subsequent force ramping experiment shows the same characteristic stretching and ripping events as seen in the initial force ramp. The experiment is remarkably robust and repeatable. We were able to perform ~20 unfolding experiments before the bead detached. Moreover, we can even wash out the bicelle solution so that the protein adopts a collapsed state in water, then re-introduce the bicelle and the protein fully refolds—something that would be impossible in bulk solution due to aggregation.
The forces where unfolding occurs are large enough to pull the protein out of the bicelle, thus the overall reaction is similar to the AFM experiments described above. Moreover, similar to original AFM unfolding experiments, we find that the protein is extracted from the bicelle in helical hairpins. Nevertheless, because we are pulling in the plane of the bilayer we can still obtain information about folding in the bicelle. By fitting unfolding rates as a function of force, we determined that the distance to the transition state is only ~15 Å. Thus, the reaction going from the folded state to the transition state must occur within a bicelle. It is only after passing through the transition state that the unfolded protein is extracted from the bicelle. At low forces where the protein refolds, the bicelle appears to reform so that folding occurs from the protein ensconced in a bicelle. If we assume the transition state is similar in the unfolding direction at high forces and the folding direction at low forces, we can build an energy landscape for GlpG as shown in Figure 5C.
The validity of the energy landscape derived from forced-unfolding experiments is supported by comparison to values obtained in bulk unfolding studies. GlpG folding has been studied by SDS denaturation assays [102–104] and by a steric trapping approach [103]. Steric trapping allows measurements of unfolding free energy under non-denaturing conditions [65,79,105–108]. In the case of GlpG, steric trapping was used to measure unfolding in dodecylmaltoside. Table I summarizes the parameters measured using magnetic tweezers and one of the other methods. For the most part, the values match reasonably well considering the different conditions and methods. The main discrepancies are in the unfolding free energies (ΔGu) measured by the SDS unfolding assay [103,104] and the unfolding rate (ku) measured by the SDS unfolding assay. The values obtained for ΔGu and ku by the SDS unfolding assay are particularly inaccurate, however, because they employed a linear extrapolation and that we now know is not applicable to SDS denaturation curves [79,103]. Folding rates measured by SDS denaturation experiments are more accurate because the extrapolation is short and these values agree well with the magnetic tweezer measurements. Moreover, ΔΔGu values for mutants involve an internal comparison and can therefore be more accurate. Thus, where reliable comparisons can be made, the results from the magnetic tweezer experiments and other methods match reasonably well considering the very different constructs and solution conditions used. We hope that the approach used for GlpG can be used to study the folding of membrane proteins that were largely impossible to study using conventional methods.
Table I.
Comparison of GlpG folding parameters measured by different methods
| SDS Assay (Trp fluorescence) [102] | SDS Assay (GlpG activity) [102] | SDS Assay (Trp fluorescence) [104] | SDS Assay (FRET) [103] | Steric Trap [103] | Magnetic Tweezers [95] | |
|---|---|---|---|---|---|---|
| ΔGu Wild Type (kcal/mol) | 4.2 | 4.5 | 8.2 * | 8.4 – 8.7 * | 4.7 – 5.8 | 3.8 |
| ΔΔGu L155A (kcal/mol) | 1.0 | 1.3 | ||||
| ΔΔGu A206G (kcal/mol) | 1.1 | 0.7 | ||||
| kf (s−1) | 3.9 × 10−2 | 2.7 × 10−2 | ||||
| ku (s−1) | 1.0 × 10−7 * | 5.6 × 10−5 | ||||
| Construct | Full-length | Full-length | Full-length | TM domain | TM domain | TM domain |
These values are likely to be in error because they involve a long linear extrapolation that later experiments suggest are invalid for SDS unfolding experiments (see text).
Ion Mobility-Mass Spectroscopy
IM-MS is a new method for studying membrane protein folding [109]. It is effectively a single-molecule method because the proteins fly in the mass spec as individual molecules. In IM-MS, the charged ions, flying solo, are forced to pass through a gauntlet of gas molecules. The rate of flow through the gas molecules is proportional to the size of the protein. Increasing the speed at which the protein passes through the gas, increases the collisional forces, eventually causing the protein to unfold. Using this technique, it is possible to observe changes in protein stability in a vacuum. Certainly a vacuum is not a membrane environment. Nevertheless, the technique can be useful for reporting on events in solution, such as specific lipid binding [110] and quaternary structure [111]. One imagines it may be possible to also use IM-MS as a reporter for the folding state of a protein in solution or in bilayers.
Conclusion
Though the applications of many single-molecule methods to membrane protein folding are relatively new, they have already shown much promise in uncovering new information about how this unique class of proteins adopts a native fold. One of the big advantages of these tools is the ability to study membrane protein folding under native conditions in lipid bilayers while obviating aggregation, thereby overcoming a major technical challenge. Future studies of single membrane proteins will hopefully focus on difficult targets that were previously challenging or outright impossible to investigate with bulk assays.
Research Highlights.
Single molecule fluorescence applications to membrane protein folding
Single molecule fluorescence methods for measuring membrane protein oligomerization
Atomic force spectroscopy of membrane protein extraction from bilayers
Forced unfolding of membrane proteins using magnetic tweezers
Unfolding of single membrane proteins in vacuo using mass spectroscopy
Acknowledgments
This work was funded by Ruth L. Kirschstein National Research Service Award GM007185 to REJ, National Research Foundation of Korea Grant NRF-2016R1A6A3A03007871 to DM, NIH Grants R01GM063919 and R01DK100482 to JUB, a Cystic Fibrosis Foundation Grant to JUB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bowie JU. Membrane proteins: A new method enters the fold. Proc Natl Acad Sci. 2004;101:3995–3996. doi: 10.1073/pnas.0400671101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dalbey RE, Wang P, Kuhn A. Assembly of Bacterial Inner Membrane Proteins. Annu Rev Biochem. 2011;80:161–187. doi: 10.1146/annurev-biochem-060409-092524. [DOI] [PubMed] [Google Scholar]
- 3.Knowles TJ, Scott-Tucker A, Overduin M, Henderson IR. Membrane protein architects: the role of the BAM complex in outer membrane protein assembly. Nat Rev Microbiol. 2009;7:206–214. doi: 10.1038/nrmicro2069. [DOI] [PubMed] [Google Scholar]
- 4.White SH, von Heijne G. The machinery of membrane protein assembly. Curr Opin Struct Biol. 2004;14:397–404. doi: 10.1016/j.sbi.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 5.White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 6.Fleming KG. Energetics of Membrane Protein Folding. Annu Rev Biophys. 2014;43:233–255. doi: 10.1146/annurev-biophys-051013-022926. [DOI] [PubMed] [Google Scholar]
- 7.Cymer F, von Heijne G, White SH. Mechanisms of Integral Membrane Protein Insertion and Folding. J Mol Biol. 2015;427:999–1022. doi: 10.1016/j.jmb.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegde RS, Keenan RJ. Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2011;12:787–798. doi: 10.1038/nrm3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatzixanthis K, Palmer T, Sargent F. A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase. Mol Microbiol. 2003;49:1377–1390. doi: 10.1046/j.1365-2958.2003.03642.x. [DOI] [PubMed] [Google Scholar]
- 10.Palmer T, Berks BC. The twin-arginine translocation (Tat) protein export pathway. Nat Rev Microbiol. 2012;10:483–496. doi: 10.1038/nrmicro2814. [DOI] [PubMed] [Google Scholar]
- 11.Popot JL, Engelman DM. Membrane protein folding and oligomerization: the two-stage model. Biochemistry (Mosc) 1990;29:4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 12.Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- 13.Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson I, White SH, von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 14.Hessa T, Meindl-Beinker NM, Bernsel A, Kim H, Sato Y, Lerch-Bader M, Nilsson I, White SH, von Heijne G. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450:1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- 15.von Heijne G. Membrane-protein topology. Nat Rev Mol Cell Biol. 2006;7:909–918. doi: 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- 16.White SH, von Heijne G. How Translocons Select Transmembrane Helices. Annu Rev Biophys. 2008;37:23–42. doi: 10.1146/annurev.biophys.37.032807.125904. [DOI] [PubMed] [Google Scholar]
- 17.Öjemalm K, Higuchi T, Jiang Y, Langel Ü, Nilsson I, White SH, Suga H, von Heijne G. Apolar surface area determines the efficiency of translocon-mediated membrane-protein integration into the endoplasmic reticulum. Proc Natl Acad Sci. 2011;108:E359–E364. doi: 10.1073/pnas.1100120108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaud S, Fernández-Vidal M, Nilsson I, Meindl-Beinker NM, Hübner NC, Tobias DJ, von Heijne G, White SH. Insertion of short transmembrane helices by the Sec61 translocon. Proc Natl Acad Sci. 2009;106:11588–11593. doi: 10.1073/pnas.0900638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ismail N, Hedman R, Schiller N, von Heijne G. A biphasic pulling force acts on transmembrane helices during translocon-mediated membrane integration. Nat Struct Mol Biol. 2012;19:1018–1022. doi: 10.1038/nsmb.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lundin C, Kim H, Nilsson I, White SH, von Heijne G. Molecular code for protein insertion in the endoplasmic reticulum membrane is similar for Nin–Cout and Nout–Cin transmembrane helices. Proc Natl Acad Sci. 2008;105:15702–15707. doi: 10.1073/pnas.0804842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engelman DM, Chen Y, Chin CN, Curran AR, Dixon AM, Dupuy AD, Lee AS, Lehnert U, Matthews EE, Reshetnyak YK, Senes A, Popot JL. Membrane protein folding: beyond the two stage model. FEBS Lett. 2003;555:122–125. doi: 10.1016/S0014-5793(03)01106-2. [DOI] [PubMed] [Google Scholar]
- 22.Daley DO. The assembly of membrane proteins into complexes. Curr Opin Struct Biol. 2008;18:420–424. doi: 10.1016/j.sbi.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Lu W, Zhong M, Wei Y. Folding of AcrB Subunit Precedes Trimerization. J Mol Biol. 2011;411:264–274. doi: 10.1016/j.jmb.2011.05.042. [DOI] [PubMed] [Google Scholar]
- 24.Woodall NB, Yin Y, Bowie JU. Dual-topology insertion of a dual-topology membrane protein. Nat Commun. 2015;6:8099. doi: 10.1038/ncomms9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seppala S, Slusky JS, Lloris-Garcera P, Rapp M, von Heijne G. Control of Membrane Protein Topology by a Single C-Terminal Residue. Science. 2010;328:1698–1700. doi: 10.1126/science.1188950. [DOI] [PubMed] [Google Scholar]
- 26.Lu Y, Turnbull IR, Bragin A, Carveth K, Verkman AS, Skach WR. Reorientation of Aquaporin-1 Topology during Maturation in the Endoplasmic Reticulum. Mol Biol Cell. 2000;11:2973–2985. doi: 10.1091/mbc.11.9.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woodall NB, Hadley S, Yin Y, Bowie JU. Complete Topology Inversion can be Part of Normal Membrane Protein Biogenesis. Protein Sci. 2017 doi: 10.1002/pro.3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bogdanov M, Xie J, Heacock P, Dowhan W. To flip or not to flip: lipid-protein charge interactions are a determinant of final membrane protein topology. J Cell Biol. 2008;182:925–935. doi: 10.1083/jcb.200803097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vitrac H, Bogdanov M, Dowhan W. In vitro reconstitution of lipid-dependent dual topology and postassembly topological switching of a membrane protein. Proc Natl Acad Sci. 2013;110:9338–9343. doi: 10.1073/pnas.1304375110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vitrac H, MacLean DM, Jayaraman V, Bogdanov M, Dowhan W. Dynamic membrane protein topological switching upon changes in phospholipid environment. Proc Natl Acad Sci. 2015;112:13874–13879. doi: 10.1073/pnas.1512994112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bogdanov M, Heacock PN, Dowhan W. A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J. 2002;21:2107–2116. doi: 10.1093/emboj/21.9.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Mol Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 33.Fleming KG, Ackerman AL, Engelman DM. The effect of point mutations on the free energy of transmembrane α-helix dimerization1. J Mol Biol. 1997;272:266–275. doi: 10.1006/jmbi.1997.1236. [DOI] [PubMed] [Google Scholar]
- 34.Fleming KG, Engelman DM. Specificity in transmembrane helix–helix interactions can define a hierarchy of stability for sequence variants. Proc Natl Acad Sci. 2001;98:14340–14344. doi: 10.1073/pnas.251367498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeGrado WF, Gratkowski H, Lear JD. How do helix–helix interactions help determine the folds of membrane proteins? Perspectives from the study of homo-oligomeric helical bundles. Protein Sci. 2003;12:647–665. doi: 10.1110/ps.0236503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cristian L, Lear JD, DeGrado WF. Use of thiol-disulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proc Natl Acad Sci. 2003;100:14772–14777. doi: 10.1073/pnas.2536751100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cristian L, Lear JD, DeGrado WF. Determination of membrane protein stability via thermodynamic coupling of folding to thiol–disulfide interchange. Protein Sci. 2003;12:1732–1740. doi: 10.1110/ps.0378603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fleming KG. Standardizing the Free Energy Change of Transmembrane Helix–Helix Interactions. J Mol Biol. 2002;323:563–571. doi: 10.1016/S0022-2836(02)00920-8. [DOI] [PubMed] [Google Scholar]
- 39.Doura AK, Fleming KG. Complex Interactions at the Helix–Helix Interface Stabilize the Glycophorin A Transmembrane Dimer. J Mol Biol. 2004;343:1487–1497. doi: 10.1016/j.jmb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Doura AK, Kobus FJ, Dubrovsky L, Hibbard E, Fleming KG. Sequence Context Modulates the Stability of a GxxxG-mediated Transmembrane Helix–Helix Dimer. J Mol Biol. 2004;341:991–998. doi: 10.1016/j.jmb.2004.06.042. [DOI] [PubMed] [Google Scholar]
- 41.Mackenzie K, Fleming K. Association energetics of membrane spanning α-helices. Curr Opin Struct Biol. 2008;18:412–419. doi: 10.1016/j.sbi.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russ WP, Engelman DM. TOXCAT: A measure of transmembrane helix association in a biological membrane. Proc Natl Acad Sci. 1999;96:863–868. doi: 10.1073/pnas.96.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gurezka R, Langosch D. In Vitro Selection of Membrane-spanning Leucine Zipper Protein-Protein Interaction Motifs Using POSSYCCAT. J Biol Chem. 2001;276:45580–45587. doi: 10.1074/jbc.M105362200. [DOI] [PubMed] [Google Scholar]
- 44.Lindner E, Langosch D. A ToxR-based dominant-negative system to investigate heterotypic transmembrane domain interactions. Proteins Struct Funct Bioinforma. 2006;65:803–807. doi: 10.1002/prot.21226. [DOI] [PubMed] [Google Scholar]
- 45.Lindner E, Unterreitmeier S, Ridder ANJA, Langosch D. An extended ToxR POSSYCCAT system for positive and negative selection of self-interacting transmembrane domains. J Microbiol Methods. 2007;69:298–305. doi: 10.1016/j.mimet.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 46.Ridder A, Skupjen P, Unterreitmeier S, Langosch D. Tryptophan Supports Interaction of Transmembrane Helices. J Mol Biol. 2005;354:894–902. doi: 10.1016/j.jmb.2005.09.084. [DOI] [PubMed] [Google Scholar]
- 47.Finger C, Volkmer T, Prodöhl A, Otzen DE, Engelman DM, Schneider D. The Stability of Transmembrane Helix Interactions Measured in a Biological Membrane. J Mol Biol. 2006;358:1221–1228. doi: 10.1016/j.jmb.2006.02.065. [DOI] [PubMed] [Google Scholar]
- 48.Su PC, Berger BW. Identifying Key Juxtamembrane Interactions in Cell Membranes Using AraC-based Transcriptional Reporter Assay (AraTM) J Biol Chem. 2012;287:31515–31526. doi: 10.1074/jbc.M112.396895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su PC, Berger BW. A Novel Assay for Assessing Juxtamembrane and Transmembrane Domain Interactions Important for Receptor Heterodimerization. J Mol Biol. 2013;425:4652–4658. doi: 10.1016/j.jmb.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 50.Gurezka R, Laage R, Brosig B, Langosch D. A Heptad Motif of Leucine Residues Found in Membrane Proteins Can Drive Self-assembly of Artificial Transmembrane Segments. J Biol Chem. 1999;274:9265–9270. doi: 10.1074/jbc.274.14.9265. [DOI] [PubMed] [Google Scholar]
- 51.Fisher LE, Engelman DM, Sturgis JN. Detergents modulate dimerization, but not helicity, of the glycophorin A transmembrane domain. J Mol Biol. 1999;293:639–651. doi: 10.1006/jmbi.1999.3126. [DOI] [PubMed] [Google Scholar]
- 52.You M, Li E, Wimley WC, Hristova K. Förster resonance energy transfer in liposomes: Measurements of transmembrane helix dimerization in the native bilayer environment. Anal Biochem. 2005;340:154–164. doi: 10.1016/j.ab.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L, Novicky L, Merzlyakov M, Hristov T, Hristova K. Measuring the Energetics of Membrane Protein Dimerization in Mammalian Membranes. J Am Chem Soc. 2010;132:3628–3635. doi: 10.1021/ja910692u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merzlyakov M, Chen L, Hristova K. Studies of Receptor Tyrosine Kinase Transmembrane Domain Interactions: The EmEx-FRET Method. J Membr Biol. 2007;215:93–103. doi: 10.1007/s00232-007-9009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li E, You M, Hristova K. FGFR3 Dimer Stabilization Due to a Single Amino Acid Pathogenic Mutation. J Mol Biol. 2006;356:600–612. doi: 10.1016/j.jmb.2005.11.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Placone J, Hristova K. Direct Assessment of the Effect of the Gly380Arg Achondroplasia Mutation on FGFR3 Dimerization Using Quantitative Imaging FRET. PLOS ONE. 2012;7:e46678. doi: 10.1371/journal.pone.0046678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sarabipour S, Del Piccolo N, Hristova K. Characterization of Membrane Protein Interactions in Plasma Membrane Derived Vesicles with Quantitative Imaging Förster Resonance Energy Transfer. Acc Chem Res. 2015;48:2262–2269. doi: 10.1021/acs.accounts.5b00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen L, Placone J, Novicky L, Hristova K. The Extracellular Domain of Fibroblast Growth Factor Receptor 3 Inhibits Ligand-Independent Dimerization. Sci Signal. 2010;3:ra86–ra86. doi: 10.1126/scisignal.2001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sarabipour S, Hristova K. Glycophorin A transmembrane domain dimerization in plasma membrane vesicles derived from CHO, HEK 293T, and A431 cells. Biochim Biophys Acta BBA - Biomembr. 2013;1828:1829–1833. doi: 10.1016/j.bbamem.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Placone J, He L, Del Piccolo N, Hristova K. Strong dimerization of wild-type ErbB2/Neu transmembrane domain and the oncogenic Val664Glu mutant in mammalian plasma membranes. Biochim Biophys Acta BBA - Biomembr. 2014;1838:2326–2330. doi: 10.1016/j.bbamem.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, Pelton JG, Shan Y, Shaw DE, Wemmer DE, Groves JT, Kuriyan J. Conformational Coupling across the Plasma Membrane in Activation of the EGF Receptor. Cell. 2013;152:543–556. doi: 10.1016/j.cell.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salaita K, Nair PM, Petit RS, Neve RM, Das D, Gray JW, Groves JT. Restriction of Receptor Movement Alters Cellular Response: Physical Force Sensing by EphA2. Science. 2010;327:1380–1385. doi: 10.1126/science.1181729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mathiasen S, Christensen SM, Fung JJ, Rasmussen SGF, Fay JF, Jorgensen SK, Veshaguri S, Farrens DL, Kiskowski M, Kobilka B, Stamou D. Nanoscale high-content analysis using compositional heterogeneities of single proteoliposomes. Nat Methods. 2014;11:931–934. doi: 10.1038/nmeth.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chadda R, Krishnamani V, Mersch K, Wong J, Brimberry M, Chadda A, Kolmakova-Partensky L, Friedman LJ, Gelles J, Robertson JL. The dimerization equilibrium of a ClC Cl−/H+ antiporter in lipid bilayers. eLife. 2016;5:e17438. doi: 10.7554/eLife.17438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hong H, Blois TM, Cao Z, Bowie JU. Method to measure strong protein–protein interactions in lipid bilayers using a steric trap. Proc Natl Acad Sci. 2010;107:19802–19807. doi: 10.1073/pnas.1010348107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hartmann A, Krainer G, Keller S, Schlierf M. Quantification of Millisecond Protein-Folding Dynamics in Membrane-Mimetic Environments by Single-Molecule Förster Resonance Energy Transfer Spectroscopy. Anal Chem. 2015;87:11224–11232. doi: 10.1021/acs.analchem.5b03207. [DOI] [PubMed] [Google Scholar]
- 67.Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE. Reversible Unfolding of Individual Titin Immunoglobulin Domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 68.del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science. 2009;323:638–641. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oesterhelt F. Unfolding Pathways of Individual Bacteriorhodopsins. Science. 2000;288:143–146. doi: 10.1126/science.288.5463.143. [DOI] [PubMed] [Google Scholar]
- 70.Janovjak H, Struckmeier J, Hubain M, Kedrov A, Kessler M, Müller DJ. Probing the Energy Landscape of the Membrane Protein Bacteriorhodopsin. Structure. 2004;12:871–879. doi: 10.1016/j.str.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 71.Kessler M, Gottschalk KE, Janovjak H, Muller DJ, Gaub HE. Bacteriorhodopsin Folds into the Membrane against an External Force. J Mol Biol. 2006;357:644–654. doi: 10.1016/j.jmb.2005.12.065. [DOI] [PubMed] [Google Scholar]
- 72.Kedrov A, Ziegler C, Janovjak H, Kühlbrandt W, Müller DJ. Controlled Unfolding and Refolding of a Single Sodium-proton Antiporter using Atomic Force Microscopy. J Mol Biol. 2004;340:1143–1152. doi: 10.1016/j.jmb.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 73.Serdiuk T, Balasubramaniam D, Sugihara J, Mari SA, Kaback HR, Müller DJ. YidC assists the stepwise and stochastic folding of membrane proteins. Nat Chem Biol advance online publication. 2016 doi: 10.1038/nchembio.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rief M, Pascual J, Saraste M, Gaub HE. Single molecule force spectroscopy of spectrin repeats: low unfolding forces in helix bundles1. J Mol Biol. 1999;286:553–561. doi: 10.1006/jmbi.1998.2466. [DOI] [PubMed] [Google Scholar]
- 75.Hummer G, Szabo A. Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc Natl Acad Sci. 2001;98:3658–3661. doi: 10.1073/pnas.071034098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hummer G, Szabo A. Free Energy Surfaces from Single-Molecule Force Spectroscopy. Acc Chem Res. 2005;38:504–513. doi: 10.1021/ar040148d. [DOI] [PubMed] [Google Scholar]
- 77.Preiner J, Janovjak H, Rankl C, Knaus H, Cisneros DA, Kedrov A, Kienberger F, Muller DJ, Hinterdorfer P. Free Energy of Membrane Protein Unfolding Derived from Single-Molecule Force Measurements. Biophys J. 2007;93:930–937. doi: 10.1529/biophysj.106.096982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Curnow P, Booth PJ. Combined kinetic and thermodynamic analysis of alpha-helical membrane protein unfolding. Proc Natl Acad Sci. 2007;104:18970–18975. doi: 10.1073/pnas.0705067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang Y-C, Bowie JU. Measuring membrane protein stability under native conditions. Proc Natl Acad Sci U S A. 2014;111:219–224. doi: 10.1073/pnas.1318576111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wimley WC, Creamer TP, White SH. Solvation Energies of Amino Acid Side Chains and Backbone in a Family of Host Guest Pentapeptides. Biochemistry (Mosc) 1996;35:5109–5124. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- 81.Kedrov A, Ziegler C, Muller DJ. Differentiating Ligand and Inhibitor Interactions of a Single Antiporter. J Mol Biol. 2006;362:925–932. doi: 10.1016/j.jmb.2006.07.049. [DOI] [PubMed] [Google Scholar]
- 82.Kedrov A, Krieg M, Ziegler C, Kuhlbrandt W, Muller DJ. Locating ligand binding and activation of a single antiporter. EMBO Rep. 2005;6:668–674. doi: 10.1038/sj.embor.7400455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zocher M, Fung JJ, Kobilka BK, Müller DJ. Ligand-Specific Interactions Modulate Kinetic, Energetic, and Mechanical Properties of the Human β2 Adrenergic Receptor. Structure. 2012;20:1391–1402. doi: 10.1016/j.str.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park PS-H, Sapra KT, Koliński M, Filipek S, Palczewski K, Muller DJ. Stabilizing Effect of Zn2+ in Native Bovine Rhodopsin. J Biol Chem. 2007;282:11377–11385. doi: 10.1074/jbc.M610341200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kedrov A, Appel M, Baumann H, Ziegler C, Muller DJ. Examining the Dynamic Energy Landscape of an Antiporter upon Inhibitor Binding. J Mol Biol. 2008;375:1258–1266. doi: 10.1016/j.jmb.2007.11.032. [DOI] [PubMed] [Google Scholar]
- 86.Bippes CA, Ge L, Meury M, Harder D, Ucurum Z, Daniel H, Fotiadis D, Müller DJ. Peptide transporter DtpA has two alternate conformations, one of which is promoted by inhibitor binding. Proc Natl Acad Sci. 2013;110:E3978–E3986. doi: 10.1073/pnas.1312959110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Serdiuk T, Madej MG, Sugihara J, Kawamura S, Mari SA, Kaback HR, Müller DJ. Substrate-induced changes in the structural properties of LacY. Proc Natl Acad Sci. 2014;111:E1571–E1580. doi: 10.1073/pnas.1404446111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zocher M, Zhang C, Rasmussen SGF, Kobilka BK, Müller DJ. Cholesterol increases kinetic, energetic, and mechanical stability of the human β2-adrenergic receptor. Proc Natl Acad Sci. 2012;109:E3463–E3472. doi: 10.1073/pnas.1210373109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Serdiuk T, Sugihara J, Mari SA, Kaback HR, Müller DJ. Observing a Lipid-Dependent Alteration in Single Lactose Permeases. Structure. 2015;23:754–761. doi: 10.1016/j.str.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Samuelson JC, Chen M, Jiang F, Möller I, Wiedmann M, Kuhn A, Phillips GJ, Dalbey RE. YidC mediates membrane protein insertion in bacteria. Nature. 2000;406:637–641. doi: 10.1038/35020586. [DOI] [PubMed] [Google Scholar]
- 91.Dalbey RE, Kuhn A. YidC family members are involved in the membrane insertion, lateral integration, folding, and assembly of membrane proteins. J Cell Biol. 2004;166:769–774. doi: 10.1083/jcb.200405161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu L, Kaback HR, Dalbey RE. YidC Protein, a Molecular Chaperone for LacY Protein Folding via the SecYEG Protein Machinery. J Biol Chem. 2013;288:28180–28194. doi: 10.1074/jbc.M113.491613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nagamori S, Smirnova IN, Kaback HR. Role of YidC in folding of polytopic membrane proteins. J Cell Biol. 2004;165:53–62. doi: 10.1083/jcb.200402067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu H, Siewny MGW, Edwards DT, Sanders AW, Perkins TT. Hidden dynamics in the unfolding of individual bacteriorhodopsin proteins. Science. 2017;355:945–950. doi: 10.1126/science.aah7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Min D, Jefferson RE, Bowie JU, Yoon T-Y. Mapping the energy landscape for second-stage folding of a single membrane protein. Nat Chem Biol. 2015;11:981–987. doi: 10.1038/nchembio.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Min D, Arbing MA, Jefferson RE, Bowie JU. A simple DNA handle attachment method for single molecule mechanical manipulation experiments. Protein Sci. 2016;25:1535–1544. doi: 10.1002/pro.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct Observation of the Three-State Folding of a Single Protein Molecule. Science. 2005;309:2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 98.Cecconi C, Shank E, Marqusee S, Bustamante C. DNA Molecular Handles for Single-Molecule Protein-Folding Studies by Optical Tweezers. In: Zuccheri G, Samorì B, editors. DNA Nanotechnol. Humana Press; 2011. pp. 255–271. [DOI] [PubMed] [Google Scholar]
- 99.Cecconi C, Shank EA, Dahlquist FW, Marqusee S, Bustamante C. Protein-DNA chimeras for single molecule mechanical folding studies with the optical tweezers. Eur Biophys J. 2008;37:729–738. doi: 10.1007/s00249-007-0247-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Czerski L, Sanders CR. Functionality of a Membrane Protein in Bicelles. Anal Biochem. 2000;284:327–333. doi: 10.1006/abio.2000.4720. [DOI] [PubMed] [Google Scholar]
- 101.Faham S, Bowie JU. Bicelle crystallization: a new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure1. J Mol Biol. 2002;316:1–6. doi: 10.1006/jmbi.2001.5295. [DOI] [PubMed] [Google Scholar]
- 102.Baker RP, Urban S. Architectural and thermodynamic principles underlying intramembrane protease function. Nat Chem Biol. 2012;8:759–768. doi: 10.1038/nchembio.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guo R, Gaffney K, Yang Z, Kim M, Sungsuwan S, Huang X, Hubbell WL, Hong H. Steric trapping reveals a cooperativity network in the intramembrane protease GlpG. Nat Chem Biol. 2016;12:353–360. doi: 10.1038/nchembio.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paslawski W, Lillelund OK, Kristensen JV, Schafer NP, Baker RP, Urban S, Otzen DE. Cooperative folding of a polytopic α-helical membrane protein involves a compact N-terminal nucleus and nonnative loops. Proc Natl Acad Sci. 2015;112:7978–7983. doi: 10.1073/pnas.1424751112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blois TM, Hong H, Kim TH, Bowie JU. Protein Unfolding with a Steric Trap. J Am Chem Soc. 2009;131:13914–13915. doi: 10.1021/ja905725n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hong H, Bowie JU. Dramatic Destabilization of Transmembrane Helix Interactions by Features of Natural Membrane Environments. J Am Chem Soc. 2011;133:11389–11398. doi: 10.1021/ja204524c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hong H, Chang Y-C, Bowie JU. Measuring Transmembrane Helix Interaction Strengths in Lipid Bilayers Using Steric Trapping. In: Ghirlanda G, Senes A, editors. Membr Proteins. Humana Press; 2013. pp. 37–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jefferson RE, Blois TM, Bowie JU. Membrane Proteins Can Have High Kinetic Stability. J Am Chem Soc. 2013;135:15183–15190. doi: 10.1021/ja407232b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Laganowsky A, Reading E, Hopper JTS, Robinson CV. Mass spectrometry of intact membrane protein complexes. Nat Protoc. 2013;8:639–651. doi: 10.1038/nprot.2013.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Laganowsky A, Reading E, Allison TM, Ulmschneider MB, Degiacomi MT, Baldwin AJ, Robinson CV. Membrane proteins bind lipids selectively to modulate their structure and function. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang SC, Politis A, Di Bartolo N, Bavro VN, Tucker SJ, Booth PJ, Barrera NP, Robinson CV. Ion Mobility Mass Spectrometry of Two Tetrameric Membrane Protein Complexes Reveals Compact Structures and Differences in Stability and Packing. J Am Chem Soc. 2010;132:15468–15470. doi: 10.1021/ja104312e. [DOI] [PubMed] [Google Scholar]
- 112.Cisneros DA, Oesterhelt D, Müller DJ. Probing Origins of Molecular Interactions Stabilizing the Membrane Proteins Halorhodopsin and Bacteriorhodopsin. Structure. 2005;13:235–242. doi: 10.1016/j.str.2004.12.005. [DOI] [PubMed] [Google Scholar]