

Abstract
Camurati-Engelmann disease (CED) is an ultrarare autosomal dominant bone dysplasia. Cortical thickening of the diaphyses of the long bones with narrowing of the medullary cavity are associated with bone pain, waddling gait, muscular weakness, easy fatigability, and a marfanoid body habitus. There is no specific treatment for CED. Nonsteroidal anti-inflammatory drugs or glucocorticoids are ineffective in improving bone lesions. A family with a mild to severe form of CED is described. Two patients received long-term bisphosphonate treatment: the 19-year-old female proband was treated with zoledronic acid for 2.2 years; the 4-year-old male proband was treated with neridronic acid for 16 months and with zoledronic acid for an additional 18 months. In both probands, zoledronic acid treatment significantly improved the clinical symptoms, bone lesions, ambulation, and body habitus. Before treatment, both probands showed a marked increase in serum levels of osteocalcin, procollagen type I N-terminal propeptide, and cross-linked carboxyterminal telopeptide of type I collagen, reflecting an increased bone turnover. Bone marker levels returned to their normal values during treatment. Zoledronic acid treatment may be an important therapeutic option in patients with severe CED. Biochemical markers of bone turnover could be considered as surrogate indexes of CED activity.
Keywords: Bone lesions, Bone turnover, Camurati-Engelmann disease, Neridronic acid, Zoledronic acid
Camurati-Engelmann disease (CED), also known as progressive diaphyseal dysplasia (MIM 131300), is an ultrarare autosomal dominant form of bone dysplasia. CED mainly affects the long bones of the upper and lower limbs and is characterized by cortical thickening of the diaphysis which affects both periosteal and endosteal bone surfaces, leading to an increased diameter of the bone with narrowing of the medullary cavity. Additional skeletal sites may be involved, such as skull, ribs, pelvis, vertebrae, and bones of the hands and feet [Janssens et al., 2006; Bartuseviciene et al., 2009; Carlson et al., 2010]. The patients may be asymptomatic, with or without radiographic hallmarks of the disease, or may show mild to severe disabling symptoms including waddling gait, diffuse and localized bone pain, muscular weakness up to severe hypotrophy, easy fatigability, and a marfanoid body habitus [Janssens et al., 2006; Bartuseviciene et al., 2009]. Bone lesions and bone marrow edema are likely main causes of bone pain in patients with CED [Ziran et al., 2002; Janssens et al., 2006].
CED is caused by domain-specific heterozygous mutations in the transforming growth factor beta 1 gene (TGFB1) on chromosome 19q13 [Janssens et al., 2000; Kinoshita et al., 2000; Wang et al., 2013]. The mutations in the signal peptide and latency-associated peptide (LAP) of TGFB1 lead to enhanced signaling by altering the activation or secretion of the protein [Janssens et al., 2003] with an impaired regulation of bone turnover [Janssens et al., 2006].
There is no specific therapy for patients with CED. Nonsteroidal anti-inflammatory drugs (NSAIDs) or glucocorticoids only have a transient effect in reducing clinical symptoms [Naveh et al., 1985; Bourantas et al., 1995; Heymans et al., 1998; Baş et al., 1999]. Bisphosphonates treatment is disputed, but a few patients have been treated in order to obtain conclusive data.
In this study, we describe a family in whom some individuals were affected by a mild to severe form of CED, and we report on 2 of them receiving long-term bisphosphonate treatment.
Case Reports
Family Report
We describe an Albanian family in whom some individuals showed bone symptoms associated with a marfanoid body habitus. In Table 1, the main clinical features of the family members are summarized. The severity of signs and symptoms varied among the individuals indicating variable expressivity and incomplete penetrance for CED: cases 1 and 2 had a mild form of CED; cases 4, 5, and 7 were affected by a severe form, whereas cases 3 and 6 did not have any signs or symptoms of CED. Case 1 and case 5 lived in Albania; case 5 was treated with NSAIDs and/or glucocorticoids with poor effectiveness on bone pain and bone lesions.
Table 1.
Clinical features of the examined subjects
| Case No./sex | Age, years | Marfanoid habitus | Bone pain | Waddling gait | Muscular weakness | Easy fatigability | Diaphyseal cortical thickening | Hyperostosis of the skull |
|---|---|---|---|---|---|---|---|---|
| 1/F | 55 | − | +* | − | +* | +* | + | NA |
| 2/F | 24 | − | +* | − | +* | +* | + | NA |
| 3/F | 23 | − | − | − | − | − | − | − |
| 4/F | 19 | + | +++ | +++ | +++ | +++ | +++ | +++ |
| 5/F | 14 | + | +++ | + + | +++ | +++ | + + | + |
| 6/F | 10 | − | − | − | − | − | − | −v |
| 7/M | 4 | + | +++ | +++ | +++ | +++ | +++ | +++ |
Case 4, female proband; case 7, male proband; cases 3 and 6 were unaffected by the Camurati-Engelmann disease.
Bone pain, muscular weakness, and easy fatigability were referred only during infancy and adolescence, but not in adulthood. NA, not available; –, absent; +, present. The number of + symbols is comparative among the individuals (+++ represents the maximum level and + the minimum level). Clinical data of the father were not available.
The family pedigree documented an autosomal dominant inheritance pattern (Fig. 1). Genetic analysis of the TGFB1 gene revealed a mutation c.505G>A (p.Glu169Lys) in exon 2, confirming the clinical diagnosis of CED in the grandmother, 3 daughters, and 1 grandson.
Fig. 1.

Pedigree of the examined family with Camurati-Engelmann disease. Arrows show female (case 4) and male (case 7) probands. The number of the other members of the family is indicated in Table 1.
Probands
Case 4
A 19-year-old Albanian female was referred to our hospital because of acute and diffuse bone pain; the numeric pain intensity score (NPIS) was 9 (score 0, no pain; score 10, worst possible pain) [Jensen and McFarland, 1993]. The history revealed the occurrence of a burning pain in the lower limbs at 12 years of age associated with difficulty in running; the intensity of bone pain had increased since she was 15, and recurrent upper limb pain also occurred. When she was living in Albania, she received high doses of indomethacin (100 mg twice a day) or glucocorticoids (prednisone 2 mg/kg body weight per day for 2 months) achieving only partial pain relief. On physical examination, she showed a marfanoid body habitus associated with proximal muscle weakness in the lower limbs and waddling gait. Her height was 172 cm (Z –score 1.4), weight 42 kg (Z –score −2.5), and BMI 14.2 (Z –score −4.3). Neurological examination, including cranial nerve assessment, was negative. No hearing loss was found. She had normal vision. Ophthalmological assessment, including fundus oculi examination, did not reveal any anomaly. She did not suffer from joint contractures or bone fractures. Menarche occurred at 11.2 years. Pubertal stage was PH5B5, and she had a normal menstrual cycle. She showed a mild form of anemia (erythrocyte count 3.720 × 106 μL; hemoglobin 9.7 g/dL; hematocrit 29.8%). Skeletal X-ray revealed cortical thickening and sclerosis of the skull and the diaphyses of the long tubular bones of the upper and lower limbs, mainly evident at tibia and femur (Fig. 2). Dual energy X-ray absorptiometry (DXA; Lunar GE, Prodigy) measurement showed normal bone mineral density (BMD) at lumbar spine (Z –score −0.9) and reduced BMD at femoral neck (Z –score −2.9). Quantitative ultrasound assessment for proximal phalanges of the hand (BP Sonic, IGEA) showed a normal value of amplitude-dependent speed of sound (AD-SoS; Z –score 1.1) and bone transmission time (BTT; Z –score 0.9). NSAIDs treatment was started, including indomethacin, paracetamol, aspirin, ibuprofen, naproxen, and diclofenac, alone or in combination for some months, achieving only partial pain relief (NPIS 7).
Fig. 2.

X-ray examination at diagnosis in the female proband. a Skull in lateral view: the calvaria and the basal bones are thickened and sclerotic. b Distal end of femurs: irregular thickening and massive cortical sclerosis of the diaphysis. c Proximal ends and diaphyses of tibiae and fibulae: thickening and massive cortical sclerosis of the diaphysis. The medullary cavity of the tibiae was not completely visualized.
Case 7
A 4-year-old Albanian male was referred to our hospital. He was unable to walk because of severe bone pain in his lower limbs, scored as 10 according to the Wong-Baker rating scale (WBS: score 0, no pain; score 10, worst possible pain) [Wong and Baker, 1988]. He wept for many hours during the day due to severe bone pain at both upper and lower limbs. He appeared dystrophic with a habitus similar to that of a patient with anorexia nervosa (Fig. 3). He showed a severe reduction in muscle mass associated with proximal muscle weakness at both upper and lower limbs. His height was 105.5 cm (Z –score 0.7), weight 13.1 kg (Z –score −2.1), and BMI 11.8 (Z –score −4.6). His body movements were extremely awkward and clumsy. Neurological examination, including cranial nerve assessment, was negative; hearing was not impaired. Vision was normal as well as the ophthalmological assessment, including fundus oculi examination. He did not suffer from joint contractures or bone fractures. Pubertal stage was PH1G1. He showed a mild form of anemia (erythrocyte count 4.050 × 106 μL; hemoglobin 10.5 g/dL; hematocrit 32.3%). Skeletal X-ray showed severe cortical thickening and sclerosis of the skull and the diaphyses of long tubular bones of the upper and lower limbs, mainly at femur (Fig. 4). A normal BMD at lumbar spine (Z –score 0.8) and a reduced BMD at femoral neck (Z –score −5.1) were found by DXA. Phalangeal AD-SoS and BTT were normal (Z –scores −0.7 and −0.2, respectively). Treatment with NSAIDs (paracetamol, aspirin, ibuprofen, and naproxen) for some months, alone or in combination, or with glucocorticoids (prednisone 1.5 mg/kg body weight per day for 3 weeks) was ineffective in reducing bone pain (WBS 7–8).
Fig. 3.
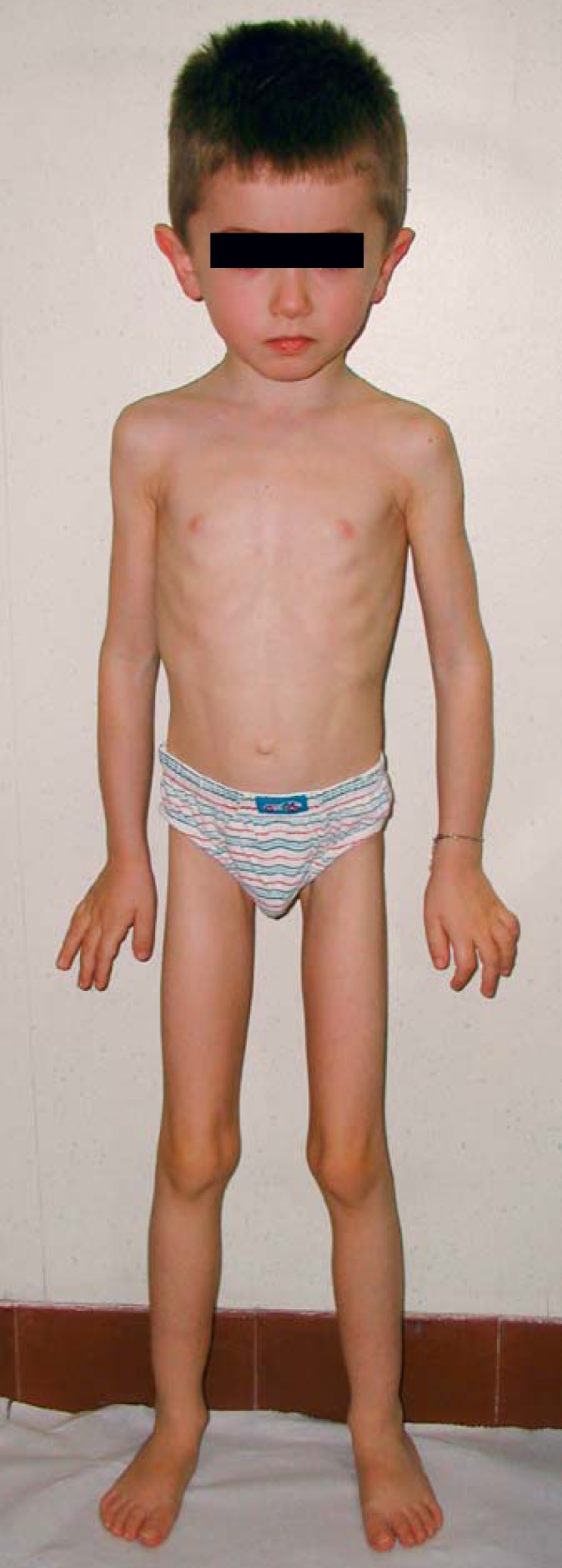
Clinical picture of the male proband. Note the dystrophic appearance, absence of subcutaneous fat (weight 13.1 kg), muscle hypotrophy, and valgus deformity of the knees.
Fig. 4.

X-ray examination at diagnosis in the male proband. a Skull in frontal view: the calvaria and the basal bones as well as the mandibular profile were markedly thickened and sclerotic. b Right arm and forearm: irregular thickening and cortical sclerosis of the diaphyses, mainly of humerus and radius. c Femurs: irregular thickening and cortical sclerosis of the diaphyses. The medullary cavity of the left femur was not completely visualized.
Methods
Bisphosphonate Treatment
The female proband received intravenous zoledronic acid (Zometa, Novartis, 0.020 mg/kg body weight, diluted in 50 mL isotonic saline solution for 40 min) every 4 months for a total of 7 cycles over a period of 2.2 years.
Our male proband was treated with intravenous neridronic acid (Nerixia, Abiogen, 1 mg/kg body weight, diluted in 250 mL isotonic saline solution for 3 h) every 4 months for a total of 4 cycles. As no significant improvement of bone pain was evident, neridronic acid was stopped, and zoledronic acid treatment was started (Zometa, Novartis, 0.015–0.018 mg/kg body weight, diluted in 50 mL isotonic saline solution for 40 min). He received 4 cycles of zoledronic acid with an interval of 4–6 months over a period of 18 months.
In both probands, each bisphosphonate infusion was followed by intravenous 10% calcium gluconate infusion (0.5 mg/kg body weight per day) diluted in 100 mL isotonic saline solution over a 1-h period in order to avoid symptomatic hypocalcemia [Baroncelli et al., 2013]. Furthermore, they received a supplement of calcium salts as carbonate (1,000 mg of elemental calcium per day for the female proband and 500 mg per day for the male proband) for 10 days after each bisphosphonate infusion. Moreover, both probands were supplemented with 1,000 IU of vitamin D per day (Dibase, Abiogen). Their serum 25-hydroxyvitamin D levels ranged from 26 to 31.5 ng/mL.
In addition, both probands underwent general clinical assessments every 1–2 months as well as dental and parodontal examination by an odontologist, and complete eye examination by an ophthalmologist every 6 months. Biochemical measurements, including a complete blood count, electrolytes, and hepatic and renal function tests, were performed at the time of each bisphosphonate infusion and every 3 months during the discontinuation of bisphosphonate treatment [Baroncelli and Bertelloni, 2014].
Assessment of Disease Severity
Disease severity was assessed by the bone pain intensity score according to age. Our female proband assessed her bone pain 3 times a day at the same time. The male proband's mother was trained to assess her son's bone pain score 3 times a day at the same time. E.F. and C.M.P. checked the bone pain score weekly. Moreover, the degree of diaphyseal cortical thickening and clinical signs, including waddling gait, muscle weakness, and easy fatigability, were assessed and compared among the members of the same family. The response to prior treatments, including NSAIDs and glucocorticoids, was also considered. Finally, a global evaluation of disease severity was established.
Results
Clinical Symptoms and Evolution of the Bone Lesions during Bisphosphonate Treatment
Our female proband showed a progressive reduction up to almost complete resolution of bone pain (NPIS 1) associated with an improvement of ambulation. At the end of zoledronic acid treatment, a marked improvement of the bone lesions, mainly at femur and tibia, was detected (Fig. 5). BMD increased at both lumbar spine (Z –score 0.8) and femoral neck (Z –score −1.2), whereas phalangeal AD-SoS and BTT did not change (Z –scores 1.2 and 1.0, respectively). Her weight and BMI also improved [49.8 kg (Z –score −1.2) and 16.8 (Z –score −2.5), respectively].
Fig. 5.

Skeletal X-ray at the end of zoledronic acic treatment in the female proband. a Distal end of femurs and proximal end of tibiae and fibulae show reduced thickening and improved cortical sclerosis. b Diaphyses and distal end of tibiae and fibulae show reduced thickening and improved cortical sclerosis. The medullary cavity of the tibiae was partially visualized. The distal end of the tibiae and fibulae was unaffected by Camurati-Engelmann disease.
The bone pain in our male proband persisted during neridronic acid treatment (WBS 5–6), and his body movements, muscle weakness, and the dystrophic appearance did not improve significantly. Bone pain relief (WBS 2) with improvement in deambulation and body movements were evident after 2 zoledronic acid cycles. After the fourth cycle of zoledronic acid, his weight and BMI increased [17.7 kg (Z –score −1.2) and 13.2 (Z –score −2.2), respectively]. At the end of zoledronic acid treatment, X-ray examination showed the persistence of severe diaphyseal thickening and cortical sclerosis with reduction of the medullary cavity of the long bones at both upper and lower limbs (Fig. 6). However, a marked improvement of the bone lesions was observed after 2 years of zoledronic acid discontinuation (Fig. 7). BMD improved at femoral neck (Z –score −3.8), whereas it did not change at lumbar spine (Z –score 0.9) by DXA. Phalangeal AD-SoS and BTT did not vary (Z –scores −0.8 and −0.5, respectively).
Fig. 6.

Skeletal X-ray at the end of zoledronic acid treatment in the male proband. a Severe diaphyseal thickening and cortical sclerosis with marked reduction of the medullary cavity of both femurs, radii and ulnae. Bilateral coxa valga with subluxation of femoral neck. b Severe diaphyseal thickening and cortical sclerosis with marked and irregular reduction of the medullary cavity of both tibiae and fibulae. c Thickening and cortical sclerosis with reduction of the medullary cavity, mainly at the distal end of the diaphysis, in the right humerus. White circles show the typical multiple zebra lines due to the intravenous bisphosphonates infusions.
Fig. 7.

X-ray examination after 2 years of suspension of zoledronic acid treatment in the male proband. Marked improvement of diaphyseal thickening and cortical sclerosis of femurs (a), tibiae and fibulae (b), and right humerus (c) is shown. The morphology of the medullary cavity was more regular, mainly at femurs and tibiae. The typical multiple zebra lines related to the previous intravenous bisphosphonates infusions were less evident and tended to disappear as they move into the diaphysis secondary to bone remodeling.
Biochemical Markers of Bone Turnover before and during Bisphosphonate Treatment
Before bisphosphonate treatment, both probands showed a marked increase in serum levels of osteocalcin, procollagen type I N-terminal propeptide (PINP; CISbio, Codolet, France) and cross-linked carboxyterminal telopeptide of type I collagen (ICTP; UniQ ORION, Espoo, Finland): female proband: 45.8 ng/mL, normal values 6.8–34.0 ng/mL; 205.2 μg/L, normal values 15.0–75.0 μg/L, and 22.3 μg/L, normal values 2.0–6.0 μg/L, respectively; male proband: 175.6 ng/mL, normal values 35–120 ng/mL; 1,667 μg/L, normal values 260–575 μg/L, and 55.1 μg/L, normal values 10.0–17.0 μg/L, respectively. Serum levels of total alkaline phosphatase (ALP) and bone alkaline phosphatase isoenzyme (BALP; Liaison BAP-Ostase, DiaSorin, Saluggia, Italy) were slightly above the normal limit in the female proband (133 IU/L, normal values 35–105 IU/L and 35 μg/L, normal values 6–30 μg/L, respectively), whereas they were in the upper normal limit in the male proband (293 IU/L, normal values 120–300 IU/L and 85 μg/L, normal values <100 μg/L, respectively).
During bisphosphonate treatment, serum osteocalcin, PINP, and ICTP levels progressively declined within their normal range in both probands (Fig. 8); the serum levels of ALP and BALP were within their normal range at term of zoledronic acid treatment (female proband: 103 IU/L and 26 μg/L, respectively; male proband: 177 IU/L and 58 μg/L, respectively).
Fig. 8.

Serum levels of osteocalcin (a), procollagen type I N-terminal propeptide (PINP) (b), and carboxyterminal telopeptide of type I collagen (ICTP) (c) before (0) and during bisphosphonate treatment. The female proband (white circles) was treated with zoledronic acid. The male proband (black circles) was treated with 4 cycles of neridronic acid (1–4) followed by 4 cycles of zoledronic acid (5–8). Gray area: normal range for female proband (reference for young adults). Lined light-gray area: normal range for male proband (reference for prepubertal children, own laboratory).
Side Effects during Bisphosphonate Treatment and Follow-up
An acute phase reaction including mild fever with myalgia, arthralgia, and bone pain exacerbation was observed in both probands during the first bisphosphonate infusion; no other symptoms were reported during the following infusions. The symptoms were self-limiting; treatment with naproxen in the female proband and paracetamol in the male proband for some days partially improved them. None of the probands showed prolonged corrected QT interval or arrhythmias after bisphosphonate infusion. However, in both probands, bisphosphonate administration was well tolerated and no side effects were evident during the follow-up period of 2.5 years after treatment discontinuation. In both probands, occasional bone pain was treated with indomethacin (female proband: 50 mg per day; male proband: 25 mg per day) with complete relief after a few days.
Discussion
The severity of clinical symptoms was variable in the family members we examined according to the penetrance of CED [Campos-Xavier et al., 2001; Wallace et al., 2004; Janssens et al., 2006; Wu et al., 2007]. However, younger members were more severely affected than their parents, suggesting anticipation. Indeed, a trend towards increased severity in successive generations has been reported in some families, even though there was amelioration of CED outcome in others [Wallace et al., 2004; Janssen et al., 2006]. Anticipation or modifying genes could explain the variable expression and reduced penetrance [Saraiva, 1997; Wallace et al., 2004].
The missense mutation we detected in the affected members was previously identified in 4 patients with CED of the same family [Wu et al., 2007]. Some studies showed that the pathogenetic mechanism underlying the sclerosing bone phenotype in patients with CED is increased TGFB1 signaling as a result of disturbed activation or secretion of the mutant protein [Janssens et al., 2006]. Most mutations in CED clustered at the LAP region of TGFB1 [Kinoshita et al., 2000; Tzakas et al., 2005]. These mutations play a crucial role in the processing of latent TGFB1 in CED; they may cause conformational abnormalities of LAP and facilitate the dissociation of the mature TGFB1 from LAP, leading to the accumulation of mature TGFB1 [Bonewald and Dallas, 1994; Campos-Xavier et al., 2001; Saito et al., 2001; Janssens et al., 2003]. Wu et al. [2007] speculated that the mutation 505G>A E169K in exon 2 of TGFB1 resulted in a change in electrostatic interactions within the peptide molecule that affected the conformation of LAP, impairing the ability of the LAP to inhibit TGFB1 bioactivity [Bonewald and Dallas, 1994; Campos-Xavier et al., 2001; Janssens et al., 2003; Tzakas et al., 2005].
Both probands had only a transient effect on bone pain relief by NSAIDs or glucocorticoids, as also reported in case 5 and in other patients with CED [Janssens et al., 2006; Babb and Carlson, 2009; Jadhav and Ghanekar, 2013; Simsek-Kiper et al., 2014]. In a prepubertal girl aged 9 years with CED treated with losartan, bone pain and muscle mass as well as function improved but waddling gait remained unchanged [Ayyavoo et al., 2014]. Losartan treatment was associated with an improvement of limb pain and muscle strength in a 13-year-old boy with CED despite a lack of radiographic improvement of the bone lesions after 1 year of therapy [Simsek-Kiper et al., 2014].
The efficacy of bisphosphonates in reducing clinical symptoms and bone lesions in patients with CED has not been clearly defined. Some studies showed that alendronate, clodronate, pamidronate, and risedronate were ineffective on bone pain relief in patients with CED [Inaoka et al., 2001; Ziran et al., 2002; Castro et al., 2005; Janssens et al., 2006]. By contrast, bone pain relief was reported in 2 adult patients with CED receiving oral pamidronate [de Rubin et al., 1997; Chérié-Lignière et al., 1999] or alendronate [Iba et al., 2008]. In one child with CED, bone pain improved after the second cycle of intravenous pamidronate [Bondestam et al., 2007].
Both probands had bone pain relief during zoledronic acid treatment. This effect was more evident in the male proband who only had a partial reduction in bone pain during neridronic acid treatment. The greater efficacy of zoledronic acid than that of neridronic acid may be related to its higher potency [Baroncelli and Bertelloni, 2014], even though we cannot exclude that the improvement of bone symptoms could be due, at least in part, to a prolonged effect of neridronic acid on bone tissue. In addition, both probands showed radiographic improvement of the bone lesions that were associated with improved body movements, walking ability, body habitus, and quality of life.
Before bisphosphonate treatment, biochemical markers of bone turnover were markedly elevated in both probands. A similar pattern was found in 2 pediatric patients with CED [Wang et al., 2013]. An uncoupled bone formation with bone resorption may be a main mechanism causing a disregulation of periosteal apposition and endosteal resorption with a concomitant broadening of diaphyses of long bones and narrowing of medullary canal. Therefore, increased serum bone marker levels likely reflected an impaired bone modeling/remodeling with upregulation of bone formation due to the mutant TGFB1. Indeed, a crucial role of TGFB1 in regulating bone turnover was demonstrated in mice [Tang et al., 2009].
In both probands, serum bone marker levels declined within their normal range during zoledronic acid treatment and were associated with the improvement of clinical symptoms and bone lesions. A reduction in cross-linked N-telopeptides of type I collagen was also observed during alendronate treatment [Iba et al., 2008]. Moreover, it has been demonstrated that the changes of bone marker levels were correlated with bone scintigraphic indices of disease activity [Hernández et al., 1997].
Trabecular osteoporosis was detected by transiliac bone biopsy in a child with CED [Bondestam et al., 2007]. At diagnosis, both probands had normal BMD at lumbar spine but reduced BMD at femoral neck which improved during bisphosphonate treatment. However, lumbar and femoral BMD values may be quite variable in patients with CED [Inaoka et al., 2001; Castro et al., 2005; Janssens et al., 2006]. The variability in BMD is likely due to increased (hyperostosis) or decreased (osteoporosis) bone mineral content at the skeletal site of measurement. Vertebrae are usually unaffected by CED, as found in our probands. In any case, thickened vertebral body margins have been observed in some patients [Bartuseviciene et al, 2009]. In both probands, the evidence of normal quantitative ultrasound measurements suggested that phalanges of the hand were not affected by the disease, as this usually occurs in patients with CED [Bartuseviciene et al, 2009].
Both prodands did not suffer from fractures confirming previous studies [Wallace et al., 2004]; however, a fracture history was reported in a child with CED [Bondestam et al., 2007].
The mechanisms of the reduced muscle and body fat mass in patients with CED and the improvement we observed during bisphosphonate treatment are unknown. It has been shown that TGFB1 may play an important role in skeletal muscle homeostasis and that sarcopenia occurring in some inherited or acquired myopathies could be due to an aberrant TGFB1 signaling [Burks and Cohn, 2011]. Moreover, overactive TGFB1 proteins are associated with a reduction in fat and muscle mass by inhibition of adipogenesis and myogenesis, respectively [Ignotz and Massagué, 1985; Massagué et al., 1986; Zhu et al., 2004]. These effects seem to be unrelated to the affection of the skeleton [Janssens et al., 2006]. Indeed, a direct pharmacological action on muscle stem cells or myocytes with proliferation of muscle cells or activation of muscle metabolism by alendronate treatment in patients with osteoporosis has been suggested [Harada et al., 2015].
Our results show that long-term zoledronic acid treatment significantly improved clinical symptoms and bone lesions as well as ambulation, body habitus, and quality of life. Zoledronic acid treatment was associated with a normalization of the biochemical markers of bone turnover suggesting that they may be considered as surrogate indexes of CED activity. The treatment was well tolerated and no side effects were evident. These data suggest that zoledronic acid treatment may be a useful therapeutic option in children and young adults with severe CED who did not have beneficial effects by NSAIDs or glucocorticoids administration.
Statement of Ethics
The study was approved by the Ethic Committee of Department of Obstetrics, Gynecology and Pediatrics of our University-Hospital. Written informed consent was obtained from the proband or the parents to perform the study and for the administration of bisphosphonates. The study was conducted according to the Declaration of Helsinki II.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
- Ayyavoo A, Derraik JGB, Cutfield WS, Hofman PL. Elimination of pain and improvement of exercise capacity in Camurati-Engelmann disease with losartan. J Clin Endocrinol Metab. 2014;99:3978–3982. doi: 10.1210/jc.2014-2025. [DOI] [PubMed] [Google Scholar]
- Babb A, Carlson WO. Orthopaedic, radiology, pathology conference: bilateral leg pain in a 4-year-old girl. Clin Orthop Relat Res. 2009;467:317–321. doi: 10.1007/s11999-008-0350-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroncelli GI, Bertelloni S. The use of bisphosphonates in pediatrics. Horm Res Paediatr. 2014;82:290–302. doi: 10.1159/000365889. [DOI] [PubMed] [Google Scholar]
- Baroncelli GI, Vierucci F, Bertelloni S, Erba P, Zampollo E, Giuca MR. Pamidronate treatment stimulates the onset of recovery phase reducing fracture rate and skeletal deformities in patients with idiopathic juvenile osteoporosis: comparison with untreated patients. J Bone Miner Metab. 2013;31:533–543. doi: 10.1007/s00774-013-0438-9. [DOI] [PubMed] [Google Scholar]
- Bartuseviciene A, Samuilis A, Skucas J. Camurati-Engelmann disease: imaging, clinical features and differential diagnosis. Skeletal Radiol. 2009;38:1037–1043. doi: 10.1007/s00256-008-0642-1. [DOI] [PubMed] [Google Scholar]
- Baş F, Darendeliler F, Petorak I, Sadikoğlu B, Bilir A, et al. Deflazacort treatment in progressive diaphyseal dysplasia (Camurati-Engelmann disease) J Paediatr Child Health. 1999;35:401–405. doi: 10.1046/j.1440-1754.1999.00381.x. [DOI] [PubMed] [Google Scholar]
- Bondestam J, Mäyränpää MK, Ikegawa S, Marttinen E, Kröger H, Mäkitie O. Bone biopsy and densitometry findings in a child with Camurati-Engelmann disease. Clin Rheumatol. 2007;26:1773–1777. doi: 10.1007/s10067-006-0511-z. [DOI] [PubMed] [Google Scholar]
- Bonewald LF, Dallas SL. Role of active and latent transforming growth factor beta in bone formation. J Cell Biochem. 1994;55:350–357. doi: 10.1002/jcb.240550312. [DOI] [PubMed] [Google Scholar]
- Bourantas K, Tsiara S, Drosos AA. Successful treatment with corticosteroid in a patient with progressive diaphyseal dysplasia. Clin Rheumatol. 1995;14:485–486. doi: 10.1007/BF02207689. [DOI] [PubMed] [Google Scholar]
- Burks TN, Cohn RD. Role of TGF-β signaling in inherited and acquired myopathies. Skelet Muscle. 2011;1:19. doi: 10.1186/2044-5040-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos-Xavier B, Saraiva JM, Savarirayan R, Verloes A, Feingold J, et al. Phenotypic variability at the TGF-beta1 locus in Camurati-Engelmann disease. Hum Genet. 2001;109:653–658. doi: 10.1007/s00439-001-0644-8. [DOI] [PubMed] [Google Scholar]
- Carlson ML, Beatty CW, Neff BA, Link MJ, Driscoll CLW. Skull base manifestations of Camurati-Engelmann disease. Arch Otolaryngol Head Neck Surg. 2010;136:566–575. doi: 10.1001/archoto.2010.68. [DOI] [PubMed] [Google Scholar]
- Castro GR, Appenzeller S, Marques-Neto JF, Bértolo MB, Samara AM, Coimbra I. Camurati-Engelmann disease: failure of response to bisphosphonates: report of two cases. Clin Rheumatol. 2005;24:398–401. doi: 10.1007/s10067-004-1056-7. [DOI] [PubMed] [Google Scholar]
- Chérié-Lignière G, Santalena G, Parafioriti A. Pamidronate in the treatment of progressive diaphyseal dysplasia (Camurati-Engelmann disease) Clin Exp Rheumatol. 1999;17:264. [PubMed] [Google Scholar]
- de Rubin ZS, Ghiringhelli G, Mansur JL. Clinical, humoral and scintigraphic assessment of a bisphosphonate as potential treatment of diaphyseal dysplasia: Ribbing and Cammurati-Engelmann diseases (in Spanish) Medicina (B Aires) 57 Suppl. 1997;1:56–60. [PubMed] [Google Scholar]
- Harada A, Ito S, Matsui Y, Sakai Y, Takemura M, et al. Effect of alendronate on muscle mass: investigation in patients with osteoporosis. Osteop Sarcop. 2015;1:53–58. [Google Scholar]
- Hernández MV, Peris P, Guañabens N, Alvarez L, Monegal A, et al. Biochemical markers of bone turnover in Camurati-Engelmann disease: a report on four cases in one family. Calcif Tissue Int. 1997;61:48–51. doi: 10.1007/s002239900293. [DOI] [PubMed] [Google Scholar]
- Heymans O, Gebhart M, Alexiou J, Sokolow Y. Camurati-Engelmann disease. Effects of corticosteroids. Acta Clin Belg. 1998;53:189–192. [PubMed] [Google Scholar]
- Iba K, Takada J, Kamasaki H, Oda T, Hatakeyama N, et al. A significant improvement in lower limb pain after treatment with alendronate in two cases of Camurati-Engelmann disease. J Bone Miner Metab. 2008;26:107–109. doi: 10.1007/s00774-007-0783-7. [DOI] [PubMed] [Google Scholar]
- Ignotz RA, Massagué J. Type β transforming growth factor controls the adipogenic differentiation of 3T3 fibroblasts. Proc Natl Acad Sci USA. 1985;82:8530–8534. doi: 10.1073/pnas.82.24.8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaoka T, Shuke N, Sato J, Ishikawa Y, Takahashi K, et al. Scintigraphic evaluation of pamidronate and corticosteroid therapy in a patient with progressive diaphyseal dysplasia (Camurati-Engelmann disease) Clin Nucl Med. 2001;26:680–682. doi: 10.1097/00003072-200108000-00003. [DOI] [PubMed] [Google Scholar]
- Jadhav A, Ghanekar J. Camurati-Engelmann disease. Indian J Clin Prac. 2013;24:171–174. [Google Scholar]
- Janssens K, Gershoni-Baruch R, Guañabens N, Migone N, Ralston S, et al. Mutations in the gene encoding the latency-associated peptide of TGF-beta 1 cause Camurati-Engelmann disease. Nat Genet. 2000;26:273–275. doi: 10.1038/81563. [DOI] [PubMed] [Google Scholar]
- Janssens K, ten Dijke P, Ralston SH, Bergmann C, Van Hul W. Transforming growth factor-beta 1 mutations in Camurati-Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J Biol Chem. 2003;278:7718–7724. doi: 10.1074/jbc.M208857200. [DOI] [PubMed] [Google Scholar]
- Janssens K, Vanhoenacker F, Bonduelle M, Verbruggen L, Van Maldergem L, et al. Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet. 2006;43:1–11. doi: 10.1136/jmg.2005.033522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MP, McFarland CA. Increasing the reliability and validity of pain intensity measurement in chronic pain patients. Pain. 1993;55:195–203. doi: 10.1016/0304-3959(93)90148-I. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Saito T, Tomita H, Makita Y, Yoshida K, et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet. 2000;26:19–20. doi: 10.1038/79128. [DOI] [PubMed] [Google Scholar]
- Massagué J, Cheifetz S, Endo T, Nadal-Ginard B. Type beta transforming growth factor is an inhibitor of myogenic differentiation. Proc Natl Acad Sci USA. 1986;83:8206–8210. doi: 10.1073/pnas.83.21.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveh Y, Alon U, Kaftori JK, Berant M. Progressive diaphyseal dysplasia: evaluation of corticosteroid therapy. Pediatrics. 1985;75:321–323. [PubMed] [Google Scholar]
- Saito T, Kinoshita A, Yoshiura K, Makita Y, Wakui K, et al. Domain-specific mutations of a transforming growth factor (TGF)-beta 1 latency-associated peptide cause Camurati-Engelmann disease because of the formation of a constitutively active form of TGF-beta 1. J Biol Chem. 2001;276:11469–11472. doi: 10.1074/jbc.C000859200. [DOI] [PubMed] [Google Scholar]
- Saraiva JM. Progressive diaphyseal dysplasia: a three-generation family with markedly variable expressivity. Am J Med Genet. 1997;71:348–352. doi: 10.1002/(sici)1096-8628(19970822)71:3<348::aid-ajmg17>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Simsek-Kiper PO, Dikoglu E, Campos-Xavier B, Utine GE, Bonafe L, et al. Positive effects of an angiotensin II type 1 receptor antagonist in Camurati-Engelmann disease: a single case observation. Am J Med Genet A. 2014;164A:2667–2671. doi: 10.1002/ajmg.a.36692. [DOI] [PubMed] [Google Scholar]
- Tang Y, Wu X, Lei W, Pang L, Wan C, et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009;15:757–765. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzakas P, Wong BYL, Logan AG, Rubin LA, Cole DEC. Transforming growth factor beta-1 (TGFβ1) and peak bone mass: association between intragenic polymorphisms and quantitative ultrasound of the heel. BMC Musculoskelet Disord. 2005;6:29. doi: 10.1186/1471-2474-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SE, Lachman RS, Mekikian PB, Bui KK, Wilcox WR. Marked phenotypic variability in progressive diaphyseal dysplasia (Camurati-Engelmann disease): report of a four-generation pedigree, identification of a mutation in TGFB1, and review. Am J Med Genet. 2004;129A:235–247. doi: 10.1002/ajmg.a.30148. [DOI] [PubMed] [Google Scholar]
- Wang C, Zhang BH, Liu YJ, Hu YQ, He JW, Zhang ZL. Transforming growth factor-β1 gene mutations and phenotypes in pediatric patients with Camurati-Engelmann disease. Mol Med Rep. 2013;7:1695–1699. doi: 10.3892/mmr.2013.1367. [DOI] [PubMed] [Google Scholar]
- Wong DL, Baker CM. Pain in children: comparison of assessment scales. Pediatr Nurs. 1988;14:9–17. [PubMed] [Google Scholar]
- Wu S, Liang S, Yan Y, Wang Y, Li F, et al. A novel mutation of TGFB1 in a Chinese family with Camurati Engelmann disease. Bone. 2007;40:1630–1634. doi: 10.1016/j.bone.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Zhu S, Goldschmidt-Clermont PJ, Dong C. Transforming growth factor-beta-induced inhibition of myogenesis is mediated through Smad pathway and is modulated by microtubule dynamic stability. Circ Res. 2004;94:617–625. doi: 10.1161/01.RES.0000118599.25944.D5. [DOI] [PubMed] [Google Scholar]
- Ziran N, Hill S, Wright ME, Kovacs J, Robey PG, et al. Ribbing disease: radiographic and biochemical characterization, lack of response to pamidronate. Skeletal Radiol. 2002;31:714–719. doi: 10.1007/s00256-002-0552-6. [DOI] [PubMed] [Google Scholar]