

Abstract
Interferon-λ (IFN-λ) is a central regulator of mucosal immunity; however, its signaling specificity relative to that of type I interferons is poorly defined. IFN-λ can induce antiviral interferon-stimulated genes (ISGs) in epithelia, while the effect of IFN-λ in non-epithelial cells remains unclear. Here we report that neutrophils responded to IFN-λ. We found that in addition to inducing ISG transcription, IFN-λ (but not IFN-β) specifically activated a translation-independent signaling pathway that diminished the production of reactive oxygen species and degranulation in neutrophils. In mice, IFN-λ was elicited by enteric viruses and acted on neutrophils to decrease oxidative stress and intestinal damage. Thus, IFN-λ acted as a unique immunomodulatory agent by modifying transcriptional and non-translational neutrophil responses, which might permit a controlled development of the inflammatory process.
Interferons have long been used to scrutinize mechanisms of inflammation, metabolism and cancer. The interferon family comprises type I interferons (represented mainly by IFN-α and IFN-β), type II interferons (IFN-γ) and type III interferons (IFN-λ). Studies of interferon activity have centered on the ability of these cytokines to regulate the transcription and translation of ISGs, which in turn contributes to the enhanced detection of, and defense against, pathogens and to the re-establishment of normal homeostasis1. In contrast to the increasing knowledge of interferon-mediated expression of ISGs, it remains unclear whether immediate (transcription-independent) interferon responses are important physiologically, or even exist. Transcription-independent activity of the innate immune system is often-associated with an acute need for rapid host responses. For example, phagocytosis, reactive oxygen species (ROS) production and the degranulation of mast cells, cytotoxic T lymphocytes and neutrophils are all transcription-independent activities that permit the rapid clearance of pathogens or infected cells.
The need for rapid immune responses is most critical during the earliest stages of infection, at barrier surfaces. At barrier sites such as the lungs, liver and intestine, epithelia are the most-prominent stromal cells, and neutrophils are the most rapidly recruited immune cells. Among the interferon family of cytokines, IFN-λ is the only class whose activity is exerted mainly on barrier epithelial cells2–9. Although the functions of some immune cells, such as phagocytes and lymphocytes10–18, have been reported to be modified by IFN-λ, the IFN-λ receptor IFNLR is expressed mainly on epithelial cells2–9. IFNLR is a heterodimer composed of the ubiquitously expressed receptor chain IL-10RB and the epithelium-specific receptor chain IFNLR1 (refs. 19–21). In contrast, the receptors for type I interferons (IFNAR) and for IFN-γ (IFNGR) are expressed by every cell type1. Due to the specific importance of IFN-λ at barrier surfaces, we hypothesized that this class of cytokines would exhibit transcription-independent activities important for barrier homeostasis and defense. In our study, we found that neutrophils were the only immune cells that promptly responded to stimulation with IFN-λ. We showed that IFN-λ, but not type I interferons, regulated a non-translational signaling pathway independent of the STAT family of transcription factors that diminished neutrophil ROS production and degranulation but did not alter other protective activities, such as cytokine production or phagocytosis. Through the use of the mouse model of dextran sodium sulfate (DSS)-induced colitis, we found that IFN-λ protected mice against intestinal inflammation; this action of IFN-λ was totally independent of signaling in epithelial cells and relied solely on signaling in neutrophils. Also, we demonstrated that the presence of intestinal viruses was important for diminishing DSS-induced inflammation and that this effect was due to signaling by IFN-λ but not by type I interferons.
RESULTS
Mouse and human neutrophils express a functional IFNLR
We investigated the ability of various types of mouse immune cells to express mRNA encoding the two chains that form the IFNLR and to respond to stimulation with IFN-λ in terms of activation of ISG transcription. Neutrophils isolated on a Percoll gradient from the bone marrow (Supplementary Fig. 1a) had higher expression of Ifnlr1 than that of primary intestinal epithelial cells (Fig. 1a) and responded to IFN-λ by upregulating expression of the ISG Rsad2 (called ‘Viperin’ here) (Fig. 1b). Next we investigated whether Ifnlr1 expression changed during neutrophil differentiation. The expression of Ifnlr1 and induction of Viperin in response to IFN-λ were lower in Ly6Gint–hiSSCint immature neutrophils sorted from the bone marrow of wild-type C57BL/6 mice than in their Ly6GhiSSChi mature counterparts (Supplementary Fig. 1b–d). Ly6GhiSSChi mature neutrophils and Ly6Gint–hiSSCint immature neutrophils had the same capacity to upregulate Viperin in response to stimulation with IFN-β (Supplementary Fig. 1d). In addition, Ly6G+CD11b+ neutrophils from the blood showed expression of Ifnlr1 similar to that of Ly6GhiSSChi mature neutrophils from the bone marrow (either sorted or purified through the use of Percoll), while Ly6G+CD11b+ neutrophils isolated from the peritoneal cavity after injection of thioglycollate had the highest expression of Ifnlr1 (Supplementary Fig. 1c). Other innate and adaptive immune cells analyzed (conventional and plasmacytoid dendritic cells (DCs), T cells and B cells, and peritoneal macrophages) did not express Ifnlr1 and did not induce Viperin expression in response to recombinant IFN-λ (Fig. 1a,b and Supplementary Fig. 1e). As expected, the cells analyzed induced Viperin expression in response to treatment with IFN-β (Fig. 1b). In vitro administration of the pro-inflammatory cytokine TNF or lipopolysaccharide (LPS) did not alter the pattern of Ifnlr1 expression in DCs, macrophages or adaptive lymphocytes but increased the expression of Ifnlr1 in neutrophils (Fig. 1c). That was in agreement with the observation that neutrophils isolated from the peritoneum after the administration of thioglycollate (which induces an inflammatory state) had higher expression of Ifnlr1 than that of blood-derived neutrophils (Supplementary Fig. 1c). Human neutrophils isolated from the blood of healthy donors via gradient centrifugation, for the isolation of human polymorphonuclear leucocytes, had low basal expression of IFNLR1 but upregulated their VIPERIN transcription when challenged with recombinant human IFN-λ2 in vitro (Fig. 1d). Similar to mouse neutrophils, human neutrophils upregulated their IFNLR1 expression following activation with LPS (Fig. 1d). These data demonstrated that mouse and human neutrophils were unique among the immune cells we assessed in their ability to respond to IFN-λ stimulation in resting and inflammatory conditions.
Figure 1.

Mouse and human neutrophils are responsive to IFN-λ. (a) qPCR analysis of Ifnlr1 mRNA (left) and Il10rb mRNA (right), which encode the two components of the IFN-λ receptor, in mouse bone-marrow-derived neutrophils (PMN), primary intestinal epithelial cells (IEC), splenic plasmacytoid DCs (pDC) and splenic conventional DCs (cDC) derived from wild-type (WT) or IFNLR1-deficient (Ifnlr1−/−) mice (key); results are presented relative to those of the control gene Gapdh. ND, not detectable. (b) qPCR analysis of Viperin mRNA in neutrophils (left), plasmacytoid DCs (middle) or conventional DCs (right) left unstimulated (US) or stimulated for 3 h with IFN-λ or IFN-β (100 U/ml each) (key); results presented as in a. (c) qPCR analysis of Ifnlr1 in cells as in a, as well as T cells, B cells and peritoneal macrophages (pM0), left unstimulated or stimulated for 3 h with LPS (10 μg/ml) or TNF (100 ng/ml) (key). (d) qPCR analysis of Ifnlr1 (left) in human neutrophils left unstimulated or stimulated for 3 h with LPS (10 μg/ml), and of Viperin mRNA (right) in human neutrophils treated for 3 h with IFN-λ2 (100 U/ml) or IFN-β (100 U/ml) (horizontal axis) and left unstimulated or stimulated for 3 h with LPS (10 μg/ml) (key). (e) Immunoblot analysis of phosphorylated (p−) STAT1 and STAT3, and β-actin (loading control throughout), in neutrophils derived from the bone marrow of wild-type mice (top) or Ifnlr1−/− mice (bottom) and stimulated for 0–60 min (above lanes) with IFN-λ or IFN-β (blots cropped to show bands of the appropriate molecular size). (f) NanoString analysis of gene expression (nCounter Mouse Inflammation v2 panel; genes modulated more than twofold, with a P value of > 0.05) in mouse neutrophils stimulated for 30 min, 1 h or 3 h (left margin) with IFN-λ or IFN-β (100 U/ml). Data are from one of three experiments (a; mean + s.e.m. of three biological replicates) or one experiment representative of three experiments (b,d; mean + s.e.m. of three biological replicates) or are representative of three independent experiments (c,e,f; mean + s.e.m. in c).
To define the function of IFN-λ in neutrophils, we compared the ability of IFN-β and IFN-λ to induce the activation of specific STAT proteins and found that both types of interferons induced phosphorylation of STAT1–STAT3 in neutrophils, even though IFN-λ-induced phosphorylation of all the STAT proteins analyzed was less robust than that induced by IFN-β (Fig. 1e and Supplementary Fig. 1f), consistent with data from epithelial cells22–24. The specificity of STAT phosphorylation in response to the administration of IFN-λ was confirmed through the use of neutrophils derived from IFNLR1-deficient mice (Fig. 1e). We next investigated by NanoString technology the effect of stimulation with IFN-β or IFN-λ on gene expression at various time points (0.5, 1 and 3 h) in neutrophils freshly isolated from the bone marrow of wild-type mice. Analysis of a NanoString probe panel containing a pool of more than 190 genes encoding products associated with the inflammatory response indicated that all the genes modulated more than twofold (P < 0.05) were co-regulated by IFN-λ and IFN-β (Fig. 1f). Overall, these data indicated that neutrophils sensed IFN-λ and induced classic IFN-β-like ISG responses.
IFN-λ regulates neutrophil function independently of transcription
To investigate whether neutrophils exhibited transcription-independent interferon responses, we assessed neutrophil ROS production and release of granule contents25 in an inflammatory context. Treatment with IFN-λ substantially reduced the capacity of human and mouse neutrophils to release ROS in response to stimulation with TNF or LPS, compared with that of neutrophils stimulated with TNF or LPS alone (without interferon treatment) (Fig. 2a,b), in a dose- and time-dependent manner (Supplementary Fig. 2a–c), but treatment with IFN-β did not. To determine if neutrophil degranulation was affected by IFN-β or IFN-λ, we assessed the enzymatic activity of the tertiary granule component MMP-9 by measuring degradation of gelatin. Treatment with IFN-λ resulted in deficient MMP-9 activity relative to that of wild-type cells stimulated with LPS alone, but not relative to that of IFNLR1-deficient neutrophils stimulated with LPS alone (Fig. 2c and Supplementary Fig. 2d), suggestive of impaired degranulation. In contrast, treatment with IFN-β resulted in more gelatin degradation than that of neutrophils treated with LPS only (Supplementary Fig. 2d), suggestive of increased degranulation. Next, we activated wild-type neutrophils from the bone marrow with LPS in the presence or absence of IFN-λ, or not, and assessed their supernatants for the presence of lactate dehydrogenase (LDH), an enzyme that is released when the integrity of the plasma membrane is altered. LDH levels were similar under all the conditions tested (Supplementary Fig. 2e), which excluded the possibility that the secretion of MMP-9 was due to passive release of cytoplasm contents. Neutrophil viability was very similar under all these conditions, as assessed by staining of the apoptosis marker annexin V and with the membrane-impermeable DNA-intercalating dye 7-AAD (Supplementary Fig. 3a), which indicated that interferon treatment did not alter in the lifespan of these neu-trophils. Neutrophils isolated from the bone marrow of wild-type mice were treated with either IFN-λ or IFN-β or no interferon and were exposed to opsonized or non-opsonized Escherichia coli expressing green-fluorescent-protein (GFP). The ‘GFP positivity’ of neutrophils was assessed by cytofluorimetry and was used as a measurement of the phagocytic capacity of neutrophils. No difference was detected in in the GFP positivity of neutrophils treated with either IFN-λ or IFN-β or no interferon (Supplementary Fig. 3b). Similarly, there were no differences in the secretion of TNF or the cytokines IL-10 or IL1-β from neutrophils treated with either IFN-β or IFN-λ or no interferon in the presence or absence of LPS (Supplementary Fig. 3c). These data suggested that treatment with IFN-λ did not lead to a general suppression of the immunological function of neutrophils. NanoString analysis of the inflammation-related gene transcripts in wild-type bone marrow neutrophils activated with LPS in the presence or absence of either IFN-λ or IFN-β showed that IFN-β induced a gene-transcription profile similar to that induced by IFN-λ in the absence of LPS (Fig. 1f) or in its presence (Supplementary Fig. 3d). These data indicated that IFN-β and IFN-λ exerted distinct effects on ROS production and degranulation by neutrophils while transcriptionally regulating the same set of genes.
Figure 2.

IFN-λ suppresses the tissue-damaging function of neutrophils in a non-translational and non-transcriptional manner. (a) Kinetic analysis of the release of superoxide (respiratory burst) from bone-marrow-derived neutrophils and left untreated (UT) or treated with various combinations (key) of stimulation with TNF (left; 100 ng/ml) or LPS (middle; 10 μg/ml) and treatment with IFN-λ (100 U/ml), assessed as the oxidized form of cytochrome C and presented as the concentration of superoxide ion (O·) per 1 × 106 cells (left and middle). Right, inhibition of ROS production in neutrophils treated for 30 min with LPS or TNF (horizontal axis) in the presence of IFN-λ or IFN-β (key; 100 U/ml each), relative to ROS production in neutrophils treated for 30 min with LPS or TNF alone. (b) Kinetic analysis of the respiratory burst in human neutrophils left untreated or treated with various combinations (key) of stimulation with TNF (100 ng/ml) and treatment with IFN-λ2 (100 U/ml), presented as in a (left), and inhibition of ROS production in neutrophils treated for 30 min with TNF in the presence of IFN-λ2 or IFN-β (horizontal axis), relative to ROS production in neutrophils treated for 30 min with TNF alone (right). (c) Inhibition of MMP-9 activity, assessing degranulation in supernatants of wild-type and IFNLR1-deficient neutrophils (key) treated with IFN-λ plus various concentrations (horizontal axis) of LPS, presented relative to that of wild type cells treated with LPS alone (left), and electrophoretic analysis of proteolytic activity (zymography) of supernatants of wild-type and IFNLR1-deficient neutrophils (left margin) left untreated or treated with IFN-λ (above gels) and left unstimulated or stimulated with LPS (above lanes) (right). (d) Kinetic analysis of the respiratory burst in neutrophils treated with cycloheximide (10 μg/ml) and stimulated as in a (key), with results presented as in a (left), and inhibition of ROS production in neutrophils treated with TNF in the presence of IFN-λ with (CHX) or without (−) cycloheximide (horizontal axis), relative to ROS production by neutrophils treated with TNF only (right). (e) Inhibition of ROS production in neutrophils obtained from wild-type (WT) mice, STAT1-deficient (Stat1−/−) mice or LysMcreStat3fl/fl (Stat3−/−) mice, in which STAT3 is deleted in myeloid cells, and treated with TNF in the presence of IFN-λ as in a; results presented as in a. (f,g) Kinetic analysis of the respiratory burst in mouse neutrophils treated for 0–30 min (horizontal axes) with Py6 (50 nM) (f) or HBC (10 μM) (g) and stimulated as in a, with results presented as in a (top), and inhibition of ROS production by neutrophils treated with TNF in the presence of IFN-λ with or without (UT) Py6 (f) or HBC (g), relative to ROS production by neutrophils treated with TNF only, assessed and presented as in a (bottom). (h) Immunoblot analysis of AKT phosphorylated at Ser473 (p-AKT 473) or Thr308 (p-AKT 308) and total AKT, phosphorylated and total STAT1, and p38 phosphorylated at Thr180 and Tyr182 (p-p38) (left margin), in mouse neutrophils treated for 30 min with IFN-λ (+) or not (−) in the presence (+) or absence (−) of HBC (10 μM), and stimulated for 30 min with LPS (10 μg/ml) (+) or not (−) (blots cropped as in Fig. 1e). NS, not significant (P > 0.05); *P < 0.05, **P < 0.01 and ***P < 0.001 (unpaired two tailed t-test). Data are representative of (a (left and middle), b,d (left), f,g (top), c (right), h) or from (a,b,d (right), f,g (bottom), c (left), e) three independent experiments (mean + s.e.m.).
To assess the dependence of the IFN-λ-induced inhibition of ROS production and degranulation on new protein synthesis, we treated wild-type bone marrow neutrophils with cycloheximide, which blocks protein synthesis. To assess the effectiveness of this treatment, we then treated neutrophils with puromycin, which binds to newly synthesized proteins and allows assessment of new protein synthesis. Immunoblot analysis of total proteins extracted from neutrophils with antibody to puromycin indicated that treatment with cycloheximide effectively blocked translation and the subsequent incorporation of puromycin (Supplementary Fig. 4a). However, the IFN-λ-induced decrease in ROS production (Fig. 2d and Supplementary Fig. 4b) and MMP-9 activity (Supplementary Figs. 2d and 4c) was not affected by cycloheximide in wild-type bone marrow neutrophils activated with TNF or LPS.
Next we focused on the transcriptional regulation of IFN-λ function. STAT1 is a master regulator of interferon-induced transcription of ISGs26. In wild-type bone marrow neutrophils treated with fludarabine, an inhibitor of STAT1, treatment with IFN-λ resulted in less ROS production than that of neutrophils treated with TNF (Supplementary Fig. 4d). Similar results were obtained with STAT1-deficient neutrophils (Fig. 2e). Increasing evidence has indicated non-transcriptional roles for STAT3 in determining several immunological functions27–29, and STAT3 was activated in wild-type bone marrow neutrophils in response to IFN-λ (Fig. 1e and Supplementary Fig. 1f). However, STAT3 deficiency did not alter the capacity of IFN-λ to diminish ROS production in neutrophils (Fig. 2e).
Jak kinases are activated following stimulation with IFN-λ, and the only difference described so far in the signaling pathway downstream of IFN-β and IFN-λ is the ability of IFNLR, but not of IFNAR, to recruit Jak2 (ref. 30). RAW264.7 mouse macrophages transfected with a construct that allows expression of the IFNLR1 chain acquired the ability to respond to IFN-λ, and stimulation with IFN-λ, or with IFN-γ, led to phosphorylation of Jak2 in these cells, but stimulation with IFN-β did not (Supplementary Fig. 4e). In wild-type bone marrow neutrophils, Py6 (pyridone-6), an inhibitor of all Jak kinases, abolished the ability of IFN-λ to inhibit ROS production following activation with either TNF or LPS (Fig. 2f and Supplementary Fig. 4f). In addition, the Jak2 inhibitors HBC (1,2,3,4,5,6-hexabromocyclohexane) and AG490 also abolished the ability of IFN-λ to down-modulate ROS production in wild-type bone marrow neutrophils activated with TNF or LPS (Fig. 2g and Supplementary Fig. 4g). These data indicated that Jak2 mediated the IFN-λ-induced inhibitory effect on ROS production in neutrophils. To address the effectiveness of Jak2 and inhibition of all Jak kinases, we analyzed the ability of neutrophils to transcribe Viperin in response to IFN-λ or IFN-β, in the presence of either HBC or Py6. As expected, inhibition of all Jak kinases by Py6 reduced the induction of Viperin in response to either interferon, while inhibition of Jak2 by HBC affected the induction of Viperin only in response to IFN-λ (Supplementary Fig. 4h). In addition, the phosphorylation of Jak2 in the IFNLR1-expressing RAW264.7 macrophages described above was impaired when these cells were stimulated with IFN-λ in the presence of HBC, relative to that of cells treated with IFN-λ alone (Supplementary Fig. 4e).
To gain further insight into the molecular mechanisms that are regulated by IFN-λ and underlie the inhibition of the Jak2- mediated tissue-damaging functions of neutrophils, we investigated the key pathways known to be involved in neutrophil activation. The production of ROS by neutrophils is dependent on assembly of the NADPH oxidase 2 (NOX2) complex31. Phosphorylation of the NOX2 component p47phox drives the translocation of p67phox, p40phox and RAC to cellular membranes, where they join NOX2 and the NOX2 component p22phox to form an active enzyme. Phosphorylation of p47phox by the kinase PKC32 or the kinase AKT33,34 is required for the activation of NADPH oxidases, while the phosphorylation of p47phox by the mitogen-activated protein kinases p38 and ERK1/2 (ref. 35) results in enhancement of this effect. The phorbol ester PMA potently activates PKC and largely bypasses the other requirements that lead to the activation of NOX2. PMA-induced production of ROS was similar in wild-type bone marrow neutrophils treated with IFN-λ and those not treated with IFN-λ (Supplementary Fig. 4i). Also, treatment with IFN-λ did not alter formation of the Rac–p67phox complex, either before or after stimulation with LPS (Supplementary Fig. 4j). Next, we analyzed by immunoblot the phosphorylation of AKT and p38, and of STAT1, as a control. We found p38 was phosphorylated similarly in the presence or absence of IFN-λ in LPS-treated neutrophils (Fig. 2h), which excluded the possibility that the activation of mitogen-activated protein kinases was altered by treatment with IFN-λ. However, treatment with IFN-λ induced a potent reduction in the phosphorylation of AKT at Ser473 and Thr308 (Fig. 2h). The phosphorylation of AKT was similar in IFN-λ-treated neutrophils and untreated neutrophils in the presence of the Jak2 inhibitor HBC (Fig. 2h), which indicated that IFN-λ diminished neutrophil function via Jak2-dependent inhibition of the AKT pathway. Together these data indicated that the effects of IFN-λ on ROS production and degranulation in neutrophils were independent of protein synthesis or transcription and were dependent on inhibition of the activation of AKT.
IFN-λ diminishes intestinal inflammation
We next investigated the effect of IFN-λ in DSS-induced colitis, a model of inflammation that depends on the activation of innate immune cells. Wild-type and IFNLR1-deficient mice co-housed 2 weeks before as well as during the experiment were given 2.5% DSS in their drinking water for 7 d. IFNLR1-deficient mice had a significantly shorter colon and lower weight, as well as a greater overall disease score, than that of wild-type mice at 7 d after the administration of DSS (Fig. 3a–c), results that we confirmed by histological analysis (Fig. 3d). Although the treatment of neutrophils with IFN-λ partially diminished their migratory ability in vitro (Supplementary Fig. 5a), in confirmation of published observations15, the number of neutrophils present in the lamina propria of IFNLR-deficient mice was similar to that of wild-type mice at all time points assessed (Supplementary Fig. 5b), which excluded the possibility that the diminished inflammatory state was caused by an inability of neutrophils to reach the inflamed tissue. Similarly, we detected no substantial difference in the amount of mRNA encoding the pro-inflammatory cytokines TNF or IL-6 in the colon of IFNLR-deficient mice and that in the colon of wild-type mice (Supplementary Fig. 5c,d), or in the frequency of live neutrophils that infiltrated the colon of these mice (Supplementary Fig. 5e). Analysis of the gene-transcription profile of the colon did not detect a significant difference between wild-type and IFNLR-deficient mice (Supplementary Fig. 5f and Supplementary Table 1). DSS-treated IFNLR1-deficient mice showed more transcription of genes encoding anti-oxidant molecules in total colon extracts than did their wild-type counterparts (Fig. 3e). In addition, wild-type mice treated with a blocking antibody to IFN-λ2 and IFN-λ3 showed a more robust increase in intestinal inflammation than that of untreated mice (Fig. 4), which indicated that the greater tissue damage and oxidative stress were not caused by differences in the microbiota composition. Overall, our data supported the conclusion that IFN-λ has a crucial role in diminishing intestinal inflammation.
Figure 3.

IFN-λ protects mice from DSS-induced colitis and diminishes oxidative stress. (a–d) Colitis progression and severity in wild-type and IFNLR1-deficient mice (key) co-housed for 2 weeks and then left untreated (UT) or treated with 2.5% DSS in the drinking water for 7 d (DSS), assessed as body weight relative to initial weight, set as 100% (a), disease activity index (DAI) (b), colon length (c) and histology (d). Original magnification (d), ×4 (left) or ×10 (right). (e) qPCR analysis of Hmox1 and Sod2 (genes induced in response to oxidative stress), assessing oxidative damage in colonic tissue obtained from wild-type and IFNLR1-deficient mice (key) at day 7 after the administration of DSS (as in a); results are presented relative to those of Gapdh. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 (two-way ANOVA (a–c) or nonparametric two-tailed t-test (d,e)). Data are pooled from three independent experiments (a,c; mean + s.e.m. of 20 mice per group) or are from one experiment representative of three independent experiments (b,d,e; mean + s.e.m. of 5 mice per group).
Figure 4.
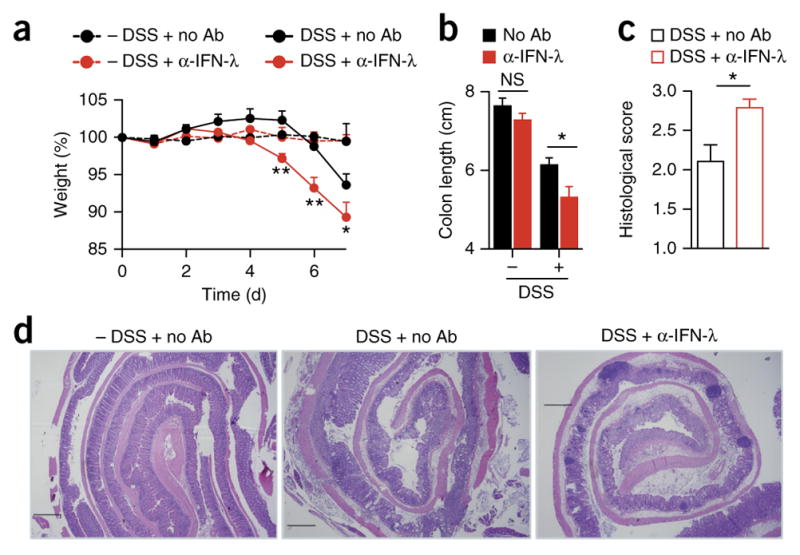
The protective effect of IFN-λ in DSS-induced colitis is independent of the genetic background or strain-specific microbiota. Colitis in wild-type mice left treated with various combinations (keys and axes) of 2.5% DSS in their drinking water (DSS) or not (− DSS) and daily intraperitoneal injection of antibody to IFN-λ (α-IFN-λ) (10 g/kg) or not (no Ab), assessed as body weight (a), colon length (b), histopathological score (c) and histology (d). Scale bars (d), 500 μm. *P < 0.05, **P < 0.01 and ***P < 0.001 (two-way ANOVA (a,b) or nonparametric two-tailed t-test (c)). Data are from one experiment representative of three independent experiments (mean + s.e.m. of five mice in a–c).
IFN-λ acts on neutrophils to curtail intestinal inflammation
We next investigated the cell type that mediated the protective effect exerted by IFN-λ during intestinal inflammation. At 7 d after the administration of DSS, only neutrophils had high expression of Ifnlr1 in the lamina propria (Supplementary Fig. 6a). We generated bone-marrow chimeras by transferring wild-type bone-marrow cells into irradiated wild-type recipient mice (WT→WT) or by transferring IFNLR1-deficient (Ifnlr1−/− (IFNLR1-KO)) bone marrow cells into irradiated wild-type recipient mice (IFNLR1-KO→WT) (chimerism analysis, Supplementary Fig. 6b,c), then co-housed the chimeras for 2 weeks and then treated them with 2.5% DSS in the drinking water for 7 d. IFNLR1-KO→WT chimeras had a significantly shorter colon, more tissue damage and weight loss and a higher pathology score when challenged with DSS than did WT→WT chimeras (Fig. 5a–c and Supplementary Fig. 6d). The intestinal epithelial cells of IFNLR1-KO→WT chimeras showed oxidative-stress-induced upregulation of gene expression and more direct oxidative damage to DNA than that of WT→WT chimeras (Fig. 5d and Supplementary Fig. 6e). Notably, even though IFNLR was expressed by epithelial cells in IFNLR1-KO→WT chimeras (data not shown), epithelial cells in those mice showed more oxidative stress than that of epithelial cells in WT→WT chimeras (Fig. 5d), which suggested that the absence of IFN-λ signaling in neutrophils had a dominant influence on tissue damage and oxidation.
Figure 5.

IFN-λ protects mice from DSS-induced colitis by inhibiting neutrophil function. (a–c) Colitis progression and severity in WT→WT and IFNLR1-KO→WT chimeras treated with 2.5% DSS in the drinking water for 7 d (or not (UT), in a), assessed as colon length (a), disease activity index (b) and histology (c). (d) qPCR analysis of Hmox1 and Sod2 in intestinal epithelial cells from mice as in a–c; results presented relative to those of Gapdh. (e–g) Colitis progression and severity in MRP8creIfnlr1fl/fl and Ifnlr1fl/fl littermates treated with 2.5% DSS in the drinking water for 7 d (or not (UT), in e), assessed as colon length (e), disease activity index (f) and histology (g). (h) qPCR analysis of Hmox1 and Sod2 in intestinal epithelial cells from mice as in e–g, presented as in d. Original magnification (c,g), ×10. *P < 0.05, **P < 0.01,***P < 0.001 and ****P < 0.0001 (two-way ANOVA (a,e) or nonparametric two-tailed t-test (b,d,f,h)). Data are representative of three experiments (mean + s.e.m. in a,b,d,e,f,h).
To further investigate whether neutrophils have a unique role in protecting mice against DSS-induced inflammation, we used MRP8creIfnlr1fl/fl C57BL/6 mice, in which expression of Cre recombinase under the control of the promoter of the human gene encoding the myeloid-cell co-regulator MRP8 selectively ablates the loxP-flanked second exon of Ifnlr1 (Ifnlr1fl/fl) in neutrophils36. Accordingly, neutrophils derived from MRP8creIfnlr1fl/fl C57BL/6 mice did not express Ifnlr1 mRNA (Supplementary Fig. 6f) and did not respond to stimulation with IFN-λ (Supplementary Fig. 6g). MRP8creIfnlr1fl/fl C57BL/6 mice recapitulated the phenotype seen in the IFNLR1-KO→WT chimeras (Fig. 5e–h and Supplementary Fig. 6h). The recruitment of neutrophils to the lamina propria of MRP8creIfnlr1fl/fl C57BL/6 mice was similar to that of Ifnlr1fl/fl C57BL/6 (control) mice (Supplementary Fig. 6i). NanoString gene-expression analysis of MRP8creIfnlr1fl/fl C57BL/6 and Ifnlr1fl/fl C57BL/6 mice 9 d after the administration of DSS excluded the possibility of major differences in gene transcription in either neutrophils or stromal cells isolated from the inflamed colons (Supplementary Fig. 6j and Supplementary Table 2), suggestive of a non-transcriptional and non-translational effect of IFN-λ. Although we cannot rule out the possibility that under inflammatory conditions, immune cells other than the ones we assessed (DCs, macrophages, natural killer cells, neutrophils, group 3 innate lymphoid cells, T cells and B cells) might acquire IFNLR1 expression and responsiveness to IFN-λ in vivo, overall our data indicated that IFN-λ signaling in neutrophils mediated the diminished colon shortening, tissue damage and oxidative stress.
Because IFN-λ has a critical role in modifying epithelial-cell responses3,4,19,36, we induced DSS-mediated colitis in irradiated IFNLR1-deficient host mice reconstituted with wild-type bone marrow (WT→IFNLR1-KO), which lack IFNLR expression in the stromal compartment (Supplementary Fig. 6k,l). We found that tissue damage, colon length, weight loss, disease score, oxidative stress mRNA response or oxidative damage to DNA were not significantly different in WT→IFNLR1 KO chimeras versus WT→WT (control) chimeras (Fig. 6a–g). Because those data did not exclude the possibility of confounding effects of other non-immune cells or radio-resistant immune cells, we induced DSS-mediated colitis in VilcreIfnlr1fl/fl C57BL/6 mice, in which Ifnlr1 is deleted in the intestinal epithelial cell compartment via Cre expressed from the promoter of the mouse gene encoding the actin-modifying protein villin-1 (Vil1)36 (Supplementary Fig. 6m). We saw no major difference between VilcreIfnlr1fl/fl C57BL/6 mice and Ifnlr1fl/fl C57BL/6 (control) mice in terms of tissue damage, colon length, weight loss, disease score or oxidative stress (Fig. 6h–k and Supplementary Fig. 6n). Together these data documented that IFN-λ signaling in neutrophils had a dominant role in diminishing tissue damage in the DSS-induced inflammatory model and favored the development of a regulated inflammatory response.
Figure 6.

The protective effect of IFN-λ in DSS-induced colitis is independent of IFNLR1 expression in epithelial cells. (a–d) Colitis progression and severity in WT→WT and WT→IFNLR1-KO chimeras co-housed for 2 weeks and then treated with 2.5% DSS in the drinking water for 7 d (or not (UT), in b), assessed as body weight (a), colon length (b), disease activity index at day 7 (c) and histology (d). (e,f) qPCR analysis of Hmox1 (e) and Sod2 (f) in intestinal epithelial cells at day 7 in mice as in a–d; results presented relative to those of Gapdh. (g) Oxidation of DNA, assessed as 8-oxoguanine (8-oxog) (per ng of DNA) at day 7 in the colon of mice as in a–d. (h–j) Colitis progression and severity in VilcreIfnlr1fl/fl and Ifnlr1fl/fl littermates treated with 2.5% DSS in the drinking water for 7 d (or not (UT), in h), assessed as colon length (h), disease activity index at day 7 (i) and histology (j). (k) qPCR analysis of Hmox1 and Sod2 in intestinal epithelial cells at day 7 in mice as in h–j, presented as in e,f. Original magnification (d,j), ×10. *P < 0.05, **P < 0.01 and ***P < 0.001 (two-way ANOVA (a,b,h) or nonparametric two-tailed t-test (c,e,f,g,I,k)). Data are from one experiment representative of three experiments (a–g; mean + s.e.m. of eight (WT→WT) or four (WT→IFNLR1 KO) mice per group in a–c,e,f) or one experiment representative of three independent experiments (h–k; mean + s.e.m. of five mice per group in h,i,k).
Enteric viruses protect against colitis via induction of IFN-λ
Enteric viruses are reported to have a protective role in the development of DSS-induced intestinal inflammation37. Because viral stimulation potently triggers the induction of IFN-λ in the intestine2–6,38, we investigated whether IFN-λ contributes to the protective role of enteric viruses. To test this hypothesis, we administered a ‘cocktail’ of anti-viral (AV) drugs to wild-type, IFNLR1-deficient or IFNAR-deficient C57BL/6 mice intragastrically for 10 d, followed by the induction of colitis via DSS. Such treatment with AV drugs reduced the number of enteric virus-like particles recovered from the stool and inhibited the induction of IFN-λ in the intestine of DSS-treated wild-type C57BL/6 mice relative to that of mice treated with DSS alone (without AV drugs) (Supplementary Fig. 7a,b). The dysbiosis caused by treatment with AV drugs significantly decreased colon length and augmented tissue damage in wild-type mice but not in IFNLR1-deficient mice (Fig. 7a–e). Like DSS-treated wild-type mice, DSS-treated IFNAR-deficient mice were sensitive to the administration of AV drugs and showed greater tissue damage (as proven by their shorter colon length and higher disease activity index and by histological analysis), as well as greater oxidative stress, relative to that of mice given DSS but not treated with AV drugs, while no substantial alteration in Tnf expression in the colon was detected (Supplementary Fig. 7c–i). These data indicated that in the absence of IFN-λ signaling, but not in the absence of IFN-α–IFN-β signaling, enteric viruses failed to protect the host against the development of intestinal inflammation.
Figure 7.

Enteric-virus-induced IFN-λ protects mice from DSS-induced colitis by diminishing oxidative stress. (a–e) Disease progression in wild-type and IFNLR1-deficient mice treated intragastrically with an AV drug ‘cocktail’ (+ AV) or PBS (control) for 10 d before the administration of 2.5% DSS in the drinking water, evaluated as weight loss (a), disease activity index (b), colon length (c), histopathological score (d) and histology (e). (f–i) Disease progression in wild-type mice treated with AV drugs or PBS, plus DSS (as in a), and given intraperitoneal injection of PBS (+ no IFN) or mouse recombinant IFN-λ to which polyethylene glycol was attached (+ IFN-λ; 4 mg per kg body weight) for 7 d, evaluated by histology (e), weight loss (f), disease activity index (g), colon length (h) and histopathological score (i). Original magnification (e), ×4 (top row) or ×10 (bottom row). *P < 0.05, **P < 0.01 and ***P < 0.001 (two-way ANOVA). Data are from one experiment representative of three independent experiments (mean + s.e.m. of eight mice per group in a–d,f–i).
The administration of mouse recombinant IFN-λ to which poly-ethylene glycol was attached (Fig. 7e–i), or commercially available recombinant IFN-λ (Supplementary Fig. 8a–c), to wild-type mice treated with AV drugs diminished the colon shortening, tissue damage, weight loss and disease activity after treatment with DSS, which indicated that the protective role of enteric viruses could be mimicked by exogenous IFN-λ. Both exogenous IFN-β and exogenous IFN-λ protected mice treated with AV drugs from DSS-induced inflammation, but IFN-λ was more efficient in this (Fig. 8a–d). In addition, the inflamed intestinal tissues showed a similar gene signature, as measured by qPCR or NanoString technology, when either IFN-β or IFN-λ was administered (Fig. 8e,g and Supplementary Table 1). However, only treatment with IFN-λ led to a robust reduction in ROS-dependent gene transcription in the epithelial cells of DSS-treated wild-type mice, relative to such transcription in their counterparts given DSS but not treated with IFN-λ (Fig. 8f). Overall, our data suggest that enteric viruses protect their hosts against the development of colitis via the induction of a translation-independent program elicited by IFN-λ (Supplementary Fig. 8d).
Figure 8.

IFN-λ displays a superior ability in suppressing DSS-induced colitis in the absence of enteric viruses, by diminishing oxidative stress. (a–d) Colitis development in wild-type mice treated intragastrically with a ‘cocktail’ of AV drugs or PBS for 10 d before the administration of 2.5% DSS in the drinking water, as well as no interferon (no IFN), mouse recombinant IFN-λ to which polyethylene glycol was attached (IFN-λ) or recombinant IFN-β (IFN-β) (200 U per gram body weight per day of either), evaluated as colon length (a), disease activity (b), weight at day 7, relative to initial weight (c), and histology (d). (e,f) qPCR analysis of Tnf in the total colon (e) and of Hmox1 in intestinal epithelial cells (measuring oxidative damage) (f) in mice as in a–d. (g) NanoString analysis of gene expression (nCounter Mouse Inflammation v2 panel; genes that generated >500 counts per treatment) in mRNA (100 ng) purified from total colon extracts of mice as in a–d. *P < 0.05, **P < 0.01 and ***P < 0.001 (two-way ANOVA.). Data are from one of three independent experiments (mean + s.e.m. of five mice per group in a–c,e,f).
DISCUSSION
Here we have described a unique ability of IFN-λ to diminish the most potent tissue-damaging functions of neutrophils in a non- transcriptional and non-translational manner and have reported that these interferons curtailed intestinal inflammation. The ability to respond to IFN-λ was not limited to terminally differentiated (Ly6ghiSSChi) bone marrow neutrophils, as blood and peritoneal neutrophils also responded to stimulation by IFN-λ. Moreover, blood-derived human neutrophils showed activation of the same processes described for mouse neutrophils. These findings are particularly relevant in the context of using IFN-λ as a potential therapeutic agent.
We found that IFN-λ regulated the activation of AKT in a JAK2-dependent manner to diminish ROS production by neutrophils. We excluded the possibility that IFN-λ altered the activation of p38, the RAC–p67phox– dependent formation of the NOX2 complex or PKC function. Instead, IFN-λ profoundly decreased the activation of AKT, and this effect depended on the unique ability of IFN-λ (not type I interferons) to activate JAK2.
The most specific effects of IFN-λ signaling regulated non-translational events in neutrophils, relative to the effects of type I interferons. We found that neutrophils treated with cycloheximide, which blocks protein synthesis, as well as neutrophils stimulated in the presence of the STAT inhibitor fludarabine, were sensitive to the anti-inflammatory effects of IFN-λ. Similarly, neutrophils deficient in STAT1, a master regulator of the interferon-dependent induction of ISGs, or STAT3, which has been shown to have non-transcriptional roles, were also sensitive to treatment with IFN-λ. Although we cannot rule out the possibility of a contribution of interferon-dependent gene transcription in vivo during DSS-induced colitis, as has been suggested in the context of viral lung infection39, our data suggest a unique, translational-independent role for IFN-λ, in contrast to the role of type I interferons.
Finally, our observations indicated that enteric viruses facilitated protection against intestinal inflammation via the production of IFN-λ. In addition, although exogenously applied type I interferons have a protective role, the newly described ability of IFN-λ to diminish oxidative stress greatly potentiated its effectiveness in decreasing intestinal inflammation.
Our data have provided a mechanism by which the IFN-λ class of interferons regulate intestinal inflammation. Published literature has addressed the role of IFN-λ in epithelial-cell responses in the context of intestinal inflammation, but little is known about the ability of this class of interferons in modulating the function of immune cells. However, mutations in the gene encoding IL-10RB, which forms the IFNLR dimer, together with IFNLR1, are associated with the early onset of inflammatory bowel disease (IBD)40,41, and loss of IL-10RB in innate immune cells results in the development of IBD42. Both observations suggest that the IFN-λ class of interferons probably participate in the development of IBDs. Indeed, it has been reported that patients with IBD show an altered profile of IFN-λ expression43. We anticipate that the previously unknown functions of the IFN-λ class of interferons and its limited cellular targets that we have documented here will inform future studies evaluating the possible relevance of these interferons in the context of the development of IBD.
ONLINE METHODS
Mouse strains
C57BL/6J (Jax 00664) (wild-type), B6.SJL-Ptprca Pepcb/BoyJ (CD45.1), IFNAR-deficient (32045-JAX), MRP8cre recombinase, and Villincre recombinase mice were purchased from Jackson Labs. B6.129S2-Ifnar1tm1Agt/Mmjax (Ifnar1−/−) mice were obtained from MMRC and purchased through Jackson Labs. C57BL/6 IL-28R−/− (IFNLR1-deficient) mice were provided by Bristol-Myers Squibb. Stat1−/− and LysMcreStat3fl/fl mice were provided by S.B. Snapper. The mutant mouse line Ifnlr1tm1a(EUCOMM)Wtsi was provided by the Wellcome Trust Sanger Institute Mouse Genetics Project (Sanger MGP) and its funders (funding and associated primary phenotypic Information, http://www.sanger.ac.uk/mouseportal). The Ifnlr1tm1a(EUCOMM)Wtsi mouse line already backcrossed to Villincre-recombinase-expressing mice43 was provided by H.W. Virgin. Mice were housed under specific pathogen-free conditions at Boston Children’s Hospital, and all the procedures were approved under the Institutional Animal Care and Use Committee (IACUC) and operated under the supervision of the department of Animal Resources at Children’s Hospital (ARCH).
Reagents and antibodies
For flow cytometry and sorting experiments, antibodies to CD4 (RM4-4), NK1.1 (PK136), CD49b (DX5), CD3 (17A2), Ly6G (1A8), B220 (RA3-6B2), CD11c (N418), CD45.2 (104), CD45.1 (A20), CX3CR1 (SA011F11), IAb (AF6-120.1), CD11b (Mac1), CD90 (G7), lineage markers (17A2/RB6-8C5/RA3-6B2/Ter-119/M1/70), and CD127 (A7R34) were purchased from BioLegend. To stimulate neutrophils in vitro and for in vivo administration, we used either recombinant mouse IFN-λ2 (purchased from Peprotech) or recombinant mouse IFN-λ2 to which polyethylene glycol was attached (provided by Bristol-Myers Squibb). Recombinant mouse IFN-β was purchased from PBL interferonsource. Recombinant human IFN-λ2 and IFN-β were purchased from Peprotech. Puromycin was purchased from Sigma. Where indicated, specific chemical inhibitors were used. To inhibit protein synthesis, cycloheximide (Sigma) was used at a concentration of 10 μg/ml. To inhibit Jak signaling, pyridone 6 (BioVision) was used. To inhibit STAT-1 signaling, fludarabine (Tocris) was used. To inhibit Jak2 signaling, either AG490 (Tocris) or 1,2,3,4,5,6-hexabromocyclohexane (HBC, Tocris) was used. To stimulate neutrophils, either E. coli LPS (Serotype O55:B5 TLR grade, purchased from Enzo) or recombinant mouse TNF (Peprotech) was used.
cDNA, RAW cell line transfection and retrovirus-mediated gene transduction
The cDNA encoding full-length mouse Ifnlr1 (Accession: BC057856; Cat. MMM1013-202767950) was purchased from openbiosystems (Darmacon). A 3xHA tag was introduced to the C terminus of Ifnlr1 by PCR.
For making RAW stable cell lines, IFNLR1-3HA was cloned into pMSCV2.2-IRES-hCD2. Retrovirus expressing the appropriate alleles was produced by 293T cells co-transfected with the packaging system composed of pVSVG, pCL-Eco and pMSCV vectors. RAW cells expressing IFNLR1-3HA were subjected for sorting by flow cytometry to an extent that 85% of the cells were positive for human CD2 in comparison to the untransduced parental cell lines.
Neutrophil isolation and functional assays
Naive neutrophils from the bone marrow of wild-type or Ifnlr1−/− mice were purified over a 62.5% Percoll gradient (GE Healthcare) in Ca2+- and Mg2+-free HBSS as previously described44. Purified neutrophils, unless specified otherwise, were pre-treated, or not, with 100 U/ml of either IFN-λ or IFN-β for 30 min in HBSS supplemented with 20 mM HEPES at 37 °C at a density of 3 × 106 cells per ml before stimulation. Where indicated, neutrophils were sorted from bone marrow of wild-type mice based on the expression of Ly6G and SSC intensity. Mature neutrophils were sorted as Ly6GhiSSChi cells, while immature neutrophils were sorted as Ly6Gint–hiSSCint cells. Blood neutrophils were sorted as Ly6G+CD11b+ cells from mouse blood, after red-blood-cell lysis. Peritoneal PMNs were sorted from peritoneal lavage of mice treated for 24 h with 1 ml 4% thioglycollate.
Human neutrophils were isolated from blood according to Boston Children’s Hospital Institutional Review Board (IRB), protocol number IRB-P00022885. In brief, blood neutrophils were purified on a gradient of Polymorphprep (Axis Shield) according to the manufacturer’s instruction. For functional assays, 3 × 105 neutrophils were plated on 96-well plates coated for 1 h at 37 °C with 150 μg/ml human fibrinogen (Millipore). Neutrophils were stimulated with 10 μg/ml LPS or 100 ng/ml TNF.
For extracellular ROS production, neutrophils were mixed with 100 mM cytochrome C (Sigma, Cat No. C-7752) before stimulation. To measure super-oxide release, the difference in absorbance of the oxidized form of cytochrome C (550 nm) and the background absorbance of the non-reduced form (540 nm) was measured every two minutes in a Fluster Omega Microplate Reader.
To evaluate degranulation, supernatants from neutrophils stimulated as described above were collected and assessed by a zymography assay for gela-tinase (MMP-9). Non-reducing sample buffer (10% glycerol (v/v), 2% SDS (w/v), 5 mg/ml bromophenol blue and 62.5 mM Tris, pH 6.8) was added to cell supernatants. Cell supernatants were then separated by SDS-PAGE in a 7.5% gel supplemented with 1 mg/ml gelatin (Sigma). Gels were subsequently renaturated in 10% Triton (Sigma) for 30 min and developed overnight in 200 mM NaCl, 5mM CaCl2 and 50 mM Tris, pH 7.4. Gels were stained with Coomassie blue and images of the gel were generated by scanning. MMP-9 activity was quantified with Image-J software.
Migration experiments were performed with Transwell inserts with 3-μm diameter pores (Corning), coated with 150 μg/ml fibrinogen. 5 × 105 neutrophils were seeded on the top of the Transwell and 1 μM fMLP was used as a chemoattractant. Migrated cells were counted on the bottom of the Transwell after 60 min of incubation at 37 °C with CasyCounter (Innovatis).
To evaluate cytokine production 3 × 105 neutrophils were plated in 96-well plates and were stimulated overnight with 10 μg/ml LPS in DMEM (GIBCO) supplemented with 10% FBS (GIBCO). Production of TNF and IL-10 was evaluated in the supernatants by ELISAmax (BioLegend) according to the manufacturer’s specifications. IL-1β production was evaluated as both cell associated and secreted IL-1β by freezing of the cells along with the supernatants twice at −80 °C. After thawing, IL-1β was measured using Mouse Ready-SET-Go ELISA kit (e Bioscience) according to themanufacturer’s instructions.
For phagocytosis experiments, E.coli (DH5α) expressing enhanced GFP (eGFP) was grown to an optical density (OD) of 1 in LB medium. E. coli was subsequently opsonized, or not, in HBSS with 10% mouse serum for 30 min at 37 °C. 1 × 105 purified neutrophils pre-treated or not with IFN-λ or IFN-β, were then incubated with E. coli at an multiplicity of infection of 1 for 30 min. Cells were then washed and analyzed by flow cytometry. The percentage of GFP+ Ly6G + neutrophils was evaluated.
For NanoString nCounter assays, 1 × 104 neutrophils were either stimulated with 100 U/ml of IFN-λ2 or IFN-β or pre-treated in the same conditions and subsequently stimulated with LPS (10 μg/ml).
At the appropriate time points, neutrophils were lysed in RLT buffer and were hybridized onto nCounter Mouse Inflammation v2 cartridges on the nCounter Prep Station in triplicates according to the manufacturer’s instructions. Hybridized cartridges were then read with the nCounter Digital Analyzer and analyzed by nSolver software (NanoString Technologies, Seattle, WA).
Flow cytometry
The absolute number of neutrophils in the lamina propria (LP) was calculated using CountBright Absolute Cell Counting Beads (Thermo Scientific) according to the manufacturer’s instructions.
To evaluate apoptosis, staining with annexin V and 7-AAD (BioLegend) was performed on purified neutrophils pre-treated or not with 100 U/ml of IFN-λ or IFN-β and stimulated with 10 μg/ml LPS for 3 h.
To evaluate apoptosis on LP neutrophils, LP cells were stained for CD45, Ly6G and CD11b (antibodies identified above (‘Reagents and antibodies’); 1:200 dilution) and were subsequently stained with annexin V and 7-AAD (Cat. No. 640930, BioLegend) according to the manufacturer’s instruction.
All samples were analyzed with a BD FACSCanto II.
Immunoblot analysis
For immunoblot analysis, 1 × 106 neutrophils from wild-type or Ifnlr1−/− mice were stimulated as indicated in the figure legends and were lysed in 100 μl of RIPA buffer supplemented with Protease and phosphatase inhibitor cocktail (Pierce) and diisopropylfluorophosphate (Sigma). Immunoblot analysis was performed using standard techniques. Blots were probed for STAT1 phosphorylated at Tyr701 (clone 14/P-STAT1, BD), STAT2 phosphorylated at Tyr690 (Cat. No. ab53132, Abcam), STAT3 phosphorylated at Tyr705 (Cat. No. 9131, Cell Signaling Technologies), β-actin (clone AC-74, Sigma), Jak2 phosphorylated at Tyr1007 and Tyr1008 (Cat. No. sc-21870, Santa Cruz Biotechnology), Jak2 (clone C20, Santa Cruz Biotechnology), AKT (Cat. No. 9272, Cell Signaling Technology), AKT phosphorylated at Thr308 (clone 244F9, Cell Signaling Technology), AKT phosphorylated at Ser473 (clone 736E11, Cell Signaling Technology), p38 phosphorylated at Thr180 and Tyr182 (clone 36/p38, BD), STAT-1 (Cat. No. 9172, Cell Signaling Technology) and anti-puromycin (clone 12D10, Millipore).
DSS-mediated colitis induction and disease evaluation
8-week-old female wild-type and Ifnlr1−/−mice were co housed for 2 weeks before DSS administration. Where indicated in the figure legends, MRP8creIfnlr1fl/fl and VilcreIfnlr1fl/fl mice were paired with age- and sex-matched littermates. To induce colitis, mice were given 2.5% (w/v) dextran sulfate sodium (DSS, Affymetrix) in the drinking water for 7 d and were killed at day 7. Where indicated in the figure legends, to deplete enteric viruses, mice were treated as previously described36. In brief, mice were given intragastric administration of a ‘cocktail’ of acyclovir (20 mg per kg body weight (mg/kg)), lamivudine (10 mg/kg) and ribavirin (30 mg/kg) (Sigma) (or same volume of PBS, for control mice) for 10 d before DSS administration. Where indicated in the figure legends, mice received intraperitoneal injection of mouse recombinant IFN-λ to which polyethylene glycol was attached (200 U/g per day) or IFN-β (200 U/g per day). Alternatively, mice received, where indicated in the figure legends, daily intraperitoneal injection of IFN-λ (1 μg/g). To deplete endogenous IFN-λ in wild-type mice, where indicated in the figure legends, mice received daily intraperitoneal injection of 1 μg/g of antibody to IFN-λ2-3 (MAB17892, R&D systems). In all experiments, control mice were given an equal volume of PBS.
Body weight, stool consistency and the presence of blood in the stool were monitored daily. Weight change was calculated as percentage of initial weight. Disease activity index was calculated as previously described45. For histology, colons were flushed with PBS, flattened and rolled into a ‘Swiss roll’. Colon rolls were fixed in 10% formalin (Fisher Scientific), dehydrated in 70% Ethanol and embedded in paraffin. Paraffin sections were stained with hematoxylin and eosin and histological features were evaluated.
Histological scoring was performed in a blinded fashion by assignment of a score of 1–5 to segments of the colon roll (1, presence of leukocyte infiltrate, loss of goblet cells; 2, bottom third of the crypt compromised; 3, two third of the crypt compromised; 4, complete crypt architecture loss; 5, complete crypt loss and lesion of the epithelial layer). Each segment was then measured with ImageJ software, and the final histological score of each sample was obtained by ‘weighting’ the score of each segment against the length of the segment and divided by the total length of the colon roll.
Evaluation of fecal virus-like particles
Fecal virus-like particles were counted as previously described36.
In brief, fecal pellets from untreated mice or mice treated with AV drugs were suspended at 0.05 g/ml in saline magnesium buffer (100 mM NaCl, 8 mM MgSO4, 50 mM Tris-HCl, pH 7.4, and 0.002% w/v gelatin). Samples were then centrifuged for 5 min at 2,000g, and supernatants were filtered with a 0.45-μm pore-size filters to remove cellular debris and bacteria. 100 μl of supernatants were then concentrated by centrifugation at 100,000g for 1 h and 30 min and were resuspended in 20 μl. Concentrated virus-like particles were then stained with either SYBR Gold (Invitrogen) to stain DNA or SYBR Green II (Invitrogen) to stain RNA and imaged at a ×40 magnification. Quantification was performed with ImageJ software on at least five fields of view per samples.
Generation of bone-marrow chimeras
To generate mice with hematopoietic-specific deletion of Ifnlr1, 8-week-old female CD45.1+ mice were exposed to lethal whole-body irradiation (950 rads per mouse) and were reconstituted with 5 × 106 bone marrow cells from 8-week-old female wild-type or IFNLR1-deficient mice. Mice were kept in autoclaved cages and were treated with sulfatrim in the drinking water for 2 weeks after reconstitution. After 2 weeks, mice were placed in cages with mixed bedding from wild-type or IFNLR1-deficient mice to replenish the microbiome and were allowed to reconstitute for 2 more weeks. A similar procedure was used to generate bone-marrow chimeras with specific deletion in stromal cells. In this case, recipient wild-type or IFNLR1-deficient mice underwent irradiation and were reconstituted with BM cells derived from CD45.1+ mice under the same conditions described above.
To evaluate the percentage of chimerism, a 30-μl sample of peripheral blood was taken from chimeric mice after 4 weeks of reconstitution. After red-blood-cell lysis was performed with ACK buffer, samples were stained for CD45.1, CD45.2 and Ly6G (antibodies identified above (‘Reagents and antibodies’)) and were analyzed by flow cytometry.
Analysis of expression of the interferon-λ receptor
Ifnlr1 expression was evaluated in immune-cell populations purified from naive mice or from the LP of colitic mice. To obtain steady-state DCs, wild-type or IFNLR1-deficient mice were given injection of 2 × 106 B16 cells overexpressing FLT3 ligand (FLT3L) to expand endogenous DC populations. After 2 weeks, the spleen was harvested and plasmacytoid DCs (pDCs) and conventional DCs (cDCs) were obtained by cell sorting with a BD FACSAria (purity >90%). pDCs were sorted as CD11c+B220+MHCII+ cells, and cDCs were sorted as CD11c+MHCII+B220− cells. CD4+ T cells were sorted from the spleen of naive mice as CD3+CD4+ lymphocytes. Peritoneal macrophages (pM0) were obtained from peritoneal lavage of mice given intraperitoneal injection of 1 ml of 4% thyoglicollate 4 d before the lavage. The peritoneal lavage was incubated for 30 min at 37 °C in a tissue-culture-treated plate, and only adhering macrophages were collected for gene expression analysis. Neutrophils were isolated from BM cells as described above. Intestinal epithelial cells were isolated from naive mouse colons. Colons were flushed with PBS, cut longitudinally, cut in ~5-mm pieces and incubated for 15 min at 37 °C under constant stirring in RPMI medium with 2% FBS, 0.5 mM EDTA and 0.08% DTT. Intestinal epithelial cells were collected from the supernatant. For colitic mice, LP cells were isolated from colon of wild-type mice that received 2.5% DSS in the drinking water for 7 d and were sorted to >90% purity with a BD FACSAria cell sorter. Colons were flushed and the epithelial layer was detached as described above. Fragments of colon devoid of the epithelial layer were incubated for 30 min at 37 °C under constant stirring in RPMI medium with 1% FBS and 200 U/ml collagenase from Clostridium histolyticum (Sigma). Liberated LP cells were collected from the supernatants and stained for cell sorting (antibodies identified above (‘Reagents and antibodies’)). Lymphocytes were selected as EPCAM−CD45+ cells and were subsequently separated based on lineage markers. B cells were selected as B220+CD3−CD11c− cell. CD4+ and CD8+ T cells were selected respectively as CD3+CD4+ cells and CD3+CD8+ cells. DCs were selected as MHCIIhiCD11c+CX3CR1− Cells. Macrophages were selected as CD11c+MHC II +CX3CR1+ cells. PMNs were selected as Ly6G+CD11b+ cells, NK1.1+ cells were selected as NK1.1+CD49b+ cells, and group 3 innate lymphoid cells were selected as CD45+CD90+CD127+Lin− cells.
The populations sorted as described above were treated with 100 U/ml IFN-λ or 100 u/ml IFN-β for 2 h or not, or were treated with 10μg/ml LPS or 100 ng/ml TNF for 1 h, or not. RNA was purified using a GeneJet RNA Purification Kit (Life Technologies). Purified RNA was analyzed for gene expression using a TaqMan RNA-to-CT 1-Step Kit (Applied Biosystems) on a CFX384 real time cycler (Bio-Rad). Gene expression was evaluated with probes from Life Technologies: Ifnlr1 (Mm00558035_m1), Il10rb (Mm00434157_m1), Viperin (Mm00491265_m1), and Gapdh (Mm99999915_g1) or 18srRNA (4308329-Applied Biosystems) as housekeeping controls.
Measure of antioxidant and cytokine gene expression in the colon
Antioxidant and cytokine gene expression was measured in colon of colitic mice treated with DSS for 7 d. 30 mg of colon samples from the distal, medial and proximal colon were placed in RNAlater RNA-stabilization solution (Ambion) and were conserved at 4 °C. The following day, samples were ground with a Tissue-Tearor Homogenizer (BIOSPEC) in Trizol (Thermo Scientific). Chloroform was added at a ratio of 20% of the homogenate volume, and after centrifugation, the aqueous phase was loaded onto GeneJet RNA Purification Columns (Life Technologies) and RNA was purified according to the manufacturer’s instructions. Where indicated, intestinal epithelial cells or LP cells purified as described above were lysed and RNA was purified with GeneJet RNA Purification Kit according to the manufacturer’s instructions (Life Technologies). Purified RNA was analyzed for gene expression using TaqMan RNA-to-CT 1-Step Kit (Applied Biosystems) with the following probes (Thermo Scientific): Hmox1 (Mm00516005), Sod2 (Mm01313000), Il6 (Mm00446190), infl3-ifnl2 (Mm04204156), TnfI (Mm00443258), Gapdh (Mm99999915_g1). Where indicated, 100 ng of mRNA purified from colonic tissue was hybridized with the nCounter Mouse Inflammation v2 gene expression kit as described above.
Evaluation of oxidative damage to DNA
DNA was extracted from 10 μg of colon tissue with a Purelink Genomic DNA mini Kit (Invitrogen) following the manufacturer’s instructions. 50 ng of DNA was then reduced to single nucleotide. After denaturation (10 min at 90 °C), DNA was digested with 20 U of nuclease P1 (30 min at 50 °C in Tris-HCl, pH 5.5) and was dephosphorylated with 20 U of alkaline phosphatase (30 min at 37 °C in Tris-HCl, pH 7).
Single nucleotides thus obtained were tested for the presence of 8-oxoguanine using a DNA/RNA Oxidative Damage ELISA kit (#589320, Cayman) following the manufacturer’s instruction.
Statistical analysis
Statistical significance for experiments with more than two groups was tested with one-way ANOVA, and Tukey’s multiple-comparison tests were performed. Two-way ANOVA with Tukey’s multiple-comparison test was used to analyze kinetic experiments. To establish the appropriate test, normal distribution and variance similarity were assessed with the D’Agostino-Pearson omnibus normality test using Prism7 (Graphpad) software.
When comparisons between only two groups were made, an unpaired two tailed t-test was used to assess statistical significance.
To determine the sample size, calculations were conducted in nQuery Advisor Version 7.0. Primary outcomes for each proposed experiment were selected for the sample size calculation and sample sizes adequate to detect differences with an 80% power were selected. For animal experiments, five to ten mice per group were used, as indicated in the figure legends.
Adjusted P values were calculated with Prism7 (Graphpad). Asterisks were used as follows (also indicated in figure legends): *P < 0.05, **P < 0.01, ***P < 0,001 and ****P < 0,0001.
A Life Sciences Reporting Summary for this paper is available.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon request (in particular, the gating strategy used for all flow cytometry, and full-length gels and blots, as well as all raw data from NanoString analyses).
Supplementary Material
Acknowledgments
We thank S.B. Snapper (Harvard Medical School) for Stat1−/− and LysMcreStat3fl/fl mice; the Wellcome Trust Sanger Institute Mouse Genetics Project (Sanger MGP) and its funders for the mutant mouse line Ifnlr1tm1a(EUCOMM)Wtsi; H.W. Virgin (Washington University) for the Ifnlr1tm1a(EUCOMM)Wtsi mouse line backcrossed to Villincre-recombinase-expressing mice; and J.C. Kagan, L.R. Marek-Kagan, the Kagan Lab, S.B. Snapper, the Snapper Lab, A. Mocsai and H.W. Virgin for discussions, help and support. Supported by the US National Institutes of Health (1R01DK115217, 1R01AI121066-01A1 to I.Z.), the Harvard Digestive Diseases Center (P30 DK034854 to I.Z.), the Harvard Medical School Milton Fund (to I.Z.), Crohn’s and Colitis Foundation of America (I.Z.), the Cariplo Foundation (I.Z. and F.G.), the Associazione Italiana per la Ricerca sul Cancro (F.G.), Fondazione Italiana di Ricerca per la SLA–Sclerosi Laterale Amiotrofica (ARISLA ) (F.G.), Fondazione Regionale per la Ricerca Biomedica (F.G.) and the Jane Coffin Child’s Memorial Fund for Medical Research (Y.T.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
A.B., F.G. and I.Z. designed the study; A.B. conducted most of the experiments; Y.T. prepared the IFNLR-expressing RAW cell line; F.G. and I.Z. conceived of the study; and I.Z. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baldridge MT, et al. Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science. 2015;347:266–269. doi: 10.1126/science.1258025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pott J, et al. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc Natl Acad Sci USA. 2011;108:7944–7949. doi: 10.1073/pnas.1100552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahlakõiv T, Hernandez P, Gronke K, Diefenbach A, Staeheli P. Leukocyte-derived IFN-α/β and epithelial IFN-λ constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. 2015;11:e1004782. doi: 10.1371/journal.ppat.1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin JD, et al. distinct roles of type I and type III interferons in intestinal immunity to homologous and heterologous rotavirus infections. PLoS Pathog. 2016;12:e1005600. doi: 10.1371/journal.ppat.1005600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernández PP, et al. Interferon-λ and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat Immunol. 2015;16:698–707. doi: 10.1038/ni.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Z, et al. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol. 2007;81:7749–7758. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doyle SE, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology. 2006;44:896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- 9.Marcello T, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 10.Souza-Fonseca-Guimaraes F, et al. NK cells require IL-28R for optimal in vivo activity. Proc Natl Acad Sci USA. 2015;112:E2376–E2384. doi: 10.1073/pnas.1424241112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin Z, et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J Immunol. 2012;189:2735–2745. doi: 10.4049/jimmunol.1102038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finotti G, Tamassia N, Calzetti F, Fattovich G, Cassatella MA. Endogenously produced TNF-α contributes to the expression of CXCL10/IP-10 in IFN-λ3-activated plasmacytoid dendritic cells. J Leukoc Biol. 2016;99:107–119. doi: 10.1189/jlb.3VMA0415-144R. [DOI] [PubMed] [Google Scholar]
- 13.Dai J, Megjugorac NJ, Gallagher GE, Yu RY, Gallagher G. IFN-lambda1 (IL-29) inhibits GATA3 expression and suppresses Th2 responses in human naive and memory T cells. Blood. 2009;113:5829–5838. doi: 10.1182/blood-2008-09-179507. [DOI] [PubMed] [Google Scholar]
- 14.Megjugorac NJ, Gallagher GE, Gallagher G. Modulation of human plasmacytoid DC function by IFN-λ1 (IL-29) J Leukoc Biol. 2009;86:1359–1363. doi: 10.1189/jlb.0509347. [DOI] [PubMed] [Google Scholar]
- 15.Blazek K, et al. IFN-λ resolves inflammation via suppression of neutrophil infiltration and IL-1β production. J Exp Med. 2015;212:845–853. doi: 10.1084/jem.20140995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Groen RA, et al. IFN-λ-mediated IL-12 production in macrophages induces IFN-γ production in human NK cells. Eur J Immunol. 2015;45:250–259. doi: 10.1002/eji.201444903. [DOI] [PubMed] [Google Scholar]
- 17.de Groen RA, Groothuismink ZM, Liu BS, Boonstra A. IFN-λ is able to augment TLR-mediated activation and subsequent function of primary human B cells. J Leukoc Biol. 2015;98:623–630. doi: 10.1189/jlb.3A0215-041RR. [DOI] [PubMed] [Google Scholar]
- 18.Wolk K, et al. IL-29 is produced by TH17 cells and mediates the cutaneous antiviral competence in psoriasis. Sci Transl Med. 2013;5:204ra129. doi: 10.1126/scitranslmed.3006245. [DOI] [PubMed] [Google Scholar]
- 19.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mordstein M, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol. 2010;84:5670–5677. doi: 10.1128/JVI.00272-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davidson S, et al. IFNλ is a potent anti-influenza therapeutic without the inflammatory side effects of IFNα treatment. EMBO Mol Med. 2016;8:1099–1112. doi: 10.15252/emmm.201606413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kotenko SV, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 23.Sheppard P, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 24.Meager A, Visvalingam K, Dilger P, Bryan D, Wadhwa M. Biological activity of interleukins-28 and -29: comparison with type I interferons. Cytokine. 2005;31:109–118. doi: 10.1016/j.cyto.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Mócsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med. 2013;210:1283–1299. doi: 10.1084/jem.20122220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wegrzyn J, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gough DJ, et al. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meier JA, et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci Signal. 2017;10:eaag2588. doi: 10.1126/scisignal.aag2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Odendall C, et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol. 2014;15:717–726. doi: 10.1038/ni.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med. 2009;41:217–225. doi: 10.3858/emm.2009.41.4.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reeves EP, et al. Direct interaction between p47phox and protein kinase C: evidence for targeting of protein kinase C by p47phox in neutrophils. Biochem J. 1999;344:859–866. [PMC free article] [PubMed] [Google Scholar]
- 33.Hoyal CR, et al. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc Natl Acad Sci USA. 2003;100:5130–5135. doi: 10.1073/pnas.1031526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Q, et al. Akt phosphorylates p47phox and mediates respiratory burst activity in human neutrophils. J Immunol. 2003;170:5302–5308. doi: 10.4049/jimmunol.170.10.5302. [DOI] [PubMed] [Google Scholar]
- 35.El Benna J, et al. Activation of p38 in stimulated human neutrophils: phosphorylation of the oxidase component p47phox by p38 and ERK but not by JNK. Arch Biochem Biophys. 1996;334:395–400. doi: 10.1006/abbi.1996.0470. [DOI] [PubMed] [Google Scholar]
- 36.Baldridge MT, et al. Expression of IFNLR1 on intestinal epithelial cells is critical to the antiviral effects of interferon λ against norovirus and reovirus. J Virol. 2017;91:e02079–16. doi: 10.1128/JVI.02079-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang JY, et al. Enteric viruses ameliorate gut inflammation via Toll-like receptor 3 and Toll-like receptor 7-mediated interferon-β production. Immunity. 2016;44:889–900. doi: 10.1016/j.immuni.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 38.Nice TJ, et al. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science. 2015;347:269–273. doi: 10.1126/science.1258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galani IE, et al. Interferon-λ mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity. 2017;46:875–890. doi: 10.1016/j.immuni.2017.04.025. [DOI] [PubMed] [Google Scholar]
- 40.Glocker EO, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kotlarz D, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143:347–355. doi: 10.1053/j.gastro.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 42.Shouval DS, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–719. doi: 10.1016/j.immuni.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiriac MT, et al. activation of epithelial signal transducer and activator of transcription 1 by interleukin 28 controls mucosal healing in mice with colitis and is increased in mucosa of patients with inflammatory bowel disease. Gastroenterology. 2017;153:123–138. doi: 10.1053/j.gastro.2017.03.015. [DOI] [PubMed] [Google Scholar]
- 44.Mócsai A, et al. Kinase pathways in chemoattractant-induced degranulation of neutrophils: the role of p38 mitogen-activated protein kinase activated by Src family kinases. J Immunol. 2000;164:4321–4331. doi: 10.4049/jimmunol.164.8.4321. [DOI] [PubMed] [Google Scholar]
- 45.Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol. 2014;104(Unit 15):25. doi: 10.1002/0471142735.im1525s104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request (in particular, the gating strategy used for all flow cytometry, and full-length gels and blots, as well as all raw data from NanoString analyses).