

Abstract
Rapid whole-exome sequencing (rWES) is used in critically ill newborn infants to inform about diagnosis, clinical management, and prognosis. Here we report a male newborn infant with hydrops, pancytopenia, and acute liver failure who was listed for liver transplantation. Given the acuity of the presentation, the procedure-related morbidity and mortality, and lack of diagnosis, we used rWES in the proband and both parents with a turnaround time of 10 business days. rWES returned one maternally inherited, likely pathogenic and one paternally inherited, likely pathogenic variant in NPC1, suggestive of a diagnosis of Niemann–Pick disease type C (NPC). Interestingly, a diagnosis of NPC was entertained prior to rWES, but deemed unlikely in light of absent cholesterol storage on liver biopsy and near-normal oxysterol levels in dried blood. The diagnosis of NPC was confirmed on filipin stain in fibroblasts demonstrating defective cholesterol trafficking. NPC is a slowly progressive neurodegenerative disorder that may also affect the liver with overall poor prognosis. It was decided to take the infant off the transplant list and transfer to palliative care, where he died after 4 wk. This case highlights the utility of rWES in an acute clinical setting for several domains of precision medicine including (1) diagnosis, (2) prognosis and outcome, (3) management and therapy, and (4) utilization of resources.
Keywords: fatal liver failure in infancy, fetal ascites, nonimmune hydrops fetalis
INTRODUCTION
Niemann–Pick disease type C (NPC; OMIM 257220) is a disorder of intralysosomal cholesterol trafficking due to compound heterozygous or homozygous mutations in either NPC1 (95%) or NPC2 (5%), respectively (Steinberg et al. 1994; Carstea et al. 1997; Naureckiene et al. 2000). NPC1 encodes a membrane protein that has several domains homologous to other integral membrane proteins that respond to cell cholesterol content (Blanchette-Mackie 2000; Davies and Ioannou 2000). NPC2 in contrast is a lysosomal glycoprotein that is secreted, although its function is poorly understood (Naureckiene et al. 2000; Vanier and Millat 2004). NPC1 and NPC2 act in concert to facilitate the intracellular transport of LDL-derived cholesterol and lipids from the lysosomes to other sites (Cruz et al. 2000; Zhang et al. 2001). The clinical phenotype of NPC follows a continuous spectrum ranging from severe, fetal-onset to mild, adult-onset disease. The prognosis of cases with fetal-onset NPC is very poor with the majority of individuals dying from liver disease within the first months of life (Spiegel et al. 2009). A subset of these will survive to later develop severe neurological disease. Classical childhood-onset NPC typically presents with asymptomatic hepatosplenomegaly and progressive neurological disease, whereas adult-onset NPC presents with neurological dysfunction at a slower rate of progression and/or with psychosis or other psychiatric illnesses. The rate of neurocognitive decline is variable depending on genotype, ancestry, and other yet-unidentified genetic modifiers. NPC is a panethnic disorder with an estimated incidence rate of between 1/150,000 and 1/1,000,000 live births (Vanier et al. 1988; Moreno et al. 2008; Imrie et al. 2015). Diagnosis of NPC is based on clinical suspicion followed by analysis of plasma oxysterol (cholestane-3β,5α,6β-triol) levels, filipin staining of fibroblasts, and molecular confirmation (Vanier et al. 2016). Liver biopsy may reveal lipid-laden macrophages (foam cells) and polymorphous cytoplasmic bodies on electron microscopy in skin (Boustany et al. 1990; Vanier et al. 2016).
The neonatal phenotype of NPC may be obscured by the acuity of its symptoms and missed unless evaluated with specific and sensitive biochemical and molecular tests. Next-generation sequencing–based gene panels, whole-exome sequencing (WES), and whole-genome sequencing (WGS) are powerful analytical tools for the diagnosis of rare genetic disorders including NPC (Bamshad et al. 2011; Wang et al. 2013). These tools are increasingly used in critically ill neonates to reduce time to diagnosis and to open a window for individualized therapy and management in a timely manner (Reardon 2014). WES may be overall cost-saving when compared with traditional diagnostic strategies, in particular when used as a first-tier test (Vissers et al. 2017).
Here we report the utility of rWES for the diagnosis of neonatal NPC presenting with fetal hydrops and acute liver failure. The diagnosis of NPC following rWES allowed informed decision-making regarding supportive therapy and withdrawal of care, thereby alleviating the need for a costly and invasive liver transplantation.
RESULTS
Clinical Presentation and Family History
The male index patient was born to a 47-yr-old G5P3 mother with a history of depression and chronic anemia due to α-thalassemia trait and to a 45-yr-old healthy father. The parents are nonconsanguineous and of mixed Armenian, Iranian, and Turkish descent. The family history is otherwise noncontributory. The mother had two prior first-trimester miscarriages. The index patient was the third child for the couple following two healthy sons, who are 6 and 9 yr old, respectively. The pregnancy was complicated by fetal hydrops including pericardial effusions and ascites, which was first identified at 29 wk of gestation. Chorionic villous sampling at 16 wk of gestation because of advanced maternal age revealed a normal 46 XY karyotype. He was delivered by emergent C-section at 30 + 6 wk of gestation with Apgar scores of 2 and 5 at 1 and 5 min, respectively, a birth weight of 2300 g (81st percentile), and birth length of 52 cm (55th percentile).
Following delivery, the infant was intubated and transferred to the neonatal intensive care unit (NICU) where he underwent peritoneal drainage of fluids, exchange transfusions for hyperbilirubinemia, and transfusions of blood products and fresh frozen plasma. He developed acute signs of bowel obstruction and renal failure that were treated accordingly. On day 12 of life he was transferred to our institution for further diagnostic workup and evaluation for liver transplantation.
Diagnostic Studies
Hemoglobin electrophoresis showed 8% Barts, 8.4% A, and 83.6% F hemoglobin, consistent with α-thalassemia trait. Molecular testing was negative, whereas deletion/duplication analysis showed the common -α20.5 deletion, which removes HBA2 and part of HBA1, producing α0-thalassemia trait. The Coombs test was negative. Metabolic testing including urine organic acids, lysosomal enzyme analysis (acid β-glucosidase, sphingomyelinase, acid α-glucosidase, galactocerebrosidase, α-galactosidase, and α-l-iduronidase), very-long-chain fatty acids, and N- and O-glycan profiling revealed unremarkable results. A plasma acylcarnitine profile showed an isolated elevation of C5–DC, whereas urine amino acids showed multiple nonspecific elevations. The C5–DC elevation was likely explained by renal insufficiency (Hennermann et al. 2009). Oxysterol levels were mildly elevated in dried blood: 0.97 nmol/mL (normal < 0.63; Mayo Clinic), which was interpreted by the laboratory as not diagnostic for NPC.
A liver biopsy (light microscopy) showed diffuse hepatocellular damage with giant cell and pseudoacinar transformation, periportal ductal metaplasia, marked canalicular and cytoplasmic cholestasis, marked interstitial fibrosis (interstitial cirrhosis), and scattered foci of extramedullary hematopoiesis, but no evidence of reticuloendothelial or hepatocellular iron deposition. No lipid storage was noted. Cytomegalovirus and Epstein–Barr virus stains were negative, and MDR3 and BSEP immunohistochemical staining showed normal bicanalicular activity. Electron microscopy identified ultrastructural changes suggestive of a congenital storage disease. Hepatocytes with glycogen accumulation and dilated mitochondria with fractured cristae and proliferation of the smooth endoplasmic reticulum were seen together with numerous lamellar deposits within lysosomal spaces.
Genomic Analyses
Rapid WES with a turnaround of 10 business days was run as part of the diagnostic workup (XomeExpress, GeneDx Laboratories Inc.). Mitochondrial DNA sequencing including deletion/duplication testing was done in parallel with an approximate turnaround time of 35 business days. Coverage information for rWES is provided in Table 1. rWES revealed two heterozygous variants in NPC1: a maternally inherited, previously reported likely pathogenic variant in exon 18, c.2728 G>A (p.G910S) (NM_000271.4) (Tarugi et al. 2002) and a paternally inherited, likely pathogenic variant in exon 9, c.1547 G>A (p.C516Y) (NM_000271.4) (Table 2). The latter variant is not listed in the Human Genome Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) (Stenson et al. 2017), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar) database, or ExAC (http://exac.broadinstitute.org) database (Lek et al. 2016).
Table 1.
Sequencing coverage information for NPC1 variants (c.1547G>A and c.2728G>A)
| Sample | Number of reads (millions) | Mean coverage | Unmapped reads (%) | Target region >20× (%) | c.1547 G>A (reads) | c.2728 G>A (reads) |
|---|---|---|---|---|---|---|
| Proband | 146 | 175 | 0.2 | 93 | 176 | 200 |
| Mother | 172 | 205 | 0.2 | 94 | 204 | 229 |
| Father | 122 | 150 | 0.2 | 93 | 141 | 215 |
Table 2.
Variant table
| Gene | Chr | HGVS DNA | HGVS Prot | Variant | Effect | ClinVar | Genotype |
|---|---|---|---|---|---|---|---|
| NPC1 | 18q | c.1547G>A | p.C516Y | Missense | Likely path | 268186 | Heterozygous |
| NPC1 | 18q | c.2728G>A | p.G910S | Missense | Likely path | 268187 | Heterozygous |
Additional variants of unknown significance were identified in MT-CO2 and HBB (Table 3). The m.7965T>C variant (p.F127S) in MT-CO2 (Complex IV, cytochrome c oxidase subunit II) has not been reported in Mitomap (www.mitomap.org), mtDB (www.genpat.uu.se/mtDB), or Mitowheel (http://mitowheel.org). The level of heteroplasmy is ∼22% in the index patient and appears lower in the mother on Sanger sequencing (GeneDx Laboratories Inc.). The conservative amino acid substitution occurs at a nonconserved site within the protein, which makes a functional impact less likely. The maternally inherited c.208G>A (p.G70S) variant in HBB (hemoglobin β) has been reported previously as the hemoglobin City of Hope (HBCH) variant. The G70S variant has an allele frequency of 0.1% in Europeans in ExAC and occurs at a position that is not conserved. There were no variants identified in NPC2.
Table 3.
Incidental findings
| Gene | Variant | Protein | Inheritance | Classification |
|---|---|---|---|---|
| MT-CO2 | m.7965T>C | p.F127S | Maternal | VUS |
| HBB | c.208G>A | p.G70S | Maternal | VUS |
Mutations in their Structural Context
The main biological function of NPC1 is to transport lipid molecules past the glycocalyx and across the lysosomal membrane (Kwon et al. 2009; Li et al. 2015). The efficiency of this process is markedly increased through the initial binding of the lipid molecule to NPC2 that is present in a soluble form in the lysosomal lumen (Infante et al. 2008). The three-dimensional structure of NPC1 (Fig. 1) shows a protein consisting of a large transmembrane unit, which includes the region formerly known as “sterol-sensing domain,” and two structural luminal units (Wang et al. 2016). The elements of these structural domains are not contiguous on the sequence (Fig. 1A). The amino-terminal regions form a lipid-binding luminal unit, distal to the membrane. Interleaved transmembrane helices and two quasi-symmetrical halves of a membrane proximal domain, the so-called NPC1-C (former NPC2-binding domain) and NPC1-I domains are located downstream from the sequence (Wang et al. 2016). The two NPC1 variants of our patient, p.C516Y and p.G910S, fall within these two domains.
Figure 1.
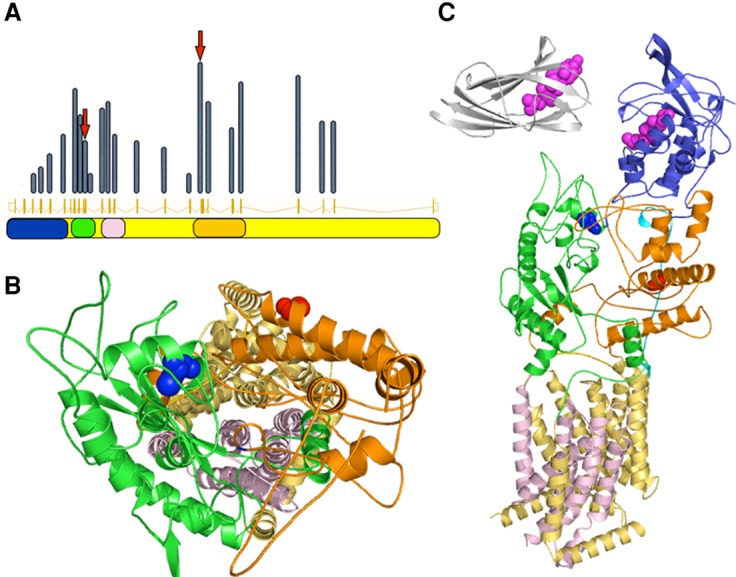
NPC1 domains are color coded consistently throughout the panels—blue, cholesterol binding domain; green, NPC1-C; orange, NPC1-I, quasi symmetric partner of NPC1-C; pink, sterol sensing domain; yellow, transmembrane region. (A) Relative exonic distribution of previously published NPC1 mutations. Middle row shows the exonic structure, whereas the height of each bar indicates the relative frequency of disease-causing mutations in each exon. Positions of the mutations described in this work are indicated with arrows. The corresponding structural domains are shown in the bottom row. (B) Structural position of the mutated amino acids Cys516 (blue spheres) and Gly910 (red spheres). A top down view of the putative lipid transport channel shows that Cys516 appears to be closer to the inner surface of the channel. The amino-terminal domain is hidden to not obstruct the view. (C) NPC1 protein domains and affected amino acids. NPC2 is also shown on the same scale (silver). Cholesterol molecules are shown in magenta.
NPC1-C plays a crucial role in facilitating cholesterol transfer from NPC2 onto NPC1's amino terminus. This domain contains four cysteine residues that are all involved in forming disulfide bonds (C468–C479, C516–C533). Disruption of a disulfide bond, as found in our patient, is expected to destabilize the entire structural domain. Accordingly, the C516–C533 pair is completely conserved across all vertebrate and insect species (Fig. 2). Judging by its location we may conjecture that this structural destabilization has an impact on the transport of lipids toward the transmembrane channel and on the transfer of cholesterol from NPC2 to NPC1-C for export from lysosomes (Fig. 1; Deffieu and Pfeffer 2011). The neighboring R518 is directly involved in NPC2 binding.
Figure 2.

Cross-species conservation of the NPC1 protein sequence in relation to the two variants p.C516Y and p.G910S, respectively.
The role of p.G910S is difficult to ascertain. It belongs to a short motif (CGG) conserved throughout vertebrates (Fig. 2). p.G910S has been reported in association with NPC, although its impact remains obscure until the mode of interaction between NPC1, NPC2, glycocalyx, and perhaps other regulators is better understood (Tarugi et al. 2002).
Biochemical Testing
Following rWES the diagnosis of NPC1 was confirmed by analysis of oxysterol and 7-ketocholesterol plasma levels and filipin staining of cultured skin fibroblasts. Plasma oxysterol and 7-ketocholesterol levels were markedly elevated: 0.71 nmol/ml (normal < 0.02) and >1.00 nmol/ml (normal < 0.05), respectively. Filipin staining of fibroblast showed massive intralysosomal accumulation of cholesterol compared with control (Fig. 3).
Figure 3.

Filipin stain of nonesterified cholesterol in fibroblasts with (A–C) and without (D–F) stimulation of LDL cholesterol uptake. (A,D) Normal control showing 100% esterification. (B,E) Known NPC 1 patient showing 1% esterification. (C,F) Index patient showing 2% esterification. Magnification, 20×.
DISCUSSION
We demonstrate the utility of rapid whole-exome sequencing for the diagnosis of prenatal-onset NPC in a young infant with acute liver failure and a history of fetal hydrops. Prenatal-onset NPC has to date only been reported in very few patients and is associated with early demise in the majority of reported cases (Maconochie et al. 1989; Manning et al. 1990; Spiegel et al. 2009; Surmeli-Onay et al. 2013; Imrie et al. 2015). The clinical course is highlighted by Maconochie et al. (1989) who reported two female infants with a history of severe fetal hydrops, hepatosplenomegaly, and acute liver failure who were born to unrelated, healthy parents. Fetal hydrops was diagnosed at 18 and 33 wk of gestational age. The diagnosis of NPC was confirmed postmortem through identification of pleomorphic lamellar cytoplasmic inclusions in neurons in one infant who died at 19 d of age and decreased intracellular esterification of exogenous cholesterol in the second infant. The second infant recovered from liver failure and was found to have mild hepatosplenomegaly and normal psychomotor development on follow-up at 17 mo of age (Maconochie et al. 1989).
The presence of nonimmune fetal hydrops and ascites in addition to hepatosplenomegaly, liver failure, anemia, and thrombocytopenia should raise the suspicion for a wide spectrum of disorders including NPC that may be indistinguishable from each other clinically (Spiegel et al. 2009). Consequently, the initial diagnostic approach needs to be broad and include analysis of disease-specific enzymes and biomarker in addition to histochemistry and electron microscopy of affected liver tissue (Norton et al. 2015). The correct diagnosis may nevertheless be elusive because of noninformative results, as demonstrated in our case where oxysterol levels in dried blood were within the borderline range and liver biopsy did not reveal changes typically observed in NPC, although there was evidence of lamellar inclusion bodies in lysosomes on electron microscopy (Vanier et al. 2016).
The advent of next-generation sequencing has moved molecular testing from a second- or third-tier confirmatory to a first-tier diagnostic tool realizing its limitations (Wang et al. 2013). WES and WGS with a rapid turnaround time are playing an increasingly important role for the diagnosis of critically ill infants and children (Smith et al. 2015). A timely diagnosis is critical to preserve optimal health outcomes, most importantly when disease-specific therapies (enzyme replacement, transplantation, or others) are available (Solomon and Muro 2017). As our patient was listed for liver transplantation, rWES was done, which revealed two likely pathogenic variants in NPC1, whose functional relevance was confirmed through analysis of oxysterol levels in plasma and cholesterol accumulation in fibroblast through filipin stain. Although it was impossible to conduct an in-depth health economic assessment, it is obvious that the direct and indirect costs for a pediatric liver transplantation far exceed those for rWES.
We cannot exclude an additional effect on the clinical course through the missense variant that was identified in MT-CO2 (22% heteroplasmy) by mitochondrial DNA sequencing. The conservative amino acid substitution occurs at a nonconserved site within MT-CO2, which makes a functional impact less likely. Deficiency of cytochrome c oxidase (COX) results in a heterogeneous spectrum of neurodevelopmental phenotypes of different severity and age of onset. To the best of our knowledge acute liver failure and fetal hydrops have not been reported in any case with confirmed COX deficiency. In contrast, our patient did not exhibit severe lactic acidosis, which is typically associated with COX deficiency (Wong et al. 2001).
Our case highlights the utility of rWES in an acute clinical setting for several domains of precision medicine including (1) diagnosis, (2) prognosis and outcome, (3) management and therapy, and (4) utilization of resources. The diagnosis of atypical presentations of otherwise well-known conditions may be delayed without WES, particularly when initial diagnostic testing is noninformative.
METHODS
Whole-Exome Sequencing
Exome sequencing at GeneDx was performed on exon targets isolated by capture with the Clinical Research Exome kit (Agilent Technologies) using Illumina HiSeq 2500 2×100 bp. The other sequencing technology and variant interpretation protocol has been previously described (Tanaka et al. 2015). The general assertion criteria for variant classification are publicly available on the GeneDx ClinVar submission page (http://www.ncbi.nlm.nih.gov/clinvar/submitters/26957/).
Protein Structure
Structures of NPC1 (Wang et al. 2016; PDB identifier 3jd8) and NPC2 (Xu et al. 2007; PDB identifier 2hka) were downloaded from the Protein Data Bank (PDB, Berman et al. 2000; http://www.rcsb.org). The sequence alignment for vertebrates (Khoo et al. 2014; http://exolocator.eopsf.org) was extended to include insects from the RefSeq database (O'Leary et al. 2016; https://www.ncbi.nlm.nih.gov/refseq/).
Skin Biopsy and Cell Culture
In brief, a 3-mm punch biopsy was done on the outer right thigh under sterile conditions following subcutaneous local anesthesia under sterile conditions. The biopsy material was transferred to sterile DMEM supplemented with 10% fetal bovine serum (Life Technologies Inc.) and stored at room temperature until the following day. Fat tissue was removed from the biopsy, which was then cut into smaller pieces and divided between T25 flasks containing sterile DMEM 10% fetal bovine serum. T25 flasks were incubated at 37°C and 5% CO2 until confluency. Skin fibroblasts were harvested and re-plated into T75 flasks for subsequent filipin staining.
Filipin Stain
The formation of (3)H-cholesterol oleate is measured against cells that are fed only (3)H-oleic acid without low-density lipoprotein. The formed (3)H-cholesterol oleate is separated from (3)H-oleic acid and its other esterified forms by thin-layer chromatography (TLC). The areas on the TLC plates corresponding to the cholesterol oleate markers are then scraped and counted on a liquid scintillation counter. Lowry proteins are done on the cell pellet to normalize the assay roughly to cell numbers. Filipin staining for cholesterol is performed on all specimens with low values (Kruth et al. 1986; Pentchev et al. 1986).
ADDITIONAL INFORMATION
Data Deposition and Access
Our patient consent does not permit patient sequence data to be uploaded to a data repository. The NPC1 variants reported have been deposited in the ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) database with accession numbers SCV000328675.3 (c.2728G>A (p.Gly910Ser) and SCV000328674.3 (c.1547G>A (p.Cys516Tyr), and the HBB (c.208G>A(p.G70S)) variant can be found under accession number SCV000599992. The MT-CO2 m.7965T>C (p.F127S) variant has been submitted to ClinVar under accession number SCV000599993 and MSeqDR under accession number MSCV_0000006.
Ethics Statement
The family was enrolled in The Manton Center for Orphan Disease Research, Gene Discovery Core under informed consent governed by the Institutional Review Board of Boston Children's Hospital (IRB number is 10-02-0053). Written informed consent was provided by both parents for research and publication.
Acknowledgments
We thank the family for their interest in this work and willingness to participate in the research study. We thank the Gene Discovery Core of The Manton Center for Orphan Disease Research for providing resources and support in patient consenting, sample collection, sequencing, and sharing of information and samples.
Author Contributions
M.R., A.O.-L., M.S., and K.L. contributed to patient recruitment and phenotyping. M.C. contributed to sequence data analysis and interpretation. I.M. and S.E. contributed to functional evaluation of the variants. M.R., S.E., and O.B. contributed to writing the initial draft of the manuscript. All authors contributed to revising the manuscript and reviewing the final draft.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Amy Brower
Anonymous
REFERENCES
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12: 745–755. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acid Res 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette-Mackie EJ. 2000. Intracellular cholesterol trafficking: role of the NPC1 protein. Biochim Biophys Acta 1486: 171–183. [DOI] [PubMed] [Google Scholar]
- Boustany RN, Kaye E, Alroy J. 1990. Ultrastructural findings in skin from patients with Niemann–Pick disease type C. Pediatr Neurol 6: 177–183. [DOI] [PubMed] [Google Scholar]
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, et al. 1997. Niemann–Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277: 228–231. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Sugii S, Yu C, Chang TY. 2000. Role of Niemann–Pick type C1 protein in intracellular trafficking of low density lipoprotein-derived cholesterol. J Biol Chem 275: 4013–4021. [DOI] [PubMed] [Google Scholar]
- Davies JP, Ioannou YA. 2000. Topological analysis of Niemann–Pick C1 protein reveals that the membrane orientation of the putative sterol-sensing domain is identical to those of 3-hydroxy-3-methylglutaryl-CoA reductase and sterol regulatory element binding protein cleavage-activating protein. J Biol Chem 275: 24367–24374. [DOI] [PubMed] [Google Scholar]
- Deffieu MS, Pfeffer SR. 2011. Niemann Pick type C1 function requires luminal domain residues that mediate cholesterol-dependent NPC2 binding. Proc Natl Acad Sci 108: 18932–18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennermann JB, Roloff S, Gellermann J, Grüters A, Klein J. 2009. False-positive newborn screening mimicking glutaric aciduria type I in infants with renal insufficiency. J Inherit Metab Dis 32Suppl 1: S355–S359. [DOI] [PubMed] [Google Scholar]
- Imrie J, Heptinstall L, Knight S, Strong K. 2015. Observational cohort study of the natural history of Niemann–Pick disease type C in the UK: a 5-year update from the UK clinical database. BMC Neurol 15: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. 2008. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci 105: 15287–15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo AA, Ogrizek-Tomaš M, Bulović A, Korpar M, Gürler E, Slijepčević I, Šikić M, Mihalek I. 2014. ExoLocator—an online view into genetic makeup of vertebrate proteins. Nucl Acid Res 42: D879–D881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruth HS, Comly ME, Butler JD, Vanier MT, Fink JK, Wenger DA, Patel S, Pentchev PG. 1986. Type C Niemann–Pick disease: abnormal metabolism of low density lipoprotein in homozygous and heterozygous fibroblasts. J Biol Chem 261: 16769–16774. [PubMed] [Google Scholar]
- Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, Infante RE. 2009. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137: 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria A, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Deffieu MS, Lee PL, Saha P, Pfeffer SR. 2015. Glycosylation inhibition reduces cholesterol accumulation in NPC1 protein-deficient cells. Proc Natl Acad Sci 112: 14876–14881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maconochie IK, Chong S, Mieli-Vergani G, Lake BD, Mowat AP. 1989. Fetal ascites: an unusual presentation of Niemann–Pick disease type C. Arch Dis Child 64: 1391–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning DJ, Price WI, Pearse RG. 1990. Fetal ascites: an unusual presentation of Niemann–Pick disease type C. Arch Dis Child 65: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno R, Lardennois C, Drouin-Garrod V, Verspyck E, Marret S, Laquerrière A. 2008. Prenatal revelation of Niemann–Pick disease type C in siblings. Acta Paediatr 97: 1136–1139. [DOI] [PubMed] [Google Scholar]
- Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. 2000. Identification of HE1 as the second gene of Niemann–Pick C disease. Science 290: 2298–2301. [DOI] [PubMed] [Google Scholar]
- Norton ME, Chauhan SP, Dashe JS. 2015. Society for maternal–fetal medicine (SMFM) clinical guideline #7: nonimmune hydrops fetalis. Am J Obstet Gynecol 212: 127–139. [DOI] [PubMed] [Google Scholar]
- O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentchev PG, Kruth HS, Comly ME, Butler JD, Vanier MT, Wenger DA, Patel S. 1986. Type C Niemann–Pick disease. A parallel loss of regulatory responses in both the uptake and esterification of low density lipoprotein-derived cholesterol in cultured fibroblasts. J Biol Chem 261: 16775–16780. [PubMed] [Google Scholar]
- Reardon S. 2014. Fast genetic sequencing saves newborn lives. Nature 514: 13–14. [DOI] [PubMed] [Google Scholar]
- Smith LD, Willig LK, Kingsmore SF. 2015. Whole-exome and whole-genome sequencing in critically Ill neonates suspected of having single-gene disorders. Cold Spring Harb Perspect Med 6: a023168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon M, Muro S. 2017. Lysosomal enzyme replacement therapies: historical development, clinical outcomes, and future perspectives. Adv Drug Deliv Rev 10.1016/j.addr.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel R, Raas-Rothschild A, Reish O, Regev M, Meiner V, Bargal R, Sury V, Meir K, Nadjari M, Hermann G, et al. 2009. The clinical spectrum of fetal Niemann–Pick type C. Am J Med Genet A 149A: 446–450. [DOI] [PubMed] [Google Scholar]
- Steinberg SJ, Ward CP, Fensom AH. 1994. Complementation studies in Niemann–Pick disease type C indicate the existence of a second group. J Med Genet 31: 317–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. 2017. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing. Hum Genet 136: 665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeli-Onay O, Yakarisik S, Korkmaz A, Akcoren Z, Yuce A, Runz H, Stampfer M, Yurdakok M. 2013. Prenatal-onset Niemann–Pick type C disease with nonimmune hydrops fetalis. Pediatr Neonatol 54: 344–347. [DOI] [PubMed] [Google Scholar]
- Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, Niyazov D, Garnica A, Gratz E, Deardorff M, et al. 2015. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet 97: 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarugi P, Ballarini G, Bembi B, Battisti C, Palmeri S, Panzani F, Di Leo E, Martini C, Federico A, Calandra S. 2002. Niemann–Pick type C disease: mutations of NPC1 gene and evidence of abnormal expression of some mutant alleles in fibroblasts. J Lipid Res 43: 1908–1919. [DOI] [PubMed] [Google Scholar]
- Vanier MT, Millat G. 2004. Structure and function of the NPC2 protein. Biochim Biophys Acta 1685: 14–21. [DOI] [PubMed] [Google Scholar]
- Vanier MT, Wenger DA, Comly ME, Rousson R, Brady RO, Pentchev PG. 1988. Niemann–Pick disease group C: clinical variability and diagnosis based on defective cholesterol esterification. A collaborative study on 70 patients. Clin Genet 33: 331–348. [DOI] [PubMed] [Google Scholar]
- Vanier MT, Gissen P, Bauer P, Coll MJ, Burlina A, Hendriksz CJ, Latour P, Goizet C, Welford RW, Marquardt T, Kolb SA. 2016. Diagnostic tests for Niemann–Pick disease type C (NP-C): a critical review. Mol Genet Metab 118: 244–254. [DOI] [PubMed] [Google Scholar]
- Vissers LE, Van Nimwegen KJ, Schieving JH, Kamsteeg EJ, Kleefstra T, Yntema HG, Pfundt R, Van der Wilt GJ, Krabbenborg L, Brunner HG, et al. 2017. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med 10.1038/gim.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu X, Yang BZ, Gelernter J. 2013. The role and challenges of exome sequencing in studies of human diseases. Front Genet 4: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Shi Y, Song J, Qi J, Lu G, Yan J, Gao GF. 2016. Ebola viral glycoprotein bound to its endosomal receptor Niemann–Pick C1. Cell 164: 258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LJ, Dai P, Tan D, Lipson M, Grix A, Sifry-Platt M, Gropman A, Chen TJ. 2001. Severe lactic acidosis caused by a novel frame-shift mutation in mitochondrial-encoded cytochrome c oxidase subunit II. Am J Med Genet 102: 95–99. [DOI] [PubMed] [Google Scholar]
- Xu S, Benoff B, Liou HL, Lobel P, Stock AM. 2007. Structural basis of sterol binding by NPC2, a lyososmal protein deficient in Niemann Pick type C2 disease. J Biol Chem 282: 23525–23531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Dwyer NK, Neufeld EB, Love DC, Cooney A, Comly M, Patel S, Watari H, Strauss JF III, Pentchev PG, et al. 2001. Sterol-modulated glycolipid sorting occurs in Niemann–Pick C1 late endosomes. J Biol Chem 276: 3417–3425. [DOI] [PubMed] [Google Scholar]