

Abstract
Marked reductions in population size can trigger corresponding declines in genetic variation. Understanding the precise genetic consequences of such reductions, however, is often challenging due to the absence of robust pre- and post-reduction datasets. Here, we use heterochronous genomic data from samples obtained before and immediately after the 2011 eruption of the Puyehue-Cordón Caulle volcanic complex in Patagonia to explore the genetic impacts of this event on two parapatric species of rodents, the colonial tuco-tuco (Ctenomys sociabilis) and the Patagonian tuco-tuco (C. haigi). Previous analyses using microsatellites revealed no post-eruption changes in genetic variation in C. haigi, but an unexpected increase in variation in C. sociabilis. To explore this outcome further, we used targeted gene capture to sequence over 2,000 putatively neutral regions for both species. Our data revealed that, contrary to the microsatellite analyses, the eruption was associated with a small but significant decrease in genetic variation in both species. We suggest that genome-level analyses provide greater power than traditional molecular markers to detect the genetic consequences of population size changes, particularly changes that are recent, short-term, or modest in size. Consequently, genomic analyses promise to generate important new insights into the effects of specific environmental events on demography and genetic variation.
Introduction
Significant reductions in population size (i.e. population bottlenecks1) can substantially impact patterns of genetic variation and thus potentially alter the evolutionary trajectories of affected organisms. Accordingly, the genetic consequences of bottlenecks have been the focus of numerous empirical and theoretical studies (e.g.1–6). Such consequences can be difficult to assess, however, particularly when reductions in population size are modest in magnitude or duration. In part, this difficulty reflects the frequent use of traditional molecular markers such as microsatellites or mitochondrial loci, both of which may have limited power to detect changes in genetic variation over small spatial or temporal scales2,3. In contrast, examination of much larger numbers of markers drawn from across the genome should increase the ability to detect temporal and spatial differences in genetic variation4,5.
Although use of genomic data to address evolutionary questions is increasing rapidly, only a limited number of studies have employed such information to study the genetic consequences of naturally occurring demographic bottlenecks. Instead, most analyses that have used genomic data to assess the impacts of reductions in population size have focused on demographic changes associated with agricultural activities such as domestication of plants and animals3,6–8. The relevance of these analyses to wild populations is unclear, as domesticated species have typically been subject to strong artificial selection that may confound signals of other evolutionary forces such as drift, mutation, migration, and gene flow3. Further, these studies have typically relied on samples taken at only a single point in time, often many generations after domestication, which may alter the ability to detect changes in genetic variation relative to studies based on samples collected shortly before and after a demographic event9,10. Thus, while genomic analyses appear to offer considerable potential, their suitability for studies of naturally occurring changes in genetic variation – particularly those occurring over relatively short time periods – remains largely unexplored.
To evaluate the immediate genetic consequences of abrupt, naturally occurring reductions in population size, we generated a genome-wide panel of single-nucleotide polymorphisms for two species of ctenomyid rodents from the Limay Valley region of southwestern Argentina. The colonial tuco-tuco (Ctenomys sociabilis) and the parapatric Patagonian tuco-tuco (C. haigi) have been the subjects of extensive behavioral, ecological, demographic, and genetic research, including comparative analyses of genetic variation over multiple spatial and temporal scales11–15. In 2011, both species were significantly affected by the eruption of the Puyehue-Cordón Caulle volcanic complex in southern Chile, resulting in deceases in population density of ~40% for C. sociabilis and ~25% for C. haigi 16 and providing a rare opportunity to compare directly levels of genetic variation before and after a natural population decline. Notably, previous analyses based on microsatellite markers16 suggested that this event was associated with a significant post-eruption increase in genetic variation in C. sociabilis but not in C. haigi.
To explore this unexpected outcome in greater detail, we employed targeted sequence capture to generate a large number of genome-wide markers for each study species. Specifically, we sought to determine if the expected greater resolution of these genomic analyses would reveal the same post-eruption increase in genetic variation in C. sociabilis. Additionally, we sought to identify potential signatures of demographic processes contributing to post-eruption changes in genetic variation in our study species. While multiple studies have explored post-bottleneck changes in demography over longer time scales (e.g.10,17–21), the more immediate impacts of these demographic parameters are not well characterized. As a result, our analyses of the effects of the Puyehue-Cordón Caulle eruption should generate important new insights into the role of demographic processes in shaping short-term genomic responses to pronounced reductions in population size.
Materials and Methods
Study system
The two species of tuco-tucos (genus Ctenomys) studied have been the subjects of long-term field research on behavior, ecology, and demography. The colonial tuco-tuco (Ctenomys sociabilis) is endemic to an approximately 1000 square kilometer area in the western Limay Valley and adjacent hills of Neuquén Province, Argentina (Fig. 1;22,23). In contrast, the Patagonian tuco-tuco (C. haigi) is much more widely distributed in the eastern Limay Valley and surrounding regions of Río Negro and Neuquén Provinces, Argentina (Fig. 1;22). A focal study population for each species was established in 1992 on Estancia Rincon Grande (C. sociabilis) and Estancia San Ramon (C. haigi); these study sites are located a few hundred meters from one another, on opposite sides of the Rio Limay24.
Figure 1.

Map of study area in southwestern Argentina. The general cone of ash fall resulting from the 2011 eruption is indicated with the dashed lines. The geographic distribution of C. sociabilis is indicated in dark gray. Figure modified with permission from Hsu et al.16.
Despite their close geographical proximity, the two species differ markedly with respect to social structure, demography, and genetic variation. Ctenomys sociabilis is group living, with burrow systems routinely shared by multiple adults25. This species has been characterized by low mitochondrial genetic diversity over the past several millennia14, including an abrupt decrease in genetic variation approximately 3,000 years ago concordant with an eruption of the Puyehue-Cordón Caulle volcanic complex26. In contrast, C. haigi is solitary, with each adult inhabiting its own burrow system28; this species has historically been characterized by higher levels of mitochondrial genetic diversity than C. sociabilis 14. Given these differences in genetic variation and the extensive pre-eruption data sets for these species, comparative studies of C. sociabilis and C. haigi should offer important insights to the immediate genomic impacts of a marked reduction in population size.
Sample collection
Pre-eruption tissue samples were collected during December 2001; post-eruption samples were collected during December 2011 and again in December 2012-January 2013. In all cases, live capture of individuals, collection of non-destructive tissue samples, and release of individuals were performed following the methods of Lacey et al.11,27,28. All field procedures were approved by the UC Berkeley Animal Care and Use Committee and followed the guidelines established by the American Society of Mammologists for the use of wild mammals in research29.
Tissue samples from 57 C. sociabilis were analyzed. Seventeen of these were pre-eruption samples from the focal population at Rincon Grande while 31 were collected post-eruption (December 2011) from the same population. No individual was included more than once in our analyses of genetic variation. To minimize potential kinship among the individuals examined, the pre-eruption samples were chosen to represent 13 different burrow systems, the majority of which (76.5%) were sampled only once. In contrast, the 31 post-eruption samples were from 9 burrows; of these, 6 of these burrows had multiple residents included in our post-eruption dataset. To add spatial perspective to our analyses, post-eruption samples were also obtained from 9 additional individuals from a second population of C. sociabilis at Estancia La Lonja, located about 3 km to the northwest of the focal Rincon Grande population. The La Lonja population had not been sampled prior to the eruption. Due to logistic challenges imposed by the substantive ash fall in the region, this was the only non-focal population of C. sociabilis that we were able to access following the eruption. Sampling of the focal study population of C. haigi at San Ramon consisted of 12 pre-eruption and 17 post-eruption (December 2012–January 2013) samples, with no material obtained from additional populations of this species.
DNA extraction
DNA was extracted from all tissue samples using the Qiagen DNEasy Blood and Tissue extraction kits. Extractions were performed immediately prior to the preparation of genomic libraries (see below) to minimize the possibility of sample contamination. Following extraction, DNA sample concentrations were measured using a Qubit fluorometer (Qubit dsDNA HS assay, Life Technologies) and Nanodrop (ThermoFisher). Samples with low yields were re-extracted using a ZR Genomic DNA Tissue Miniprep kit (Zymo); this process, followed by ethanol precipitation, increased the concentrations of DNA in these extracts.
Genomic library preparation
DNA libraries for targeted gene capture were prepared following the dual index protocol of Kircher et al.30, as modified by Crawford31. In brief, 580 ng of DNA from each individual was sheared using a BioRuptor Sonicator (Diagenode), after which a small quantity of the resulting fragmented DNA was visualized on a 1.2% agarose gel to verify fragment size distribution. Sonication was performed for four rounds at 90-second intervals to optimize fragment sizes of 450 bp. We then performed blunt-end repair using T4 DNA polymerase as per Meyer and Kircher32, after which reactions were cleaned using Ampure XP beads. We then ligated P5 and P7 adapters to each fragment and performed adapter fill-in with Bst polymerase. Following this, we measured DNA concentration using a Nanodrop and then conducted two rounds of indexing PCR reactions (11 cycles each) using a Phusion High Fidelity kit and standard Illumina indices. Every sample was then divided into two pools, each of which was indexed separately with a unique combination of P5 and P7 indices in order to reduce errors during sequencing30. We used experimental assays of indexing PCRs with different numbers of cycles, followed by analysis of the resulting fragment length distribution with a Bioanalyzer assay, to determine the number of cycles per reaction that would minimize the number of PCR duplicates and thus maximize read mapping33. Once indexing PCRs were complete, we quantified DNA concentrations again using Nanodrop, and then performed automated size selection (Pippen Prep, Sage Science) to isolate DNA fragments ranging in size from 580 to 610 bp. Fifty nanograms of each size-selected library were then aggregated in to a single pool in preparation for the targeted gene capture.
Targeted gene capture and sequencing
Custom in-solution probes were designed using the NimbleGen SeqCap EZ Target Enrichment System; the details of this procedure are given in Crawford (2016)31. We identified 2,027 target segments of nuclear DNA based on analyses of C. sociabilis and C. haigi transcriptomes31. Targeted regions were approximately 1,200 bp long and putatively neutral. Capture of these target regions was completed using the NimbleGen SeqCap protocol for hybridization, with several modifications. In brief, we performed an in-solution hybridization of our pooled libraries with the custom probes. After incubation at 47 °C for 70 hours, we washed and recovered the captured multiplex DNA sample using M-270 Streptavidin Dynabeads. We then amplified the captured DNA via ligator-mediated PCR using the HiFi HotStart ReadyMix (KAPA Biosystems). We performed three parallel rounds of ligator-mediated PCR, checking the resulting DNA concentrations after each PCR reaction (using Qubit) and adjusting the number of cycles in subsequent reactions in order to reduce PCR stochastic drift34. The first PCR reaction was performed for 13 cycles, while the second and third PCR reactions were run for 14 cycles. Amplification products were then pooled in equimolar amounts, cleaned with Ampure XP beads, and subjected to Bioanalyzer analysis to gauge the concentration and distribution of product sizes. Finally, to assess the success of hybridization, we performed a series of quantitative PCR reactions using five different primer pairs: three primer pairs had been designed to amplify regions targeted by our probes (MATR F/R: 5′TCCTAGTCTCAACCCAGTGCT 3′/5′GTTATGCGAGGTCTCACCAA 3′; MR1 F/R: 5′AATGTGGCTCTCATCACCAA 3′/5′AATCTCTTGAGCCAGGCAAT3′; RNF130 F/R: 5′TGGATTGCCTTGCTACAGAG 3′/5′TTGTGACTGGCTCCTCTTTG3′), while the remaining two primer pairs had been designed to amplify “control” regions not targeted by these probes (Mat2A F/R: 5′TTGTGGATACTTATGGCGGTT3′/5′AAGAACCCTCCTGCACAGAC3′; CS1 F/R: 5′CCTGGCAAGTGTACTTCCGT/5′ GTCAGCAGGGTCAATCCAGT 3′). These qPCR reactions allowed us to quantify the rate of enrichment for target sequences, thereby confirming that our capture procedure was successful prior to submitting samples for sequencing. Finally, samples were sequenced with 300 bp paired-end reads on an Illumina HiSeq. 2500 RAPID platform at the UC Berkeley QB3 core facility; our samples were pooled with those from other projects for a total of 240 samples run across three sequencing lanes.
Sequence cleanup, assembly, and variant calling
We cleaned and verified the quality of the Illumina sequences obtained following the protocol of de Wit et al.35. Specifically, we used the FastX toolkit36 to trim and remove regions of sequence with quality scores below 20, after which we removed reads that were <20 bp in length. Quality distributions were visualized using FastQC37 and Galaxy38. We then merged all reads for the same individual using custom Bash and Python scripts, after which we used Bowtie239 to align these merged reads to the original transcriptome sequences used to design our bait capture probes. Following alignment, reads were processed with SAMtools40, which generated summary statistics for average sequence depth and cover for each individual. Based on these distributions, we used a custom Python script and SNPCleaner41 to further filter the sequences to ensure high confidence in our reads. Using these scripts, we allowed a maximum of one mismatched base per read and applied quality thresholds derived from those used by Bi et al.34 for working with both historical and modern DNA. Specifically, at the individual level, we eliminated any individuals with extremely low or high coverage (less than one-third or more than three times the average coverage across all individuals). We then filtered out reads below the 5th or above the 95th percentiles of coverage depth for all samples, which was approximately 3X and 100X, respectively. Finally, we removed sites with a root mean square mapping quality below 10. Single nucleotide polymorphisms (SNPs) in the remaining reads were identified using the software package ANGSD42.
Population genomics analyses
We generated multiple summary statistics to assess post-eruption changes in genetic variation. Prior to these analyses, we used LOSITAN43 to identify and remove any loci that appeared to be under directional selection. This program, which uses an FST-outlier method to identify signs of selection, was run for 50,000 simulations with a false discovery rate of 0.1. Additionally, we used a custom Python script to identify and remove any loci that were not in Hardy-Weinberg equilibrium. Collectively, these procedures ensured that the variation contained in our final data set reflected genome-wide consequences of demographic changes driven by neutral processes, rather than changes driven by directional selection.
Genetic differences among populations
To quantify genetic variation within populations, we used a custom Python script to calculate mean heterozygosity for each population sampled. To account for potential differences in heterozygosity resulting from the variable number of individuals sampled per population, we conducted a bootstrap analysis in which we subsampled an equal number of randomly selected individuals from each population; this process was repeated 100 times, after which we calculated mean heterozygosity for each population based on these subsamples. Similarly, to minimize the potential effects of loci with extremely high or low levels of heterozygosity, we randomly selected a subsample of 100 loci and then estimated heterozygosity based on this panel of SNPs; this process was repeated 100 times, after which mean heterozygosity was calculated across all iterations. To examine additional measures of genetic diversity, we plotted per-site distributions of Watterson’s theta (θW) and nucleotide diversity (π) for each population. Finally, to determine if estimates of genetic diversity were influenced by the demographic structure of the population, we partitioned the data by sex and calculated mean heterozygosity for randomly selected subsets of males and females. For C. sociabilis, we also calculated mean heterozygosity and inbreeding coefficients using data from only a single randomly selected individual per burrow system in both pre- and post-eruption populations.
Next, we examined patterns of genetic diversity among populations using ngsTools44 and custom Python and R scripts. First, we calculated pairwise FST values for all populations of conspecifics. Then, to assess differentiation among populations visually, we generated principal component analyses (PCA) plots for both species using a custom Python script. We also generated PCA plots in which data for each species were subdivided by 1) sex, 2) age (juvenile or adult), and 3) burrow system (for C. sociabilis) to determine if any of those factors influenced estimates of genetic differentiation among populations. As another measure of genetic differentiation among populations, we conducted admixture analyses using a range of values for K, the number of putative populations. We ran these analyses using the a priori hypotheses of K = 2 (accounting for the two spatially distinct populations) and K = 3 for C. sociabilis (accounting for the three populations sampled that were either temporally or spatially distinct), and K = 2 for C. haigi (accounting for the two populations sampled that were temporally distinct). We also explored outcomes for values up to K = 6 for both species to determine if a greater number of putative populations revealed unanticipated evidence of genetic differentiation.
Effects of number of loci examined
To explore the effects of the number of loci analyzed on estimates of genetic variation, we used a custom Python script to conduct a simulation in which we varied the number of loci used to characterize variation in the pre- and post-eruption populations of C. sociabilis from Rincon Grande, as well as the post-eruption population of this species from La Lonja. We randomly subsampled loci, starting with n = 50 and incrementing by 50 loci per iteration to a total of n = 1000. Following this, we then subsampled n = 1000 to n = 10,000 loci in 500 locus increments. In these simulations, subsampling was repeated 100 times for each value of n, with observed heterozygosity determined for each subsample. We then calculated the mean and associated standard deviation for estimates of heterozygosity obtained for each value of n.
Inferring signals of demographic history
To identify demographic processes that may have contributed to changes in pre- versus post-eruption patterns of genetic variation, we used custom Python scripts to generate folded site frequency spectra (SFS) for each species. SFS provide an overview of the distribution of allelic frequencies, which can reveal evidence of past demographic events such as reductions in population size45–47. Because differences in the numbers of individuals or alleles sampled can affect such distributions, we conducted SFS analyses using equal numbers of randomly selected pre- and post-eruption individuals; this subsampling was repeated 20 times for each time period, after which allele frequency distributions were averaged across iterations and pre- and post-eruption data sets were compared for each focal study population.
Because patterns of allele sharing among populations may also reflect demographic factors such as bottlenecks, we determined the number of private alleles – alleles unique to the populations in which they were detected – for each population sampled, as well as the number of alleles shared between two, but not all three, of the populations of C. sociabilis sampled. For both sets of analyses, we completed 100 iterations of random subsampling with equal numbers of individuals per population to control for any effects due to differences in the actual number of individuals genotyped per population. In addition, to control for any potential effects due to sampling of multiple individuals per burrow system in C. sociabilis, we conducted 100 iterations of subsampling in which only one randomly chosen individual per burrow system was included in each population. To confirm that these analyses were not biased by outlier loci, we also randomly subsampled 100 loci; we repeated this procedure 100 times, after which we assessed the mean occurrence of private and shared alleles across iterations.
To explore further potential signals of demographic change related to the volcanic eruption, we plotted distributions of Tajima’s D for each population sampled. This statistic, which can reflect both past demographic events and selection, was estimated across 100-bp sliding window regions using ngsTools; use of a distribution of values for Tajima’s D allows for more robust assessments of demographic history than a single mean estimate of this parameter48,49. We also generated pairwise mismatch distributions and their associated raggedness indices50 for each population using a custom Python script. The shapes of mismatch distributions can provide insights into recent demographic history, while raggedness indices can be used to compare such distributions quantitatively51. Finally, to determine if levels of inbreeding changed after the eruption, we calculated coefficients of inbreeding (F) for each individual using Plink52, after which we calculated mean values of f for each pre- and post-eruption population.
Demographic simulations
To explore the potential impacts of demographic processes on post-eruption genetic variation in our study populations, we conducted a series of simulations. First, to quantify the expected effects of drift resulting from post-eruption declines in population size, we simulated an approximately 50% reduction in each of our pre-eruption study populations; this level of decline was largely consistent with that observed for C. sociabilis immediately following the 2011 eruption16. Specifically, we randomly designated 50% of individuals in each pre-eruption sample as survivors, after which we calculated heterozygosity for both the entire pre-eruption population and for the “surviving” post-eruption population. This procedure was repeated 100 times to generate a mean and standard deviation for heterozygosity in the simulated populations. Although this procedure did not take into account other demographic processes that may have affected post-eruption genetic variation, it provides a general estimate of expected changes in post-eruption heterozygosity due solely to stochastic loss of individuals from a population.
To examine the potential impacts of other demographic processes on genetic variation, we used MIGRATE-N53 to examine post-eruption data from the Rincon Grande and La Lonja populations. This program allows the testing of different models of demographic history by generating Bayes Factors, derived from marginal likelihoods for different historical scenarios. We tested the following four hypotheses regarding post-eruption population structure at Rincon Grande and La Lonja: (1) panmixia (2) bi-directional movement (immigration and emigration) between the populations, (3) immigration only (La Lonja to Rincon Grande), and (4) emigration only (Rincon Grande to La Lonja). Although the power of our analyses may have been somewhat limited due to the assumption of constant population sizes and the failure to incorporate observed post-eruption demographic changes, tests of these hypotheses provide an important first step toward determining whether different patterns of migration may have shaped genetic variation in post-eruption populations of C. sociabilis.
Results
Our sequence alignment resulted in a mean per-individual depth of coverage of 11.46 for C. sociabilis and 11.41 for C. haigi, with greater than 99.9% cover per individual of targeted areas for both species. No individuals had less than one-third or greater than three times the mean coverage, indicating that all animals fell within the suggested thresholds for filtering samples with extremely low or high coverage34. After examining all SNPs for evidence of departures from Hardy-Weinberg equilibrium, we excluded two sites from analyses for C. sociabilis and six sites from analyses for C. haigi. Similarly, we excluded an additional 24 loci for C. sociabilis and 4 for C. haigi that were identified by LOSITAN as potentially being under directional selection. Thus, the final data set consisted of 531 SNPs for C. sociabilis and 449 SNPs for C. haigi. Despite the overall greater number of variant sites analyzed for C. sociabilis, the mean number of variant sites per individual for this species (N = 16.35) was less than that for C. haigi (N = 34.55).
Pre- and post-eruption genetic differentiation
Mean heterozygosity in both focal study populations was reduced following the 2011 eruption. In C. sociabilis, mean heterozygosity across all sites decreased from 0.00167 pre-eruption to 0.00130 post-eruption; for C. haigi, these values were 0.00310 and 0.00274, respectively. For both species, this difference in variation was significant (two sample Kolmogorov–Smirnov test; both p < 0.0001). The same outcome was obtained when comparing estimates of pre- and post-eruption heterozygosity for a randomly selected subsample of 100 loci per species (C. sociabilis: pre = 0.00164, post = 0.00131; C. haigi: pre = 0.00273, post = 0.00231; two sample Kolmogorov-Smirnov tests, both p < 0.0001), indicating that this outcome was not impacted by interspecific differences in the number of loci examined. This pattern of post-eruption reduction in genetic variation was also evident in the distributions of the per-site values of θW and π, both of which showed a shift toward lower values after the eruption (Supplemental Figs 1 and 2).
A reduction in post-eruption variation was also detected for C. sociabilis for subsamples based on one randomly chosen individual per burrow system (pre = 0.00169; post = 0.00124; two sample Kolmogorov-Smirnov test, p < 0.0001). In contrast, there was no apparent difference in heterozygosity between males and females in the post-eruption population of this species at Rincon Grande (males: 0.00130, females: 0.00129; two sample Kolmogorov-Smirnov test, p < 0.0001); this was the only population of C. sociabilis for which sample sizes were sufficient for comparisons of known-sex individuals. Among populations of C. sociabilis, post-eruption heterozygosity at La Lonja (0.00122) was significantly lower than either pre- or post-eruption heterozygosity at Rincon Grande (two sample Kolmogorov-Smirnov test, both p < 0.0001). Pre-eruption samples from La Lonja were not available for analysis and thus we could not determine if the lower post-eruption variation in this population reflects consistent differences between La Lonja and Rincon Grande or if the impacts of the 2011 eruption were greater at La Lonja.
The relatively modest reductions in heterozygosity detected for both study species were associated with limited evidence of genetic differentiation between the pre- and post-eruption samples from these animals. For example, FST values across time periods were low for both C. sociabilis (0.00348) and C. haigi (−0.00042), suggesting little temporal differentiation between pre- and post-eruption populations. Similarly, principal component analyses failed to reveal strong temporal differentiation in either study species (Supplemental Fig. 3). Further, admixture plots for C. sociabilis (Fig. 2A and B) provided no clear indication of temporal segregation between populations of this species; outcomes did not differ as the number of putative populations varied from K = 3 to K = 6. Similarly, admixture analyses revealed no evidence of temporal differentiation between pre- and post- eruption samples of C. haigi (Fig. 2C). Thus, although genetic variation was reduced in both study species, the 2011 eruption of Puyehue-Cordón Caulle complex did not appear to result in immediate temporal differentiation of the pre- and post-eruption populations.
Figure 2.

Admixture plots for C. sociabilis and C. haigi. For each panel, the populations and temporal periods (pre- versus post-eruption) examined are indicated. For C. sociabilis, the number of putative populations was set at (A) K = 2 and (B) K = 3. For C. haigi (C), analyses were run with K = 2.
Effects of number of loci examined
Varying the number of loci used to characterize genetic variation revealed that increasing the number of loci subsampled reduced the standard deviations around estimates of mean heterozygosity (Fig. 3). Indeed, there was a sharp decline in standard deviation as the number of loci examined increased from the initial value of 50 to approximately 1,000 loci, after which the impact of number of loci on estimates of standard deviation of mean heterozygosity appeared to stabilize.
Figure 3.

Relationship between the number of randomly subsampled loci (100 iterations each) and the standard deviation of mean heterozygosity. Data are shown for three C. sociabilis populations: (A) pre-eruption Rincon Grande, (B) post-eruption Rincon Grande, and (C) post-eruption La Lonja.
Signatures of demographic processes
Folded site frequency spectra (SFS; Supplemental Fig. 4) were similar in shape for pre- and post-eruption populations of both study species, providing no evidence of recent differences in demographic history between C. sociabilis and C. haigi. Subsampling of the data set to control for differences in the numbers of individuals and alleles examined resulted in a slight post-eruption increase in the proportion of low-frequency polymorphisms in each species, contrary to expectations following a reduction in population size. These changes, however, were not significant (two sample t-tests; p > 0.05 for both species), and thus SFS analyses failed to reveal evidence of post-eruption changes in demography.
Within the focal population of each study species, the number of private alleles differed significantly between pre- and post-eruption samples (two-sample T test; both p < 0.0001). In both species, post-eruption populations were characterized by fewer private alleles (Fig. 4), suggesting that one consequence of the reported reductions in population size was the loss of unique alleles. We also compared the distribution of alleles shared between any two (but not all three) populations of C. sociabilis. Of the three pairwise comparisons conducted (pre- and post-eruption Rincon Grande; pre-eruption Rincon Grande and post-eruption La Lonja; and post-eruption Rincon Grade to post-eruption La Lonja), the pre- and post-eruption Rincon Grande populations shared the greatest number of alleles, while the post-eruption Rincon Grande and post-eruption La Lonja populations shared the fewest alleles (Fig. 4C; two-sample T tests; p < 0.0001 for each pairwise comparison). The same general result was obtained for comparisons conducted using (1) a randomly selected subset of individuals per population, (2) a subsample consisting of only one randomly chosen individual per burrow system, and (3) a randomly selected subset of loci. These findings indicate that overall patterns of allele sharing among populations of C. sociabilis were not biased by differences in the number of animals or number of loci sampled per population.
Figure 4.
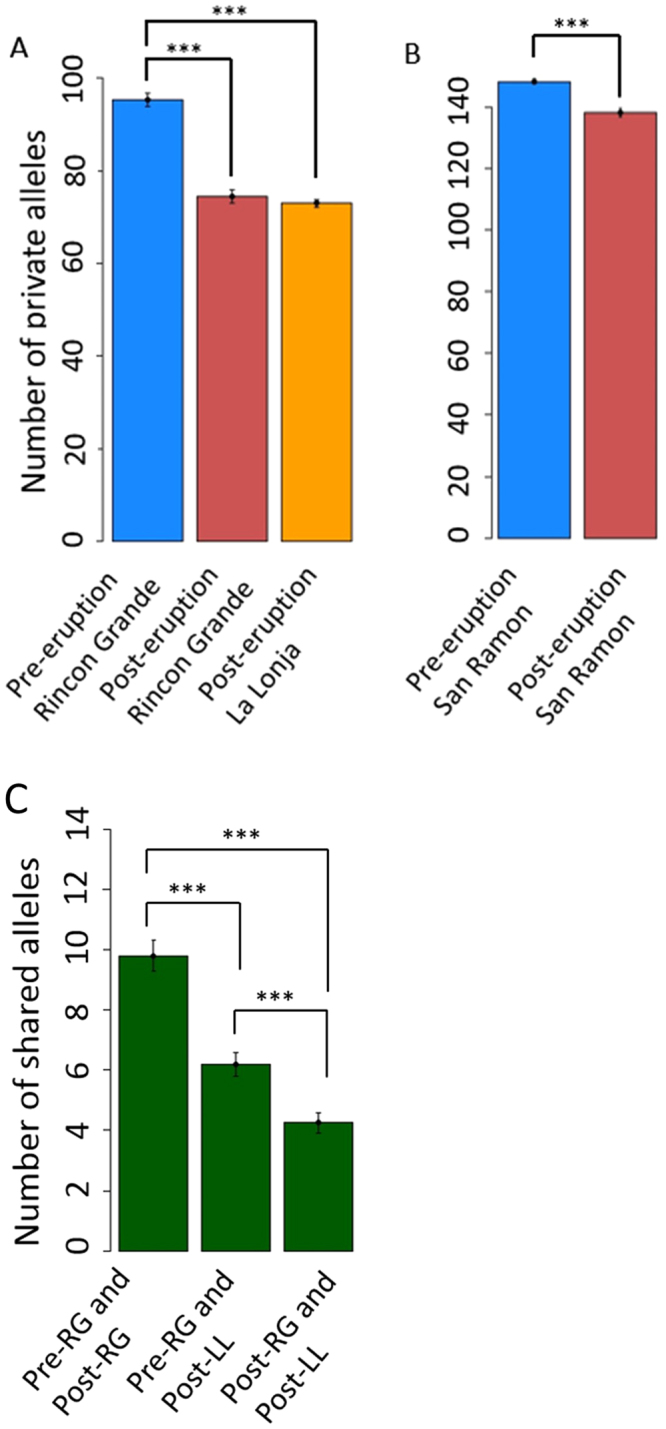
Comparisons of the mean (±SE) number of private alleles per population for (A) C. sociabilis and (B) C. haigi. For both species, values for pre- and post-eruption populations are indicated. In (C), the number of all alleles shared between two (but not three) populations is shown for all pairwise combinations of populations of C. sociabilis. Populations are abbreviated as following: pre-eruption Rincon Grande (Pre-RG), post-eruption Rincon Grande (Post-RG), and post-eruption La Lonja (Post-LL). ***Indicates significant contrasts (all p < 0.001).
Both pre- and post-eruption values of Tajima’s D (calculated across 100-bp sliding windows) were generally negative for each study species, indicating an excess of low frequency alleles. Contrary to expectation, modal values of D decreased following the eruption in both study species (mean change from pre- to post-eruption C. sociabilis: −0.429; C. haigi: −0.892; two sample Kolmogorov–Smirnov tests; both p < 0.0001; Fig. 5), suggesting that low frequency alleles were more abundant after the 2011 eruption.
Figure 5.

Smoothed kernel density distribution of values of Tajima’s D for (A) C. sociabilis and (B) C. haigi based on analyses of 100-bp sliding windows. Density denotes the relative frequency of individual values of Tajima’s D across the 100-bp sliding windows.
Inspection of mismatch distributions revealed that the pre-eruption focal populations of each study species were characterized by a single primary peak (Figs 6A and 7A), with Harpending’s raggedness index values of r = 0.0102 for C. sociabilis and r = 0.0156 for C. haigi. Post-eruption, mismatch distributions for these populations remained unimodal (Figs 6B and 7B) with raggedness indices of r = 0.0244 for C. sociabilis and r = 0.0230 for C. haigi. In contrast, the mismatch distribution for the post-eruption samples of C. sociabilis at La Lonja was more ragged, with an index value of r = 0.0887 (Fig. 6C). No significant change in mismatch distributions, however, was detected for either species when comparing the centered, re-scaled mismatch distributions from pre- and post-eruption populations (two sample Kolmogorov-Smirnov test; both p > 0.05). Collectively, these analyses provide no clear evidence of recent demographic changes in either study species.
Figure 6.

Pairwise mismatch distributions for C. sociabilis depicting the frequency of the number of sequence differences across all pairs of individuals in a given population: (A) pre-eruption Rincon Grande, (B) post-eruption Rincon Grande, and (C) post-eruption La Lonja.
Figure 7.

Pairwise mismatch distributions for C. haigi depicting the frequency of number of the number of sequence differences across all pairs of individuals in a given population: (A) pre-eruption San Ramon and (B) post-eruption San Ramon.
Finally, mean values for individual coefficients of inbreeding (F) in the focal population of C. sociabilis at Rincon Grande were greater after the eruption, even when the data were restricted to one randomly subsampled individual per burrow system (pre-eruption: −0.204, post-eruption: 0.127; two-sample t-test, p < 0.0001). The mean value of F for the post-eruption population of C. sociabilis at La Lonja was −0.0143, which fell within the range of values reported for this species at Rincon Grande. In the absence of pre-eruption samples from La Lonja, we could not assess the impacts of the eruption on levels of inbreeding in this population. There was no significant difference between pre- and post-eruption values of F in the focal population of C. haigi (pre-eruption, 0.159; post-eruption, 0.0580; two-sample t-test, p = 0.087). Thus, our analyses suggest that the 2011 eruption resulted in increased inbreeding within the focal study population of C. sociabilis but not the focal study population of C. haigi.
Simulations of demographic parameters
The simulated 50% reduction in size of the pre-eruption population of C. sociabilis at Rincon Grande did not result in a significant change in heterozygosity (pre: 0.03337; post: 0.03338; two-sample T test, p = 0.29). Our Bayesian modeling analyses revealed that the most strongly supported demographic hypothesis was that of panmixia between the Rincon Grande and La Lonja populations (model probability = 0.986), with the hypotheses of full migration (model probability = 0.00467), immigration from La Lonja to Rincon Grande (model probability = 0.00667), and emigration from Rincon Grande to La Lonja (model probability = 0.00271) receiving considerably less support. Thus, we found little evidence to suggest that post-eruption changes in genetic variation in C. sociabilis were influenced by migration between populations.
Discussion
Our results indicate that in both C. sociabilis and C. haigi the reduction in population size documented immediately following the 2011 eruption of the Puyehue-Cordón Caulle volcanic complex16 was associated with a decrease in genetic variation, as revealed by measures of mean heterozygosity across the multiple loci examined. In the more extensively sampled C. sociabilis, this decrease in genetic variation was evident throughout the focal population, with no apparent differences in response to the eruption based on the sex of the animals or the number of burrow systems sampled. Despite the post-eruption decrease in heterozygosity detected in each study species, we found little evidence of temporal or spatial genetic differentiation between populations of conspecifics, suggesting that the eruption did not lead to increased genetic structure. Because these results are based on analyses of a large number of putatively neutral markers distributed throughout the genome, we believe that our findings are indicative of the impact of the eruption on genetic variation in the study animals. Intriguingly, the decrease in genetic variation reported here for C. sociabilis contradicts previous findings regarding the impacts of the Puyehue-Cordón Caulle eruption; our prior analyses based on a much more limited suite of microsatellite loci16 had revealed an increase in post-eruption genetic variation in the focal study population for this species. This difference in outcomes raises intriguing questions regarding the abilities of different types of molecular markers, including associated differences in numbers of loci surveyed, to detect the genetic consequences of recent demographic changes.
Loss of genetic variation following population decline
Classical population genetics theory suggests that reductions in genetic variation associated with demographic bottlenecks are dependent on the magnitude and duration of the reduction in population size34,54,55. Due to the relatively modest declines in population size (~50%) observed16 and the short duration of this decline (<2 generations) prior to the collection of our post-eruption samples, we had not expected to find significant changes in post-eruption genetic variation in the focal study populations. As noted above, however, previous analyses based on microsatellite loci16 revealed a significant post-eruption increase in genetic variation in C. sociabilis, an unexpected result that, based on demographic modeling, appeared to reflect post-eruption changes in migration and gene flow. In contrast, the data set considered here revealed small but significant decreases in genetic variation in both study species, a finding that is more consistent with theoretical expectations (e.g. Nei et al. 1). Given the considerably larger molecular data sets employed in this study, we expect that the reductions in post-eruption genetic variation reported here are more indicative of the genetic changes experienced by our study populations.
Factors affecting post-eruption genetic variation
Although reductions in population size are typically expected to lead to declines in genetic variation1, the exact processes by which such declines occur may vary. We found little evidence to suggest that selection was acting on the regions of the genome targeted for analysis. Very few of the loci examined (<5%) revealed departures from Hardy-Weinberg expectations or signals of directional selection. Further, because loci demonstrating departures from neutral expectations were excluded from analyses of genetic variation in our study populations, we do not believe that the post-eruption changes in variation reported here resulted from strong selective forces acting on these animals.
Similarly, it is unlikely that the observed post-eruption changes in genetic variation were the result of altered mutation rates following the eruption. Although mutation rates can be affected by external factors (e.g. Ellegren et al.56), we are not aware of any demonstrated or suggested links between ash produced by volcanic eruptions and increases in genomic mutation rates. Further, the average rate of mutation for mammalian genomes has been estimated at 2.2 × 10−9 per base pair per year57. Thus, given the limited temporal interval between our pre- and post-eruption sampling, this rate of mutational change could not have produced the observed post-eruption changes in genetic variation.
Although Hsu et al.16 reported increases in post-eruption genetic variation and suggested that increased migration and gene flow contributed to these changes, our analyses failed to reveal evidence of significant changes in these demographic parameters following the 2011 eruption. Populations at equilibrium58 as well as populations which have undergone a recent bottleneck51,59–61 are expected to display a multimodal mismatch distribution with relatively high values of raggedness. In contrast, pairwise mismatch distributions for both the pre- and post-eruption focal populations of C. sociabilis and C. haigi were roughly unimodal with low levels of raggedness, findings that are more typical of populations that are expanding or that are characterized by high levels of immigration and gene flow50,62,63. More importantly, these distributions were not significantly different between pre- and post-eruption samples, providing no evidence of demographic change in this interval. Thus, the results of our mismatch analyses suggest that (1) neither focal study population was in demographic equilibrium prior to the 2011 eruption and (2) the eruption did not substantially alter patterns of migration and gene flow in these populations.
Comparisons of molecular markers
Our analyses offer important insights into the genomic consequences of an abrupt reduction in size in natural populations of mammals. While apparent population bottlenecks have been reported for other species of Ctenomys based on analyses of microsatellite and allozyme data16,64–67, our study offers the first direct comparison of pre- and post-bottleneck samples using genomic-level data. Accordingly, our analyses employed a much larger number of loci to detect changes in genetic variation. The presumably greater resolution offered by our data set is important given the relatively modest reductions in size and density detected in our focal study populations following the 2011 eruption. While studies of other ctenomyid species have examined the genetic consequences of decreases in population size of >90%26,64,65, our focal study populations experienced declines of only ~25–50%, suggesting that associated changes in genetic diversity would also be relatively modest. As a result, the use of a large panel of markers located throughout the genome should have increased our likelihood of detecting genetic signatures of these reductions in population size.
Particularly intriguing is the contrast between our results and those of Hsu et al.16, who reported a post-eruption increase in microsatellite heterozygosity in C. sociabilis. Although the same post-eruption samples were used in both studies, the pre-eruption samples examined differed: the samples analyzed here were collected in 2001, while those genotyped by Hsu et al.16 were collected during 1993–1998. It is possible that this difference in data sets contributed to the contrasting outcomes for these studies. However, given the generally low levels of genetic variation in both sets of pre-eruption samples (see also Lacey11), it seems unlikely that the temporal difference between these data sets underlies the reported differences post-eruption changes in genetic variation. Indeed, a marked reduction in population size in 1999 (Lacey, pers. comm.) should have reduced pre-eruption variation among the individuals sampled here compared to those analyzed by Hsu et al.16.
Instead, we posit that this difference in outcomes is due primarily to the greater power of our genomic-level markers to detect modest differences in genetic variation arising over short time periods. The number of markers employed in this study was more than two orders of magnitude greater than that used by Hsu et al.16 and this difference alone is likely to have affected the reliability of the post-eruption estimates of genetic diversity generated by each study2. Consistent with this, our analyses of the effects of number of loci on measures of genetic diversity revealed that the variance in estimates of mean heterozygosity decreased markedly as the number of loci employed increased, suggesting that larger numbers of these markers generate more robust metrics of genetic variation. Consequently, studies relying on a small number of markers likely show greater variance and more potential for summary statistics that are not representative of the true variation across the genome. In addition, microsatellite markers have been found to be unreliable for estimating genomic diversity, with marked differences in estimates of genetic variation generated with microsatellites versus SNPs68,69. Further, microsatellites have been found to have limited ability to detect complex demographic histories, including bottlenecks followed by rapid population growth70, and models demonstrating consistent tempo and mode in the evolution of microsatellites are lacking71. These limitations, combined with the presumptively greater analytical power of a large panel of genomic markers leads us to expect that estimates of genetic variation based on the latter provide a more accurate picture of pre- versus post-eruption changes in genetic variation in our study populations. If this supposition is correct, our analyses indicate that both C. sociabilis and C. haigi experienced decreases in genetic variation following the 2011 volcanic eruption.
Conclusion
Our analyses of genetic variation in two species of ctenomyid rodents impacted by the 2011 eruption of the Puyehue-Cordón Caulle volcanic complex indicate that abrupt, naturally occurring reductions in population size can impact genetic variation over even brief time scales. For both C. sociabilis and C. haigi, analyses of genome-wide markers revealed small but significant reductions in post-eruption heterozygosity. Multiple lines of evidence supported the hypothesis that these reductions in genetic variation were associated with post-eruption declines in population size, indicating that even modest (~50%) reductions occurring over a span of 1–2 generations are sufficient to generate signals in genomic variation. Characterizing the impacts of such demographic changes has critical implications for understanding and for predicting how species will respond genetically to altered environmental conditions and thus may generate important insights into the conservation of species affected by such events.
Electronic supplementary material
Acknowledgements
We thank Hunter Fraser, Noah Rosenberg, Steve Palumbi, Stefan Prost, and Heather Machado for invaluable feedback and advice on the genomic sequence analyses. Lydia Smith provided technical advice and support during the library preparation and sequence capture. We also thank members of the Hadly lab for helpful feedback on this manuscript. This work was funded by NSF RAPID grant DEB-1201541 (Elizabeth Hadly and Eileen Lacey) and a Stanford Center for Computational, Evolutionary, and Human Genomics trainee grant (Jeremy Hsu). This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303. Jeremy Hsu was supported by a NSF Graduate Research Fellowship and a Stanford Graduate Fellowship, Mauro Tammone by a CONICET Postdoctoral Fellowship, and Jeremy Chase Crawford by an NSF Graduate Research Fellowship, an NSF Doctoral Dissertation Improvement Grant (IOS-1309436), and the Jerry O. Wolff Dissertation Fellowship from the Museum of Vertebrate Zoology. We also thank the Delegacion Tecnica Patagonia (Administracion de Parques Nacionales, Argentina) in Bariloche and the Secretaria de Ambiente y Desarrollo Sustentable, Rio Negro Province for issuing the permits required for field work, as well as Alain Thouyaret for permission to work on Estancia Rincon Grande and a series of administrators for permission to work on Estancia San Ramon.
Author Contributions
E.A.H. and E.A.L. proposed and supervised the experiments, E.A.L. and M.N.T. conducted the fieldwork, J.C.C. and J.L.H. performed the experiments, J.L.H. and U.R. analyzed the data, and J.L.H. wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-16430-1.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nei M, Maruyama T, Chakraborty R. The bottleneck effect and genetic variability in populations. Evolution. 1975;29:1–10. doi: 10.1111/j.1558-5646.1975.tb00807.x. [DOI] [PubMed] [Google Scholar]
- 2.Peery MZ, et al. Reliability of genetic bottleneck tests for detecting recent population declines. Mol. Ecol. 2012;21:3403–18. doi: 10.1111/j.1365-294X.2012.05635.x. [DOI] [PubMed] [Google Scholar]
- 3.Pilot M, et al. Genome-wide signatures of population bottlenecks and diversifying selection in European wolves. Heredity. 2014;112:428–42. doi: 10.1038/hdy.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morin PA, Martien KK, Taylor BL. Assessing statistical power of SNPs for population structure and conservation studies. Mol. Ecol. Resources. 2009;9:66–73. doi: 10.1111/j.1755-0998.2008.02392.x. [DOI] [PubMed] [Google Scholar]
- 5.McCoy RC, Garud NR, Kelley JL, Boggs CL, Petrov DA. Genomic inference accurately predicts the timing and severity of a recent bottleneck in a nonmodel insect population. Mol. Ecol. 2014;23:136–150. doi: 10.1111/mec.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyten DL, et al. Impacts of genetic bottlenecks on soybean genome diversity. Proc Natl Acad Sci USA. 2006;103:16666–71. doi: 10.1073/pnas.0604379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caicedo AL, et al. Genome-wide patterns of nucleotide polymorphism in domesticated rice. PLoS Genetics. 2007;3:1745–56. doi: 10.1371/journal.pgen.0030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Axelsson E, et al. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495:360–4. doi: 10.1038/nature11837. [DOI] [PubMed] [Google Scholar]
- 9.Hadly EA, et al. Genetic response to climatic change: insights from ancient DNA and phylochronology. PLoS Biology. 2004;2:e290. doi: 10.1371/journal.pbio.0020290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramakrishnan U, Hadly EA, Mountain JL. Detecting past population bottlenecks using temporal genetic data. Mol. Ecol. 2005;14:2915–22. doi: 10.1111/j.1365-294X.2005.02586.x. [DOI] [PubMed] [Google Scholar]
- 11.Lacey EA. Microsatellite variation in solitary and social tuco-tucos: molecular properties and population dynamics. Heredity. 2001;86:628–637. doi: 10.1046/j.1365-2540.2001.00881.x. [DOI] [PubMed] [Google Scholar]
- 12.Hadly EA, Van Tuinen M, Chan Y, Heiman K. Ancient DNA evidence of prolonged population persistence with negligible genetic diversity in an endemic tuco-tuco (Ctenomys sociabilis) J Mammalogy. 2003;84:403–417. doi: 10.1644/1545-1542(2003)084<0403:ADEOPP>2.0.CO;2. [DOI] [Google Scholar]
- 13.Chan YL, Lacey EA, Pearson OP, Hadly EA. Ancient DNA reveals Holocene loss of genetic diversity in a South American rodent. Biology Letters. 2005;1:423–426. doi: 10.1098/rsbl.2005.0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan YL, Hadly EA. Genetic variation over 10 000 years in Ctenomys: comparative phylochronology provides a temporal perspective on rarity, environmental change and demography. Mol. Ecol. 2011;20:4592–4605. doi: 10.1111/j.1365-294X.2011.05295.x. [DOI] [PubMed] [Google Scholar]
- 15.Tammone M. N., Pardiñas U. J. F. & Lacey E. A. Contrasting patterns of Holocene genetic variation in two parapatric species of Ctenomys from Northern Patagonia, Argentina. Biological Journal of the Linnean Society, 10.1093/biolinnean/blx118 (2017).
- 16.Hsu J, Kam S, Tammone M, Lacey EA, Hadly EA. Rapid increase in genetic diversity in an endemic Patagonian tuco-tuco following a recent volcanic eruption. J Mammalogy. 2017;98:779–792. doi: 10.1093/jmammal/gyx008. [DOI] [Google Scholar]
- 17.Keller LF, Arcese P, Smith JN, Hochachka WM, Stearns SC. Selection against inbred song sparrows during a natural population bottleneck. Nature. 1994;372:356–7. doi: 10.1038/372356a0. [DOI] [PubMed] [Google Scholar]
- 18.Berthier K, Charbonnel N, Galan M, Chaval Y, Cosson J-F. Migration and recovery of the genetic diversity during the increasing density phase in cyclic vole populations. Mol. Ecol. 2006;15:2665–76. doi: 10.1111/j.1365-294X.2006.02959.x. [DOI] [PubMed] [Google Scholar]
- 19.Pinsky ML, et al. Dispersal provided resilience to range collapse in a marine mammal: insights from the past to inform conservation biology. Mol. Ecol. 2010;19:2418–29. doi: 10.1111/j.1365-294X.2010.04671.x. [DOI] [PubMed] [Google Scholar]
- 20.Price MR, Sischo D, Pascua M-A, Hadfield MG. Demographic and genetic factors in the recovery or demise of ex situ populations following a severe bottleneck in fifteen species of Hawaiian tree snails. PeerJ. 2015;3:e1406. doi: 10.7717/peerj.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osborne, A. J. et al. Genetic evidence of a population bottleneck and inbreeding in the endangered New Zealand sea lion, Phocarctos hookeri. J Heredity, esw015 (2016). [DOI] [PubMed]
- 22.Pearson OP, Christie MI. Los tuco-tucos (genero Ctenomys) de los parques nacionales Lanin y Nahuel Huapi, Argentina. Historia Natural. 1985;5:337–343. [Google Scholar]
- 23.Tammone MN, Lacey EA, Relva MA. Habitat use by colonial tuco-tucos (Ctenomys sociabilis): specialization, variation, and sociality. J Mammalogy. 2012;93:1409–1419. doi: 10.1644/11-MAMM-A-266.1. [DOI] [Google Scholar]
- 24.Lacey EA, Wieczorek JR. Ecology of sociality in rodents: A ctenomyid perspective. J Mammalogy. 2003;84:1198–1211. doi: 10.1644/BLe-014. [DOI] [Google Scholar]
- 25.Lacey EA, Wieczorek JR. Kinship in colonial tuco-tucos: evidence from group composition and population structure. Behavioral Ecology. 2004;15:988–996. doi: 10.1093/beheco/arh104. [DOI] [Google Scholar]
- 26.Chan YL, Anderson CNK, Hadly EA. Bayesian estimation of the timing and severity of a population bottleneck from ancient DNA. PLoS Genetics. 2006;2:451–460. doi: 10.1371/journal.pgen.0020059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lacey EA, Braude SH, Wieczorek JR. Solitary burrow use by adult Patagonian tuco-tucos (Ctenomys haigi) J Mammalogy. 1998;79:986–991. doi: 10.2307/1383106. [DOI] [Google Scholar]
- 28.Lacey EA, Braude SH, Wieczorek JR. Burrow sharing by colonial tuco-tucos (Ctenomys sociabilis) J Mammalogy. 1997;78:556–562. doi: 10.2307/1382907. [DOI] [Google Scholar]
- 29.Sikes RS. and the Animal Care and Use Committee of the American Society of Mammalogists. 2016 Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. J Mammalogy. 2016;97:663–688. doi: 10.1093/jmammal/gyw078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Research. 2012;40:e3. doi: 10.1093/nar/gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crawford, J. C. C. Elucidating the evolutionary consequences of sociality through genome-wide analyses of social and solitary rodents. PhD dissertation, University of California, Berkeley (2016).
- 32.Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protocols. 2010;2010:pdb.prot5448. doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 33.Kozarewa I, et al. Amplification-free Illumina sequencing-library preparation faiclitates improved mapping and assembly of GC-biased genomes. Nat Methods. 2009;6:291–295. doi: 10.1038/nmeth.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bi K, et al. Unlocking the vault: next-generation museum population genomics. Mol. Ecol. 2013;22:6018–32. doi: 10.1111/mec.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Wit P, et al. The simple fool’s guide to population genomics via RNA-Seq: an introduction to high-throughput sequencing data analysis. Mol. Ecol. Resources. 2012;12:1058–1067. doi: 10.1111/1755-0998.12003. [DOI] [PubMed] [Google Scholar]
- 36.Gordon A. FastX-Toolkit, http://hannonlab.cshl.edu/fastx_toolkit/index.html (2011).
- 37.Andrews S. FastQC: A quality control tool for high throughput sequence data (2010).
- 38.Giardine B, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Research. 2005;15:1451–5. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vieira F. SNPCleaner, https://github.com/fgvieira/ngsClean (2015).
- 42.Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinformatics. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antao T, et al. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics. 2008;9:323. doi: 10.1186/1471-2105-9-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fumagalli M, Vieira FG, Linderoth T, Nielsen R. ngsTools: methods for population genetics analyses from next-generation sequencing data. Bioinformatics (Oxford, England) 2014;30:1486–7. doi: 10.1093/bioinformatics/btu041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen R. Molecular signatures of natural selection. Annual Review of Genetics. 2005;39:197–218. doi: 10.1146/annurev.genet.39.073003.112420. [DOI] [PubMed] [Google Scholar]
- 46.Keightley PD, Halligan DL, Begun D. Inference of site frequency spectra from high-throughput sequence data: quantification of selection on nonsynonymous and synonymous sites in humans. Genetics. 2011;188:931–940. doi: 10.1534/genetics.111.128355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bustamante CD, Wakeley J, Sawyer S, Hartl DL. Directional selection and the site-frequency spectrum. Genetics. 2001;159:1779–1788. doi: 10.1093/genetics/159.4.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ronald J, Akey JM. Genome-wide scans for loci under selection in humans. Human Genomics. 2005;2:113. doi: 10.1186/1479-7364-2-2-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carlson CS, et al. Genomic regions exhibiting positive selection identified from dense genotype data. Genome Research. 2005;15:1553–65. doi: 10.1101/gr.4326505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harpending HC, Sherry ST, Rogers AR, Stoneking M. The genetic structure of ancient human populations. Current Anthropology. 1993;34:483–496. doi: 10.1086/204195. [DOI] [Google Scholar]
- 51.Harpending HC. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Human Biology. 1993;66:591–600. [PubMed] [Google Scholar]
- 52.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J Human Genetics. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beerli P. MIGRATE-N: estimation of population sizes and gene flow using the coalescent (2002).
- 54.Hoezel AR. Impact of population bottlenecks on genetic variation and the importance of life-history; a case sudy of the northern elephant seal. Biological J Linnean Soc. 1999;68:23–39. doi: 10.1111/j.1095-8312.1999.tb01156.x. [DOI] [Google Scholar]
- 55.Zhang W, et al. Impact of population structure, effective bottleneck time, and allele frequency on linkage disequilibrium maps. Proc Natl Acad Sci USA. 2004;101:18075–18080. doi: 10.1073/pnas.0408251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ellegren H, Lindgren G, Primmer CR, Møller AP. Fitness loss and germline mutations in barn swallows breeding in Chernobyl. Nature. 1997;389:593–6. doi: 10.1038/39303. [DOI] [PubMed] [Google Scholar]
- 57.Kumar S, Subramanian S. Mutation rates in mammalian genomes. Proc Natl Acad Sci USA. 2002;99:803–8. doi: 10.1073/pnas.022629899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dadi H, et al. Effect of population reduction on mtDNA diversity and demographic history of Korean cattle populations. Asian-Australasian J Animal Sciences. 2012;25:1223–8. doi: 10.5713/ajas.2012.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weber DS. Genetic consequences of a severe population bottleneck in the Guadalupe fur seal (Arctocephalus townsendi) J Heredity. 2004;95:144–153. doi: 10.1093/jhered/esh018. [DOI] [PubMed] [Google Scholar]
- 60.Johnson JA, Dunn PO, Bouzat JL. Effects of recent population bottlenecks on reconstructing the demographic history of prairie-chickens. Mol. Ecol. 2007;16:2203–22. doi: 10.1111/j.1365-294X.2007.03285.x. [DOI] [PubMed] [Google Scholar]
- 61.Liao P-C, et al. Historical spatial range expansion and a very recent bottleneck of Cinnamomum kanehirae Hay. (Lauraceae) in Taiwan inferred from nuclear genes. BMC Evolutionary Biology. 2010;10:124. doi: 10.1186/1471-2148-10-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ray N, Currat M, Excoffier L. Intra-deme molecular diversity in spatially expanding populations. Molecular Biology and Evolution. 2003;20:76–86. doi: 10.1093/molbev/msg009. [DOI] [PubMed] [Google Scholar]
- 63.Excoffier L. Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol. Ecol. 2004;13:853–864. doi: 10.1046/j.1365-294X.2003.02004.x. [DOI] [PubMed] [Google Scholar]
- 64.Gallardo MH, Kohler N, Araneda C. Bottleneck effects in local populations of fossorial Ctenomys (Rodentia, Ctenomyidae) affected by vulcanism. Heredity. 1995;74:638–646. doi: 10.1038/hdy.1995.87. [DOI] [Google Scholar]
- 65.Gallardo MH, Kohler N. Demographic changes and genetic losses in populations of a subterranean rodent (Ctenomys maulinus brunneus) affected by a natural catastrophe. Zeitschrift fur Saugetierkunde. 1994;59:358–365. [Google Scholar]
- 66.Gallardo MH, Kohler N, Araneda C. Loss of genetic variation in Ctenomys coyhaiquensis (Rodentia, Ctenomyidae) affected by vulcanism. Mastozoologia Neotropical. 1996;3:7–13. [Google Scholar]
- 67.Fernandez-Stolz GP, Stolz JFB, de Dreitas TRO. Bottlenecks and dispersal in the tuco-tuco das dunas, Ctenomys flamarioni (Rodentia: Ctenomyidae), in southern Brazil. J Mammalogy. 2007;88:935–945. doi: 10.1644/06-MAMM-A-210R1.1. [DOI] [Google Scholar]
- 68.Väli U, Einarsson A, Waits L, Ellegren H. To what extent do microsatellite markers reflect genome-wide genetic diversity in natural populations? Mol. Ecol. 2008;17:3808–17. doi: 10.1111/j.1365-294X.2008.03876.x. [DOI] [PubMed] [Google Scholar]
- 69.Lozier JD. Revisiting comparisons of genetic diversity in stable and declining species: assessing genome-wide polymorphism in North American bumble bees using RAD sequencing. Mol. Ecol. 2014;23:788–801. doi: 10.1111/mec.12636. [DOI] [PubMed] [Google Scholar]
- 70.Hundertmark KJ, Van Daele LJ. Founder effect and bottleneck signatures in an introduced, insular population of elk. Conservation Genetics. 2009;11:139–147. doi: 10.1007/s10592-009-0013-z. [DOI] [Google Scholar]
- 71.Buschiazzo E, Gemmell NJ. The rise, fall and renaissance of microsatellites in eukaryotic genomes. BioEssays: Mol., Cell., and Developmental Bio. 2006;28:1040–50. doi: 10.1002/bies.20470. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.