

Abstract
Using whole-exome sequencing, we identified seven unrelated individuals with global developmental delay, hypotonia, dysmorphic facial features, and an increased frequency of short stature, ataxia, and autism with de novo heterozygous frameshift, nonsense, splice, and missense variants in the Early B-cell Transcription Factor Family Member 3 (EBF3) gene. EBF3 is a member of the collier/olfactory-1/early B-cell factor (COE) family of proteins, which are required for central nervous system (CNS) development. COE proteins are highly evolutionarily conserved and regulate neuronal specification, migration, axon guidance, and dendritogenesis during development and are essential for maintaining neuronal identity in adult neurons. Haploinsufficiency of EBF3 may affect brain development and function, resulting in developmental delay, intellectual disability, and behavioral differences observed in individuals with a deleterious variant in EBF3.
Keywords: autism; central hypotonia; intellectual disability, mild; moderate global developmental delay; neurogenic bladder
INTRODUCTION
Early B-cell factor 3 (EBF3) is located on Chromosome 10q26.3 and is a member of the collier/olfactory-1/EBF (COE) family of transcription factors required for the development and differentiation of various cell types across species, from planarians to humans (Bu and Su 2003; Siponen et al. 2010; Cowles et al. 2014). The COE family of proteins has low sequence similarity to other protein families, but all COE proteins share a conserved amino-terminal DNA-binding domain with a unique zinc-finger binding motif, an immunogolubulin Ig-like, plexins, transcription factors (IPT/TIG) domain, and an atypical helix–loop–helix domain with a dimerization motif (Fig. 1A; Liberg et al. 2002; Siponen et al. 2010). COE transcription factors play an important role in the nervous system and brain development, including neuronal differentiation, migration, axon guidance, and dendritogenesis, and are also necessary for the specification and maintenance of neuronal identity in adult neurons (Wang et al. 2004). Mice and humans have four COE paralogs, EBF1-4 (Daburon et al. 2008), which may have complementary and redundant roles (Dubois and Vincent 2001; Liberg et al. 2002). Among the COE paralogs, EBF3 is highly expressed throughout the brain during development in mice and humans (Zhao et al. 2006; Tao et al. 2015) and is required for the migration of Cajal–Retzius cells during corticogenesis and the terminal differentiation and maintenance of specialized neurons in the adult brain (Dubois and Vincent 2001; Garcia-Dominguez et al. 2003; Thuret et al. 2004; Chiara et al. 2012). EBF3 also acts as a putative tumor suppressor by activating cell cycle arrest and inducing apoptosis (Zhao et al. 2006).
Figure 1.

(A) De novo variants in EBF3. Missense variants in EBF3 (NP_001005463.1) are shown above the protein diagram and likely gene-disrupting mutations are below. The variants identified in our patients are in black and ones in individuals reported by others are in gray. COE1, collier/olfactory-1/EBF. DBD, DNA-binding domain. IPT, Ig-like, plexins, transcription factors domain. HLHLH, atypical helix–loop–helix (HLH) domain. TAD, transactivation domain. Light blue region in COE1 DBD represents unique zinc finger binding motif. (B) Sequence alignment of EBF3 and its homologs across species with residues mutated in our patients in red. Macmu, rhesus monkey, Xentr, Xenopus tropicalis, Danre, zebrafish, Drome, Drosophila melanogaster, Tribr, parasitic roundworm.
Previous reports have identified approximately 100 patients with terminal 10q deletions, who demonstrate a wide range of clinical features frequently including delayed development, intellectual disability, and craniosynostosis (Faria et al. 2015). EBF3 is within a shared deletion of Chromosome 10q26.3 in many of these patients. Four recent reports identified a total of 20 unrelated individuals and two siblings who have de novo variants in EBF3 and a distinct neurodevelopmental syndrome (Blackburn et al. 2017; Chao et al. 2017; Harms et al. 2017; Sleven et al. 2017). Here we add further evidence for the role of EBF3 in brain development and expand the phenotype of this syndrome caused by pathogenic variants in EBF3. We describe seven unrelated individuals who have heterozygous de novo variants in EBF3 that are predicted to be deleterious and who share common features of global developmental delay, intellectual disability (ID), hypotonia, and dysmorphic features.
RESULTS
Clinical WES was performed on 8057 individuals with developmental delay and/or intellectual disabilities in a single clinical laboratory. In 11 affected individuals from 11 unrelated families, we identified de novo variants in EBF3 as potentially causative for the neurodevelopmental phenotype. Candidate disease-causing variants in the EBF3 gene were confirmed by Sanger sequencing to be de novo. Only seven of the 11 de novo predicted pathogenic variants we observed are reported in the manuscript based on permission of the referring physicians to report the results. Some of the patients who were diagnosed were included in other publications and were therefore not included in this series. The variants include one nonsense, two frameshift deletions, one splice, and three missense variants (Fig. 1). All seven variants identified are novel, located in highly evolutionarily conserved regions across species, and predicted to be deleterious by SIFT (http://sift.jcvi.org/), CADD (http://cadd.gs.washington.edu/), MetaSVM (https://omictools.com/meta-svm-tool), and MutationTaster (http://www.mutationtaster.org/) (Table 1; Fig. 1B). No likely gene-disrupting EBF3 variants were detected in ExAC (http://exac.broadinstitute.org/), 1000 Genomes (http://www.internationalgenome.org/), ESP (http://evs.gs.washington.edu/EVS/), or our own internal database of 24,709 exomes of unaffected parents of children referred for testing. Additionally, the non-TCGA ExAC v0.3.1 gene tolerance scores of misZ = 4.89 and p(LI) = 1.00 indicate that the gene is highly constrained against both missense and loss-of-function (LOF) variation across humans, respectively. Using TADA (He et al. 2013) default parameters for ID/DD and the expected mutation rate of 2.331847 × 10−05 for missense variants and 8.693607 × 10−07 for LOF variants (Samocha et al. 2014), we calculate a false-discovery corrected q-value of 3.84 × 10−08 for observing six de novo LOF and five de novo missense variants in EFB3 within our cohort.
Table 1.
Predicted pathogenicity and allele frequencies of EBF3 variants
| Patient | Chromosome 10 coordinates (GRCh37/hg19) | Nucleotide change | Amino acid change | SIFT | PROVEAN | PolyPhen2 | Mutation-Taster | CADD Phred | Allele frequency in ExAC and GeneDx database of 49,418 alleles |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 131761731:T>G | c.191A>C | K64T | Damaging | Deleterious (−4.3) | Probably damaging (0.99) | Disease-causing | 19.3 | 0 |
| 2 | 131761679:C>- | c.244delG | V82WfsX50 | N/A | N/A | N/A | N/A | N/A | 0 |
| 3 | 131757212:G>T | c.471C>A | H157Q | Damaging | Deleterious (−6.2) | Possibly damaging (0.9) | Disease-causing | 18.3 | 0 |
| 4 | 131755591:C>T | c.486-1G>A | IVS5-1G>A | N/A | N/A | N/A | Disease-causing | 32 | 0 |
| 5 | 131676052:G>A | c.616C>T | R206X | N/A | N/A | N/A | Automatic disease-causing | 18.1 | 0 |
| 6 | 131676042:C>T | c.626G>A | R209Q | Damaging | Deleterious (−3.4) | Possibly damaging (0.9) | Disease-causing | 20.4 | 0 |
| 7 | 131639239GTACT GCTGGGGA>- | c.1402_1414d eI13 | T464PfsX10 | N/A | N/A | N/A | N/A | N/A | 0 |
Clinical Presentation and Family History
The seven unrelated individuals include five females and two males who range in age from 1 to 24 yr (see Table 2; Supplemental Table S1). They all share similar clinical features of global developmental delay or intellectual disabilities and of hypotonia or rarely hypertonia. Four of the five individuals over the age of three were verbal. Many of the individuals also demonstrated autism and/or attention deficit hyperactivity disorder (ADHD) and behavioral differences including poor eye contact, self-injurious behavior, or altered pain sensitivity. The only adult in our series also had tics and auditory and visual hallucinations. Notably, none had seizures nor were there consistent brain malformations, and head circumferences ranged from relatively small to relatively large. Most of the individuals have distinctive physical features including triangular face, synophrys, small ears, highly arched palate and crowded teeth, and single palmar crease (Fig. 2). All of our patients have moderate-to-severe neurodevelopmental impairment. Approximately half of the patients have autism and half of them have motor coordination problems including ataxia. Six of the seven individuals had neurological findings, including coordination issues, insensitivity to pain, muscle weakness, and ataxic gait. Abnormal brain MRIs were observed, including vermis hypoplasia, delayed myelination, or irregular frontal cerebral white matter and protuberance of optic papillae in three separate patients (Fig. 3). Additional notable features in the minority of individuals included altered pain sensitivity and sleep issues. Ophthalmologic problems were common and included strabismus, esotropia, amblyopia, and delayed visual maturation. Short stature was common and height was in the lower quartile for all but one individual. Skeletal abnormalities were less common and included pectus excavatum, severe scoliosis, pronation, and hallux valgus. Genitourinary issues included microphallus in two, urinary tract infections and hydronephrosis in one, and unilateral renal duplication, and vesicoureteral reflux in another.
Table 2.
Clinical details of patients with EBF3 variants
| Patient | Age | Sex | Variant | Age at sitting | Age at walking | Verbal skills | Muscle tone | Additional neurological findings | Vision | Seizure | Abnormal behavior | ADD/ADHD | Brain MRI | Renal or urogenital issues | Gl |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 30 mo | M | c.191A>C p.Lys64Thr; | 18 mo | Cannot walk on own, able to stand without support | Nonverbal; able to communicate with sign language | Hypotonia | Coordination issues; insensitivity/decreased sensitivity to pain; fine and gross motor difficulties | N/A | None | Moderate eye contact; did not smile until 11 mo old | N/A | N/A | Small penis | History of poor weight gain |
| 2 | 24 yo | F | c.244delG, p.Val82TrpfsX50 | 9 mo | 2 yo | Talked at 9 mo; Full sentences, normal mild articulation problems | Hypotonia | None | Strabismus, surgically repaired | None | Autistic features; tics; hallucinations (auditory and visual); emotional fragility; self- injurious behavior; hair pulling | No | Normal | Normal | Normal |
| 3 | 10 yo | F | c.471C>A, p.His157Gln | 1 yo | 3 yo | Talked at 5 yo; normal vocabulary for age with articulation differences | Hypotonia | Fine motor difficulties; decreased pain sensitivity | Strabismus | None | Autism; tantrums and melt-downs | Hyperactivity | Normal | Vesicostomy surgery for vesicoureteral reflux and poor bladder emptying; left renal duplication; urine retention with neurogenic bladder, needs nocturnal catheter | Normal |
| 4 | 11 yo | F | c.486-1G>A, IVS5-1G>A | N/A | Cannot walk on own, needs assistance or walker | Nonverbal, no words but hums tunes | Hypotonia | Muscle weakness; ataxic gait | Strabismus, surgically repaired | None | Autistic features; rubs face on hard objects to point of bruising | No | Vermis hypoplasia | Normal | Constipation; requires G- tube feeds; takes only baby foods orally |
| 5 | 11 yo | F | c.616C>T, p.Arg206X | 4 mo | 14 mo; Wears leg braces | Talked at 10 mo; Articulation defect | Hypotonia | Hyporeflexia or difficult to elicit DTRs; mild dysmetria and gait incoordination; inconsistent sensory exam, mild proximal muscle weakness | Refractive error | None | Autism; oppositional defiant disorder | ADHD | Small schizencephalic cleft in right temporal lobe and delayed myelination at 2 yo, later resolved | History of UTIs and hydronephrosis, resolved with growth | Excess weight gain, BMI 33.5 (≫95th percentile) |
| 6 | 15 mo | M | c.626G>A p.Arg209Gln | 9 mo | Cannot walk at 12 mo | Babbling at 8 mo; No clear first words at 12 mo | Hypotonia | Muscle weakness; ataxic gait | Strabismus | None | Some eye contact, but no social smile | N/A | Curvilinear tract-like signal abnormality in left frontal cerebral white matter; protuberance of right optic papillae | Small penis; testes high in scrotum | Normal |
| 7 | 4 yo | F | c.1402_1414del13, p.Thr464ProfsX10 | 8 mo | 2 yo | Talked at 2 yo; Speaking in full sentences at 4 yo | Hypertonia in hips | Muscle weakness; ataxic gait | Normal | None | Oral fixation, awakens at night | No | Normal | Recurrent urinary tract infections | Normal |
See Supplemental Table S1 for additional details of clinical presentations.
yo, years old; ADD, attention deficit disorder; ADHD attention deficit hyperactivity disorder; N/A, not available.
Figure 2.
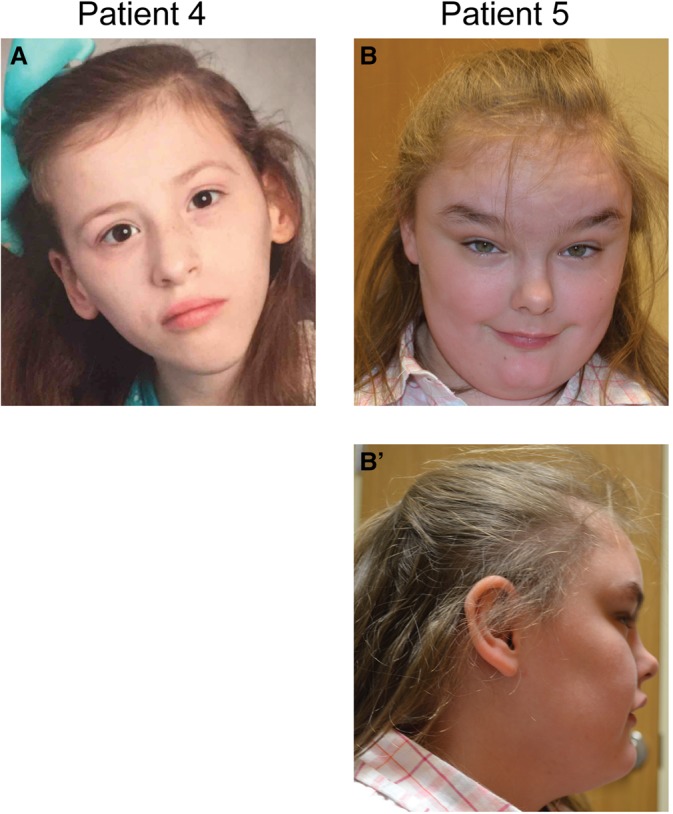
Photographs of patients with de novo variants in EBF3. (A) Patient 4 at 11 yr old. Note triangular face. (B,B′) Patient 5 at 11 yr old. Note round, mildly coarse face, mild maxillary hypoplasia and prominent mandible, highly arched eyebrows and synophrys, long palpebral fissures, short philtrum and thin upper lip, and anteverted nares.
Figure 3.
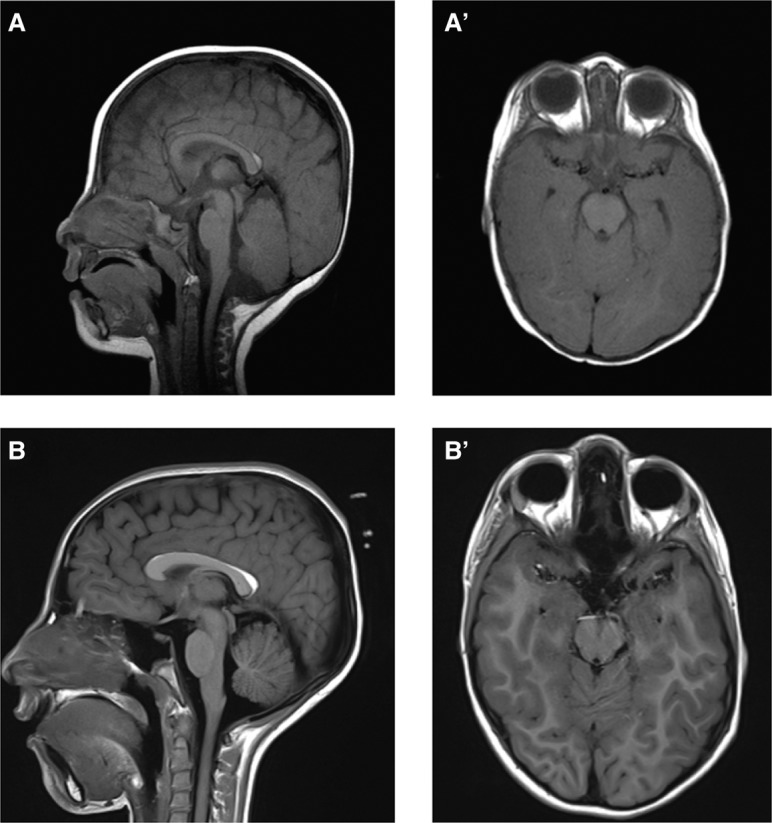
Brain imaging data of individual with EBF3 variant. (A,B) Sagittal and (A′,B′) axial images of Individual 4 at 9 mo (A,A′) and 9 yr (B,B′) of age show a mild hypoplasia of the cerebellar vermis, mildly dysplastic corpus callosum, and small pericallosal lipoma without evidence of significant progression.
DISCUSSION
We add to the emerging evidence for a clinical syndrome characterized by global developmental delay/intellectual disability, autism, hypotonia, and dysmorphic features due to heterozygous de novo predicted pathogenic variants in EBF3. Although not observed in all patients, ataxia/coordination problems, strabismus, skeletal abnormalities, short stature, and urogenital anomalies are also common features. The allelic spectrum we observed includes several likely gene-disrupting variants: one nonsense, two frameshift, and one splice variant. In addition there are three de novo missense variants, (p.(Lys64Thr), p.(His157Gln), and p.(Arg209Gln), which are all in the COE1 DNA-binding domain (DBD) (Fig. 4; Siponen et al. 2010). The p.(His157Gln) mutation lies in the “EBF zinc knuckle” motif located in the COE1 DBD, which is crucial for zinc coordination and positioning of residues for DNA binding. The splice site variant, c.486-1G>A, may also cause the loss of the splice acceptor site and induce skipping of exon 6, which encodes the conserved zinc knuckle motif in the COE1 DBD. Molecular modeling of EBF3 mutations at the zinc knuckle has demonstrated decreased DNA affinity resulting in aberrant DNA binding (Blackburn et al. 2017). Altered expression or binding of EBF3 is likely responsible for the predominance of neurological symptoms seen in our patients. Mutations in Ebf3 in the brains of adult mice result in a modest reduction in olfactory bulb size and defective olfactory axon projections to the dorsal and lateral surfaces of the olfactory bulb (Wang et al. 2004). Jin et al. (2014) further determined that Ebf3 is strongly expressed in the midbrain, cerebellum, and the mantle layer of the spinal cord and generated Ebf3-deficient mice that died from respiratory failure due to dysfunctional diaphragm relaxation within 12 h following birth (P0.5) (Jin et al. 2014). Detailed investigation of the brains of Ebf3 mutant mice and the generation of conditional knockout mice will likely reveal additional functions for EBF3 in regulating the genetic programs involved in developing and maintaining neuronal function. In humans, reports of deletions of a common 1-Mb region on 10q26.3 including EBF3 have been associated with severe neurodevelopmental delay and intellectual disability (Faria et al. 2015), although additional genes in the interval including MGMT, PPP2R2D, and BNIP3 may also contribute to the phenotype in 10q26.3 deletion patients.
Figure 4.

Deleterious de novo missense variants in the EBF DNA-binding domain (DBD). (A) Structural representation of the EBF DBD based on original 3D representation of EBF3 in Siponen et al. (2010) with the three residues affected by missense variants in our patients in green. The H157 residue is part of the EBF zinc knuckle (box), which coordinates EBF binding to DNA. (B) Top view of residues involved in zinc coordination are indicated.
Four groups independently reported a total of 22 unique individuals who have EBF3 nonsense, missense, and splice variants with clinical features shared by our patients (Blackburn et al. 2017; Chao et al. 2017; Harms et al. 2017; Sleven et al. 2017), including one individual with a diagnosis of ADHD (Harms et al. 2017), three with autistic-like behavior (Sleven et al. 2017), and one with a family history of ADHD and sleep problems (Blackburn et al. 2017). Several of our patients exhibited behavioral differences including repetitive hair pulling, self-injurious face rubbing, and lack of eye contact or social smile. Three of the individuals with autism in our study also had abnormal brain imaging, including vermis hypoplasia, delayed myelination, and small structural defects in the right temporal lobe (Patient 5) and optic papillae (Patient 6) (Table 2; Supplemental Table S1). We provide additional evidence for the role of EBF3 in brain development and hypothesize that decreased or aberrant binding of EBF3 to its targets lead to abnormal gene regulation during brain development and may be responsible for the global developmental delay and altered brain function consistently observed in individuals with mutations in EBF3. We suggest the variants we describe result in loss of function and haploinsufficiency of EBF3 and are a cause of autism and intellectual disabilities.
The missense variants identified in the individuals in this study are all amino acid substitutions that result in a change in charge, which may alter the DNA binding of EBF3 to its binding partners, including aristaless related homeobox (ARX) (Friocourt and Parnavelas 2011; Olivetti et al. 2014). Genes regulated by ARX include ones that are involved in or have been linked to CNS disorders with intellectual disability and autism (Fulp et al. 2008). ARX regulates the transcription of genes involved in cell differentiation, migration, and maturation during interneuron development in the brain and has been shown to repress EBF3 and its putative tumor-suppressing properties, such as cell cycle arrest and apoptosis (Fulp et al. 2008; Olivetti et al. 2014). ARX is predominantly expressed in the fetal and adult brain, testis, skeletal muscle, and pancreas (Shoubridge et al. 2010; Uhlén et al. 2015). Patients with pathogenic variants in ARX exhibit heterogeneous clinical features resulting in a number of different developmental disorders including Ohtahara (OMIM ID: 308350), Partington (OMIM ID: 309510), and Proud (OMIM ID: 300004) syndromes, X-linked lissencephaly and ambiguous genitalia (XLAG; OMIM ID: 3000215), X-linked infantile spasms syndrome (ISSX; OMIM ID: 308350), and syndromic and nonsyndromic mental retardation (Shoubridge et al. 2010; Olivetti and Noebels 2012).
The phenotypes and clinical outcomes of individuals with ARX-related disorders resemble those with mutations in EBF3, and disruptions in both genes are associated with intellectual disability with various neurological symptoms and are often associated with urogenital malformations. Recent reports of individuals with mutations in EBF3 have documented genitourinary abnormalities, including several cases of undescended testes as well as micropenis and hypospadias (Blackburn et al. 2017; Chao et al. 2017; Harms et al. 2017; Sleven et al. 2017). The male individuals in our study with missense variants, p.Lys64Thr (Patient 1) and p.Arg209Gln (Patient 6), each had a small penis and undescended testes perhaps because of aberrant binding of ARX to EBF3, which could result in the disruption of genitourinary patterning during development. It is thus possible that dysregulation of EBF3 caused by defective binding by ARX may be part of the mechanism leading to the phenotype observed in these patients. Additional studies of EBF3 are required to elucidate the molecular mechanisms of EBF3-associated neurodevelopmental deficiency and genitourinary abnormalities.
METHODS
Studies were approved by the Institutional Review Board of Columbia University.
Whole-Exome Sequencing (WES)
Genomic DNA was extracted from whole blood from the affected children and their parents. Exome sequencing at GeneDx was performed on exon targets captured using the Agilent SureSelect Human All Exon V4 (50 Mb) or the Clinical Research Exome kit (Agilent Technologies) according to the manufacturer's instructions. Libraries were sequenced using the Illumina HiSeq 2000 or 2500 sequencing system with 100-bp paired-end reads (Illumina). Whole-exome sequence data for all sequenced family members was analyzed using GeneDx's XomeAnalyzer (a variant annotation, filtering, and viewing interface for WES data) as described previously (Table 3; Tanaka et al. 2016). Identified sequence variants of interest were confirmed in each proband and both parents by conventional di-deoxy DNA sequence analysis using an ABI3730 (Life Technologies).
Table 3.
Sequencing results
| Patient | WES 10× cov | Mean cov | Kit | EBF3 10× cov | EBF3 mean CDS cov |
|---|---|---|---|---|---|
| 1 | 97.32% | 153 | Agilent CRE | 100.00% | 137 |
| 2 | 92.88% | 47 | Agilent SSv4 | 96.77% | 42 |
| 3 | 96.36% | 142 | Agilent SSv4 | 100.00% | 118 |
| 4 | 91.52% | 103 | Agilent SSv4 | 100.00% | 98 |
| 5 | 97.00% | 113 | Agilent CRE | 100.00% | 101 |
| 6 | 94.85% | 113 | Agilent CRE | 100.00% | 125 |
| 7 | 94.69% | 69 | Agilent SSv4 | 98.03% | 48 |
| Mean | 94.95% | 105 | N/A | 99.26% | 95 |
Results from individuals identified at GeneDx. cov, coverage; CDS, coding sequence.
EBF3 Primer Sequences
Patient 1: Forward ACAGTGTCCAGGAACACGTGA, Reverse CAGGAGGTCTGACCAAGGGC
Patient 2: Forward CATGTTTGGGATTCAGGAGAA, Reverse CCTGCCTCCCGCTTCTA
Patient 3: Forward TAGCCCAGCTCCGAGGGTGA, Reverse CAATCGATGCCCTTCCCGGA
Patient 4: Forward ACCGCTTCATTGTCAGACGT, Reverse TTTTGTTGCTGCTGCGGTTT (This primer set has been discontinued and is not recommended.)
Patient 5: Forward TCTGATAACCCTAATAAATATCAT, Reverse GATTACTTCCTGAACAGTTGC
Patient 6: Forward TCTGATAACCCTAATAAATATCAT, Reverse GATTACTTCCTGAACAGTTGC
Patient 7: Forward GGCGCTAGAGCAGGTGGAAA, Reverse GAGATCACATCGGGCCCGTT
ADDITIONAL INFORMATION
Data Deposition and Access
Whole-exome sequencing data are not publicly available because patient consent could not be obtained. The EBF3 variants found in this study have been deposited in ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV000584020.1–SCV000584025.1 and SCV000570295.2.
Ethics Statement
The study was approved by the Institutional Review Board of Columbia University. Written informed consent was obtained from the probands or probands’ parents for publication and accompanying images.
Acknowledgments
We thank the patients and their families for their generous participation. The Simons Foundation provided funding to W.K.C.
Author Contributions
A.J.T. and M.T.C. analyzed the data and drafted and critically reviewed the manuscript. R.W. and M.J.G.S. analyzed the data and critically reviewed the manuscript. K.R. generated and analyzed the data and critically reviewed the manuscript. Y.A.Z., K.B., V.S., K.O., A.W., G.N.W., A.B., T.B.-D., L.O., S.S., M.K.G., I.J.A., and R.E.S. provided the clinical data and critically reviewed the manuscript. W.K.C. conceived of the study, analyzed the data, and drafted and clinically reviewed the manuscript.
Funding
This work was supported in part by a grant from the Simons Foundation.
Competing Interest Statement
M.C., R.W., K.R., M.G.S., and R.E.S. are employees of GeneDx. W.C. is a former employee of GeneDx and a member of the Scientific Advisory Board of Regeneron Genetics Center.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Blackburn PR, Barnett SS, Zimmerman MT, Cousin MA, Kaiwar C, Pinto E Vairo F, Niu Z, Ferber MJ, Urrutia RA, Selcen D, et al. 2017. Novel de novo variant in EBF3 is likely to impact DNA binding in a patient with a neurodevelopmental disorder and expanded phenotypes: patient report, in silico functional assessment, and review of published cases. Cold Spring Harb Mol Case Stud 3: a001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu W, Su LK. 2003. Characterization of functional domains of human EB1 family proteins. J Biol Chem 278: 49721–49731. [DOI] [PubMed] [Google Scholar]
- Chao HT, Davids M, Burke E, Pappas JG, Rosenfeld JA, McCarty AJ, Davis T, Wolfe L, Toro C, Tifft C, et al. 2017. A syndromic neurodevelopmental disorder caused by de novo variants in EBF3. Am J Hum Genet 100: 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara F, Badaloni A, Croci L, Yeh ML, Cariboni A, Hoerder-Suabedissen A, Consalez GG, Eickholt B, Shimogori T, Parnavelas JG, et al. 2012. Early B-cell factors 2 and 3 (EBF2/3) regulate early migration of Cajal–Retzius cells from the cortical hem. Dev Biol 365: 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles MW, Omuro KC, Stanley BN, Quintanilla CG, Zayas RM. 2014. COE loss-of-function analysis reveals a genetic program underlying maintenance and regeneration of the nervous system in planarians. PLoS Genet 10: e1004746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daburon V, Mella S, Plouhinec JL, Mazan S, Crozatier M, Vincent A. 2008. The metazoan history of the COE transcription factors. Selection of a variant HLH motif by mandatory inclusion of a duplicated exon in vertebrates. BMC Evol Biol 8: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois L, Vincent A. 2001. The COE—Collier/Olf1/EBF—transcription factors: structural conservation and diversity of developmental functions. Mech Dev 108: 3–12. [DOI] [PubMed] [Google Scholar]
- Faria ÁC, Rabbi-Bortolini E, Rebouças MR, de S Thiago Pereira AL, Frasson MG, Atique R, Lourenço NC, Rosenberg C, Kobayashi GS, Passos-Bueno MR, et al. 2015. Craniosynostosis in 10q26 deletion patients: a consequence of brain underdevelopment or altered suture biology? Am J Med Genet A 170A: 403–409. [DOI] [PubMed] [Google Scholar]
- Friocourt G, Parnavelas JG. 2011. Identification of Arx targets unveils new candidates for controlling cortical interneuron migration and differentiation. Front Cell Neurosci 5: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulp CT, Cho G, Marsh ED, Nasrallah IM, Laboskey PA, Golden JA. 2008. Identification of Arx transcriptional targets in the developing basal forebrain. Hum Mol Genet 17: 3740–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Dominguez M, Poquet C, Garel S, Charnay P. 2003. Ebf gene function is required for coupling neuronal differentiation and cell cycle exit. Development 130: 6013–6025. [DOI] [PubMed] [Google Scholar]
- Harms FL, Girisha KM, Hardigan AA, Kortüm F, Shukla A, Alawi M, Dalal A, Brady L, Tarnopolsky M, Bird LM, et al. 2017. Mutations in EBF3 disturb transcriptional profiles and cause intellectual disability, ataxia, and facial dysmorphism. Am J Hum Genet 100: 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, Schellenberg GD, Gibbs RA, Daly MJ, Buxbaum JD, et al. 2013. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet 9: e1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Kim J, Willert T, Klein-Rodewald T, Garcia-Dominguez M, Mosqueira M, Fink R, Esposito I, Hofbauer LC, Charnay P, et al. 2014. Ebf factors and MyoD cooperate to regulate muscle relaxation via Atp2a1. Nat Commun 5: 3793. [DOI] [PubMed] [Google Scholar]
- Liberg D, Sigvardsson M, Akerblad P. 2002. The EBF/Olf/Collier family of transcription factors: regulators of differentiation in cells originating from all three embryonal germ layers. Mol Cell Biol 22: 8389–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivetti PR, Noebels JL. 2012. Interneuron, interrupted: molecular pathogenesis of ARX mutations and X-linked infantile spasms. Curr Opin Neurobiol 22: 859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivetti PR, Maheshwari A, Noebels JL. 2014. Neonatal estradiol stimulation prevents epilepsy in Arx model of X-linked infantile spasms syndrome. Sci Transl Med 6: 220ra212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnström K, Mallick S, Kirby A, et al. 2014. A framework for the interpretation of de novo mutation in human disease. Nat Genet 46: 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoubridge C, Fullston T, Gécz J. 2010. ARX spectrum disorders: making inroads into the molecular pathology. Hum Mutat 31: 889–900. [DOI] [PubMed] [Google Scholar]
- Siponen MI, Wisniewska M, Lehtiö L, Johansson I, Svensson L, Raszewski G, Nilsson L, Sigvardsson M, Berglund H. 2010. Structural determination of functional domains in early B-cell factor (EBF) family of transcription factors reveals similarities to Rel DNA-binding proteins and a novel dimerization motif. J Biol Chem 285: 25875–25879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleven H, Welsh SJ, Yu J, Churchill ME, Wright CF, Henderson A, Horvath R, Rankin J, Vogt J, Magee A, et al. 2017. De novo mutations in EBF3 cause a neurodevelopmental syndrome. Am J Hum Genet 100: 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka AJ, Cho MT, Retterer K, Jones JR, Nowak C, Douglas J, Jiang YH, McConkie-Rosell A, Schaefer GB, Kaylor J, et al. 2016. De novo pathogenic variants in CHAMP1 are associated with global developmental delay, intellectual disability, and dysmorphic facial features. Cold Spring Harb Mol Case Stud 2: a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao YF, Xu LX, Lu J, Hu SY, Fang F, Cao L, Xiao PF, Du XJ, Sun LC, Li ZH, et al. 2015. Early B-cell factor 3 (EBF3) is a novel tumor suppressor gene with promoter hyperrmethylation in pediatric acute myeloid leukemia. J Exp Clin Cancer Res 34: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuret S, Bhatt L, O'Leary DD, Simon HH. 2004. Identification and developmental analysis of genes expressed by dopaminergic neurons of the substantia nigra pars compacta. Mol Cell Neurosci 25: 394–405. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. 2015. Proteomics. Tissue-based map of the human proteome. Science 347: 1260419. [DOI] [PubMed] [Google Scholar]
- Wang SS, Lewcock JW, Feinstein P, Mombaerts P, Reed RR. 2004. Genetic disruptions of O/E2 and O/E3 genes reveal involvement in olfactory receptor neuron projection. Development 131: 1377–1388. [DOI] [PubMed] [Google Scholar]
- Zhao LY, Niu Y, Santiago A, Liu J, Albert SH, Robertson KD, Liao D. 2006. An EBF3-mediated transcriptional program that induces cell cycle arrest and apoptosis. Cancer Res 66: 9445–9452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.