

Abstract
Autism Spectrum Disorder (ASD) is a behaviorally defined condition that manifests in infancy or early childhood as deficits in communication skills and social interactions. Often, restricted and repetitive behaviors (RRBs) accompany this disorder. ASD is polygenic and genetically complex, so we hypothesized that focusing analyses on intermediate core component phenotypes, such as RRBs, can reduce genetic heterogeneity and improve statistical power. Applying this approach, we mined Caucasian GWAS data from two of the largest ASD family cohorts, the Autism Genetics Resource Exchange (AGRE) and Autism Genome Project (AGP). Of the twelve RRBs measured by the Autism Diagnostic Interview-Revised (ADI-R), seven were found to be significantly familial and substantially variable, and hence, were tested for genome-wide association in 3 104 ASD affected children from 2 045 families. Using a stringent significance threshold (p < 7.1×10-9), GWAS in the AGP revealed an association between ‘the degree of the repetitive use of objects or interest in parts of objects’ and rs2898883 (p < 6.8×10-9), which resides within the sixth intron of PHB. To identify the candidate target genes of the associated SNPs at that locus, we applied chromosome conformation studies in developing brains and implicated three additional genes: SLC35B1, CALCOCO2 and DLX3. Gene expression, brain imaging, and fetal brain eQTL studies prioritize SLC35B1 and PHB. These analyses indicate that GWAS of single heritable features of genetically complex disorders followed by chromosome conformation studies in relevant tissues can be successful in revealing novel risk genes for single core features of ASD.
Introduction
Autism Spectrum Disorders (ASD), first described by Kanner (1) in 1943, are lifelong neurobehavioral syndromes identified by the key features of marked deficits in communication skills and social interactions that are accompanied by restricted interests and repetitive behaviors (RRBs), with an onset prior to the age of three (2). Diagnoses are made by an expert's opinion of the child's behavior, using instruments such as the Autism Diagnostic Interview–Revised (ADI-R) (3). Currently, the estimate of ASD incidence is 1 in 68, with a ratio of affected males to females of 4:1 (4), and an incidence in males of 1 in 42, making it compelling to identify the etiologies of this disorder. Although genetics is currently central to investigations of ASD etiologies, its importance was not immediately clear. Heritability analyses to establish a genetic component began in the 1970s with the availability of appropriate ASD study samples to estimate and compare the concordance rates in monozygotic (MZ) versus dizygotic (DZ) twin pairs. Although previous heritability estimates were as high as 90% (5), implicating the etiological impact of genes almost exclusively, current heritability estimates of approximately 50%- 60% (6) indicate that both genes and environmental factors, and possibly their interactions, are important. Not surprisingly, given its complexity as a phenotype and our emerging knowledge of its genetic heterogeneity, the specific patterns of genetic and environmental risk factors that lead to ASDs have not been easy to identify (7). A salient outcome of a decade of intensive genetic analysis of ASD has been a realization that the genetics of this disorder is extremely complex, and that as many as 1 000 genes, in various combinations with each other and with environmental factors, are likely to be involved (8).
One powerful approach to reducing genetic complexity is to focus on identifying the genes that predispose to a single phenotype related to ASD. Such phenotypes may have less complex genetic etiologies than the diagnosis of ASD itself (9). Successful studies will reveal genes predisposing to the individual phenotypes, and possibly provide insight into the etiologies of ASD. This approach has been successful in studying the genetics of a number of ASD related phenotypes. In early studies of the Autism Genetics Resource Exchange (AGRE) sample (10), linkage analyses of a single component of ASD, language delay, identified a locus on chromosome 7q as harboring one or more risk genes for that feature (11). In follow-up association analyses, Contactin Associated Protein 2 (CNTNAP2) was identified (12) as a risk gene in that 7q region. In another study of the AGRE sample, linkage to deficits in nonverbal communication was found in four chromosome regions (13), and follow-up association analyses at 1p13-q12 revealed Nerve Growth Factor (NGF) as a risk gene for these deficits (14). More recently, using the expanded AGRE sample, a quantitative assessment of deficits in social behavior in those with ASD (the Social Responsiveness Scale (SRS)) revealed two linked loci on chromosome 8 (8p21.3 and 8q24.22) (15). A very recent study shows, as predicted, (16) that genome-wide risk loci contributing to variation in core ASD features of social cognition and adaptive functioning also contribute to variation in the same traits in the general population (17). This supports the notion that common variation contributes to individual core features of ASD. The analyses reported here use GWAS of common variants to find risk genes for RRBs.
Investigations into the genetics of RRBs have been less aggressive than genetic studies of ASD. However, similar to the study designs used to investigate the broader ASD diagnosis, early genetic studies of RRBs examined familiality (18, 19) genome-wide linkage (18), and the association of candidate genes (19). The aggregate of these genetic studies of RRBs, along with studies in model organisms, implicate dopaminergic, glutamatergic and serotonergic genes, as reviewed by Lewis and Kim (20). Factor (21) and principal components (22) analyses of the ADI-R items assessing RRBs in those with ASD support the hypothesis that RRBs do not reflect a single underlying trait. These analyses of the correlation structure of the ADI-R RRB items consistently reveal that there are likely to be two major independent factors contributing to RRBs. The first represents repetitive sensory motor actions and the second represents insistence on sameness. Previous linkage analysis of the two RRB factors in large pedigrees implicate chromosome regions 2q37 and 15q13-14 (23).
Recently, a study was conducted in the AGRE (10) sample (24) where a GWAS of the two RRB principal components, were analyzed to identify risk loci. The Genome-wide Efficient Mixed Model Analysis (GEMMA) software (25) that conducts a multivariate GWAS was applied to the bivariate principal components. A standard p<5×10-8 level of significance resulted in no significant associations; however, the authors reported 8p21.2-8p21.1 as harboring the top signal in the AGRE sample. The Simons Simplex Collection (SSC) (26) was used for replication, but this was not achieved. One might expect this, as the SSC is designed primarily to address the role of de novo variants in ASD, because the families used to ascertain the parent/affected child trios do not have multiple affected individuals. That is, the ascertainment scheme of the SSC depletes the supply of inherited risk alleles, making this sample statistically underpowered to identify familial inherited contributions.
Here, we report GWAS analyses to identify RRB risk genes using existing ASD GWAS data. We made five choices in the study design in order to increase statistical power. 1)We studied RRBs, hypothesizing that because they exhibit less phenotypic complexity than ASD, they are more likely to have reduced genetic complexity and greater locus specific heritabilities. 2) We screened the RRBs for those with significant familiality and substantial variability. 3) To find common inherited risk alleles, we analyzed the AGRE and AGP study samples. 4) To reduce allelic and locus heterogeneity, we selected Caucasian AGRE and AGP sub-samples because it was the most frequent ethnicity in both samples. 5) For the GWAS analysis, we used a variance components analytic approach that corrects for the non-independence of siblings and exhibits superior statistical power over family-based approaches.
Some of our study design choices may not appear straightforward; our reasoning follows. Studying RRBs in those with ASD promises to reduce phenotypic heterogeneity, because, although the composite diagnosis of ASD has a high heritability, we hypothesize that the individual RRBs are less genetically complex. We studied the individual RRBs rather than the two factors that capture their phenotypic correlations because they are likely to be less complex and because significant phenotypic correlations do not necessarily reflect the presence of genotypic correlations.
We began by screening the RRBs for those most likely to be heritable and have adequate variability. Although ASD heritability is 50-60%, RRB heritabilities are not limited by this value. Significant heritability is an essential feature of a genetic trait, and we make the most precise estimates by comparing the trait correlations or concordance rates of MZ and DZ twin pairs. Since the AGRE sample does not contain an adequate number of MZ and DZ pairs, we use the approximately 1500 phenotyped sibling pairs in this sample to estimate RRB familialities with their Spearman correlations. Because the sample is composed of siblings, these estimates reflect both genetic and environmental contributions, making significant familiality necessary, but not sufficient, for significant heritability. An upper bound of heritability is twice the correlation. We also screened the RRBs for sufficient variability in order to achieve adequate statistical power for the GWAS. We dichotomized the response for each item using the criterion that each of the two groups contain between 40 and 60% of the individuals. We reasoned that the RRBs are continuous traits that are coded as ordinal in the ADI-R and could be re-coded as binary without compromising their genetic interpretation.
Ethnicities were restricted to only those individuals who are Caucasian, because those of Asian and African descent currently comprise only 15 and 10 percent of the AGRE and AGP samples, respectively. Samples from different ethnicities are very likely to introduce allelic and locus heterogeneity, a concern not addressed adequately using standard GWAS approaches. We reasoned that the potential increase in power achieved by including small samples from other ethnic groups would be compromised by the likely introduction of locus and allelic heterogeneity.
We chose a statistically powerful variance components approach to test RRB association in the children with ASD. Although the AGRE and AGP samples include the parents of the ASD affected children, transmission disequilibrium tests that assess allelic transmission to affected offspring, are much less powerful, as reported by Eu-Ahsunthornwattana and colleagues (27). Their power analyses indicate that, in pedigrees, the FBAT family based association test has 2% percent of the power consistently exhibited by the variance component approaches, using a standard p-value of 5×10-8. We analyzed seven familial and variable RRBs that and conducted the GWAS in the AGRE and AGP samples separately. Although this design required us to apply a very stringent level of significance to account for multiple testing, possibly decreasing our statistical power, we hypothesized that the selected study samples and filtered traits would compensate for this stringent criterion.
Given the recent development of molecular technologies and bioinformatics databases, we reasoned that it is possible, straightforward and compelling to use these resources to interpret any SNP/trait associations resulting from a GWAS. We were fortunate to find significant SNP associations, and extended these to identify the genes most likely to be interacting with them. This was achieved using an analytic approach to detect the most ‘credible’ SNPs in the associated chromosome region, followed by the perusal of a database designed to detecting SNP/gene interactions using chromosome interaction studies of fetal brains. Prioritization of the target genes derived from studies of 1) gene expression in RRB relevant brain regions during development, 2) SNP associations with RRB relevant brain regions, and 3) expression quantitative trait locus (eQTL) studies in fetal brains.
Methods
Overview of the Study
This study was designed to identify genes predisposing to RRBs in individuals with ASD by analyzing GWAS from the well-characterized AGRE (10) and AGP (28) samples. We conducted GWAS in the uncombined study samples of Caucasian AGRE and AGP families testing the individual familial and variable ADI-R RRBs for association with genotyped and imputed common SNPs. We present descriptions of the AGRE and AGP samples, SNP genotyping, SNP imputation and selection of Caucasian families in Supplementary Methods. SNPs in the associated chromosome region was tested to identify the ‘credible SNPs’ and these were queried in our database of Hi-C fetal brain SNP-gene interactions (29) to identify candidate risk genes. These genes were then prioritized using bioinformatics databases reporting gene expression in developing brains, brain region volume associations with SNPs, and eQTL studies of fetal brains.
Identifying Familial and Variable RRBs
The ADI-R is an interview interrogating caregivers of potential ASD cases regarding the defining features of ASD and providing the information used to make an ASD diagnosis in the AGRE and AGP samples. Data from these interviews are stored in the AGRE and AGP databases, and items 67-78 measure the degrees of severity of the RRBs. Responses are divided into three ordinal categories, ranging from not exhibiting the RRB to the RRB being extremely disruptive. It also asks whether the individual currently has the trait, or has ever exhibited the trait, and since we wished to capture any manifestations of each RRB, we scored the RRBs according to the degree of severity in the category of ‘ever’. We estimated RRB familiality with Spearman correlations in the affected siblings. We tested the twelve ADI-R RRBs for non-zero Spearman sibling correlations with a 0.05/12 significance threshold, which is approximately p < 0.004, and omitted the RRBs with non-significant correlations. Seven RRBs queried by ADI-R questions 68 - 72, 76 and 78, met these criteria. To screen for adequate variability, the dichotomy was set between 0 and the more severe categories of 1,2 and 3, however ror item 71, those receiving 0 and 1 were in the first category, and for item 69, those receiving a score of 0,1, or 2 were in the first category. We summarize these significantly familial and adequately variable RRBs in Table 1.
Table 1. Familial and Variable RRBs.
| RRB (ADI-R Item Number) | Number of Sibling Pairs | Sibling Correlation, Maximum Heritability, (p-value) | % of Individuals in Category 1 |
|---|---|---|---|
| Circumscribed interests (68) | 1287 | 0.29, .58 (<.0001) | 54 |
| Repetitive use of objects or interest in parts of objects (69) | 1382 | 0.13, .36 (<.0001) | 40 |
| Compulsions/rituals (70) | 1412 | 0.15, .30 (<.0001) | 45 |
| Unusual sensory interests (71) | 1414 | 0.10, .20 (0.0001) | 49 |
| Undue general sensitivity to noise (72) | 1361 | 0.14, .28 (<.0001) | 56 |
| Unusual attachments to objects (76) | 1196 | 0.15, .30 (<.0001) | 40 |
| Other complex mannerisms or stereotyped body movements (78) | 1413 | 0.16, .32 (<.0001) | 58 |
GWAS to Identify SNPs Associated with RRBs
The GWAS were conducted separately in the AGRE (1 996 ASD affected children from 1 053 families) and AGP (1 108 ASD affected children from 1 092 families) samples for the seven RRBs. To increase genome-wide SNP coverage, imputation was conducted, providing 5 755 879 AGRE and 3 972 813 AGP SNPs. We conducted the association analyses using a linear mixed model approach that captures structure within the data that violates the assumption of independence. That is, the assumption of standard case/control GWAS using Fisher's Exact Test or a logistic regression analysis is that all individuals in the study are unrelated and therefore independent. Data from multiplex families analyzed here introduce a clear violation of this assumption. We selected the Efficient Mixed-Model Association eXpedited (EMMAX) (30) software for association testing. Similar to other linear mixed model software, EMMAX accounts for the structure within the study sample by modeling that structure using the variance components term in the linear mixed model. EMMAX estimates the relatedness matrix empirically using the SNP data. By assuming that the phenotype under analysis is polygenic, with a large number of alleles each having a small effect, the computational algorithm avoids time-consuming repetitive variance components estimation procedures. EMMAX association tests the SNP genotypes with either quantitative or binary phenotypes. Type 1 statistical errors are as expected, and power is consistent with other linear mixed model approaches. We set a conservative level of significance of 5×10-8/7, or 7.14×10-9, to account for the seven familial traits tested for association in the two independent samples.
Interpreting the Associated SNPs
We analyzed SNPs in the associated chromosome region to find interacting genes regulated by that locus and evaluated these SNPs and genes using data on gene expression in relevant brain structures at relevant periods of development, SNP associations with relevant brain structures volumes, and SNP/gene eQTL in fetal brains. First, the CAusal Variants Identification in Associated Regions (CAVIAR) software (31, 32) was applied to select a minimal set of putative causal SNPs (credible SNPs) in the associated chromosome region. By analyzing the association p-values and the linkage disequilibrium estimates among the tested SNPs at the associated locus and jointly modeling and conditioning on these p-values, the software generated a set of variants that includes all of those classified as causal with 95% probability. CAVIAR identified five credible SNPs for RRBs. These SNPs were classified as nonsense, missense, or splice site variants or residing in the gene promoter, and assigned to their target genes. For the remaining un-annotated SNPs, chromatin contact matrices from mid-gestation human developing brains were leveraged to find genes that physically interact with them and are predicted regulatory targets (29). We describe this SNP/gene interaction database in Supplementary Methods.
We prioritized the ‘credible’ SNPs and their target genes using data from three sources. They are: 1) the Enhancing NeuroImaging Genetics Through Meta-Analysis (ENIGMA) database (30, 33) that associates SNPs and brain volumes, 2) the Allen Brain Atlas (36-38), which assesses gene expression levels in brain regions at key stages during fetal and childhood development, and 3) a database of expression quantitative trait loci (eQTL) in fetal brain tissue that is currently under development. The latter is based on gene expression and genotyped and imputed SNP data from 200 abortuses.
Results
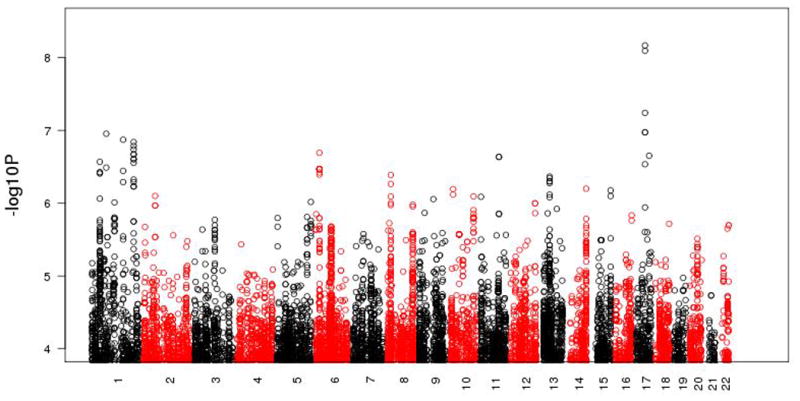
We filtered 12 ADI-R RRBs for those with significant familiality and adequate variability. Table 1 presents the sibling correlation, upper bound of heritability and the degree of variability for the seven RRBs passing this filter. To illustrate, in the first line of Table 1, 1 287 sibling pairs have data assessing circumscribed interests, as queried by item 68 on the ADI-R. The significant sibling correlation of 0.29 provides a maximum heritability of 0.58, and the measure of variability where 54% of individuals fall in to the first category of the dichotomy. As reported in Table 2, these familial RRBs exhibit only moderate pairwise correlations, thus providing support for conducting GWAS separately on the individual RRBs. We conducted GWAS using a stringent Bonferroni correction for multiple testing and identified one locus on 17q21.33. ADI-R item 69, querying the degree of ‘repetitive use of objects or interest in parts of objects,’ is significantly associated with two SNPs in this region in the AGP sample. One genotyped SNP, rs2898883, meets the criterion for association (p < 6.79×10-9 with an empirical p < 4×10-9), and a second genotyped SNP in the region, rs7502499, provides additional support (p < 5.73×10-8 with an empirical p < 3.1×10-8). Between them, an imputed SNP, rs2233667, meets the criterion for significant association (p < 8.02×10-9 with an empirical p < 4×10-9). These SNPs are in moderate pairwise linkage disequilibrium and are located within the sixth intron of the Prohibitin gene (PHB).
Table 2. Correlations Among Familial and Variable RRB ADI-R Questions in the AGRE Sample.
| 68 | 69 | 70 | 71 | 72 | 76 | 78 | |
|---|---|---|---|---|---|---|---|
| 68 | 1.00 | 0.07 | 0.16 | 0.02 | 0.14 | 0.12 | 0.02 |
| 69 | 1.00 | 0.19 | 0.43 | 0.16 | 0.14 | 0.27 | |
| 70 | 1.00 | 0.15 | 0.14 | 0.16 | 0.12 | ||
| 71 | 1.00 | 0.13 | 0.13 | 0.27 | |||
| 72 | 1.00 | 0.09 | 0.15 | ||||
| 76 | 1.00 | 0.09 | |||||
| 78 | 1.00 |
In the AGRE sample, there are no SNP/trait association tests meeting the 7.14×10-9 criterion for significance. The p-values for the three SNPs that have a significant association with ‘repetitive use of objects or interest in parts of objects’ in the AGP sample range between 0.49 and 0.51 for that RRB in AGRE. Meta-analyses result in p-values of 2.0 × 10-8 for rs2898883, 2.0 × 10-8 for rs2233667, and 6.7 × 10-7 for rs7502499, as reported in Supplementary Table 1. In early studies of the AGRE families, the 17q21 chromosome region was linked to ASD in a much smaller AGRE sample where all affected children were male, (34). That finding was replicated in an independent set of ‘male only’ AGRE families (35). Association analyses of the three significant SNPs at 17q21.33 and ADI-R item 69 in the families where all affected children are male in this current expanded AGRE sample results in p-values ranging between .78 and .92
As with almost all complex traits, none of the significantly associated SNPs is located in a protein-coding region of the genome. Identifying genes interacting with such SNPs has been a challenge, however, the availability of software and a database allowed us to address this. The CAVIAR software identified a set of ‘credible’ SNPs that has a greater than 95% likelihood of containing the causal SNPs. This analysis added two SNPs (chr17_47494782_D, p < 1.06×10-7 and rs71379361, p < 1.06×10-7) to the three identified by the GWAS. These five credible SNPs have minor allele frequencies ranging between 0.16 and 0.33 in Caucasians, indicating they are very common. Among the five, one (rs7502499) is in the PHB promoter. PHB encodes a mitochondrial scaffolding protein (36), and its role as a key regulator of neuronal survival is emerging (37, 38).
The other four credible SNPs are in non-annotated non-coding regions. We therefore used the SNP/gene fetal brain interaction database to identify them. Hi-C interactions in the germinal zone, GZ, indicate that a genomic region that is 10kb in size containing two ‘credible’ SNPs (rs2898883 and rs2233667) interacts with Solute Carrier Family 35 Member B1 (SLC35B1), a gene that encodes a nucleotide sugar transporter whose function in neurodevelopment is unknown. Figure 2a, where rs2898883 shows a significant interaction with the 10kb bin containing SLC35B1, at a false discovery rate (FDR) < .01), illustrates this interaction. The bin that containing the other two credible SNPs (chr17_47494782_D and rs71379361) interacts with Calcium Binding and Coiled-Coil Domain 2 (CALCOCO2) in CP and Distal-Less Homeobox 3 (DLX3) in GZ, as illustrated in Figure 2b. CALCOCO2 encodes an autophagy receptor and is involved in the clearance of phosphorylated tau in Alzheimer's disease (39), while DLX3 encodes a homeodomain transcription factor. Notably, although the function of DLX3 in brain development remains to be studied, Distal-Less Homeobox 5 (DLX5) and Distal-Less Homeobox 6 (DLX6) are implicated in the development of GABAergic interneurons (40). Loss of function mutations in SLC35B1 and CALCOCO2 have been identified in ASD probands (41), while there are no published reports of DLX3 and PHB being associated with ASD or RRBs.
Figure 2.
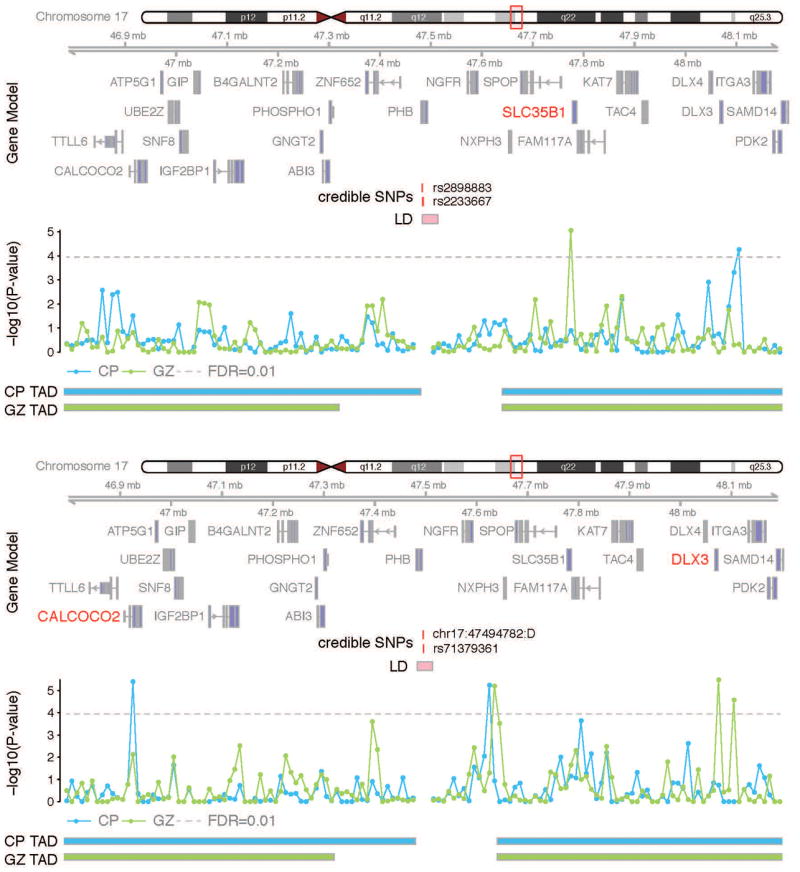
Representative interaction map for the ‘credible’ SNPs rs2898883 and rs2233667 (top) and chr17: 47494782: D and rs71379361 (bottom). Chromosome ideogram and genomic axis are across the top. Gene Model and possible target genes are in red. Horizontal axis is credible SNPs and their genomic coordinates. Genomic coordinates for credible SNPs and linkage disequilibrium (LD) region for the index SNP are indicated. Vertical axis is -log10(P-value) for the significance of the interaction between credible SNPs and each 10kb bin. Grey dotted line is for FDR=0.01. TAD is the topologically associating domain borders in CP and GZ.
Since ASD is a neurodevelopmental disorder often diagnosed before the age of three, and previous studies have demonstrated that ASD risk genes are expressed primarily during fetal development, genes with a prenatal-to-early-postnatal expression trajectory are excellent candidates, particularly within brain regions that are consistent with the development of RRBs. Thus, we examined gene expression trajectories over periods of development in different brain regions using the Allen Brain Atlas (42). Expression trajectories for the genes we identified are illustrated in Figure 3. SLC35B1 shows a very distinct regional expression pattern in the mediodorsal thalamic nucleus across development. CALCOCO2 displays a developmental stage-specific expression trajectory at mid-childhood, between ages 6 and 12 years. PHB shows a global expression pattern throughout cortical and subcortical regions except amygdala and cerebellum in childhood through adolescence (between ages 6 and 20 years) and a developmental expression trajectory in the mediodorsal thalamic nucleus. DLX3 expression was not detected in the brain (42).
Figure 3.
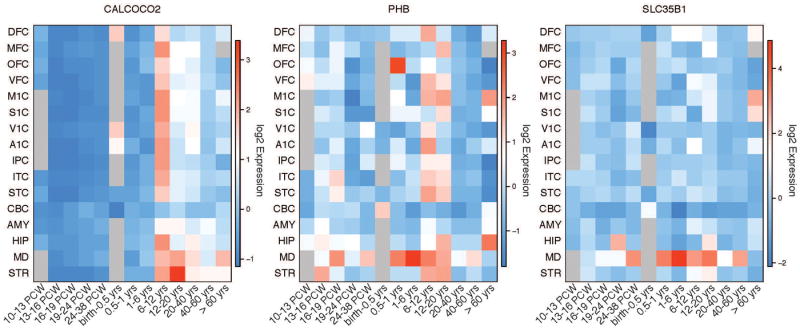
Spatiotemporal maps depicting the expression trajectories of candidate genes. Expression is given across multiple brain regions (vertical axis) and developmental stages (horizontal axis) spanning from the prenatal to postnatal adult stage. Cortical regions include: DFC, dorsolateral prefrontal cortex; MFC, medial prefrontal cortex; OFC, orbital prefrontal cortex; VFC, ventrolateral prefrontal cortex; M1C, primary motor cortex; S1C, primary somatosensory cortex; V1C, primary visual cortex; A1C, primary auditory cortex; IPC, posterior inferior parietal cortex; ITC, inferior temporal cortex; STC, superior temporal cortex. Subcortical regions include CBC, cerebellum; AMY, amygdala; HIP, hippocampus; MD, mediodorsal nucleus of the thalamus; STR, striatum.
Most notably, both SLC35B1 and PHB are highly expressed in the thalamus, which receives the input from the basal ganglia and its frontal cortical circuitry, brain regions implicated in motor activity and repetitive behaviors (20, 43). Since alterations in thalamic volume have been observed in ASD patients (48-50), we evaluated whether the credible SNPs that interact with SLC35B1 (rs2898883 and rs2233667) or are located in the PHB promoter (rs7502499) are associated with the thalamic volume (33, 44) using the ENIGMA database. Notably, rs2898883 (p < 0.008) and rs7502499 (p < 0.01) are significantly associated with thalamic volume. Moreover, another SNP in strong linkage disequilibrium with rs2898883 (r2=0.75), rs4987082, is also associated with thalamic volume (p < 0.0005) and also shows a suggestive association with RRBs (p < 1.14×10-6), providing additional support for the credible SNPs and their interacting genes in the etiology of RRBs in those with ASD.
We also examined our developing database of eQTL in fetal brains and were very encouraged to find that credible SNPs rs2898883 (p < 0.009) and rs2233667 (p < 0.01) are significant eQTL for their target gene SLC35B1. Table 3 presents the ‘credible’ associated SNPs, their interacting genes, and the evidence supporting these SNPs and genes from the Allen Brain Atlas, ENIGMA database and our prenatal eQTL database. We order the genes in this table according to their degree of prioritization, which is derived from known facts and supportive evidence. Clearly, SLC35B1, which exhibits an appropriate expression pattern in thalamus, and SNPs rs2898883 an rs233667, because both are its eQTL in fetal brains, with the latter also associated with thalamus volume, is the strongest.
Table 3. Credible SNPs for RRBs at 17q21, Candidate Target Genes and Spatiotemporal Expression pattern, Association with Subcortical Volume.
| Credible SNP (Candidate Gene eQTL) | Candidate Gene (Function) | Spatiotemporal Expression Pattern | SNP Association with Subcortical Volume |
|---|---|---|---|
| rs2898883 (P<0.009) | SLC35B1 (Hi-C) | Thalamus, prenatal to postnatal stages | Thalamus, P=0.008 |
| rs2233667 (P<0.01) | * | ||
|
| |||
| rs7502499 (P>.05) | PHB (Promoter) | Thalamus, prenatal to postnatal stages Pan brain, childhood to adolescence (6-20 years) | Thalamus, P=0.01 |
|
| |||
| rs71379361 (P>.05) | CALCOCO2 (Hi-C) | Pan brain, childhood (6-12 years) | Thalamus, P=0.01 |
| chr17_47494782_D (*) | DLX3 (Hi-C) | * | * |
data not available,
Discussion
ASD is a genetically complex disorder with etiologies that continue to be poorly understood. A recent report on its genetic architecture shows that common variation explains a substantial proportion of the risk (45); however, a decade of analyses reveals there may be as many as 1 000 predisposing genes. Our overarching hypothesis is that analyzing a single feature of ASD that reduces phenotypic heterogeneity will also reduce genetic heterogeneity, and our aim is to identify genome-wide significant RRB loci through genetic mapping, assigning their potential genes by leveraging chromatin interaction maps derived from the developing human brain.
In mice, RRBs derive from three etiologies: 1) targeted insults to the CNS, 2) pharmacological agents, and 3) restricted environments and experience (46). Although these studies do not implicate any genes directly, they suggest that those genes involved in developing and maintaining cortical-basal ganglia circuitry in the fetal and childhood brain may be critical to the development of RRBs. Thus, genes that are highly expressed in brain regions in the development and maintenance of cortical-basal ganglia circuitry may be central to the development of RRBs in ASD. Support for these brain regions is also derived from brain morphology studies of RRBs in children with ASD. These studies provide evidence that RRBs may be associated with decreased volumes of the basal ganglia and thalamus.
Phenotypic heterogeneity can be reduced in two ways. The first is to stratify individuals in the ASD study sample based on a selected single feature and conduct genetic analyses only in those exhibiting that feature. This approach is designed to capture ASD genes, as the ‘control’ sample will not have ASD. Although this approach reduces heterogeneity, it also can decrease the sample size, and is likely to reduce the statistical power to detect associated SNPs. However, sufficiently large genetic effects can compensate for the reduced sample size. The second approach, taken here, is to retain the full study sample and analyze an ASD related trait. Because the ‘cases’ and ‘controls’ both have ASD, analyses will capture variation of a single feature of ASD with possibly greater genetic homogeneity and larger effect sizes to identify genes contributing to that trait in those with ASD. The relative effect sizes of both approaches will depend upon the specific disorder and the trait under analysis, indicating that neither approach is better in all situations. In previous studies, we have used the second approach successfully to identify chromosome loci and genes for language delay, deficits in nonverbal communication, and deficits in social responsiveness in those with ASD. Here we examined the genetics of an additional core feature of ASD: RRBs.
Applying this approach, we conducted individual GWAS of seven familial and variable RRBs. Using a conservative Bonferroni-corrected p-value, we detected association in the AGP sample at 17q21.33, with the lead SNP located in the sixth intron of PHB for the RRB described in the ADI-R as ‘repetitive use of objects or interest in parts of objects.’ The association is not replicated in the AGRE sample, and a meta-analysis of the lead SNP and the RRB ‘repetitive use of objects or interest in parts of objects’ in the AGRE and AGP samples results in a p-value of 2.0 × 10-8, which does not meet the 8 × 10-9 stringent p-value that corrects for the seven GWAS conducted initially. However, it does meet the 5 × 10-8 criterion set for a single GWAS. Interpreting the discrepancy between these two samples is not straightforward, and it illustrates a quandary faced by other investigators who analyze these ASD cohorts. Because these samples are small when compared to those ascertained for other complex disorders such as Schizophrenia and Coronary Artery Disease, replication, while welcomed, has not yet become an expectation. Small samples are more vulnerable to the effects of stochastic differences in their composition, and combined with genotypic and phenotypic heterogeneity of the traits and the small effect sizes with polygenetic disorders, we interpret this lack of replication as a challenge for future studies in larger samples. We also interpret our functional associations, as summarized below, as providing additional credibility to this association.
Since the lead SNP in this analysis is not necessarily the causal variant, we obtained a set of five ‘credible’ causal variants, all of which reside in the non-coding region of the genome. Non-coding variants have been previously assigned to their nearest genes; however, interrogation of chromatin structure and eQTL studies have shown that this approach is often not correct. Here, we leveraged Hi-C data, which interrogates actual physical 3D chromatin contacts from the developing brain to functionally annotate common variants with their physically interacting genes, and detected three additional genes, each within 1 Mb of the ‘credible’ SNPs. The expression trajectories and imaging GWAS show that PHB and SLC35B1 are expressed in developing thalamus and influence thalamic volume in adults, prioritizing them among the four. SLC35B1 is further prioritized by our eQTL studies in fetal brains.
In conclusion, these analyses 1) identify four novel genes predisposing those with ASD to exhibit a behavior involving a ‘repetitive use of objects or interest in parts of objects,’ 2) prioritize two of these genes, PHB and SLC35B1, and 3) illustrate that a reduction in phenotypic heterogeneity is a powerful approach to reduce genetic heterogeneity.
Supplementary Material
Supplementary Table 1: Meta-Analysis of AGP Associated SNPs
Figure 1. Manhattan plot of 14 GWAS for 7 familial and variable RRBs as measured by the ADI-R in the AGRE and AGP Samples.
Acknowledgments
The Autism Genetic Resource Exchange (AGRE) database is available at Autism Speaks. Approved researchers can obtain the AGRE population dataset by applying at http://www.agre.org.
The Autism Genome Project (AGP) consortium collection and genotyping was supported by Autism Speaks. Genotype imputations were carried out on the Genetic Cluster Computer (http://www.geneticcluster.org) hosted by SURFsara and financially supported by the Netherlands Scientific Organization (NWO 480-05-003), along with a supplement from the Dutch Brain Foundation and the VU University Amsterdam.
This work was supported by Autism Center of Excellence grant MH100027 to DHG and the Glenn/AFAR Postdoctoral Fellowship Program (20145357) and Basic Science Research Program through the National Research Foundation of Korea (2013024227) to HW.
Footnotes
Conflicts of Interest Statement: The authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
References
- 1.Kanner L. Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- 2.Association AP, editor. DSM-5. Diagnostic and statistical manual of mental disorders. 5th ed. American Psychiatric Association; 2013. [Google Scholar]
- 3.Lord C, Rutter M, Couteur AL. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. Journal of Autism and Developmental Disorders. 1994;24(5):659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 4.CDC CfDCaP. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2010. Morbidity and Mortality Weekly Report. 2014;63(2):1–21. [PubMed] [Google Scholar]
- 5.Bailey AJ, Couteur AL, Gottesman I, Bolton P, Simonoff E, Yuzda E, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25(1):63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 6.Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68(11):1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantor RM. Molecular Genetics of Autism. Current Psychiatry Reports. 2009;11:137–142. doi: 10.1007/s11920-009-0021-1. [DOI] [PubMed] [Google Scholar]
- 8.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506(7487):185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantor RM. Autism Endophenotypes and Quantitative Trait Loci. In: Amaral D, Dawson G, Geschwind DH, editors. Autism Spectrum Disorders. Oxford University Press; New York: 2011. [Google Scholar]
- 10.Geschwind DH, Sowinski J, Lord C, Iversen P, Shestack J, Jones P, et al. The autism genetic resourse: a resource for the study of autism and related neuropsychiatric conditions. Human Genetics. 2001;69(2):463–466. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alarcon M, Cantor RM, Liu J, Gilliam TC, Geschwind DH. Evidence for a Language Quantitative Trait Locus on Chromosome 7q in Multiplex Autism Families. Am J Hum Genet. 2002;70(1):60–71. doi: 10.1086/338241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, et al. Linkage, Association, and Gene-Expression Analyses Identify CNTNAP2 as an Autism-Susceptibility Gene. Am J Hum Genet. 2008;82(1) doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen GK, Kono N, Geschwind DH, Cantor RM. Quantitative trait locus analysis of nonverbal communication in autism spectrum disorder. Molecular Psychiatry. 2006;11:214–220. doi: 10.1038/sj.mp.4001753. [DOI] [PubMed] [Google Scholar]
- 14.Lu A-H, Yoon J, Geschwind DH, Cantor RM. QTL replication and targeted association highlight the nerve growth factor gene for nonverbal communication deficits in autism spectrum disorders. Molecular Psychiatry. 2011;18(2):226–235. doi: 10.1038/mp.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowe JK, Werling DW, Constantino JN, Cantor RM, Geschwind DH. Quantitative linkage analysis to the autism endophenotype social responsiveness identifies genome-wide significant linkage to two regions on chromosome 8. Am J Psychiatry. 2014;172(3):266–275. doi: 10.1176/appi.ajp.2014.14050576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geschwind DH. Autism: many genes, common pathways? Cell. 2008;135(3):391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson EB, Pourcain BS, Anttila V, Kosmicki JA, Bulik-Sullivan B, Grove J, et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nature Genetics. 2015;48:552–555. doi: 10.1038/ng.3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shao Y, Cuccaro M, Hauser E, Raiford K, Menold M, Wolpert C, et al. Fine Mapping of Autistic Disorder to Chromosome 15q11-q13 by Use of Phenotypic Subtypes. Am J Hum Genet. 2003;72(3):539–548. doi: 10.1086/367846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brune CW, Kim SJ, Salt J, Leventhal BL, Lord C, Cook EH. 5-HTTLPR Genotype-Specific Phenotype in Children and Adolescents With Autism. The American Journal of Psychiatry. 2006;163(12):2148–2156. doi: 10.1176/ajp.2006.163.12.2148. [DOI] [PubMed] [Google Scholar]
- 20.Lewis M, Kim SJ. The pathophysiology of restricted repetitive behavior. J Neurodevelop Disord. 2009;1(2):114–132. doi: 10.1007/s11689-009-9019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuccaro ML, Shao Y, Grubber J, Slifer M, Wolpert CM, Donnelly SL, et al. Factor Analysis of Restricted and Repetitive Behaviors in Autism Using the Autism Diagnostic Interview-R. Child Psychiatry and Human Development. 2003;34(1):3–17. doi: 10.1023/a:1025321707947. [DOI] [PubMed] [Google Scholar]
- 22.Bishop SL, Hus V, Duncan A, Huerta M, Gotham K, Pickles A, et al. Subcategories of restricted and repetitive behaviors in children with autism spectrum disorders. Journal of Autism and Developmental Disorders. 2013;43(6):1287–1297. doi: 10.1007/s10803-012-1671-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cannon DS, Miller JS, Robison RJ, Villalobos ME, Wahmhoff NK, Allen-Brady K, et al. Genome-wide linkage analyses of two repetitive behavior phenotypes in Utah pedigrees with autism spectrum disorders. Molecular Autism. 2010;1(3) doi: 10.1186/2040-2392-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tao Y, Gao H, Ackerman B, Guo W, Saffen D, Shugart YY. Evidence for contribution of common genetic variants within chromosome 8p21.2-8p21.1 to restricted and repetitive behaviors in autism spectrum disorders. BMC Genomics. 2016;17 doi: 10.1186/s12864-016-2475-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou X, Stephens M. Genome-wide efficient mixed model analysis for association studies. Nat Genet. 2012;44(7):821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simons Simplex Colletion. 2011 [cited; Available from: https://sfari.org/resources/autism-cohorts/simons-simplex-collection.
- 27.Eu-ahsunthornwattana J, Miller EN, Fakiola M, Jeronimo SMB, Blackwell JM, Cordell HJ. Comparison of Methods to Account for Relatedness in Genome-Wide Association Studies with Family-Based Data. PLOS Genetics. 2014;10(7) doi: 10.1371/journal.pgen.1004445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu X-Q, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39(3):319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Won H, Torre-Ubieta Ldl, Stein JL, Parikshak NN, Huang J, Opland CK, et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. doi: 10.1038/nature19847. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang HM, Sul JH, Service SK, Zaitlen NA, Kong Sy, Freimer NB, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42(4):348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hormozdiari F, Kichaev G, Yang WY, Pasaniuc B, Eskin E. Identification of causal genes for complex traits. Bioinformatics. 2015;31(12):206–213. doi: 10.1093/bioinformatics/btv240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hormozdiari F, Kostem E, Kang EY, Pasaniuc B, Eskin E. CAVIAR: Causal Variants Identication in Associated Regions. 2014 doi: 10.1534/genetics.114.167908. [cited; Available from: http://genetics.cs.ucla.edu/caviar/ [DOI] [PMC free article] [PubMed]
- 33.Novak NM, Stein JL, Medland SE, Hibar DP, Thompson PM, Toga AW. EnigmaVis: online interactive visualization of genome-wide association studies of the Enhancing NeuroImaging Genetics through Meta-Analysis (ENIGMA) consortium. Twin Res Hum Genet. 2012;15(3):414–418. doi: 10.1017/thg.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stone JL, Merriman B, Cantor RM, Geschwind DH, Nelson SF. High density SNP association study of a major autism linkage region on chromosome 17. Human Molecular Genetics. 2007;16(6):704–715. doi: 10.1093/hmg/ddm015. [DOI] [PubMed] [Google Scholar]
- 35.Cantor RM, Kono N, Duvall JA, Alvarez-Retuerto A, Stone JL, Alarcon M, et al. Replication of Autism Linkage: Fine-Mapping Peak at 17q21. Human Genetics. 2005;76(6):1050–1056. doi: 10.1086/430278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, et al. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22(4):476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merkwirth C, Martinelli P, Korwitz A, Morbin M, Bronneke HS, Jordan SD, et al. Loss of Prohibitin Membrane Scaffolds Impairs Mitochondrial Architecture and Leads to Tau Hyperphosphorylation and Neurodegeneration. PLOS Genetics. 2012;8(11) doi: 10.1371/journal.pgen.1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou P, Qian L, D'Aurelio M, Cho S, Wang G, Manfredi G, et al. Prohibitin reduces mitochondrial free radical production and protects brain cells from different injury modalities. J Neurosci. 2012;32(2):583–592. doi: 10.1523/JNEUROSCI.2849-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GVW. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun. 2014;5 doi: 10.1038/ncomms4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Dye CA, Sohal V, Long JE, Estrada RC, Roztocil T, et al. Dlx5 and Dlx6 Regulate the Development of Parvalbumin-Expressing Cortical Interneurons. J neurosci. 2010;30(15):5334–5345. doi: 10.1523/JNEUROSCI.5963-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rubeis SD, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional, and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatiotemporal transcriptome of the human brain. Nature. 2011;478(7370):483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ting JT, Feng G. Neurobiology of obsessive-compulsive disorder: insights into neural circuitry dysfunction through mouse genetics. Curr Opin Neurobiol. 2011;21(6):842–848. doi: 10.1016/j.conb.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N, et al. Common genetic variants influence human subcortical brain structures. nature. 2015;520(7546):224–229. doi: 10.1038/nature14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46(8):881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lewis MH, Tanimura Y, Lee LW, Bodfish JW. Animal models of restricted repetitive behavior in autism. Behav Brain Res. 2006;176(1):66–74. doi: 10.1016/j.bbr.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Estes A, Shaw DW, Sparks BF, Friedman S, Giedd JN, Dawson G, et al. Basal ganglia morphometry and repetitive behavior in young children with autism spectrum disorder. Autism Res. 2011;4:212–220. doi: 10.1002/aur.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hollander E, Anagnostou E, Chaplin W, Esposito K, Haznedar MM, Licalzi E, et al. Striatal volume on magnetic resonance imaging and repetitive behaviors in autism. Biol Psychiatry. 2005;58:226–232. doi: 10.1016/j.biopsych.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 49.Whalen S, Truty RM, Pollard KS. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet. 2016;48:488–496. doi: 10.1038/ng.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Meta-Analysis of AGP Associated SNPs