

Abstract
Synchronous endometrial and ovarian (SEO) carcinomas involve endometrioid neoplasms in both the ovary and uterus at the time of diagnosis. Patients were traditionally classified as having independent primary SEO lesions or as having metastatic endometrioid carcinoma. Recent studies have supported that SEO tumors result from the dissemination of cells from one organ site to another. However, whether this can be considered a “metastasis” or “dissemination” remains unclear. In this report, we performed whole-exome sequencing of tumor samples from a woman with well-differentiated endometrioid SEO tumors and a clinical “recurrent” poorly differentiated peritoneal tumor that was diagnosed 8 years after the complete resection of the SEO tumors. Somatic mutation analysis identified 132, 171, and 1214 nonsynonymous mutations in the endometrial, ovarian, and peritoneal carcinomas, respectively. A unique mutation signature associated with mismatch repair deficiency was observed in all three tumors. The SEO carcinomas shared 57 nonsynonymous mutations, whereas the clinically suspected recurrent carcinoma shared only eight nonsynonymous mutations with the SEO tumors. One of the eight shared somatic mutations involved PTEN; these shared mutations represent the earliest genetic alteration in the ancestor cell clone. Based on analysis of the phylogenetic tree, we predicted that the so-called recurrent peritoneal tumor was derived from the same endometrial ancestor clone as the SEO tumors, and that this clone migrated and established benign peritoneal endometriosis where the peritoneal tumor later arose. This case highlights the usefulness of next-generation sequencing in defining the etiology and clonal relationships of synchronous and metachronous tumors from patients, thus providing valuable insight to aid in the clinical management of rare or ambiguous tumors.
Keywords: endometrial carcinoma, endometriosis, ovarian neoplasm
INTRODUCTION
Synchronous endometrial and ovarian (SEO) carcinomas, defined as two tumors detected at the same time during surgery, occur in ∼5%–10% of women diagnosed with ovarian or endometrial tumors (Zaino et al. 2001). Patients are often of a younger age, present with early-stage disease, and have a more favorable prognosis than do patients who present with only endometrial or ovarian carcinoma at the same clinical stage (Zaino et al. 2001; Soliman et al. 2004; Chiang et al. 2008; Signorelli et al. 2008; Williams et al. 2009; Lim et al. 2011). Endometrial and ovarian carcinomas are typically divided into type I and type II carcinomas (Kurman and Shih 2016), with type I referring to low-grade, genetically stable, and relatively indolent cancers (Bokhman 1983; Sherman 2000; Kurman and Shih 2010). The majority of endometrioid carcinomas in both organ sites fall into the type I category (particularly when synchronous) and are characterized by mutations of genes in the PTEN and β-catenin signaling pathways, as well as by deficiency in DNA mismatch repair (MMR). However, type I tumors rarely harbor TP53 mutations (Sohaib et al. 2007; Cho and Shih 2009; Djordjevic et al. 2012, 2013; O'Hara and Bell 2012). SEO tumors frequently meet these criteria and can thus be classified as type I cancers. Over the years, research groups such as Ulbright and Roth (1985) and Scully et al. (1998) have delineated criteria in an attempt to distinguish SEO tumors from metastatic ovarian or endometrial disease on the basis of histological, genetic, and clinicopathological features. In general, patients are classified as having (1) two independent carcinomas, (2) ovarian carcinoma with metastasis to the endometrium, or (3) endometrial carcinoma with metastasis to the ovary. These distinctions are often vague, however, and diagnosis can be difficult. Results from recent studies have suggested that what were previously categorized as SEO carcinomas may, in fact, represent dissemination from one site to the other (e.g., the spread of cells from the ovary to the endometrium or vice versa without the involvement of the lymphatic or vascular system [Anglesio et al. 2016; Chao et al. 2016; Schultheis et al. 2016]). In this event, it is critical to understand the extent to which such dissemination occurs in order to provide an accurate diagnosis and thereby provide better clinical care for patients. Here, we report a case of a woman presenting with a “recurrent” peritoneal carcinoma discovered 8 years after complete resection of SEO tumors. Comprehensive exome-sequence examination demonstrated that the peritoneal carcinoma did not result from metastasis from the SEO tumors; instead, it was likely a second primary that developed from a benign endometriotic lesion.
RESULTS
Clinical Presentation
A 56-yr-old woman with a history of endometriosis-related infertility experienced 2 years of irregular uterine bleeding. The endometrial biopsy showed atypical endometrial hyperplasia with areas of worrisome histological appearance suggestive of endometroid carcinoma. She then underwent a total abdominal hysterectomy and bilateral salpingo-oophorectomy with peritoneal washing. During the operation, synchronous tumor masses involving the endometrium and right ovary were observed, measuring 2.0 × 1.1 cm (endometrium) and 2.0 cm (ovary). In addition, a ruptured hemorrhagic ovarian cyst and peritoneal lesions suggestive of endometriosis were specifically noted at the right uterosacral ligament and right broad ligament. No other grossly visible peritoneal lesions were detected. Histological examination showed that the endometrial tumor was a FIGO grade 1 endometrioid carcinoma arising in a background of atypical complex hyperplasia without myometrial or cervical invasion (Fig. 1). The right ovarian tumor was diagnosed as a FIGO grade 1 endometrioid carcinoma, associated with an endometriotic cyst. The peritoneal washing cytological results were negative for malignancy. She was clinically assessed as having a FIGO stage IA endometrial carcinoma and a stage IC ovarian carcinoma. She declined further surgical staging and elected to be treated with six cycles of postoperative adjuvant chemotherapy with carboplatin and paclitaxel. The patient was followed up with Pap smear, clinical examination, computed tomography scan, and serum CA-125 test. No evidence of recurrence was noted until 8 yr after the primary surgery, when she experienced severe right pelvic pain. A pelvic magnetic resonance imaging examination revealed a 5.2 × 4.5 × 3.8 cm mass at the right pelvic wall. The patient then underwent a secondary tumor resection. This revealed an isolated tumor, 7.0 × 5.0 × 5.0 cm in size, at the right pelvic sidewall involving the sigmoid mesentery, right ureter, and upper vagina. Different from the original SEO tumors, the pelvic wall peritoneal tumor histologically presented as a poorly differentiated carcinoma, characterized by sheets of nondescript tumor cells and abundant tumor-infiltrating lymphocytes (Fig. 1). From the presentation of focal glandular proliferation with squamous differentiation, this tumor was classified as a high-grade endometrioid carcinoma. The patient has completed additional courses of chemotherapy and pelvic irradiation and is currently disease-free, having been followed for 13 mo since resection.
Figure 1.

Histological appearance of the SEO carcinomas and the “recurrent” peritoneal carcinoma.
Immunohistochemistry
Immunostaining was performed with antibodies against a panel of DNA mismatch repair proteins, including MSH2, MSH6, MLH1, and PMS2, as well as CD8 and PD-1. The data showed loss of MLH1 and PMS2 expression in the synchronous uterine and ovarian endometrioid carcinomas, as well as in the metachronous peritoneal carcinoma (Fig. 2). In addition, the peritoneal carcinoma contained approximately four to five times more CD8+ tumor-infiltrating lymphocytes than did the SEO tumors (Fig. 2; Supplemental Table 1). PD-1 expression was elevated in the peritoneal tumor compared with that in the SEO tumors, though it was still relatively weak (Supplemental Table 1). The patient's ovarian endometrioid carcinoma was associated with an endometriotic cyst (endometrioma), and the tissue was available for immunostaining. We found that the normal-appearing endometriotic cyst epithelium manifested an identical staining pattern to that of the ovarian endometrioid carcinoma, the results being negative for MLH1 and PMS2 and positive for MSH2 and MSH6 (Fig. 2; Supplemental Table 1).
Figure 2.

Immunohistochemical staining performed on SEO and peritoneal tumors by using antibodies against a panel of mismatch repair (MMR) gene products, CD8, and PD-1. The complete loss of PMS2 and MLH1 immunoreactivity in tumor cells was observed in all three lesions. The positivity in the stromal cells serves as an internal positive control. MSH2 and MSH6 staining is retained in all lesions. Immunohistochemical staining revealed abundant tumor-infiltrating CD8 cytotoxic T cells as well as PD-1 positivity in the peritoneal tumor.
Genomic Analyses
Single-Nucleotide Variants and Insertion/Deletions
To investigate the clonal relationship between the synchronous and metachronous tumors, we performed whole-exome sequencing on the tumors and on saliva-swab normal specimens obtained from the same patient. After filtering the list against germline sequence variants, we identified 165 (132 nonsynonymous) somatic mutations in the endometrial carcinoma, 217 (171 nonsynonymous) somatic mutations in the ovarian carcinoma, and 1573 (1214 nonsynonymous) somatic mutations in the peritoneal carcinoma (somatic variant list shown in Supplemental Table 2). The SEO carcinomas shared 72 (57 nonsynonymous) somatic mutations, among which are well-known driver genes in endometrioid cancer, including KRAS_12G>D, ARID1A_Q1327Afs*11, and PTEN_130R>G (Supplemental Table 3). Based on the significant number of shared somatic mutations, we concluded that the SEO tumors evolved from a common tumor origin. The precursor lesion of endometrial carcinoma (atypical endometrial hyperplasia) was observed in the index endometrial biopsy sample, suggesting that the endometrial tumor likely represented the primary tumor, and that the ovarian tumor likely derived from a disseminated clone that metastasized from the endometrial tumor at some point.
Surprisingly, the peritoneal carcinoma, diagnosed 8 years later and initially considered a recurrent tumor, displayed a disproportionally high number of somatic mutations (1573 mutations, 1214 nonsynonymous) but shared only 10 mutations (8 nonsynonymous) with the SEO carcinomas. This suggests that the founder of the peritoneal carcinoma (i.e., the most recent common ancestor) had migrated to the peritoneum long before the endometrial carcinoma disseminated to the ovary, as depicted in the phylogenetic tree (Fig. 3). This ancestor clone carried a PTEN_130R>G mutation (Chr10_89692904-89692904_C_G), which was the earliest driver gene and was shared among the three tumors in this patient. A distinct mutation in ARID1A, D1850Tfs*33, was observed in the peritoneal tumor (Supplemental Table 2), indicating that this mutation was acquired independently from the ovarian and endometrial carcinomas.
Figure 3.
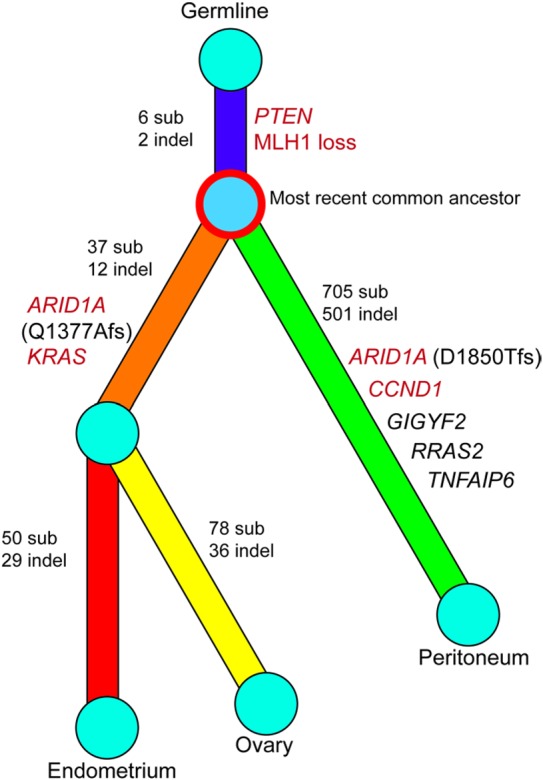
Phylogenetic tree illustrating the clonal evolution history among the endometrial, ovarian, and peritoneal tumors. The colored branches indicate divergent evolution, distinguishing each clone from the ancestral clone. Genes highlighted in red are well-known cancer driver genes in endometrial carcinomas (The Cancer Genome Atlas Research Network et al. 2013). MLH1 loss is based on immunostaining results. sub, substitution; indel, small insertions and deletions.
Mutational Signature Indicative of Mismatch Repair Deficiency
Analysis of a mutation signature revealed that all three tumors have enriched C > A transversions and C > T transitions at GpC loci, a pattern highly resembling mutation signature 6, which is commonly identified in tumors with DNA mismatch repair deficiency (http://cancer.sanger.ac.uk/cosmic/signatures) (Fig. 4; Alexandrov et al. 2013). The bioinformatics tool deconstructSigs, which reconstructs the mutational profile of a tumor by determining the linear combination of predefined signatures, also assigned the highest weight to mutation signature 6, suggesting that DNA mismatch repair deficiency contributed the most to the mutational patterns of all three tumors (Fig. 4; Rosenthal et al. 2016). Consistently, these tumors all harbored many small insertions and deletions (indels) in mono/oligonucleotide repeats (499 of 503 repeats in peritoneal tumors, 41 of 43 repeats in endometrial tumors, and 48 of 50 repeats in ovarian tumors), another hallmark of DNA mismatch repair deficiency. Two of the eight founder mutations displayed indels at A/T homopolymeric stretches, suggesting that DNA mismatch repair deficiency occurred before peritoneal or ovarian spreading of the most recent common ancestor (Fig. 3). Notably, germline mutations were not detected in MLH1, MSH2, MSH6, or PMS2, suggesting that this patient does not have a genetically inherited deficiency in mismatch repair, as observed in patients with Lynch syndrome. Therefore, the molecular genetic evidence supports the conclusion that the peritoneal carcinoma represents neither recurrence nor metastasis of the primary endometrial or ovarian carcinoma; instead, this cancer represents a primary peritoneal carcinoma that likely developed from preexisting endometriosis.
Figure 4.

Mutational signatures of the peritoneal, endometrial, and ovarian carcinomas. On the basis of 96 potential substitution classes (six substitution classes multiplied by 16 combinations of immediate 5′ and 3′ nucleotides) (Alexandrov et al. 2013), all three tumors from this patient displayed a high fraction of C > A transversions and C > T transitions, particularly in the GpC context, and were highly correlated with mutation signature 6 (bottom panel), which defines deficiency in DNA mismatch repair (Alexandrov et al. 2013).
DISCUSSION
In this case study, the SEO carcinomas shared a significant number of somatic mutations, several of which are well-known cancer driver genes of endometrioid carcinomas, such as PTEN, ARID1A, and KRAS. This finding supports the view that SEO carcinomas are clonally related and likely represent tumor dissemination or metastasis from one site to the other. In contrast, only 8 (0.6%) of the 1214 somatic mutations in the “recurrent” peritoneal carcinoma were shared with the prior SEO carcinomas. Based on the paucity of common somatic mutations between the SEO tumors and the peritoneal tumor as well as only one cancer driver gene that is shared, PTEN, we make the following conclusions. First, the ancestor clone of the peritoneal carcinoma probably diverged from the ancestor of SEO tumors very early on, even prior to the fully transformed stage of all three tumors (Fig. 3). Second, our study indicates that PTEN mutation is likely the earliest molecular genetic aberration in this patient, and that it provides a survival advantage to the endometrial epithelial cells at a distant site (Mutter et al. 2000). This finding is in agreement with our recent study demonstrating that somatic mutations of genes in the PIK3 pathway or mutations of ARID1A can be detected in endometriosis (Anglesio et al. 2017), a benign lesion of ectopic endometrial tissue in women of reproductive age. In addition to the genomic events, we examined the expression pattern of mismatch repair genes and found that all three carcinomas, as well as the endometriotic cyst associated with the ovarian endometrioid carcinoma, lost both MLH1 and PMS2. The data indicate that loss of MLH1 expression, likely due to methylation or deletion, occurred at a very early stage before transformation of endometrial epithelial cells.
At least two clinical implications can be derived from this study. First, the fact that the recurrent peritoneal tumor from this patient is more likely to be a primary tumor indicates that the patient is at a relatively low risk of recurrence following complete excision of the peritoneal tumor. Second, the significantly increased mutation load and increased number of prominent tumor-infiltrating lymphocytes in the recurrent peritoneal tumor suggest that the patient may have an increased neoantigen load and would benefit from immune checkpoint blockage-based therapy (Pico de Coaña et al. 2015).
This report also highlights that endometriosis is the likely precursor lesion of tumors outside the uterus in this patient. Endometriosis is a relatively common chronic inflammatory disease that affects ∼10% of women at reproductive age (Bulletti et al. 2010). Although nearly 25% of women with endometriosis are asymptomatic (Bulletti et al. 2010), it is responsible for up to 50% of cases of pelvic pain and/or infertility (Giudice and Kao 2004). Although endometriosis does not often undergo full-blown malignant transformation, it is an established risk factor for gynecologic malignancies, including uterine and ovarian endometrioid carcinoma, clear-cell carcinoma, and seromucinous neoplasms (Melin et al. 2006; Pearce et al. 2012; Kok et al. 2015; Wilbur et al. 2017). Predicting which endometriotic lesions will progress to carcinoma represents a clinical challenge. Therefore, a deeper understanding of the exposure factor(s) contributing to the evolution of endometriosis and endometriosis-related tumors represents an unmet need, as does the identification of molecular markers that can predict the risk of malignant transformation. Attaining this knowledge will impact future clinical management of this common disease.
In conclusion, we report a case of peritoneal carcinoma that was initially diagnosed as a recurrent tumor, as it was discovered 8 years after the resection of SEO tumors. Analysis of the tumor exomes revealed that the peritoneal tumor in fact descended from the same ancestor clone as did the SEO tumors, but that it diverged at a much earlier time than initially suspected. Because the peritoneal tumor emerged after chemotherapy and displayed reactive lymphocytes and a high mutation load, future investigation is warranted to evaluate whether platinum-based chemotherapy causes or primes tumor cells to acquire additional somatic mutations, as well as whether these tumor-specific neoantigens could be targeted by the immune system and exploited clinically to increase the effectiveness of immunotherapy.
METHODS
Immunohistochemical Staining and Scoring System
Immunohistochemistry was performed at the Johns Hopkins Immunopathology Laboratory with the following antibodies: MLH1 (Ventana; Cat # 790-4535), MSH2 (Cell Marque; Cat # 760-4265), MSH6 (Ventana, Cat # 790-4455), PMS2 (Cell Marque, Cat # 760-4531), CD8 (Cell Marque, Cat # 108M-98), PD-1 (Cell Marque, Cat # 315M-96), ER (Ventana, Cat # 790-4325), and PR (Ventana, Cat # 790-4296). All tissue sections were automatically immunostained by using either Ventana Benchmark Ultra (Ventana Medical Systems, Inc.) or Ventana Benchmark XT (Ventana Medical Systems, Inc.). Antigen retrieval was performed with Ultra CC1 (Ventana, Cat# 950-224), except for CD8 staining, which was performed with CC1 (Ventana, Cat# 950-124). Antibodies against MLH1, MSH2, MSH6, PMS2, CD8, ER, and PR were prediluted, and PD-1 antibody was diluted (1:100) according to the manufacturer's instructions. Immunoreactivity was detected with the UltraView DAB system (Ventana, Cat # 760-500).
The expression of MLH1, MSH2, MSH6, and PMS2 was determined qualitatively to be retained or lost, as is standard. PD-1 and CD8 expression was scored semiquantitatively by two independent pathologists (AA and IMS). For PD-1, the intensity of staining (between 1 and 3) was multiplied by the area of positive cells (between 0% and 100%). CD8 expression was scored according to the mean number of positive cells per 40× HPF area (averaged across 10 areas).
Sample Preparation and Next-Generation Sequencing
Tumor tissues were cored from formalin-fixed, paraffin-embedded (FFPE) blocks. Two pathologists (E.V. and I.-M.S.) evaluated H&E sections and selected areas with 40%–70% tumor purity for coring and further analysis. Genomic DNA isolation, library construction, exome and targeted capture, next-generation sequencing, and bioinformatics analyses of tumor and normal samples were performed at Personal Genome Diagnostics. Briefly, gDNA was extracted from frozen or FFPE tissue with matched saliva samples by using the QIAGEN DNA FFPE tissue kit or QIAGEN DNA blood mini kit (QIAGEN). Genomic DNA from tumor and normal samples was fragmented and used for library construction (Illumina) according to the manufacturer's instructions or as previously described (Sausen et al. 2013). Genomic DNA (50 ng to 3 µg) in 100 µl of TE was fragmented in a Covaris sonicator (Covaris) to a size of 150–450 bp. To remove fragments smaller than 150 bp, we purified the DNA twice by using Agencourt AMPure XP beads (Beckman Coulter) at a ratio of 1.0 to 0.9 of PCR product to beads and washed it with 70% ethanol according to the manufacturer's instructions. Purified, fragmented DNA was mixed with 36 µl of H2O, 10 µl of End Repair Reaction Buffer, and 5 µl of End Repair Enzyme Mix (NEB; cat# E6050). The 100-µl end-repair mixture was incubated for 30 min at 20°C, purified with Agencourt AMPure XP beads (Beckman Coulter) at a ratio of 1.0 to 1.25 of PCR product to beads, and washed with 70% ethanol per the manufacturer's instructions. To generate an A-tail, we mixed 42 µl of end-repaired DNA with 5 µl of 10× dA Tailing Reaction Buffer and 3 µl of Klenow (exo-) (NEB, Cat # E6053). The 50-µl mixture was incubated at 37°C for 30 min, purified by using Agencourt AMPure XP beads (Beckman Coulter) at a ratio of 1.0 to 1.0 of PCR product to beads, and washed with 70% ethanol per the manufacturer's instructions. For adaptor ligation, 25 µl of A-tailed DNA was mixed with 6.7 µl of H2O, 3.3 µl of PE-adaptor (Illumina), 10 µl of 5× Ligation Buffer, and 5 µl of Quick T4 DNA ligase (NEB, cat# E6056). The ligation mixture was incubated for 15 min at 20°C, purified twice with Agencourt AMPure XP beads (Beckman Coulter), and subsequently washed with 70% ethanol per the manufacturer's instructions. To obtain an amplified library, we set up 12 PCRs of 25 µl each, each including 15.5 µl of H2O, 5 µl of 5× Phusion HF Buffer, 0.5 µl of a dNTP mix containing 10 mM of each dNTP, 1.25 µl of DMSO, 0.25 µl of Illumina PE primer #1, 0.25 µl of Illumina PE primer #2, 0.25 µl of Hotstart Phusion polymerase, and 2 µl of the DNA. The PCR program was run for 2 min at 98°C, followed by 12 cycles of 15 sec at 98°C, 30 sec at 65°C, and 30 sec at 72°C, with a final step of 5 min at 72°C. DNA was purified with Agencourt AMPure XP beads (Beckman Coulter) at a ratio of 1.0 to 1.0 of PCR product to beads and washed with 70% ethanol per the manufacturer's instructions. Exonic regions were captured in solution by using the Agilent SureSelect v.4 kit according to the manufacturer's instructions (Agilent). The captured library was subsequently purified by using a QIAGEN MinElute column purification kit and eluted in 17 µl of 70°C elution buffer to obtain 15 µl of the captured DNA library. The captured DNA library was amplified as follows: eight 30-µl PCR reactions were set up, each containing 19 µl of H2O, 6 µl of 5× Phusion HF buffer, 0.6 µl of 10 mM dNTP, 1.5 µl of DMSO, 0.30 µl of Illumina PE primer #1, 0.30 µl of Illumina PE primer #2, 0.30 µl of Hotstart Phusion polymerase, and 2 µl of captured exome library. The PCR program was run for 30 sec at 98°C, followed by 14 cycles (exome) or 16 cycles (targeted) of 10 sec at 98°C, 30 sec at 65°C, and 30 sec at 72°C, with a final step of 5 min at 72°C. To purify the PCR products, we used a NucleoSpin Extract II purification kit (Macherey-Nagel) according to the manufacturer's instructions. Paired-end sequencing, resulting in 100 bases from each end of the fragments, was performed by using an Illumina HiSeq 2500 instrument (Illumina). A minimum average coverage of 100-fold per base pair was achieved for each tumor sample, and the coverage for germline (normal) specimens was 20-fold per base pair. Table 1 summarizes the total reads, mapped reads, and sequence coverage of each sample.
Table 1.
Total reads, mapped reads, and sequence coverage of each sample
| Sequencing statistics | Endometrial tumor | Ovarian tumor | Recurrent tumor | Germline |
|---|---|---|---|---|
| Sequenced bases mapped to genome | 23,965,396,300 | 15,021,998,300 | 16,596,549,300 | 12,726,336,300 |
| Sequenced bases mapped to target regions (exome) | 13,277,690,574 | 7,877,300,760 | 9,237,133,936 | 6,721,137,245 |
| Bases in target regions with at least 10 reads | 46,678,905 | 46,375,246 | 46,582,694 | 46,783,511 |
| Fraction of bases in target regions with at least 10 reads | 93% | 92% | 93% | 93% |
| Average number of total high-quality sequences at each base | 251 | 148 | 171 | 129 |
| Average number of distinct high-quality sequences at each base | 127 | 102 | 148 | 112 |
Primary Processing of Next-Generation Sequencing Data and Identification of Putative Somatic Mutations
Somatic mutations were identified with VariantDx custom software for identifying mutations in matched tumor and normal samples. Before mutation calling, primary processing of sequence data for both tumor and normal samples was performed with Illumina CASAVA software (v1.8). The sequence reads were aligned against the human reference genome (version hg19) by using ELAND. Candidate somatic mutations, comprising point mutations, insertions, and deletions, were subsequently identified with VariantDx across the whole exome. VariantDx examines sequence alignments of tumor samples against a matched normal sample while applying filters to exclude alignment and sequencing artifacts. Briefly, an alignment filter was applied to exclude quality failed reads, unpaired reads, and poorly mapped reads in the tumor. A base quality filter was applied to limit the inclusion of bases with reported Phred quality scores of >30 for the tumor and >20 for the matched normal samples (http://www.phrap.com/phred/). A mutation in the tumor was identified as a candidate somatic mutation only when (i) distinct paired reads contained the mutation in the tumor; (ii) the number of distinct paired reads containing a particular mutation in the tumor was at least 10% of the total distinct read pairs; (iii) the mismatched base was not present in >1% of the reads in the matched normal sample and not present in a custom database of common germline variants derived from dbSNP; and (iv) the position was covered in both tumor and normal tissues. Mutations arising from misplaced genome alignments, including paralogous sequences, were identified and excluded by searching the reference genome. The full list of somatic variants is shown in Supplemental Table 2.
Candidate somatic mutations were further filtered based on gene annotation to identify mutations occurring in protein-coding regions. Functional consequences were predicted by using snpEff and a custom database of CCDS, RefSeq, and Ensembl annotations, with the latest transcript versions available on hg19 from UCSC (https://genome.ucsc.edu/). Predictions were ordered to prefer transcripts with canonical start and stop codons and CCDS or RefSeq transcripts over Ensembl when available. A manual visual inspection step was used to further remove artifacts.
Mutation Signature and Phylogenetic Tree Analyses
To understand the composition of the mutation signatures of the three tumors, we used R package deconstructSigs to determine the linear combination of mutation signatures cataloged at the Wellcome Trust Sanger Institute (Forbes et al. 2008; Rosenthal et al. 2016). All optional parameters of the main function, whichSignatures( ), were set to default values.
To reconstruct the evolutionary history, we depicted the phylogenetic tree with the trunk denoting somatic mutations shared by all three tumors, subtrunks denoting mutations shared by two tumors, and terminal branches denoting mutations exclusive to individual tumors. Eight mutations shared by the peritoneal and endometrial tumors and three by the peritoneal and ovarian tumors were excluded when the phylogenetic tree was built because they were incongruent with the hypothesized phylogenetic tree. The data are limited, however, by the tumor cellularity of each specimen and should be interpreted with caution. We also attempted to evaluate the subclonal structure in these tumors but failed to identify any separate subclones, most likely because of the narrow distribution of the allelic fraction of most variants (10%–25%).
ADDITIONAL INFORMATION
Data Deposition and Access
Somatic mutation variants (SNV and indels) are deposited in The Catalogue of Somatic Mutations in Cancer (COSMIC) with the identifier COSP43748. Because our institutional review board (IRB) does not currently allow deposition of sequencing data in public repositories for case studies, requests for sequencing data can be directed to Tian-Li Wang (tlw@jhmi.edu), the corresponding author.
Ethics Statement
Research was carried out in accordance with the Federal Policy for the Protection of Human Subjects 45C.F.R.46. The patient has reviewed this manuscript and given her permission (consent) via email correspondence for both the analysis and publication of her data. She has also signed an institutional consent form to participate in this study.
Acknowledgments
This research was supported in part by the following National Cancer Institute grants, UO1 CA200469 and P30 CA006973. We greatly appreciate the participation and kind donation of the patient in this study.
Competing Interest Statement
The authors have declared no competing interest.
Referees
G. Steven Bova
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415– 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglesio MS, Wang YK, Maassen M, Horlings HM, Bashashati A, Senz J, Mackenzie R, Grewal DS, Li-Chang H, Karnezis AN, et al. 2016. Synchronous endometrial and ovarian carcinomas: evidence of clonality. J Natl Cancer Inst 108: djv428. [DOI] [PubMed] [Google Scholar]
- Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Horlings HM, Noe M, Lum A, Jones S, Senz J, Seckin T, et al. 2017. Cancer-associated mutations in endometriosis without cancer. N Eng J Med 376: 1835–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhman JV. 1983. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol 15: 10–17. [DOI] [PubMed] [Google Scholar]
- Bulletti C, Coccia ME, Battistoni S, Borini A. 2010. Endometriosis and infertility. J Assist Reprod Genet 27: 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao A, Wu RC, Jung SM, Lee YS, Chen SJ, Lu YL, Tsai CL, Lin CY, Tang YH, Chen MY, et al. 2016. Implication of genomic characterization in synchronous endometrial and ovarian cancers of endometrioid histology. Gynecol Oncol 143: 60–67. [DOI] [PubMed] [Google Scholar]
- Chiang YC, Chen CA, Huang CY, Hsieh CY, Cheng WF. 2008. Synchronous primary cancers of the endometrium and ovary. Int J Gynecol Cancer 18: 159–164. [DOI] [PubMed] [Google Scholar]
- Cho KR, Shih IM. 2009. Ovarian cancer. Annu Rev Pathol Mech Dis 4: 287–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djordjevic B, Barkoh BA, Luthra R, Broaddus RR. 2013. Relationship between PTEN, DNA mismatch repair, and tumor histotype in endometrial carcinoma: retained positive expression of PTEN preferentially identifies sporadic non-endometrioid carcinomas. Mod Pathol 26: 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djordjevic B, Hennessy BT, Li J, Barkoh BA, Luthra R, Mills GB, Broaddus RR. 2012. Clinical assessment of PTEN loss in endometrial carcinoma: immunohistochemistry outperforms gene sequencing. Mod Pathol 25: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. 2008. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet 57: 10.11.1–10.11.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudice LC, Kao LC. 2004. Endometriosis. Lancet 364: 1789–1799. [DOI] [PubMed] [Google Scholar]
- Kok VC, Tsai HJ, Su CF, Lee CK. 2015. The risks for ovarian, endometrial, breast, colorectal, and other cancers in women with newly diagnosed endometriosis or adenomyosis: a population-based study. Int J Gynecol Cancer 25: 968–976. [DOI] [PubMed] [Google Scholar]
- Kurman RJ, Shih IM. 2010. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol 34: 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurman RJ, Shih IM. 2016. The dualistic model of ovarian carcinogenesis: revisited, revised, and expanded. Am J Pathol 186: 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YK, Padma R, Foo L, Chia YN, Yam P, Chia J, Khoo-Tan H, Yap SP, Yeo R. 2011. Survival outcome of women with synchronous cancers of endometrium and ovary: a 10 year retrospective cohort study. J Gynecol Oncol 22: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melin A, Sparen P, Persson I, Bergqvist A. 2006. Endometriosis and the risk of cancer with special emphasis on ovarian cancer. Hum Reprod 21: 1237–1242. [DOI] [PubMed] [Google Scholar]
- Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, Weng LP, Eng C. 2000. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst 92: 924–930. [DOI] [PubMed] [Google Scholar]
- O'Hara AJ, Bell DW. 2012. The genomics and genetics of endometrial cancer. Adv Genomics Genet 2012: 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce CL, Templeman C, Rossing MA, Lee A, Near AM, Webb PM, Nagle CM, Doherty JA, Cushing-Haugen KL, Wicklund KG, et al. 2012. Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case-control studies. Lancet Oncol 13: 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico de Coaña Y, Choudhury A, Kiessling R. 2015. Checkpoint blockade for cancer therapy: revitalizing a suppressed immune system. Trends Mol Med 21: 482–491. [DOI] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. 2016. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 17: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, Blackford A, Parmigiani G, Diaz LA Jr, Papadopoulos N, et al. 2013. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 45: 12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheis AM, Ng CK, De Filippo MR, Piscuoglio S, Macedo GS, Gatius S, Perez Mies B, Soslow RA, Lim RS, Viale A, et al. 2016. Massively parallel sequencing-based clonality analysis of synchronous endometrioid endometrial and ovarian carcinomas. J Natl Cancer Inst 108: djv427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully RE, Young RH, Clement PB. 1998. Tumors of the ovary, maldeveloped gonads, fallopian tube, and broad ligament. In Atlas of tumor pathology, 3rd series, facsicle 23 Armed Forces Institute of Pathology, Washington, DC. [Google Scholar]
- Sherman ME. 2000. Theories of endometrial carcinogenesis: a multidisciplinary approach. Mod Pathol 13: 295–308. [DOI] [PubMed] [Google Scholar]
- Signorelli M, Fruscio R, Lissoni AA, Pirovano C, Perego P, Mangioni C. 2008. Synchronous early-stage endometrial and ovarian cancer. Int J Gynaecol Obstet 102: 34–38. [DOI] [PubMed] [Google Scholar]
- Sohaib SA, Houghton SL, Meroni R, Rockall AG, Blake P, Reznek RH. 2007. Recurrent endometrial cancer: patterns of recurrent disease and assessment of prognosis. Clin Radiol 62: 28–34; discussion 35–36. [DOI] [PubMed] [Google Scholar]
- Soliman PT, Slomovitz BM, Broaddus RR, Sun CC, Oh JC, Eifel PJ, Gershenson DM, Lu KH. 2004. Synchronous primary cancers of the endometrium and ovary: a single institution review of 84 cases. Gynecol Oncol 94: 456–462. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. 2013. Integrated genomic characterization of endometrial carcinoma. Nature 497: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbright TM, Roth LM. 1985. Metastatic and independent cancers of the endometrium and ovary: a clinicopathologic study of 34 cases. Hum Pathol 16: 28–34. [DOI] [PubMed] [Google Scholar]
- Wilbur MA, Shih IM, Segars JH, Fader AN. 2017. Cancer implications for patients with endometriosis. Semin Reprod Med 35: 110–116. [DOI] [PubMed] [Google Scholar]
- Williams MG, Bandera EV, Demissie K, Rodriguez-Rodriguez L. 2009. Synchronous primary ovarian and endometrial cancers: a population-based assessment of survival. Obstet Gynecol 113: 783–789. [DOI] [PubMed] [Google Scholar]
- Zaino R, Whitney C, Brady MF, DeGeest K, Burger RA, Buller RE. 2001. Simultaneously detected endometrial and ovarian carcinomas—a prospective clinicopathologic study of 74 cases: a gynecologic oncology group study. Gynecol Oncol 83: 355–362. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.