

Abstract
Perinatal insults, including fetal undernutrition and hypoxia, are associated with an increased susceptibility to several adult-onset metabolic disorders. These include cardiovascular disease, insulin resistance, and obesity. However, the mechanisms driving the long-term phenotypic consequences have only recently begun to be elucidated. A primary mechanism accounting for perinatal adaptation is the epigenetic modification of chromatin. In this context, epigenetic modifications to chromatin are thought to arise in response to a perinatal insult in an effort to modulate gene expression and maximize fetal survival. In this symposium report, we discuss epigenetics as a mechanism by which perinatal adaptations can be made by the developing fetus. We examine the benefits of using multiple in vivo models to understand the interrelation of signals that come together and result in perinatal adaptation. Epigenetic effects on IGF-1 arising from a perinatal insult are discussed, as are the difficulties and challenges associated with this complex field. In conclusion, epigenetics provides a means of modulating gene transcription, thus allowing fetal adaptation to a broad variety of conditions.
Keywords: epigenetics, intrauterine growth retardation, perinatal adaptation, rat, uteroplacental insufficiency
INTRODUCTION
The ability to adapt to the in utero environment provides a means of increasing the odds of fetal survival under a variety of conditions. However, this short-term gain appears to be accompanied by a long-term cost, and it is now well established that fetal insults are associated with an increased susceptibility to several adult-onset metabolic disorders, including cardiovascular disease, insulin resistance, and obesity (Eriksson et al., 2002; Barker, 2004; Fattal-Valevski et al., 2005). Insults generally tend to involve an interruption of nutrients to the fetus, with subsequent in utero growth restriction. Common insults include fetal undernutrition, hypoxia, or both, resulting from maternal malnutrition or uteroplacental insufficiency (UPI). Although the phenotypic outcome of these perinatal events is well characterized, the mechanisms behind fetal adaptability have only recently begun to be elucidated.
The search for a mechanism explaining fetal adaptation has been accelerated by the knowledge that many of the adult-onset morbidities are accompanied by changes in the expression of genes regulating, among other things, metabolic processes, apoptosis, and cellular signaling. The idea that a perinatal insult could have lasting effects on gene expression has opened the door to epigenetics as a mechanism for fetal adaptation. Perhaps most important, epigenetic regulation of gene expression provides a mechanism that allows for adaptation to a variety of conditions that may be encountered during development. Further, the response to these varied conditions and the need for particular responses from particular genes requires that the adaptation be both specific and precise. The epigenetic regulation of gene expression provides the fidelity, or reliability, of gene-specific responses to diverse conditions.
This report discusses epigenetics as a mechanism by which perinatal adaptations can be made by the developing fetus. Consideration is given to the importance of the use of multiple models to define mechanisms that contribute to specific vs. general adaptations. The effect of intrauterine growth restriction (IUGR) on the epigenetic characteristics of several genes is discussed, with particular emphasis on IGF-1.
EPIGENETICS IN THE CONTEXT OF PERINATAL ADAPTATION
A general definition of epigenetics is “heritable changes in gene expression that occur without a change in DNA sequence.” It may, however, be more useful to think of epigenetics as a means of selectively using the large array of information contained within the genome, particularly in the context of tissue specificity and developmental timing. The imprinting and gene-silencing aspects of epigenetics have been well studied and are discussed in several comprehensive reviews (Laprise, 2009; Shiota and Yamada, 2009; Wilkins-Haug, 2009). However, another important role of epigenetics has recently been identified, namely, its role in fetal adaptation (Mathers and McKay, 2009; Zeisel, 2009). In this context, epigenetic regulation occurs later in development and can be considered separate from the role of epigenetics during early embryogenesis and implantation. Another important concept is that traditional epigenetics tends to involve gene expression in terms of “on” or “off”; that is, genes are either silent or activated. In contrast, epigenetic mechanisms regulating perinatal adaptation tend to be associated with an adjustment in the expression of genes that are already being transcribed, somewhat akin to a rheostat or volume effect.
The regulation of gene expression via epigenetics involves modifications to chromatin, the DNA-protein complex that contains all genetic information. The simplest unit of chromatin is the nucleosome, consisting of 146 bp of DNA wrapped around a core octomer of histone proteins (Luger et al., 1997). Individual nucleosomes are then compacted into higher order structures that eventually form chromosomes. In the context of chromatin, epigenetic modifications can be read collectively as an epigenetic code. This epigenetic code of chromatin regulates the transcription of genes by affecting DNA interactions with the transcription machinery and other regulatory moieties. This provides a means of adjusting the transcriptional levels over the long-term.
One of the best characterized modifications to chromatin is that of DNA methylation. The methylation of the 5′ position of cytosine that precedes a guanosine (also referred to as CpG) is performed by a member of the DNA methyltransferase family. A second major group of modifications is the posttranslational covalent modifications made to the histone proteins. For the most part, the most explored modifications are to the N-terminal histone tails, particularly to histone 3 (H3). However, several covalent modifications on the core of the histone proteins have also been characterized. The range of modifications is vast and includes acetylation and methylation (Kouzarides, 2007). The enzymes responsible for modifying histone proteins are equally diverse and include the histone acetylase and methyltransferase families (Marmorstein and Trievel, 2009). In addition, histone deacetylase (HDAC) and demethylase enzymes are well studied and highlight the reversible nature of these modifications (Marmorstein and Trievel, 2009). It is also interesting to consider that DNA methylation and histone modifications appear to act in a coordinated fashion (Cedar and Bergman, 2009).
In summary, epigenetics provides a means of modulating gene transcription, thus allowing fetal adaptation to a broad variety of conditions. This will elicit subtle modifications in the phenotype by providing the fidelity necessary for the fetal genotype to respond to a variety of conditions. The dynamic nature of epigenetics and the complexities of whole-organism biology make it necessary to examine the epigenetic mechanisms in vivo. This in vivo approach, however, also adds to the difficulties of studying the complex area of epigenetics. In our laboratory, we have taken an integrative approach to elucidating the complex role of epigenetics in fetal adaptation. We have used multiple in vivo models in an effort to understand the interrelation of signals that come together and result in perinatal adaptation.
UNCOVERING THE MECHANISTICALLY IMPORTANT ASPECTS
A difficult problem to overcome is the determination of which aspects of epigenetic phenomena are important in achieving perinatal adaptation. Each different perinatal insult will be associated with unique target genes, unique tissue-specific changes in gene expression, and unique patterns of DNA and histone modifications. However, perinatal insults will have some characteristics in common. The identification of common phenomena will enable a more complete understanding of the important epigenetic contributions to perinatal adaptation and will allow identification of specific interventions that may improve the mature phenotype. In our group, we have taken the approach of looking at representative “candidate” genes in multiple disparate models of perinatal insult. We have used animal models ranging from IUGR, produced by UPI (Lueder and Ogata, 1990; Lane et al., 1996) and maternal malnutrition (Desai et al., 2008), to mechanical ventilation (Albertine et al., 1999) to test the hypothesis that significant changes in the perinatal environment can 1) increase the risk of associated adult morbidities, such as diabetes; 2) affect perinatal and postnatal gene expression of the same candidate genes; and 3) modify the chromatin structure of relevant candidate genes.
We have strategically chosen disparate models to maximize our chances of identifying the most relevant epigenetic modifications that function as independent variables. In other words, those modifications that are not critical will vary greatly among the models (Figure 1). We have also focused on genes that are likely to be particularly important in the context of perinatal adaptation. Several genes have emerged as important candidate genes, one of which is IGF-1. In multiple models of perinatal disease, IGF-1 displays epigenetic modifications that are common, critical, and correlated with IGF-1 expression. These are likely to be mechanistically important in the epigenetic regulation of perinatal adaptability and represent a good place to begin investigations.
Figure 1.
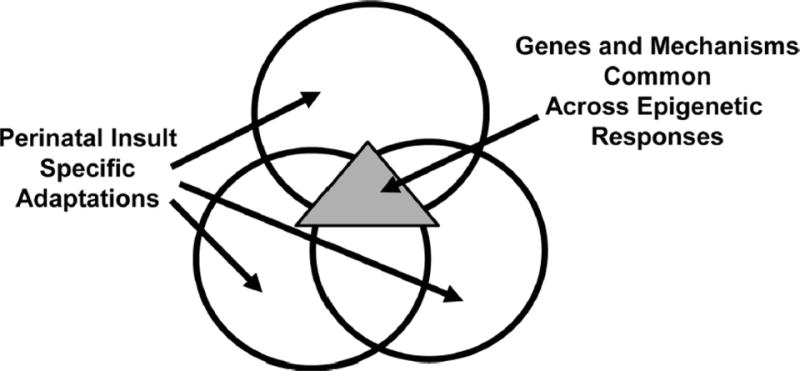
Epigenetic events in multiple models of perinatal insult. The consideration of epigenetic events in the context of multiple models of perinatal insult, represented by the large circles, allows separation of those events that are specific to one particular model from those that are common to all, represented by the triangle. The events common to multiple models are likely to be mechanistically important.
IGF-1: A PARADIGM GENE
The common theme of many models of perinatal insult is that they disrupt growth and predispose the individual to adult-onset metabolic disease. Because IGF-1 is integrally involved in both these aspects, IGF-1 is relevant as a lifetime marker of perinatal insult. Insulin-like growth facor-1 is an intriguing player in the mechanisms relating early malnutrition and subsequent growth restriction to adult disease for several reasons (Aagaard-Tillery et al., 2008b). First, IGF-1 plays a key role in feto-placental growth throughout gestation. In humans, IGF-1 gene deletion results in severe prenatal growth failure (Woods et al., 1996), and null mutations of IGF-1 in mice reduce fetal size by approximately 40% (Rajkumar et al., 1995). Second, IGF-1 mediates many of the anabolic and mitogenic actions of GH in postnatal life. Short children have lesser IGF-1 concentrations than tall children. Third, a recent nested case-control study found that reduced IGF-1 concentrations in adulthood predicted increased risk for developing ischemic heart disease (Juul et al., 2002). Finally, IGF-1 regulates or moderates insulin sensitivity in adulthood (Isaksson et al., 2001). Hepatic IGF-1 is of vital importance for normal carbohydrate metabolism in both humans and mice. In humans, recombinant IGF-1 is approximately 6% as potent as insulin in the production of hypoglycemia, and severe human IGF-1 deficiency leads to insulin resistance, which can be reversed with recombinant IGF-1 (Guler et al., 1987). In mice, elimination of hepatic IGF-1 production by using a Cre/loxP recombination system increases serum concentrations of insulin without significantly affecting glucose elimination (Yakar et al., 2004). Finally, birth weight, postnatal BW, and postnatal height correlated directly with IGF-1 concentrations. These and other studies have only begun to examine the complex relationship between early life exposures and IGF-1 biology. However, it is clear that IGF-1 expression is a relevant marker of these early life exposures, and expression of IGF-1 is likely to be altered (either increased or decreased) as part of fetal adaptation.
Characteristics of the gene architecture of IGF-1 make it exquisitely susceptible to the effects of perinatal adaptation by epigenetic mechanisms. The organization of the rat IGF-1 gene, shown in Figure 2, is remarkably similar to that of the human IGF-1 gene (Shimatsu and Rotwein, 1987). Both human and rat IGF-1 genes are large and contain 6 exons separated by 5 introns. Extensive nucleotide and AA conservation exists between the IGF-1 genes of both species, as does comparable expression of multiple messenger RNA (mRNA) variants. These mRNA variants are useful markers of altered transcriptional regulation and may function to modulate translational efficiency and cell growth. In the rat, exon 1 and exon 2 encode 2 different leader sequences and involve multiple transcription start sites with multiple in-frame start codons. Either exon 1 or exon 2 is spliced to exon 3, which encodes the N-terminus of the mature peptide. Exon 1-derived transcripts (P1) predominate in every tissue expressing the IGF-1 gene, with the exception of the liver, in which relatively large quantities of exon 2-derived transcripts (P2) are produced (Adamo et al., 1989).
Figure 2.
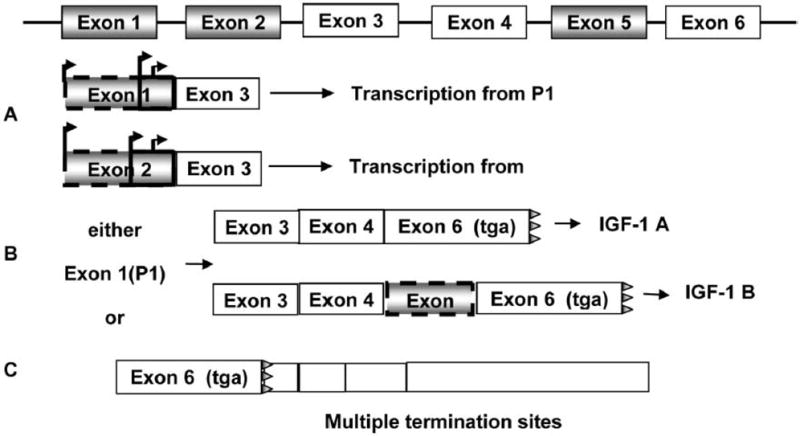
Simplified structure of the rat IGF-1 gene. Panel A: IGF-1 is transcribed from either the promoter 1 (P1; includes exon 1) or promoter 2 (P2; includes exon 2). Both have multiple initiation start sites and can be part of IGF-1 A or B. Panel B: IGF-1 A vs. IGF-1 B is differentiated by the absence or presence of exon 5. Panel C: IGF-1 has multiple termination sites. In the liver, the shorter transcripts are most common. The stop codon is represented by tga.
Another variable that occurs with hepatic IGF-1 mRNA transcripts involves the inclusion or exclusion of exon 5, which changes the translational reading frame of the peptide coding region of exon 6 (Adamo et al., 1989). The IGF-1A transcript lacks exon 5, whereas IGF-1B contains exon 5. Unfortunately, little evidence exists to tie specific promoter use to a specific IGF-1A or IGF-1B transcript, respectively. Finally, multiple polyadenylation sites in the 3′ untranslated region (UTR) of the IGF-1 gene generate mRNA of different lengths, ranging from 0.8 to 7.5 kb. The high-molecular-weight species represents <50% of total IGF-1 mRNA in liver, in contrast to most other tissues.
In support of the concept that IGF-1 expression is regulated by epigenetic mechanisms, we have performed extensive studies of IGF-1 in rat liver under normal conditions, as well as secondary to IUGR resulting from UPI (Fu et al., 2009). We have also recently begun to characterize IGF-1 in the lungs of preterm sheep undergoing mechanical ventilation or high-frequency nasal continuous positive airway pressure, alternative forms of respiratory support (Albertine et al., 2008). Transcripts of IGF-1 present in the lungs of sheep, term and preterm, receiving various forms of postnatal respiratory support include transcripts from P1 and P2, as well as the IGF-1A transcript. Of interest, however, is the fact that the IGF-1B transcript (i.e., the transcript containing exon 5) is absent from the sheep lung under all conditions. On the other hand, rat liver IGF-1 is characterized by the expression of transcripts from P1 and P2 as well as by both the IGF1A and IGF-1B transcripts. This tissue and species-specific regulation of splice variants exemplifies the epigenetic complexity of IGF-1.
An appreciation of the IGF-1 mRNA transcripts and the pattern of histone modifications across the gene, or histone code, of the normal rat liver sets the stage for an examination of the effects of a perinatal insult, that is, IUGR secondary to UPI. We found that IUGR decreased hepatic and serum IGF-1. Concurrently, IUGR modified epigenetic characteristics, particularly the histone code, along the length of the hepatic IGF-1 gene in a persistent and sex-dependent manner (Fu et al., 2009). Figure 3 shows the changes to the histone code of IGF-1, in each case compared with the age- and sex-matched control. Of note, whereas the histone code of IGF-1 was not different between the control male and female rat liver, each responded differently to the UPI insult. Another important concept is that of persistence, or the continuation of histone modifications well beyond the period of insult. The histone code of IGF-1 remains different from that of control animals at postnatal d 21, and again the difference is sex dependent. It is likely that IUGR alters the initial histone code (i.e., that observed at birth) as part of a perinatal adaptation, and this new code is then built upon with subsequent development. It may be that the initial epigenetic modifications alter gene expression to favor fetal survival, whereas the evolving and persistent changes produce metabolically detrimental patterns of gene expression later on. The final thing to note is that the entire IGF-1 gene is subject to epigenetic changes as a result of IUGR. Figure 3 shows that the histone code is modified in the regions of both P1 and P2, the alternatively spliced exon 5, and the 3′UTR region. This indicates that the epigenetic changes associated with a perinatal insult can potentially affect transcription initiation, elongation, and termination.
Figure 3.
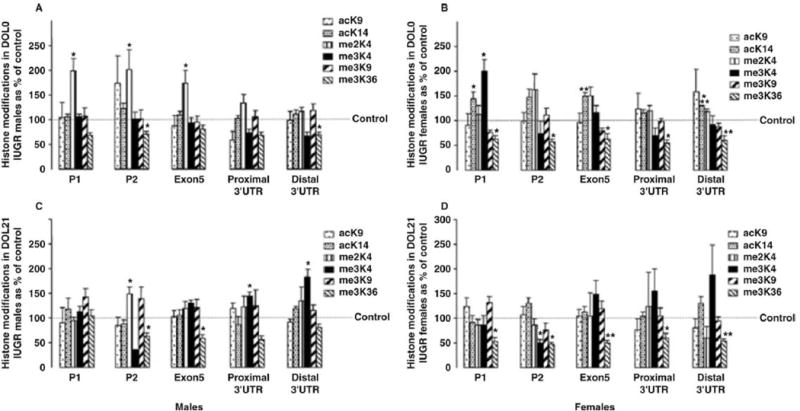
Histone modifications along the IGF-1 gene at birth (DOL0) and at 21 d of age (DOL21) in rat intrauterine growth restricted (IUGR) livers relative to control. Six histone modifications at 5 sites, including P1, P2, exon 5, and proximal and distal 3′ untranslated regions (UTR) on the IGF-1 locus, were analyzed by chromatin immunoprecipitation and real-time PCR. Values for IUGR at each site were compared with their equivalent control values (set to 100%). Panel A: DOL0 male. Panel B: DOL0 female. Panel C: DOL21 male. Panel D: DOL21 female. ac = acetylation; K = lysine; me = methylation. Asterisks above bars indicate that means differ (*P < 0.05; **P < 0.01; ***P < 0.001) from control means. Reproduced from Fu et al. (2009) with permission of The Federation of American Societies for Experimental Biology.
These findings highlight several important facts. First, IGF-1 is an epigenetically regulated gene evident by the multiple splice variants, different promoter use, and different 3′UTR. Second, this epigenetic regulation has a tissue-specific component. Third, the effects of a perinatal insult are both sex dependent and persistent. Finally, the whole gene is affected, not only the promoter regions. These important concepts are not limited to IGF-1; in fact, these are observations that we and others have made in several important candidate genes.
OTHER EPIGENETIC PHENOMENA ASSOCIATED WITH GROWTH RESTRICTION
Other examples of genes that are “epigenetically susceptible” being affected by perinatal insults have been reported by our group and others (Fu et al., 2006; Ke et al., 2006; Lillycrop et al., 2007; Park et al., 2008; Raychaudhuri et al., 2008). Interestingly, these examples continue to demonstrate that, within the context of a perinatal adaptation, epigenetics acts as a rheostat associated with alterations in mRNA variant expression not necessarily on or off. The dual-specificity phosphatase 5 (DUSP5) is an example of an epigenetically susceptible gene that displays epigenetic changes in response to a perinatal insult. One target of DUSP5 is the extracellular-related kinases 1 and 2 that ultimately target the insulin receptor substrate 1, a major component of the insulin signaling pathway (Mothe and Van Obberghen, 1996). In rats, IUGR alters the expression and DNA methylation of DUSP5, all in the context of a phenotype of mature insulin resistance (Lane et al., 2002; Barker, 2005). The methylation content of the CpG contained within the promoter region of liver DUSP5 is unaffected by IUGR. Interestingly, we have observed that it is the CpG within exon 2 of DUSP5 that is altered by IUGR. In addition, contrary to established opinion, IUGR decreased methylation of 5 CpG contained within exon 2 of DUSP5, in conjunction with decreased mRNA abundance of hepatic DUSP5 (Fu et al., 2006). This apparent paradox of decreased transcription being associated with decreased methylation is contrary to the general idea that increased DNA methylation is associated with decreased transcription and is supported by evidence that downstream methylation may be involved in promoting transcription (Murrell et al., 2001).
In addition to the gene-specific chromatin modifications discussed, we have also shown that IUGR is accompanied by global differences in chromatin modifications as well as changes in the quantity and activity of enzymes responsible for making the modifications to chromatin. Of note, it is likely that global changes imply the state of a cell and its ability to replicate, as opposed to the propensity of a specific gene to be up or downregulated.
Liver chromatin modifications affected by IUGR include persistent increases in acetylation of H3 lysine 9 (K9) and K14 (Fu et al., 2006). These changes are accompanied by decreased nuclear protein concentrations, and activity of HDAC isoform 1 in the IUGR rat liver (Fu et al., 2004). Liver glucocorticoid receptors, involved with chromatin modifications at the transcriptional level, are also known to be altered in the IUGR rat (Baserga et al., 2005). In the IUGR rat brain at birth, global decreases in DNA methylation and increases in H3 acetylation on K9 and K14 have been observed (Ke et al., 2006). Consistent with the gene-specific observations, the modifications to chromatin in IUGR rat brains are sex dependent, with a divergence in global acetylation occurring at postnatal d 21 when female brains continue to be characterized by increased site-specific acetylation, whereas male brains become characterized by decreased acetylation at K9 and K14 of H3 (Ke et al., 2006). The IUGR rat brain is also characterized by altered expression of HDAC1, DNA methyltransferase 1, and methyl-CpG binding protein (Ke et al., 2006).
Interestingly, modified histones are not only associated with limited fetal nutrition. In a nonhuman primate model of maternal obesity, we have also demonstrated hyperacetylation of specific lysines of H3 and reduced mRNA, protein concentrations, and activity of HDAC1 (Aagaard-Tillery et al., 2008a). The availability of methyl donors provides yet another aspect of fetal adaptation. The elegant and well-described studies of Waterland and Jirtle (2003) demonstrate that methylation status of the Agouti gene promoter in the viable yellow agouti mouse varies greatly with maternal dietary methyl availability at the time of conception. In addition, the methylation status of the Agouti gene has been demonstrated to be hypermethylated when maternal mice are supplemented with genistein (Dolinoy et al., 2006), but not soy protein isolates (Badger et al., 2008), and to be hypomethylated when maternal mice are given bisphenol A, an effect that can be reversed by the simultaneous addition of dietary methyl donors (Dolinoy et al., 2007).
Although the global changes discussed above are produced in the context of perinatal adaptation, it is important to remember that a global change in levels of a specific modification, for example, H3 hyperacetylation, is not necessarily a reflection of the acetylation status of a particular gene. Likewise, it is likely that methylation differences are gene specific and the methylation status of a particular gene, for example the Agouti gene, may not be representative of the methylation status of other metabolically important genes.
Conclusions
The conditions experienced by the developing fetus are not always conducive to optimal development and survival. Epigenetic mechanisms of perinatal adaptation allow the fetus to fine-tune its response to a variety of conditions. Although this prepares the fetus for optimal survival in the short term, it also has the potential to compromise later aspects of metabolic health. One challenge faced by researchers in this field is to develop the technology to modify a specific gene epigenetically and produce an effect later in life. Such experiments will provide the definitive link between epigenetics and adult-onset disease.
The use of global epigenetic-modifying agents, for example, HDAC inhibitors, to change the epigenetics of a particular gene present risks in that these agents will modify the epigenetics of many genes, possibly with unfavorable results. A complete understanding of the epigenetic events that constitute fetal adaptation will allow the development of gene-specific approaches to intervention. A carefully orchestrated gene-specific approach to intervention is important in perinatal adaptations because the epigenetic effect is different from that of imprinting and cancer epigenetics. Finally, an important future goal in the field will be to understand how to moderate the consequences of the epigenetic perinatal adaptations to minimize the predisposition toward postnatal morbidities.
Footnotes
Based on the presentation titled “Challenges and Opportunities Facing Livestock Reproduction in the 21st Century” at the Triennial Reproduction Symposium during the joint annual meeting of the American Society of Animal Science, American Dairy Science Association, and Canadian Society of Animal Science in Montreal, Canada, July 12 to 16, 2009.
LITERATURE CITED
- Aagaard-Tillery K, Grove K, Bishop J, Ke X, Fu Q, McKnight R, Lane RH. Developmental origins of disease and determinants of chromatin structure: Maternal diet modifies the primate fetal epigenome. J Mol Endocrinol. 2008a;41:91–102. doi: 10.1677/JME-08-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aagaard-Tillery K, Mitchell N, desRoberts C, Lane R. Adult consequences of neonatal and fetal nutrition: Mechanisms. In: Neu J, editor. Gastroenterology and Nutrition: Neonatal Questions and Controversies. Saunders Elsevier; Philadelphia, PA: 2008b. Pages 318–352. [Google Scholar]
- Adamo M, Lowe WL, Jr, LeRoith D, Roberts CT., Jr Insulin-like growth factor I messenger ribonucleic acids with alternative 5′-untranslated regions are differentially expressed during development of the rat. Endocrinology. 1989;124:2737–2744. doi: 10.1210/endo-124-6-2737. [DOI] [PubMed] [Google Scholar]
- Albertine K, Metcalfe D, McKnight R, Dong L, Dahl M, Callaway C, Moyer-Mileur L, Null D, Lane R. Ventilation mode alters expression of an IGF-1 transcript in preterm lamb lung. 2008 Page 634040.6 in E-PAS2008. [Google Scholar]
- Albertine KH, Jones GP, Starcher BC, Bohnsack JF, Davis PL, Cho SC, Carlton DP, Bland RD. Chronic lung injury in preterm lambs. Disordered respiratory tract development. Am J Respir Crit Care Med. 1999;159:945–958. doi: 10.1164/ajrccm.159.3.9804027. [DOI] [PubMed] [Google Scholar]
- Badger TM, Ronis MJ, Wolff G, Stanley S, Ferguson M, Shankar K, Simpson P, Jo CH. Soy protein isolate reduces hepatosteatosis in yellow Avy/a mice without altering coat color phenotype. Exp Biol Med (Maywood) 2008;233:1242–1254. doi: 10.3181/0802-RM-60. [DOI] [PubMed] [Google Scholar]
- Barker DJ. The developmental origins of chronic adult disease. Acta Paediatr Suppl. 2004;93:26–33. doi: 10.1111/j.1651-2227.2004.tb00236.x. [DOI] [PubMed] [Google Scholar]
- Barker DJ. The developmental origins of insulin resistance. Horm Res. 2005;64(Suppl 3):2–7. doi: 10.1159/000089311. [DOI] [PubMed] [Google Scholar]
- Baserga M, Hale MA, McKnight RA, Yu X, Callaway CW, Lane RH. Uteroplacental insufficiency alters hepatic expression, phosphorylation, and activity of the gluco-corticoid receptor in fetal IUGR rats. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1348–R1353. doi: 10.1152/ajpregu.00211.2005. [DOI] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Desai M, Guang H, Ferelli M, Kallichanda N, Lane RH. Programmed upregulation of adipogenic transcription factors in intrauterine growth-restricted offspring. Reprod Sci. 2008;15:785–796. doi: 10.1177/1933719108318597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci USA. 2007;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinoy DC, Weidman JR, Waterland RA, Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect. 2006;114:567–572. doi: 10.1289/ehp.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JG, Forsen T, Tuomilehto J, Jaddoe VW, Osmond C, Barker DJ. Effects of size at birth and childhood growth on the insulin resistance syndrome in elderly individuals. Diabetologia. 2002;45:342–348. doi: 10.1007/s00125-001-0757-6. [DOI] [PubMed] [Google Scholar]
- Fattal-Valevski A, Toledano-Alhadef H, Golander A, Leitner Y, Harel S. Endocrine profile of children with intrauterine growth retardation. J Pediatr Endocrinol Metab. 2005;18:671–676. doi: 10.1515/jpem.2005.18.7.671. [DOI] [PubMed] [Google Scholar]
- Fu Q, McKnight RA, Yu X, Callaway CW, Lane RH. Growth retardation alters the epigenetic characteristics of hepatic dual specificity phosphatase 5. FASEB J. 2006;20:2127–2129. doi: 10.1096/fj.06-6179fje. [DOI] [PubMed] [Google Scholar]
- Fu Q, McKnight RA, Yu X, Wang L, Callaway CW, Lane RH. Uteroplacental insufficiency induces site-specific changes in histone H3 covalent modifications and affects DNA-histone H3 positioning in day 0 IUGR rat liver. Physiol Genomics. 2004;20:108–116. doi: 10.1152/physiolgenomics.00175.2004. [DOI] [PubMed] [Google Scholar]
- Fu Q, Yu X, Callaway CW, Lane RH, McKnight RA. Epigenetics: Intrauterine growth retardation (IUGR) modifies the histone code along the rat hepatic IGF-1 gene. FASEB J. 2009;23:2438–2449. doi: 10.1096/fj.08-124768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guler HP, Zapf J, Froesch ER. Short-term metabolic effects of recombinant human insulin-like growth factor I in healthy adults. N Engl J Med. 1987;317:137–140. doi: 10.1056/NEJM198707163170303. [DOI] [PubMed] [Google Scholar]
- Isaksson OG, Jansson JO, Sjogren K, Ohlsson C. Metabolic functions of liver-derived (endocrine) insulin-like growth factor I. Horm Res. 2001;55(Suppl 2):18–21. doi: 10.1159/000063468. [DOI] [PubMed] [Google Scholar]
- Juul A, Scheike T, Davidsen M, Gyllenborg J, Jorgensen T. Low serum insulin-like growth factor I is associated with increased risk of ischemic heart disease: A population-based case-control study. Circulation. 2002;106:939–944. doi: 10.1161/01.cir.0000027563.44593.cc. [DOI] [PubMed] [Google Scholar]
- Ke X, Lei Q, James SJ, Kelleher SL, Melnyk S, Jernigan S, Yu X, Wang L, Callaway CW, Gill G, Chan GM, Albertine KH, McKnight RA, Lane RH. Uteroplacental insufficiency affects epigenetic determinants of chromatin structure in brains of neonatal and juvenile IUGR rats. Physiol Genomics. 2006;25:16–28. doi: 10.1152/physiolgenomics.00093.2005. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lane RH, Dvorak B, MacLennan NK, Dvorakova K, Halpern MD, Pham TD, Philipps AF. IGF alters jejunal glucose transporter expression and serum glucose levels in immature rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1450–R1460. doi: 10.1152/ajpregu.00172.2002. [DOI] [PubMed] [Google Scholar]
- Lane RH, Flozak AS, Ogata ES, Bell GI, Simmons RA. Altered hepatic gene expression of enzymes involved in energy metabolism in the growth-retarded fetal rat. Pediatr Res. 1996;39:390–394. doi: 10.1203/00006450-199603000-00003. [DOI] [PubMed] [Google Scholar]
- Laprise SL. Implications of epigenetics and genomic imprinting in assisted reproductive technologies. Mol Reprod Dev. 2009;76:1006–1018. doi: 10.1002/mrd.21058. [DOI] [PubMed] [Google Scholar]
- Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr. 2007;97:1064–1073. doi: 10.1017/S000711450769196X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueder FL, Ogata ES. Uterine artery ligation in the maternal rat alters fetal tissue glucose utilization. Pediatr Res. 1990;28:464–468. doi: 10.1203/00006450-199011000-00009. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Marmorstein R, Trievel RC. Histone modifying enzymes: Structures, mechanisms, and specificities. Biochim Biophys Acta. 2009;1789:58–68. doi: 10.1016/j.bbagrm.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathers JC, McKay JA. Epigenetics—Potential contribution to fetal programming. Adv Exp Med Biol. 2009;646:119–123. doi: 10.1007/978-1-4020-9173-5_13. [DOI] [PubMed] [Google Scholar]
- Mothe I, Van Obberghen E. Phosphorylation of insulin receptor substrate-1 on multiple serine residues, 612, 632, 662, and 731, modulates insulin action. J Biol Chem. 1996;271:11222–11227. doi: 10.1074/jbc.271.19.11222. [DOI] [PubMed] [Google Scholar]
- Murrell A, Heeson S, Bowden L, Constancia M, Dean W, Kelsey G, Reik W. An intragenic methylated region in the imprinted IGF2 gene augments transcription. EMBO Rep. 2001;2:1101–1106. doi: 10.1093/embo-reports/kve248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of. Pdx1 J Clin Invest. 2008;118:2316–2324. doi: 10.1172/JCI33655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar K, Barron D, Lewitt MS, Murphy LJ. Growth retardation and hyperglycemia in insulin-like growth factor binding protein-1 transgenic mice. Endocrinology. 1995;136:4029–4034. doi: 10.1210/endo.136.9.7544274. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri N, Raychaudhuri S, Thamotharan M, Devaskar SU. Histone code modifications repress glucose transporter 4 expression in the intrauterine growth-restricted offspring. J Biol Chem. 2008;283:13611–13626. doi: 10.1074/jbc.M800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimatsu A, Rotwein P. Mosaic evolution of the insulin-like growth factors. Organization, sequence, and expression of the rat insulin-like growth factor I gene. J Biol Chem. 1987;262:7894–7900. [PubMed] [Google Scholar]
- Shiota K, Yamada S. Intrauterine environment-genome interaction and children’s development (3): Assisted reproductive technologies and developmental disorders. J Toxicol Sci. 2009;34(Suppl 2):SP287–SP291. doi: 10.2131/jts.34.sp287. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins-Haug L. Epigenetics and assisted reproduction. Curr Opin Obstet Gynecol. 2009;21:201–206. doi: 10.1097/GCO.0b013e32832d7b95. [DOI] [PubMed] [Google Scholar]
- Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335:1363–1367. doi: 10.1056/NEJM199610313351805. [DOI] [PubMed] [Google Scholar]
- Yakar S, Setser J, Zhao H, Stannard B, Haluzik M, Glatt V, Bouxsein ML, Kopchick JJ, LeRoith D. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J Clin Invest. 2004;113:96–105. doi: 10.1172/JCI200417763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel SH. Epigenetic mechanisms for nutrition determinants of later health outcomes. Am J Clin Nutr. 2009;89:1488S–1493S. doi: 10.3945/ajcn.2009.27113B. [DOI] [PMC free article] [PubMed] [Google Scholar]