

Abstract
There are currently no effective targeted therapies for KRAS mutant cancers. Therapeutic strategies that combine MEK inhibitors with agents that target apoptotic pathways may be a promising therapeutic approach. We investigated combining MEK and MDM2 inhibitors as a potential treatment strategy for KRAS mutant non-small cell lung cancers and colorectal carcinomas that harbor wild-type TP53. The combination of pimasertib (MEK inhibitor) + SAR405838 (MDM2 inhibitor) was synergistic and induced the expression of PUMA and BIM, led to apoptosis and growth inhibition in vitro, and tumor regression in vivo. Acquired resistance to the combination commonly resulted from the acquisition of TP53 mutations, conferring complete resistance to MDM2 inhibition. In contrast, resistant clones exhibited marked variability in sensitivity to MEK inhibition, which significantly impacted sensitivity to subsequent treatment with alternative MEK inhibitor-based combination therapies. These results highlight both the potential promise and limitations of combining MEK and MDM2 inhibitors for treatment of KRAS mutant NSCLC and CRC.
Introduction
Mutations that lead to MAP kinase pathway activation are commonly observed in cancer. Clinically effective targeted therapies have been developed for non-small cell lung cancers (NSCLC) bearing activating EGFR mutations (1, 2) and melanomas with activating BRAF mutations (3, 4). In contrast, there are no approved targeted therapies for cancers that harbor KRAS mutations, which occur in 15–20% of NSCLCs, 30–35% of colorectal cancers (CRC) and the majority of pancreatic cancers (5). MEK inhibitors, which target the primary downstream signaling pathway activated by mutant KRAS, have shown disappointing clinical activity when used as monotherapy (6), partly due to inability to induce robust apoptosis. This has prompted evaluation of MEK inhibitor-based combination therapies designed to induce apoptosis in KRAS mutant cancers (7–9).
The tumor suppressor p53 acts as a master cellular regulator, integrating multiple stress signals and activating transcription of genes regulating cell cycle arrest and apoptosis (10). Inactivation of p53 function is an almost universal feature of human cancer cells. While loss of the tumor suppressive function of p53 is often due to TP53 mutations or deletions, approximately half of all tumors still harbor wild-type TP53 (11, 12). In TP53 wild-type cells, the function of p53 is restrained by the murine double minute 2 protein (MDM2) (13). Direct interaction of p53 with the N-terminal region of MDM2 inhibits p53 transcriptional activity, while the MDM2 ring finger E3-ubiquitin ligase activity maintains p53 at a low baseline level in normal cells by targeting it for proteasomal degradation (14, 15). MDM2-mediated suppression of p53 activity in TP53 wild-type cancers may result from genomic MDM2 amplification (16) or by loss of CDKN2A (17), which encodes the MDM2 antagonist p14ARF (18). Therefore, disruption of the interaction between p53 and MDM2, with subsequent reactivation of p53, represents an attractive targeted therapy strategy for TP53 wild-type tumors. Indeed, striking pre-clinical activity of MDM2 inhibitors has been observed in models exhibiting genomic amplification of the MDM2 gene (19–24). However, MDM2 amplified tumors represent only a small proportion of the TP53 wild-type tumor population, and single agent responses may be limited outside of the MDM2 amplified tumor population.
Combining MDM2 inhibitors with other targeted agents such as kinase inhibitors may lead to improved responses of TP53 wild-type cancers that do not respond to MDM2 inhibitors alone. In prior studies, the combination of MEK or BRAF inhibitors with the MDM2 inhibitor nutlin-3 exhibited synergistic activity in BRAF mutant melanoma in vitro, in which CDKN2A is frequently lost (25, 26). Another recent study reported synergy between MDM2 inhibitors and wide range of other targeted agents, but no clear correlation between drug combination and genotype was observed (27). While these studies suggest that combining MDM2 inhibitors with agents that target oncogenic signaling pathways may hold clinical promise, it remains unclear which MDM2 inhibitor combinations should be prioritized for specific cancer sub-types.
Our prior study investigating the combination of MEK and PI3K inhibitors demonstrated that induction of apoptosis by up-regulation of the BH3-only proteins BIM and PUMA is critical for the response of KRAS mutant NSCLC in vitro and in vivo (28). Since PUMA (p53-upregulated mediator of apoptosis) can be induced by p53 activation, we hypothesized that combining MDM2 and MEK inhibitors might provide an alternative strategy for promoting apoptosis of KRAS mutant cancer cells that harbor wild-type TP53. In this study, we demonstrate that MDM2 inhibitors synergize with MEK inhibitors by upregulating BIM and PUMA and induce apoptosis in KRAS mutant NSCLC and CRC models, establishing rationale for clinical investigation of this combination for KRAS mutant cancers. Efficacy of combined MDM2 + MEK inhibition was ultimately limited by the development of acquired drug resistance commonly associated with acquisition of TP53 mutations, rendering cells fully resistant to MDM2 inhibition. In contrast, acquired resistance to the MEK inhibitor was variable, leading to heterogeneous response to subsequent treatment with a MEK + BCL-XL inhibitor combination.
Results
MDM2 and MEK inhibitors exhibit synergistic activity against KRAS mutant NSCLC and CRC cells that harbor wild-type TP53
Our prior work suggested that MEK inhibitor-based combination therapies that up-regulate pro-apoptotic BIM and PUMA or inhibit anti-apoptotic BCL-XL proteins may have potential for treatment of KRAS mutant cancers (8, 28). Increased expression of PUMA positively correlated with induction of apoptosis for TP53 wild-type KRAS mutant NSCLC cells, unlike TP53 mutant cells for which BIM expression appeared more predictive (Sup. Figure 1). We hypothesized that activating p53 by inhibiting MDM2 might provide an alternative approach to induce PUMA expression and stimulate apoptosis in KRAS mutant cancers that harbor wild-type TP53, which account for approximately 20% of all lung and colorectal adenocarcinomas (Sup. Figure 2). Similar to treatment with the combination of MEK+PI3K inhibitors, the MDM2 inhibitor SAR405838(24) induced PUMA mRNA and protein expression in KRAS mutant TP53 wild-type NSCLC cells (Sup. Figure 3).
We next investigated whether MDM2 inhibitors might synergize with MEK inhibitors in KRAS mutant, TP53 wild-type NSCLC and CRC cells. Using a modified ray design experiment, we treated A427 (NSCLC), DV-90 (NSCLC), GP5d (CRC) and LoVo (CRC) cells with SAR405838 and the allosteric MEK inhibitor pimasertib (AS703026) in fixed dose combinations and evaluated cell viability (Sup. Figure 4A, B). Calculation of Hewlett synergy λ values indicated synergistic drug activity (λ>2) in all four cell lines (Sup. Figure 4C). To more broadly profile the activity of the MEK + MDM2 inhibitor combination, we assessed the effects of SAR405838 and pimasertib alone or in combination on cell proliferation of 25 KRAS mutant cell lines (14 NSCLC, 11 CRC) that harbored either wild-type, mutated or deleted TP53. The TP53 status of each cell line was confirmed by sequencing as well as evaluation of p53 protein expression and function (Sup. Table 1, Sup. Figure 5). Whereas pimasertib exerted inhibitory effects on cell proliferation regardless of TP53 mutation status, the activity of SAR405838 was observed only for wild-type TP53 cells as expected (Figure 1A). As a group, KRAS mutant cell lines harboring wild-type TP53 were significantly more sensitive to combined MDM2+MEK inhibition compared to those with mutated or deleted TP53, and in most cases, the combination of SAR405838 + pimasertib showed greater inhibition of cell proliferation than either single agent alone (Figure 1B).
Figure 1. Sensitivity of KRAS mutant NSCLC and CRC cell lines to SAR405838, pimasertib or combination.
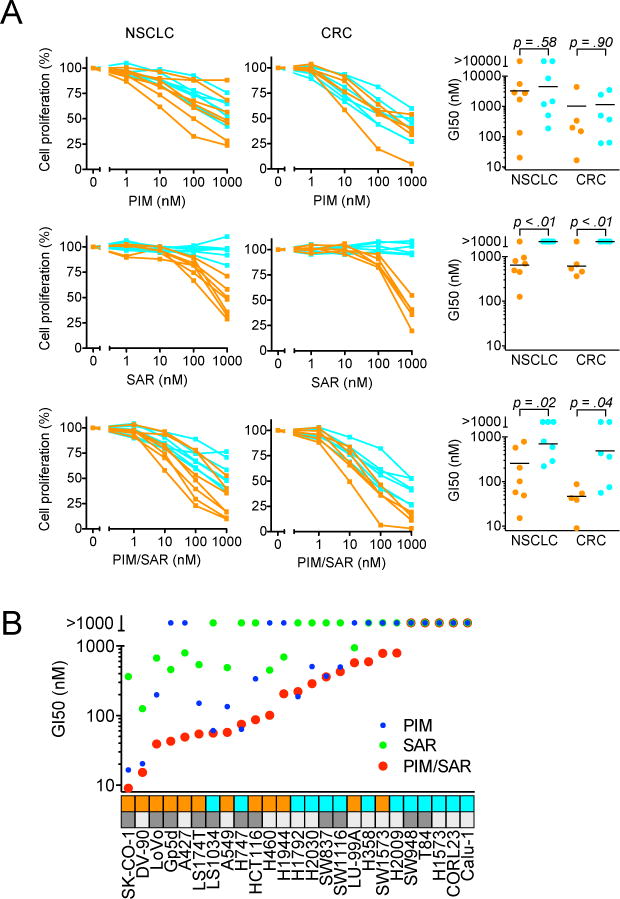
A, TP53 wild-type (orange) and TP53 mutated/deleted (cyan) were treated with SAR405838, pimasertib or the combination for 72 hours and cell viability determined. For combination dose response, equimolar concentrations of each drug were used. Data are mean of three independent experiments. P values were calculated using t-test. B, Comparison of GI50 values for SAR405838, pimasertib or combination for individual cell lines in Figure 1A. TP53 wild-type and mutated/deleted cell lines are denoted by orange and cyan boxes, respectively. NSCLC and CRC cell lines are denoted by light and dark gray boxes, respectively.
Activation of p53 and inhibition of MEK lead to cell cycle arrest, induction of apoptosis and cellular senescence (29–31). We compared the effects of SAR405838 and pimasertib alone or in combination on cell cycle progression and apoptosis. The combination of SAR405838+pimasertib induced greater cell cycle arrest in TP53 wild-type NSCLC cells, largely due to the lack of activity of SAR405838 on TP53 mutated cells (Figure 2A, Sup. Figure 6). Likewise, the combination induced caspase 3/7 activation and apoptosis in TP53 wild-type but not TP53 mutant NSCLC and CRC cells, and to a greater extent than either single agent alone (Figure 2B, Sup. Figure 7). In long term viability assays, the combination of SAR405838+pimasertib led to an absolute decrease in cell number over time (Figure 2C). To determine whether induction of cellular senescence might also contribute to the efficacy of this combination, we stained for senescence-associated beta-galactosidase activity in drug treated cells (Sup. Figure 8). We observed heterogeneous beta-galactosidase staining in DV-90 and HCT116 cells over time with SAR405838 and pimasertib. However, no beta-galactosidase staining was observed after either drug treatment in A427 cells, which exhibited the greatest overall sensitivity. Together, these results indicate that induction of cell cycle arrest, apoptosis, and in some cases cellular senescence all contribute to the response to combination MEK and MDM2 inhibitor treatment.
Figure 2. Pimasertib + SAR405838 induces cell cycle arrest and apoptosis, leading to anti-tumor activity in vitro and in vivo.
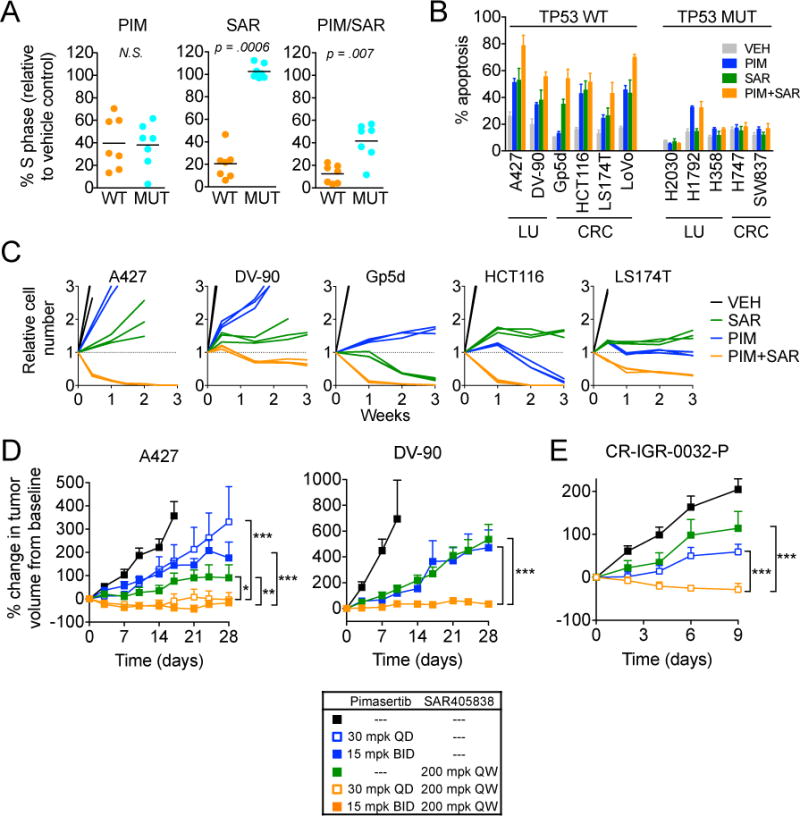
A, Cell cycle analysis of TP53 wild-type versus mutant KRAS mutant NSCLC cell lines after treatment with pimasertib, SAR405838 or combination. Cells were treated with 1 μM pimasertib, SAR405838 or combination, and cell cycle populations were determined. % S phase was calculated by dividing the percentage of cells in S phase after drug treatment with that of vehicle treated cells. P values were calculated using t-test. B, Cells were treated with 1 μM pimasertib, SAR405838 or combination for 72 hours, and apoptosis determined by annexin V staining. Data is mean and standard error of 3–4 independent experiments. C, Cells were treated with 1 μM pimasertib, SAR405838 or combination and relative cell viability determined by RealTime-Glo viability assay at 0.5, 1, 2 and 3 weeks. Each line represent a replicate pool of cells. D, Mice bearing established A427 or DV-90 xenograft tumors were treated with pimasertib, SAR405838 or combination (animals per treatment arm: A427 N=7, DV-90 N=6–9). Tumor response is expressed as the percentage change from the baseline tumor volume at the time of treatment initiation (Day 0). Baseline tumor volumes are listed in Methods. ***P < 0.001, **P < 0.01, *P < 0.05. E, Response of patient-derived colorectal carcinoma xenograft CR-IGR-0032-P tumors after treatment with pimasertib, SAR405838 or combination (N=8 animals per treatment arm). Abbreviations: QD = daily, BID = twice daily, QW = once weekly, mpk = mg/kg.
To evaluate the anti-tumor activity of SAR405838+pimasertib in vivo, we established A427 and DV-90 NSCLC sub-cutaneous xenograft tumors in immunocompromised mice. Treatment with either SAR405838 (200 mg/kg once weekly) or pimasertib (30 mg/kg daily or 15 mg/kg BID) alone modestly slowed the growth rate of tumors compared with control animals. In both tumor models, however, we observed significantly greater anti-tumor activity and induction of apoptosis with the combination (Figure 2D, Sup. Figure 9). Finally, in a TP53 wild-type patient-derived xenograft model of colorectal cancer harboring KRAS and PIK3CA mutations (CR-IGR-0032-P), SAR405838+pimasertib induced tumor regression (Figure 2E). Taken together, these results demonstrate that the combination of MEK+MDM2 inhibitors exhibits synergistic activity to induce cell cycle arrest and apoptosis in KRAS mutant TP53 wild-type NSCLC and CRC cells leading to tumor regression in vivo.
Relative expression of BCL-2 family proteins correlates with apoptotic sensitivity to MEK + MDM2 inhibitor combination
To determine mediators of drug response, we performed mRNA-Seq on six KRAS mutant NSCLC cell lines (A427, DV-90, H1944, LU99A, SW1573 – TP53 wild-type; H358 – TP53 mutant) before and after treatment with 10 nM, 100 nM or 1 μM concentration of pimasertib, SAR405838 or combination. As expected, treatment with SAR405838 or pimasertib led to up or down-regulation of genes regulated by p53 (e.g. CDKN1A, MDM2) and ERK (e.g. DUSP6, SPRY4, ETV4), respectively (Sup. Figure 10). We identified genes synergistically up-regulated by the combination compared with single agents alone for each cell line (Sup. Figure 11A) and performed Ingenuity Pathway Analysis to determine upstream regulators. Interestingly, in all cell lines, the synergistic gene set was highly enriched for genes regulated by p53 and p21 (CDKN1A), suggesting that MEK inhibition synergistically enhances the effects of MDM2 inhibition on p53 gene targets in KRAS mutant cells (Sup. Figure 11B). We also observed a weaker synergistic effect on down-regulation of FOXM1 gene targets, consistent with a prior report (27). Quantitative PCR demonstrated that induction of p21 and down-regulation of FOXM1 transcript correlated strongly with induction of cell cycle arrest and only weakly with induction of apoptosis (Sup. Figure 12A). Indeed, upregulation of p21 and downregulation of FOXM1 transcript was observed both in cells that had minimal and robust apoptotic responses upon treatment with SAR405838+pimasertib (Sup. Figure 12B).
We had originally hypothesized that MEK+MDM2 inhibition would induce BIM and PUMA expression to stimulate apoptosis; however, p53 has also been reported to regulate expression of other pro-apoptotic BCL-2 family proteins such as NOXA and BAX (32, 33). To determine whether other BCL-2 family proteins may be contributing, we examined the expression levels of BCL-2 family proteins after treatment with SAR405838+pimasertib. Comparison of PUMA and BIM mRNA expression relative to BCL-XL expression determined by RNA-seq revealed that the most sensitive A427 and DV-90 cell lines had increased expression of PUMA and BIM relative to BCL-XL at baseline and after drug treatment compared to the less sensitive H1944, LU99A and SW1573 cells and TP53 mutant H358 cells (Figure 3A). In contrast, NOXA/BCL-XL and BAX/BCL-XL ratios did not correlate with sensitivity (Sup. Figure 13). In TP53 wild-type cells, activation of p53 by SAR405838 and inhibition of MEK by pimasertib resulted in accumulation of PUMA and BIM, respectively, whereas only BIM accumulated in TP53 null cells (Sup. Figure 14, Figure 3B). Consistent with the observation that MEK inhibition synergistically enhanced the effect of MDM2 inhibition on p53 target genes, greater induction of PUMA mRNA and protein was observed after treatment with SAR405838+pimasertib compared with SAR405838 alone (Figure 3C, D). siRNA and shRNA mediated knockdown of BIM and PUMA reduced the degree of caspase-3/7 activation following treatment with SAR405838+pimasertib (Figure 3E, Sup. Figure 15A) and resulted in increased cell viability (Sup. Figure 15B), demonstrating that induction of PUMA- and BIM-mediated apoptosis plays a centrol role in the response to MDM2 + MEK inhibition.
Figure 3. Relative expression of BCL-2 family proteins correlates with apoptotic sensitivity to MEK + MDM2 inhibitor combination.
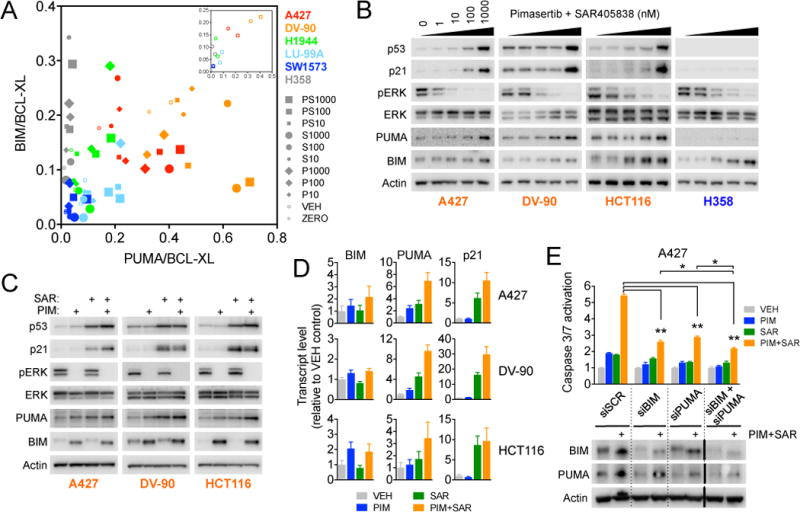
A, BIM/BCL-XL (x-axis) and PUMA/BCL-XL (y-axis) expression ratios were determined from RNA-seq data for KRAS mutant NSCLC cell lines treated with vehicle (VEH), pimasertib, SAR405838 or combination (10 nM, 100 nM, 1000 nM) for 24 hours. “ZERO” = control cells harvested at the time of addition of either drug or vehicle. Inset, Baseline expression ratios replotted showing only untreated (ZERO) and vehicle (VEH) cells. B, Cells were treated with the indicated equimolar concentrations of pimasertib + SAR405838 for 24 hours and harvested for western blotting. For BIM blots, BIMEL (MW ≈ 25 kDa) is shown. C, Cells were treated with 1 μM pimasertib, SAR405838 or combination for 24 hours and harvested for western blotting. D, Cells were treated with 1 μM pimasertib, SAR405838 or combination for 24 hours and mRNA transtript levels were determined by quantitative RT-PCR. Data shown are mean and standard error of three independent experiments. E, A427 cells were transfected with siRNA targeting BCL-2 family proteins and caspase-3/7 activation and protein expression levels were determined 48 and 24 hours after drug treatment, respectively. *P < 0.05, **P < 0.01 t-test.
Complexity of mechanisms of acquired drug resistance to MDM2 + MEK inhibition
It is now well established that the effectiveness of targeted therapies is ultimately limited by the emergence of acquired drug resistance. Prior studies have demonstrated that acquisition of TP53 mutations can lead to resistance to single agent MDM2 inhibitors (34, 35). However, the extent to which mechanisms of acquired resistance to combination therapies overlap with resistance mechanisms to the respective single agents has been largely unexplored. To model acquired drug resistance to combination MDM2+MEK inhibitor therapy, we generated resistant KRAS mutant NSCLC and CRC resistant cell lines by prolonged culturing in the presence of SAR405838+pimasertib (Figure 4A). We employed two different dosing strategies: continuous exposure to both agents, and to more closely mimic in vivo dosing, continuous pimasertib with intermittent exposure to SAR405838 (3 days on, 4 days off). For each experimental condition, multiple resistant colonies were isolated and expanded. Overall, we were able to generate resistant clones (denoted “PSR”) in one out of three NSCLC cell lines (DV-90) and three out of four CRC cell lines (GP5d, LoVo, LS174T). Two cell lines (A427, SK-CO-1) proved too highly sensitive in vitro, and even slowly escalating intermittent drug treatment led to complete cell killing (data not shown). One NSCLC cell line (A549) yielded surviving cells after drug treatment, but these did not develop into proliferating resistant clones even after 9 months of drug exposure.
Figure 4. Acquisition of TP53 mutations is a frequent mechanism of acquired resistance to the MEK+MDM2 inhibitor combination.
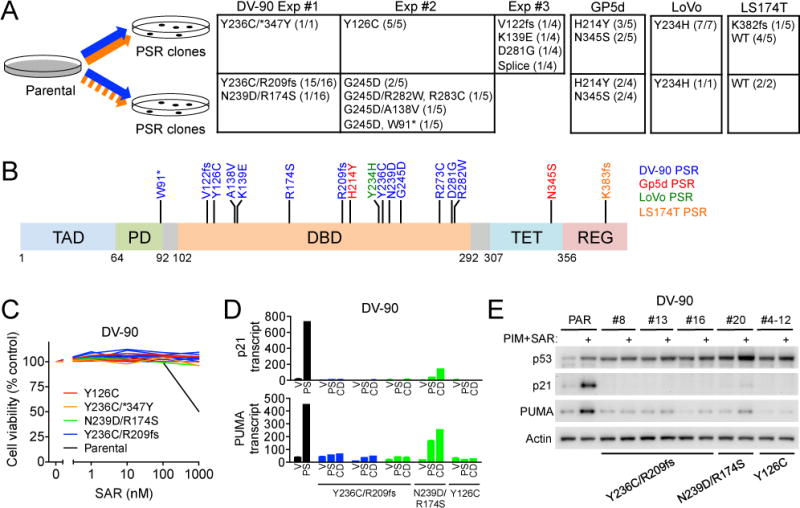
A, KRAS mutant NSCLC (DV-90) and CRC (GP5d, LoVo, LS174T) cell lines were made resistant to SAR405838 (orange, continuous or intermittent exposure) + pimasertib (blue, continuous exposure). Frequency and identity of TP53 mutations in resistant (PSR) clones isolated from each experiment are indicated (WT = wild-type). B, Mutations observed in resistant PSR clones mapped to the TP53 gene structure. C, DV-90 PSR clones (N=13) were treated with SAR405858 for 72 hours and cell viability was determined. D, PUMA and p21 mRNA expression after treatment with vehicle or SAR405838 + pimasertib DV-90 PSR clones. CD denotes cells that were cultured continuously in the presence of SAR405838 + pimasertib. E, PSR clones were treated with vehicle or pimasertib + SAR405838 for 24 hours and lysates harvested for western blotting.
For three out of four cell lines, all resistant PSR clones harbored TP53 mutations (Figure 4A). In some cases, PSR clones that arose from a given parental cell line shared the same TP53 mutation (e.g. LoVo – 8/8 clones harbored Y234H). For others, a small number of different TP53 mutations were consistently observed (e.g. GP5d – 5/9 clones harbored H214Y and 4/9 clones harbored N345S). Conversely, DV-90 cells gave rise to PSR clones with a wide diversity of TP53 mutations that varied both within and between independent experiments. In contrast to the three aforementioned cell lines, the majority (6/7) of LS174T PSR clones retained wild-type TP53. Most acquired TP53 mutations localized to the DNA binding domain, however, occasional mutations were observed in the tetramerization, regulatory and proline-rich domains (Figure 4B). These TP53 mutations were associated with resistance to single agent SAR405838 (Figure 4C, Sup. Figure 16A, B) due to loss of p53 transcriptional activity and decreased p21 and PUMA expression (Figure 4D, E, Sup. Figure 16C, D). In LS174T PSR clones that retained wild-type TP53, SAR405838 induced transient p21 and PUMA expression indicating a functional MDM2-TP53 axis. This was not maintained, however, during chronic exposure to drug (Sup. Figure 16D). These data indicate that acquired resistance to combined MDM2+MEK inhibitors is associated with loss of p53 activity frequently resulting from the acquisition of loss-of-function TP53 mutations.
We next examined whether the PSR clones with TP53 mutations would maintain sensitivity to alternative MEK-based combination therapies. We treated GP5d PSR cells with the combination of pimasertib + navitoclax (ABT263, BCL-XL/BCL-2 inhibitor), which we recently showed was effective for inducing apoptosis in GP5d cells (8). Surprisingly, some GP5d PSR clones were sensitive to pimasertib + navitoclax, while others were resistant (Figure 5A). This differential sensitivity mirrored the sensitivity to single agent pimasertib (Figure 5B), indicating that acquired resistance to MEK inhibition may also contribute to resistance of some PSR clones to the MEK+MDM2 combination. To define the degree of heterogeneity in acquired resistance to MEK inhibition, we compared 20 DV-90 PSR clones that harbored a variety of TP53 mutations. In contrast to their uniform and complete resistance to SAR405838 (Figure 4C), DV-90 PSR clones exhibited variable sensitivity to pimasertib ranging from that of parental cells to complete resistance (Figure 5C). Consistent with this, in the presence of SAR405838+pimasertib, DV-90 PSR clones that maintained sensitivity to pimasertib grew more slowly than pimasertib insensitive cells (Figure 5D), but still exhibited net proliferation.
Figure 5. Differential MEK inhibitor resistance underlies differential response to other MEK-based combination therapies.
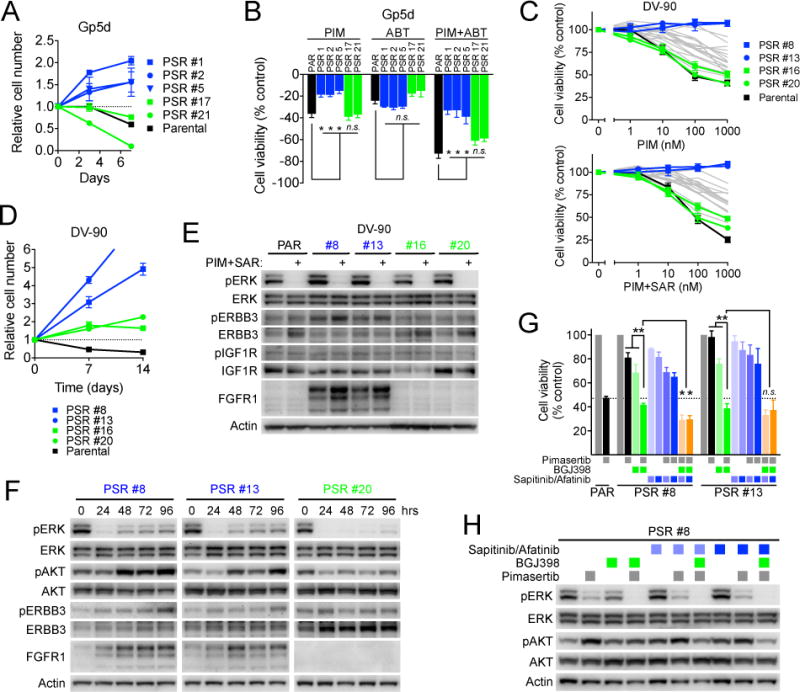
A, GP5d PSR clones were treated with pimasertib + navitoclax and cell viability monitored over time with RealTime-glo assay. Blue and green indicate resistant and sensitive clones, respectively. B, GP5d PSR clones were treated with 1 μM pimasertib, navitoclax (ABT) or combination for 72 hours and cell viability was determined. Data are normalized to vehicle control and are the mean and standard error of three independent experiments. *P < 0.05 t-test. C, DV-90 PSR clones (N=20) were treated with pimasertib or SAR405838 + pimasertib for 72 hours and cell viability was determined. Colored lines highlight clones depicted in panels D–G, gray lines indicate remaining clones. D, DV-90 PSR clones were treated with 1 μM SAR405838 + pimasertib and cell viability monitored over time with RealTime-glo assay. E, DV-90 PSR clones were cultured in the absence of drug for 72 hours and then treated with vehicle or 1 μM SAR405838 + pimasertib for 24 hours and lysates harvested for western blotting. F, Cells were cultured in the absence of drug for 72 hours and then treated with 1 μM pimasertib + SAR405838 and harvested for western blotting at the indicated time points. Fresh media and drug were exchanged 24 hours prior to final 96 hour time point. G, Cells were treated with 1 μM pimasertib, BGJ398, sapitinib (light blue), afatinib (dark blue) or combinations for 3–5 days to achieve equivalent cell doublings and cell viability was determined. Data are mean and standard error of three independent experiments. *P < 0.05, **P < 0.01 t-test. H, DV-90 PSR #8 cells were treated 1 μM concentrations of the indicated inhibitors (sapitinib, light blue; afatinib, dark blue) for 24 hours and lysates harvested for western blotting.
To understand the basis for the differential sensitivity to MEK inhibition, we examined downstream suppression of MEK signaling after drug treatment. After acute treatment with pimasertib, phospho-ERK was suppressed in both pimasertib-sensitive PSR 16 and 20 and resistant PSR 8 and 13 clones (Figure 5E). but after prolonged pimasertib exposure, PSR clones 8 and 13 exhibited rebound of phospho-ERK and phospho-AKT (Figure 5F). Consistent with these findings, resistant PSR clones cultured continuously in the presence of drug (i.e. no drug washout prior to the experiment) had incomplete suppression of ERK transcriptional targets such as DUSP6, ETV4 and ETV5 (Sup. Figure 17A). We next examined whether this might be due to engagement of receptor tyrosine kinase (RTK) feedback loops, which have been shown to underlie intrinsic resistance to MEK inhibitors in KRAS mutant cancer cells (9, 36–38). FGFR1 was dramatically up-regulated in the pimasertib-resistant clones (PSR 8 and 13), whereas little FGFR1 expression was detected in the pimasertib-sensitive clones (PSR 16 and 20) or in parental DV-90 cells (Figure 5E). We also observed modest up-regulation of phospho-ERBB3 in PSR 8 and 13 clones, whereas levels of phospho-IGF1R were unchanged. Addition of the FGFR inhibitor BGJ398 fully suppressed pERK rebound and resensitized resistant clones to pimasertib (Figure 5G, H, Sup. Figure 17B). Addition of the pan-HER inhibitors saptitinib or afatinib to pimasertib weakly suppressed cell proliferation, and further suppressed proliferation of the PSR 8 clone when combined with pimasertib and BGJ398. These findings demonstrate that differential rewiring of RTK feedback loops may contribute to heterogeneity of acquired resistance to MEK inhibition. Interestingly, we did not observe any change in vimentin or E-cadherin expression (data not shown), indicating that the increased FGFR1 expression was not associated with EMT. This highlights the potential complexity of acquired resistance to combination therapies, for which loss of sensitivity to one or the other partner may be sufficient to cause acquired resistance to the combination. Moreover, these studies demonstrate that this heterogeneity of acquired resistance may disparately affect responsiveness to subsequent targeted therapy strategies.
Discussion
Due to the central role of p53 in regulating cell cycle progression and apoptosis, pharmacologic restoration of p53 function remains an attractive therapeutic strategy for cancers that harbor wild-type TP53. Although experience is limited, the clinical responses to single agent MDM2 inhibitors have been disappointing, even for MDM2 amplified cancers such as dedifferentiated liposarcoma (39). Combination strategies pairing MDM2 inhibitors with various targeted therapies (MEK, BRAF, PI3K inhibitors) have been explored for various cancer types (melanoma, NSCLC, acute myelogenous leukemia) in pre-clinical studies (25–27, 40), however it is unclear which combinations should be prioritized for specific disease sub-types. In this study, we evaluated the potential for the combination of MDM2 + MEK for KRAS mutant NSCLC and CRC cancers. We observed synergistic activity in vitro due to induction of both apoptosis and cell cycle arrest. Moreover, we observed efficacy of the combination in vivo, suggesting that the clinical evaluation of this compound is warranted for these cancers that have limited targeted therapy options (NCT01985191, www.clinicaltrials.gov).
Our prior studies investigating MEK inhibitor combinations for KRAS mutant cancers have revealed that induction of a robust apoptotic response is a critical, but often elusive goal (8, 28). MEK + MDM2 inhibitors led to the induction of the pro-apoptotic BCL-2 family proteins BIM and PUMA in sensitive KRAS mutant cells, consistent with a prior study using AML models (40). We also observed that induction of p53 target genes was enhanced by the combination relative to MDM2 inhibitor alone. This enhancement of p53 activity may explain prior observations that survivin and FOXM1 were synergistically induced in melanoma and NSCLC cells, respectively, after treatment with combined MDM2 and RAF or MEK inhibitors (26, 27). Our results suggest that downregulation of survivin or FOXM1 does not fully explain apoptotic sensitivity, as a similar suppression was also observed in cell lines that had minimal apoptotic response to MEK + MDM2 inhibition. Rather, similar to other targeted therapy strategies, the ability to engage BH3 signaling at the level of the mitochondria may be the primary determinant of drug efficacy. It is worth noting that SAR405838 does not inhibit MDMX (24), which may cooperate with MDM2 to suppress p53 in some contexts (41). Although inhibition of MDM2 by SAR405838 was sufficient for activation of p53 in all TP53-wild type cell lines tested, it is possible that strategies that target both MDM2 and MDMX might lead to more potent p53 activation.
Acquired drug resistance is a major problem limiting clinical efficacy of targeted therapies. The mechanisms of resistance to drug combinations are potentially complex, as tumor progression might result from resistance to either drug alone, or both. We observed TP53 mutations in the majority of models made resistant to SAR405838+pimasertib. This is consistent with prior studies that reported that the acquisition of TP53 mutations conferred resistance to single agent nutlin-3 (34, 35) or SAR405838 (42). Emerging data from the clinic suggest that emergence of tumor clones with TP53 mutations may be a significant mechanism of acquired resistance in patients, as a recent trial of single agent SAR405838 for de-differentiated liposarcoma revealed acquired TP53 mutations in 20% of patients, including multiple independent TP53 clones in some patients(43). We did not observe any overlap between acquired TP53 mutations in resistant PSR clones derived from different cell lines, but for a given cell line, multiple clones were observed to harbor the identical mutation, suggesting that these may have arisen from pre-existing TP53 mutated sub-clones. The development of TP53 mutations was not universal, however, as the majority of LS174T PSR clones examined harbored wild-type TP53 and retained at least partial sensitivity to SAR405838. Whether emergence of TP53 mutations will also be observed in patients treated with combination MDM2+MEK inhibitors remains to be determined.
In contrast to the complete resistance of TP53 mutant PSR clones to SAR405838, we observed variable resistance to MEK inhibition. Although pimasertib acutely inhibited downstream phospho-ERK signaling in PSR clones, we observed greater rebound activation of MEK-ERK and PI3K-AKT signaling after longer exposure to pimasertib in clones that were more resistant to pimasertib. Consistent with this, transcriptional outputs of MEK-ERK signaling such as DUSP6 and ETS family transcription factors (ETV4, ETV5) were not fully suppressed during chronic drug exposure in these clones. Prior studies have demonstrated that treatment of KRAS mutant cells with MEK inhibitors causes de-repression of negative feedback loops that lead to re-activation of RTK and downstream signaling pathways (9, 36–38). Recently, FGFR signaling has been implicated in intrinsic resistance of mesenchymal KRAS mutant NSCLC cells to MEK inhibition (37, 44). While we observed that FGFR1 was up-regulated in PSR clones with acquired resistance to MEK inhibition, this was not accompanied by any change in epithelial-mesenchymal status (DV-90 cells are epithelial). Resistant PSR clones also had greater up-regulation of ERBB3 phosphorylation compared with sensitive PSR clones, albeit less dramatic than the difference in FGFR1 expression. This suggests that KRAS mutant cells may can engage similar mechanisms to achieve intrinsic and acquired resistance to MEK inhibition, but that this may be associated with different features (e.g. epithelial-mesenchymal phenotype) in different contexts.
These studies highlight the potential complexity of mechanisms of acquired resistance that develop in response to combination therapies, and suggest that defining the relative contribution of each drug to the resistance phenotype may be important for making decisions about subsequent therapy. This may be especially relevant for KRAS mutant cancers, for which a number of MEK-based combinations are being developed. For instance, if a tumor develops resistance to MDM2 + MEK inhibitors by acquiring a TP53 mutation, determining whether the tumor is also resistant to the MEK inhibitor would inform whether a subsequent MEK inhibitor-based combination such as trametinib+navitoclax might be warranted (8) (NCT02079740, www.clinicaltrials.gov). On the other hand, if no TP53 mutations are acquired, subsequent treatment with a different MDM2 inhibitor-based combination might be considered (27).
Materials and Methods
Cell lines, antibodies and reagents
Human KRAS mutant NSLCLC and CRC cell lines were obtained from the Center of Molecular Therapeutics at the MGH Cancer Center who performed STR verification. TP53 genotype was independently confirmed for each cell line (see below). Cell lines were tested for mycoplasma infection every 2 months during experimental use. DV-90, A549, H460, H1944, Lu-99A, H1792, H2030, COR-L23, H358, Calu-1, HCT116, SK-CO-1, SW948, and H747 cells were maintained in RPMI supplemented with 5% FBS; A427, SW1573, H2009, H1573, GP5d, LS174T, LS1034, LoVo, SW116, SW837 and T84 cells were maintained in DMEM/F12 with 5% FBS. For cell culture studies, pimasertib and SAR405838 were provided by Sanofi and dissolved in DMSO to a final concentration of 10 mmol/l and stored at −20°C. BGJ398, afatinib and sapitinib were purchased from Selleckchem. For western blotting, the following antibodies were used: Actin (4970, Cell Signaling), p-AKT S473 (9271, Cell Signaling), BIM (2933, Cell Signaling), p-ERBB3 Y1289 (4791, Cell Signaling), ERBB3 (4754, Cell Signaling), p-ERK1/2 T202/204 (9101, Cell Signaling), ERK1/2 (9102, Cell Signaling), FGFR1 (9740, Cell Signaling), p-IGF1R Y1135/1136 (3024, Cell Signaling), IGF1R (9750, Cell Signaling), p21 (556431, BD), p53 (SC-126, Santa Cruz), PUMA (SC-374223, Santa Cruz).
Cell viability and proliferation analysis
Cell lines were seeded into 96 well plates 24 hours before addition of drug. For dose response experiments, drugs were added to cells and cell proliferation determined by CellTiter-Glo assay (Promega) after 72 hours. For experiments on DV-90 PSR resistant clones, which proliferate more slowly than parental cells, cells were incubated for 5 days to allow for the same number of cell doublings of vehicle treated cells. For long-term viability assays, cell number was determined 24 hours after seeding and prior to drug addition, and at indicated time points by RealTime-Glo assay (Promega).
Annexin/PI staining by flow cytometry
Cells were seeded at low density 24 hours prior to drug addition. After 72 hours floating and adherent cells were collected and stained with propidium iodide and Cy5-Annexin V and analyzed by flow cytometry. % apoptotic cells was calculated by subtracting percentage of annexin positive cells in vehicle control from percentage of annexin positive cells with drug treatment.
Caspase 3/7 activity assay
Cells were seeded into 96 well plates 24 hours prior to addition of drug. Drugs were added to cells for 48 hours and cell viability and caspase 3/7 activation were determined by Caspase 3/7 activity. Values were normalized to viability reading.
Cell cycle analysis
Cells were seeded 24 hours prior to experiment to give a confluency of 30–50%. Drugs were added for 24 hours and cells harvested, stained with propidium iodide and analyzed by flow cytometry. Cell cycle sub-populations were calculated using the cell cycle analysis package of FloJo software.
Cellular Senescence analysis
In vitro cellular senescence was determined using the Senescence β-Galactosidase Staining Kit (Cell Signaling) according to the manufacturers recommendations. Briefly, vehicle or drug treated cells were washed with PBS and fixed in Fixative Solution for 10 minutes at room temperature. Fixed cells were washed twice with PBS and then incubated with β -Galactosidase Staining Solution (pH 6.0) overnight at 37°C.
Quantitative RT-PCR
Cells were seeded 24 hours prior to give a confluency of 50%. Cells were treated with drugs for 24 hours and RNA was extracted using the RNeasy Kit (Qiagen). cDNA was prepared using the First Strand Synthesis Kit (Invitrogen) with oligo-dT primers. Quantitative PCR was performed on a Lightcycler 480 using FastStart Sybr Green (Roche). mRNA expression relative to GAPDH was calculated using the Delta-Delta threshold cycle (Ct) method. Human primers used: BCL-XL F 5′-agccttggatccaggagaa-3′, R 5′-agcggttgaagcgttcct-3′; BIM F 5′-gatccttccagtgggtatttctctt-3′, R 5′-actgagatagtggttgaaggcctgg-3′; DUSP6 F 5′-cgactggaacgagaatacgg-3′, R 5′- ttggaacttactgaagccacct-3′; ETV4 F 5′-gcagtttgttcctgatttcca-3′, R 5′-actctggggctccttcttg-3′; ETV5 F 5′-cctacatgagagggggttatttc-3′, R 5′-cgtcaaagtataatcggggatct-3′; GAPDH F 5′-aacagcgacacccatcctc-3′, R 5′-cataccaggaaatgagcttgacaa-3′; MCL-1 F 5′-aagccaatgggcaggtct-3′, R 5′-tgtccagtttccgaagcat-3′; PUMA F 5′-gacctcaacgcacagtacga-3′, R 5′-gagattgtacaggaccctcca-3′; p21 F 5′-ccgaagtcagttccttgtgg-3′, R 5′-catgggttctgacggacat-3′; FOXM1 F 5′-actttaagcacattgccaagc-3′, R 5′-cgtgcagggaaaggttgt-3′; BIRC5 F 5′-gcccagtgtttcttctgctt-3′, R 5′-aaccggacgaatgcttttta-3′; CLASPN F 5′-gaagagaaagaggaagaactagagga-3′, R 5′-cactactaagaaggaattctgcagtc-3′.
RNAi gene knockdown
A427 cells with shRNA knockdown of BIM and PUMA were previously described(28). A427 cells with stable TP53 knockdown were generated by infecting with lentivirus harboring pLKO TP53 shRNA (TRCN0000003756) followed by selection in puromycin. Knockdown of TP53 mRNA and p53 protein was determined by quantitative RT-PCR and western blotting, respectively. siRNA gene knockdown was performed by transfection of cells with the following siRNAs (Qiagen): BIM (Hs_BCL2L11_5), PUMA (Hs_BBC3_2).
Mouse xenograft studies
All mouse studies were conducted through Institutional Animal Care and Use Committee–approved animal protocols in accordance with institutional guidelines. For A427 and DV-90 xenograft studies, cell line suspensions were prepared in 1:1 matrigel:PBS and 5x106 cells were injected unilaterally into the subcutaneous space on the flanks of athymic nude (Nu/Nu) mice (6–8 weeks). Xenograft tumors formed within 2–3 weeks and were allowed to grow to approximately 350 mm3. CR-IGR-0032-P was obtained from Lariboisière Hospital, France upon patient consent for providing surgical tumor samples to CReMEC and for HIV1, HIV2, Hepatitis B virus and Hepatitis C virus, HCV serological status testing. The PDX models was established in CB17/lCR-Prkdc severe combined immunodeficiency (SCID)/Crl mice (Charles River). Animals were implanted unilaterally and solid tumors were allowed to grow for 28 days, at which point they had reached approximately 150–200 mm3. Tumors were measured with electronic calipers and the tumor volume was calculated according to the formula V = 0.52 × L × W2. Mice with established tumors were randomized to drug treatment groups using covariate-adaptive randomization to minimize differences in baseline tumor volumes. Tumor numbers and volumes at the time of treatment initiation were: A427 – vehicle (N=7) 369+/−24 mm3, pimasertib 15 BID (N=7) 377+/−77 mm3, pimasertib 30 QD (N=7) 358+/−38 mm3, SAR405838 200 QW (N=7) 341+/−45 mm3, pimasertib 15 BID + SAR405838 200 QW (N=7) 379+/−53 mm3, pimasertib 30 QD + SAR405838 200 QW (N=7) 354+/−29 mm3; DV-90 vehicle (N=9) 313+/−42 mm3, pimasertib 15 BID (N=6) 354+/−35 mm3, SAR405838 200 QW (N=8) 364+/−58 mm3, pimasertib 15 BID + SAR405838 200 QW (N=7) 373+/−42 mm3; CR-IGR-0032-P – vehicle (N=8)161+/−25 mm3, pimasertib 30 QD (N=8) 185+/−19 mm3, SAR405838 200 QW (N=8) 177+/−25 mm3, pimasertib 30 QD + SAR405838 200 QW (N=8) 181+/−21 mm3. Pimasertib was dissolved in 0.5% carboxymethyl cellulose/0.25% Tween-20. SAR405838 was prepared in PEG200/TPGS (98%/2%). Drug treatments were administered by oral gavage. Investigators performing tumor measurements were not blinded to treatment group. Sample size (minimum N=7 per treatment group) was chosen to verify satisfactory inter-animal reproducibility.
Cleaved caspase-3 immunohistochemistry
Formalin fixed paraffin embedded xenograft tumors were sectioned, de-paraffinized and re-hydrated through xylene and alcohol washes. Antigen retrieval was performed. Slides were blocked in goat serum and Avidin D. Slides were incubated with cleaved caspase-3 antibody (Cell Signaling) overnight at 4°C. Slides were blocked with biotin, incubated with rabbit biotinylated secondary antibody for 1 hour, washed in PBS, incubated with an Avidin D, horseradish peroxidase, and developed with DAB coloring reagent. Slides were counter-stained in hemotoxylin.
In vivo cellular senescence
Snap frozen xenograft tumor tissue was embedded in OCT, sectioned into 5 micron sections and stained using the Senescence β-Galactosidase Staining Kit (Cell Signaling) with an adapted protocol. Frozen tissue slides were placed in Fixative Solution for 10 minutes at room temperature. Fixed cells were washed three times with PBS for 60 second and then incubated with β -Galactosidase Staining Solution (pH 6.0) overnight at 37°C. Cells were then counter-stained in hemotoxylin.
Generation of resistant cell lines
DV-90, GP5d, LS174T and LoVo PSR resistant cell lines were generated by continuous treatment of parental cells with 1 μM pimasertib + 1μM SAR405838 or continuous pimasertib with intermittent SAR405838 (3 days on, 4 days off). Resistant colonies were selected with cloning rings and expanded independently. After resistant colonies were established, all resistant cell lines were cultured in the continuous presence of pimasertib and SAR405838. For drug sensitivity studies, western blot and gene expression studies, cells were removed from drug for 48–72 hours prior to experiment, except when indicated (continuous drug (CD) samples).
RNA-Sequencing
Total RNA was isolated using the RNeasy Mini Kit (Qiagen). 200ng of total RNA was used for each sample for a strand-specific RNA-seq library preparation using the Ovation Universal RNA-Seq System 1–96 (NuGEN Technologies, 0402-A01) and implemented on the Agilent Bravo liquid handling system according to the manufacturer’s recommended procedures. Resulting libraries were pooled at an equimolar concentration and 50bp single-end sequencing data was generated with an Illumina HiSeq 2500 sequencer. RNA-Seq data processing was performed using Sanofi’s NGS data analysis framework called Synapse. RNA-Seq reads were initially mapped and aligned to GRCh38 using STAR (45) aligner (v.2.4.0h1). Then mapped reads were assembled into possible transcripts by using Cufflinks (46) (v2.2.1) with the reference gene annotation GENCODE v21. A read summarization tool, featureCounts (47) was used to count the number of reads mapped to each of genes annotated in GENCODE v21. The data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE99015. Differentially expressed transcripts were identified using EdgeR (48). Synergy-associated genes were defined as those whose expression increased or decrease by 2-fold comparing to the vehicle control and 1.5-fold comparing to both single-agent groups. Ingenuity pathway analysis was used to perform functional analysis on synergy-associate genes. IPA identified the over-representation in a define set of genes, which could be known targets of a regulator, using a Fisher’s exact test. Based on the gene expression pattern, it predicted the activation states of upstream regulator with a confidence z-score. A linear regression model was used to calculate the correlation between gene expression log fold change after drug treatment and inhibition of proliferation, induction of apoptosis and cell cycle arrest data (p < 0.01).
Drug synergy evaluation
Cells were seeded into 96 well plates and treated with varying concentrations of pimasertib, SAR405838 or combination according to a modified ray design. Cell proliferation/viability was determined at time zero and after 72 hours using the CellTiter-Glo assay. Each data point was determined in triplicate, with individual replicates performed on separate plates to account for inter-plate variability. We estimated synergy in each cell line with a Hewlett model, using the methods of Ritz and Streibig (49) in R (50). The Hewlett model assesses the curvature of an isobologram for 2 compounds with an exponent λ, in the following way:
where fA and fB denote the mixture fraction of each compound respectively. Fitted values of λ >> 1 denote synergy, and fitted values of λ << 1 denote antagonism. We assessed the statistical significance of λ with a 1-tailed t-test The results were always statistically significant, with resulting p-values on the order of 10−12 – 10−25, corresponding to λ fitted values ranging from +2 to +5, which are strong effect sizes.
Targeted cell line genotyping
A custom sequencing panel was developed using xGEN® Lockdown® Probes (Integrated DNA Technologies) including entire coding sequences (CDS) and untranslated regions (UTR) of 7 genes, TP53, KRAS, NRAS, HRAS, BRAF, PTEN and MDM2. Whole genome sequencing (WGS) libraries were made using the KAPA Hyper Prep Kits (KAPA Biosystems, Catalog # KK8504) using 100 ng of gDNA, following the Rapid Protocol Version 2.1 from IDT using a total of 1 μg of WGS library; the hybridization reaction was incubated at 65°C (lid 80°C) for 18–24 hours. Sequencing was performed the Illumina Hi-Seq platform. In order to retain high-quality base calls, sequencing reads were trimmed down to 75bp and adapter sequences were removed by using Trimmomatic(51). Then the sequencing reads were mapped to the human reference genome hg19 with the Novoalign (v3.0205 – http://www.novocraft.com/). The alignment was further refined using the indel realigner, ABRA (52), to increase mapping/alignment accuracy and consequently performance for detection of insertion and deletion (INDEL) mutations. MuTect (53), LoFreq (54) and Pindel (55) were used for single nucleotide variant (SNV) and INDEL detection. ONCOTATOR (56) was used for annotating the SNVs and INDELs. Silent mutations or variants affecting protein non-coding sequences were excluded from the final analysis.
Data and statistical analysis
Data were analyzed using GraphPad Prism software (GraphPad Software). GI50 values were calculated using a four parameter variable slope model. Western blot band quantitation was performed using SynGene Gene Tools Software. Correlation calculations comparing gene expression levels were determined by quantitative RT-PCR, and apoptosis and cell cycle arrest were determined using the Spearman’s correlation test. For RNA-seq linear regression correlation, p < 0.01 was considered significant. Unless otherwise specified, data displayed are mean and standard error. Comparisons between groups (e.g., experimental versus control) were made using paired or unpaired t-tests, unless otherwise noted. Variance between comparison groups were verified to be equivalent. P values below 0.05 were considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank all members of the Engelman Lab for helpful discussions and feedback. We thank MercK KGaA, Darmstadt, Germany for provision of pimasertib for our research and for scientific review of this manuscript. This study was funded by support from the NIH R01CA14059408 (J.A.E.), Uniting Against Lung Cancer (A.N.H), Conquer Cancer Foundation of ASCO (A.N.H.)
Footnotes
Author Contributions
A.N.H., J.W. and J.A.E. designed the study, analyzed the data and wrote the paper. A.N.H, F.M.S., H.L.A and M.G.C. performed cell line and biochemical studies. M.G.C. performed cell line tumor xenograft studies. S.R. performed drug synergy analysis. J.J., S.R., F.J. and R.I.S. performed RNA-Sequencing and analysis. M.L., J.S.L and J.J. performed TP53 genotyping of cell lines. L.D. and S.S. generated the CRC PDX model and performed in vivo studies. All authors discussed the results and commented on the manuscript.
The authors declare competing financial interests: J.A.E. is a consultant for Novartis, Sanofi, Genentech and Amgen; has research agreements with Novartis, Sanofi, and Amgen. A.N.H. has provided consulting services for Amgen; has research agreements with Amgen and Novartis. S.R., M.L., J.S.L, J.J., S.S., L.D., and J.W. are employees of Sanofi, as noted in the affiliations.
Conflict of Interest:
No other conflicts of interest were reported by the other authors.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 2.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–8. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 3.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 4.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blumenschein GR, Jr, Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Ann Oncol. 2015;26:894–901. doi: 10.1093/annonc/mdv072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic Lethal Interaction of Combined BCL-XL and MEK Inhibition Promotes Tumor Regressions in KRAS Mutant Cancer Models. Cancer Cell. 2013;23:121–8. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014;7:86–93. doi: 10.1016/j.celrep.2014.02.045. [DOI] [PubMed] [Google Scholar]
- 10.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nature reviews Cancer. 2009;9:862–73. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 11.Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Advances in cancer research. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- 12.Muller PA, Vousden KH. p53 mutations in cancer. Nature cell biology. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 13.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes & development. 1993;7:1126–32. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 14.Bond GL, Hu W, Levine AJ. MDM2 is a central node in the p53 pathway: 12 years and counting. Current cancer drug targets. 2005;5:3–8. doi: 10.2174/1568009053332627. [DOI] [PubMed] [Google Scholar]
- 15.Momand J, Wu HH, Dasgupta G. MDM2–master regulator of the p53 tumor suppressor protein. Gene. 2000;242:15–29. doi: 10.1016/s0378-1119(99)00487-4. [DOI] [PubMed] [Google Scholar]
- 16.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–3. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 17.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17:5001–14. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3933–8. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annual review of pharmacology and toxicology. 2009;49:223–41. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer research. 2013;73:2587–97. doi: 10.1158/0008-5472.CAN-12-2807. [DOI] [PubMed] [Google Scholar]
- 22.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends in molecular medicine. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 24.Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014;74:5855–65. doi: 10.1158/0008-5472.CAN-14-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji Z, Njauw CN, Taylor M, Neel V, Flaherty KT, Tsao H. p53 rescue through HDM2 antagonism suppresses melanoma growth and potentiates MEK inhibition. J Invest Dermatol. 2012;132:356–64. doi: 10.1038/jid.2011.313. [DOI] [PubMed] [Google Scholar]
- 26.Ji Z, Kumar R, Taylor M, Rajadurai A, Marzuka-Alcala A, Chen YE, et al. Vemurafenib synergizes with nutlin-3 to deplete survivin and suppresses melanoma viability and tumor growth. Clin Cancer Res. 2013;19:4383–91. doi: 10.1158/1078-0432.CCR-13-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saiki AY, Caenepeel S, Yu D, Lofgren JA, Osgood T, Robertson R, et al. MDM2 antagonists synergize broadly and robustly with compounds targeting fundamental oncogenic signaling pathways. Oncotarget. 2014;5:2030–43. doi: 10.18632/oncotarget.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hata AN, Yeo A, Faber AC, Lifshits E, Chen Z, Cheng KA, et al. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers. Cancer Res. 2014;74:3146–56. doi: 10.1158/0008-5472.CAN-13-3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 30.Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15:577–92. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- 31.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–96. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 32.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 33.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 34.Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene. 2011;30:4678–86. doi: 10.1038/onc.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michaelis M, Rothweiler F, Barth S, Cinatl J, van Rikxoort M, Loschmann N, et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011;2:e243. doi: 10.1038/cddis.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ebi H, Corcoran RB, Singh A, Chen Z, Song Y, Lifshits E, et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121:4311–21. doi: 10.1172/JCI57909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitai H, Ebi H, Tomida S, Floros KV, Kotani H, Adachi Y, et al. Epithelial-to-Mesenchymal Transition Defines Feedback Activation of Receptor Tyrosine Kinase Signaling Induced by MEK Inhibition in KRAS-Mutant Lung Cancer. Cancer Discov. 2016;6:754–69. doi: 10.1158/2159-8290.CD-15-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012;72:3228–37. doi: 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. The lancet oncology. 2012;13:1133–40. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W, Konopleva M, Burks JK, Dywer KC, Schober WD, Yang JY, et al. Blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase and murine double minute synergistically induces Apoptosis in acute myeloid leukemia via BH3-only proteins Puma and Bim. Cancer Res. 2010;70:2424–34. doi: 10.1158/0008-5472.CAN-09-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffman-Luca CG, Yang CY, Lu J, Ziazadeh D, McEachern D, Debussche L, et al. Significant Differences in the Development of Acquired Resistance to the MDM2 Inhibitor SAR405838 between In Vitro and In Vivo Drug Treatment. PLoS One. 2015;10:e0128807. doi: 10.1371/journal.pone.0128807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jung J, Lee JS, Dickson MA, Schwartz GK, Le Cesne A, Varga A, et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun. 2016;7:12609. doi: 10.1038/ncomms12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manchado E, Weissmueller S, Morris JPt, Chen CC, Wullenkord R, Lujambio A, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534:647–51. doi: 10.1038/nature18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 48.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ritz C, Streibig J. From additivity to synergism – A modelling perspective. Synergy. 2014;1:22–9. [Google Scholar]
- 50.R_Core_Team R. A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 51.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mose LE, Wilkerson MD, Hayes DN, Perou CM, Parker JS. ABRA: improved coding indel detection via assembly-based realignment. Bioinformatics. 2014;30:2813–5. doi: 10.1093/bioinformatics/btu376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic acids research. 2012;40:11189–201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, et al. Oncotator: cancer variant annotation tool. Hum Mutat. 2015;36:E2423–9. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.