

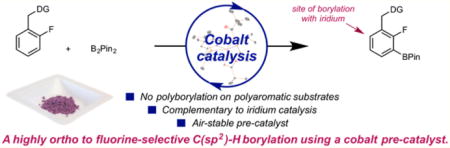
Abstract
Cobalt catalysts with electronically enhanced site selectivity have been developed, as evidenced by the high ortho-to-fluorine selectivity observed in the C(sp2)–H borylation of fluorinated arenes. Both the air-sensitive cobalt(III) dihydride boryl 4-Me-(iPrPNP)Co(H)2BPin (1) and the air-stable cobalt(II) bis(pivalate) 4-Me-(iPrPNP)Co(O2 CtBu)2 (2) compounds were effective and exhibited broad functional group tolerance across a wide range of fluoroarenes containing electronically diverse functional groups, regardless of the substitution pattern on the arene. The electronically enhanced ortho-to-fluorine selectivity observed with the cobalt catalysts was maintained in the presence of a benzylic dimethylamine and hydrosilanes, overriding the established directing-group effects observed with precious-metal catalysts. The synthetically useful selectivity observed with cobalt was applied to an efficient synthesis of the anti-inflammatory drug flurbiprofen.
INTRODUCTION
The direct, selective C–H functionalization of organic molecules in the absence of directing groups is a grand challenge in modern catalysis. Fluorinated arenes are prominent targets given the prevalence of this subunit in pharmaceuticals,1 agrochemicals,2 and organic materials (Figure 1).3 Efficient methods for the synthesis of o-fluorinated aryl boronate esters are attractive, given the versatility of the boron substituent for elaboration by Suzuki–Miyuara cross-coupling, Chan–Lam– Evans coupling, and a host of other methods.4
Figure 1.
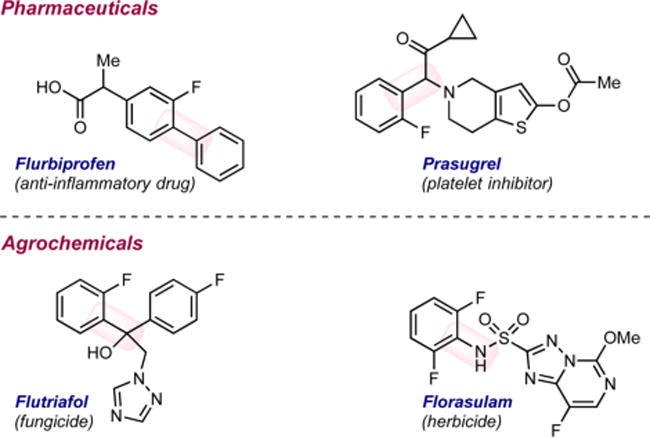
Examples of o-fluoroaryl motifs in pharmaceuticals and agrochemicals.
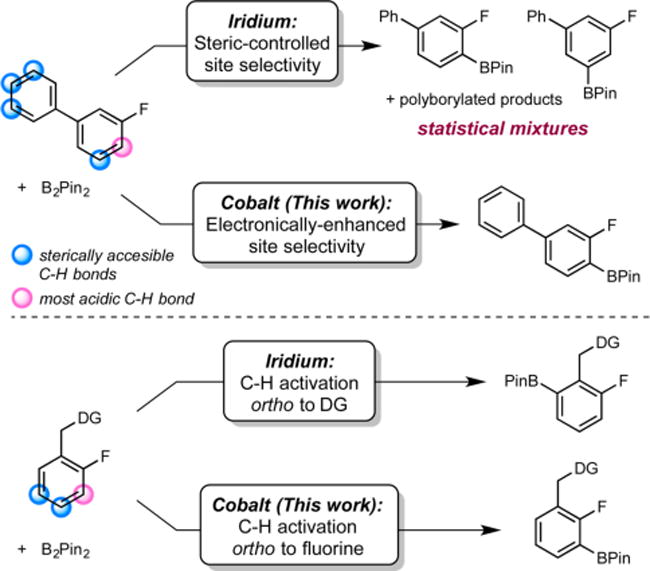
Iridium-catalyzed C–H borylation has emerged as one of the most widely used C–H functionalization methods due to its high efficiency and complementary selectivity to traditional electrophilic aromatic substitution.5,6 Iridium complexes containing bipyridine or phenanthroline ligands are the most widely used and mechanistically well understood7–9 and exhibit predictable site selectivity that is typically controlled by the steric accessibility of the C–H bond.
Distortion interaction analysis established that the regioselectivity in these reactions is largely controlled by the interaction of the arene carbon with the iridium catalyst, although fluorinated arenes were not thoroughly addressed in this study.9 It is well-established that the C–H bond ortho to fluorine in fluoroarenes is more acidic relative to the meta and para C–H bonds;10 however, selective, catalytic C–H borylation of this position in the presence of other sterically accessible C–H bonds still remains a challenge.11 Alternative strategies for the preparation of single regioisomers of fluorinated aryl boronate esters have been developed, including installation and removal of blocking groups to increase the selectivity of the iridium-catalyzed reaction.12 Use of NHC-13a and PSiN-ligated platinum catalysts,13b as well as phosphine-14a and POP-supported rhodium catalysts,14b have all been explored to increase the ortho-to-fluorine selectivity in fluoroarene borylation. While these were important advances, the requirement of excess arene, elevated temperatures, and multiple fluorines in the arene substrate detract from the general utility of these methods.13,14
In a recent patent application, specific electron-poor bidentate ligands, such as 4,4′-bis(trifluoromethyl)-2,2′-bipyridine (btfbpy) and 4,4′,5,5′-tetrakis(trifluoromethyl)-2,2′-bipyridine (ttfbpy), have been claimed to enable selective iridium-catalyzed C–H borylation of 1-chloro-3-fluoro-2-substituted benzenes.15 Up to 82:18 ortho:meta selectivity was reported for the iridium-catalyzed C–H borylation of 1-chloro-2,3-difluorobenzene using ttfbpy as the ligand. Monodentate pyridine ligands were also claimed to be effective ligands to achieve high ortho-to-fluorine selectivity in the iridium-catalyzed C–H borylation of 3-fluorotoluene. Specifically, an 82:18 (4.7:1) ortho:meta selectivity was reported with 2-methoxy pyridine (2-OMe-Py) as the ligand.
First-row transition-metal catalysts for C–H borylation are attractive not only for their potential cost and environmental advantages but also for the opportunity for new reactivity and selectivity over known precious-metal catalysts.16 Among the base-metal examples reported, [(iPrPNP)Co]-based catalysts are the most active for the C(sp2)–H borylation of arenes and heteroarenes.17 Mechanistic studies support a Co(I)–Co(III) pathway, in which a cobalt(I)–boryl is responsible for C–H activation. Substitution of the 4-position of the pincer prevented catalyst deactivation by C–H borylation of the ligand and inspired the preparation of improved, second-generation 4-methyl- and 4-pyrrolidinyl-substituted catalysts.18 With the first-generation cobalt alkyl, (iPrPNP)CoCH2SiMe3, unprecedented 89:11 ortho:meta selectivity for the borylation of fluorobenzene with B2Pin2 (Pin = pinacolate) was observed.17 Here, we describe a more general cobalt-catalyzed method for the ortho-to-fluorine-selective borylation of a wide range of fluorinated arenes (Scheme 1). The cobalt precatalysts, including an air-stable variant, offer distinct selectivity enhancements over known precious-metal catalysts, enabling an efficient synthesis of the anti-inflammatory drug flurbiprofen.
Scheme 1. Cobalt-catalyzed C(sp2)–H Borylation with Enhanced ortho Site Selectivity with Fluoroarenes.
RESULTS AND DISCUSSION
Synthesis of 4-Me-(iPrPNP)Co(O2CtBu)2 (2)
The 4-methyl-substituted pincer 4-Me-iPrPNP was selected for these studies due to its relative ease of synthesis, electron-donating properties, and resistance to deactivation by borylation during turnover.18 The cobalt(III) dihydride boryl (1) and cobalt(II) bis(pivalate) (2) complexes were used as precatalysts (Figure 2). Complex 1 was selected due to the precedent for cobalt(III) precursors as effective precatalysts for C–H borylation,18 while complex 2 was prepared due to its relative ease of synthesis and its air stability. Recent studies from our laboratory19 and others20 have demonstrated both the air stability and utility of cobalt19a–e,20 and nickel19f carboxylates as catalyst precursors.
Figure 2.
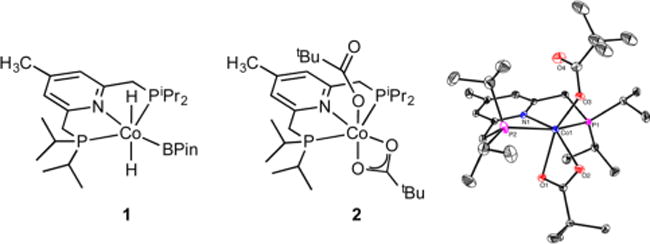
Cobalt precatalysts 1 and 2 and the solid-state molecular structure of 2 at 30% probability ellipsoids. Hydrogen atoms omitted for clarity.
Cobalt complex 2 was synthesized by the straightforward addition of the free ligand to anhydrous cobalt pivalate21 and was isolated in 49% yield as a purple powder with an S = 3/2 ground state [μeff = 4.1(1) μB at 23 °C, solid state]. Structural characterization (Figure 2) established a six-coordinate cobalt complex with κ1 and κ2 carboxylate ligands. A single paramagnetically shifted tert-butyl resonance was observed by 1H NMR spectroscopy, suggesting rapid interconversion of κ1 and κ2 carboxylate ligands on the NMR time scale, similar to related pyridine diimine19c and terpyridine cobalt complexes.19d,e It is also possible that the κ1 and κ2 forms are indistinguishable by NMR spectroscopy. Compound 2 exhibited excellent bench stability, as no change in the 1H NMR spectrum of the compound was observed after exposure of the solid to air for 5 days [Figure S1, Supporting Information (SI)].
Substrate Scope Using 1 as a Precatalyst
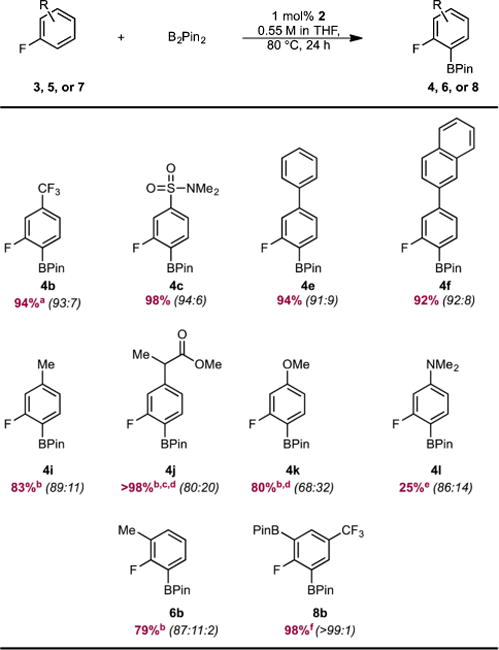
The site selectivity of cobalt-catalyzed C(sp2)–H borylation was explored in a variety of arenes with various substitution patterns (Table 1). Precatalyst 1 was evaluated initially with a series of 3-substituted fluoroarenes. Efficient borylation was observed over the course of 24 h at 50 °C with B2Pin2. The arylboronate products were obtained in high yields and ortho-to-fluorine selectivity with arenes containing ester (4a), trifluoromethyl (4b), sulfonamide (4c), and amide (4d) functional groups. Recrystallization of the regioisomeric mixture of 4d yielded regiochemically pure 4d in 69% isolated yield. With polyaromatic substrates (4e, 4f, 4g, 4h), exclusive borylation of the ring containing the fluorine atom was observed despite the presence of multiple sterically accessible C(sp2)–H bonds. Fluoroarenes containing electron-donating groups (4i, 4j, 4k, 4l) were borylated with reduced ortho-to-fluorine selectivity. These observations support the hypothesis that the site selectivity of the catalytic borylation reaction of fluoroarenes is determined by the relative acidity of the C–H bond in addition to the steric factors that typically dictate selectivity with precious-metal catalysts.5,6
Table 1.
Substrate Scope of the ortho-to-Fluorine C(sp2)–H Borylationg of 3-Substituted Fluoroarenes Catalyzed by 1
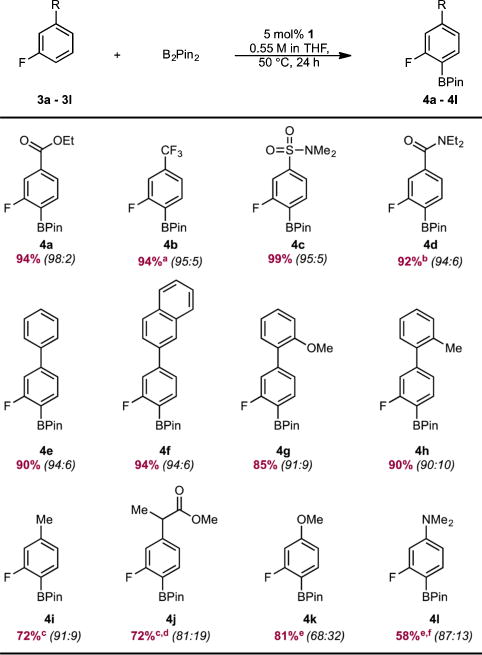
|
With 1 mol % of 1.
Yield of 69% (>99:1 ortho:meta) after recrystallization.
Reacted for 48 h.
On 2.74 mmol scale.
Reacted for 72 h.
Conditions: 25 mol % of 1, 80 °C, 1.1 M in THF, 5 equiv of 3l.
Typical reaction conditions: arene (0.55 mmol), B2Pin2 (0.55 mmol), 1 (0.0275 mmol, 5 mol %), THF (1 mL), 50 °C. Reported numbers are isolated yields after column chromatography. Numbers in parentheses correspond to the regioselectivities (ortho:meta ratio) determined by 19F NMR spectroscopy.
Fluoroarenes containing substituents at the 2-position were also suitable substrates for cobalt-catalyzed borylation (Table 2). With this class of substrates, where three sterically accessible C–H bonds are present, selective borylation of the C–H bond ortho to fluorine was observed with negligible borylation of the C–H bond para to fluorine. As in the case of 3-substituted fluoroarenes, no polyborylation of the polyaromatic substrates (6c, 6d, 6e, 6f) was detected.
Table 2.
Substrate Scope of the ortho-to-Fluorine C(sp2)–H Borylation of 2-Substituted Fluoroarenes Catalyzed by 1a
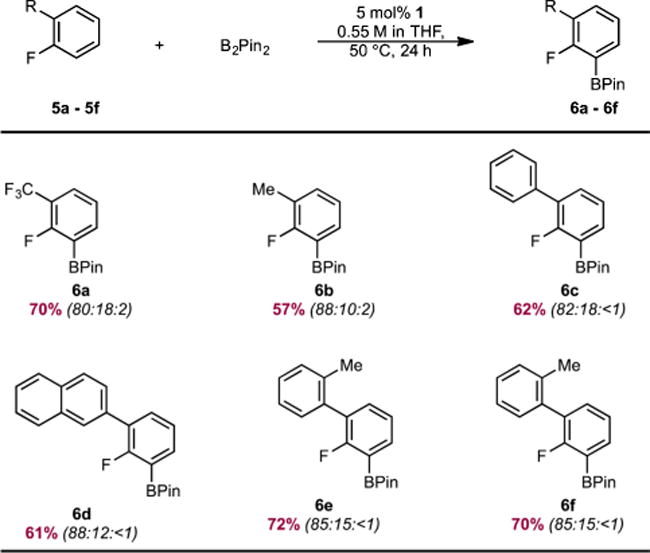
|
Reaction conditions: arene (0.55 mmol), B2Pin2 (0.55 mmol), 1 (0.0275 mmol, 5 mol %), THF (1 mL), 50 °C. Reported numbers are isolated yields after column chromatography. Numbers in parentheses correspond to the regioselectivities (ortho:meta:para ratio) determined by 19F NMR spectroscopy.
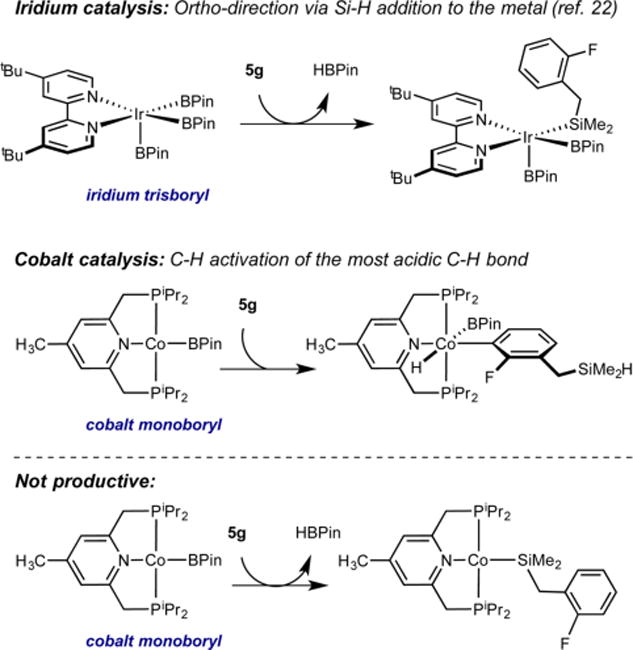
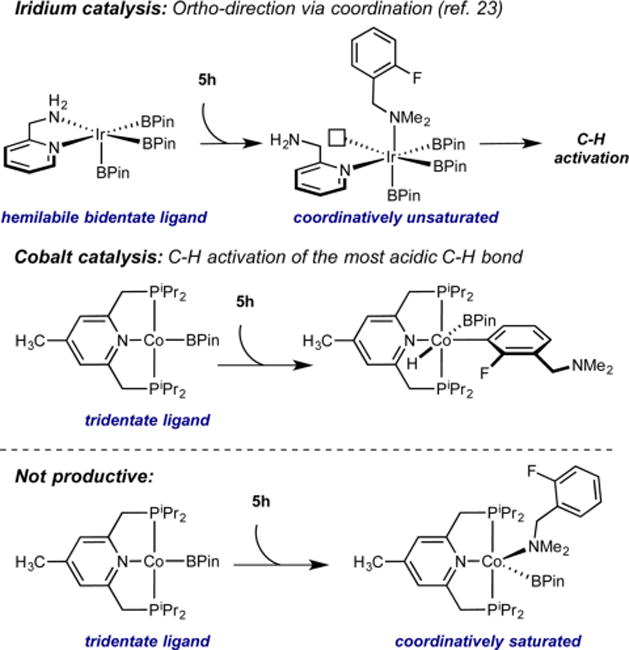
Fluoroarenes containing a benzylic hydrosilane group and a benzylic dimethylamine, well-known ortho-directing groups in iridium-catalyzed C–H borylation,22,23b were selectively borylated ortho to fluorine rather than ortho to these functional groups to yield fluoroarenes 6g and 6h (Scheme 2). In iridium catalysis, benzylic hydrosilanes are proposed to direct ortho-borylation from formation of a putative iridium bis(boryl) silyl intermediate arising from reaction of the silane Si–H bond with the iridium tris(boryl) followed by selective C–H activation (Scheme 3). If a similar sequence was operative with cobalt, the intermediate cobalt silyl complex obtained from reaction of 5g with the cobalt(I) boryl18 lacks an additional boryl ligand to promote C–H activation and C–B bond formation and likely accounts for the lack of the directing effect with the first-row transition metal. Instead, borylation of the most acidic C(sp2)– H bond is observed, consistent with the enhanced electronic selectivity imparted by cobalt.
Scheme 2. Complementary Selectivity in Cobalt- and Iridium-Catalyzed C(sp2)–H Borylation of 5g and 5ha.
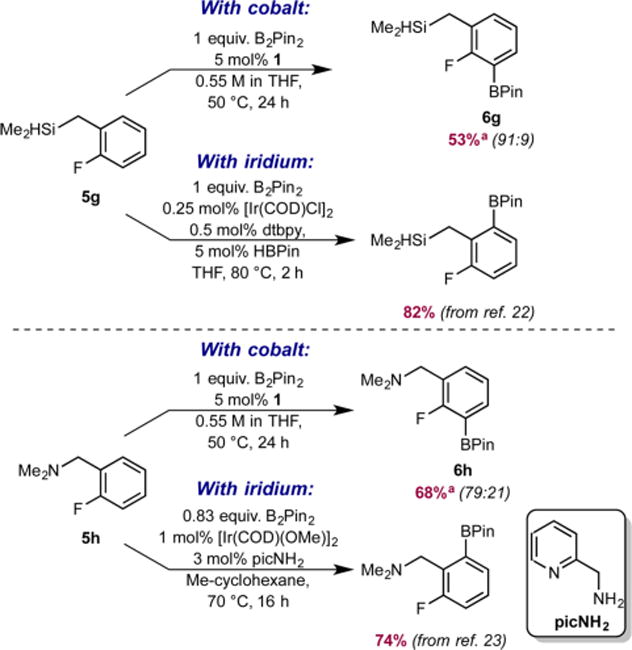
aReported numbers are combined NMR yield of ortho and meta monoborylated products (crude mixture) determined by 19F NMR spectroscopy using 4-F-toluene as the internal standard, and numbers in parentheses are the ortho:meta ratios.
Scheme 3. Proposed Origin of the Complementary Selectivity in Cobalt- and Iridium-Catalyzed C(sp2)–H Borylation of 5g.
Iridium catalysts containing hemilabile N,N ligands23a promote ortho-borylation using a benzylic dimethylamine as a directing group.23b The ortho selectivity is proposed to arise from coordination of the [NMe2] group to the metal followed by dissociation of an amine nitrogen from the supporting N,N chelate, opening a site for C–H activation (Scheme 4).23b With cobalt, the benzylic dimethylamino group has proven ineffective for directing ortho selectivity, likely due to the tridentate pincer. While dissociation of one of the phosphine arms is possible, coordination of the amine does not influence the outcome of the C(sp2)–H borylation, and the first-row metal maintains its preference for the most acidic C–H bond. This outcome arises from reversible coordination of the amine without concurrent C–H activation, perhaps due to the formation of a coordinatively saturated intermediate, or lack of coordination of the [NMe2] group.
Scheme 4. Proposed Origin of the Complementary Selectivity in Cobalt- and Iridium-Catalyzed C(sp2) –H Borylation of 5h.
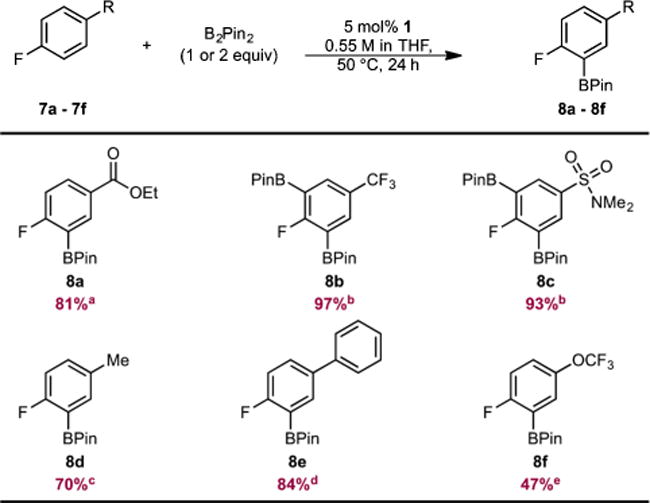
Exclusive ortho-to-fluorine selectivity was observed in the C(sp2)–H borylation of 4-substituted fluoroarenes containing a variety of functional groups (Table 3). These findings are similar to what was observed in the iridium-catalyzed C–H borylation to furnish 8d24 and 8f,25 with slightly higher ortho-to-fluorine selectivity for 8f. As with 3- and 2-substituted polyaromatic fluoroarenes, exclusive borylation of the fluorine-containing ring was observed in p-fluorobiphenyl (7e) to yield a 94:6 ratio of mono:diborylated products, where the BPin groups are located ortho to fluorine.
Table 3.
Substrate Scope of the ortho-to-Fluorine C(sp2)–H Borylation of 4-Substituted Fluoroarenes Catalyzed by 1f
|
Isolated as a 91:9 mixture of mono- and diborylated products. Reported numbers correspond to % yield of monoborylated product.
Used 2 equiv of B2Pin2.
Isolated as a 93:7 mixture of mono- and diborylated products. Reported numbers correspond to % yield of monoborylated product.
Isolated as a 94:6 mixture of mono- and diborylated products. Reported numbers correspond to % yield of monoborylated product.
NMR yield of 8f determined by 19F NMR spectroscopy using 4-F-toluene as the internal standard.
Typical reaction conditions: arene (0.55 mmol), B2Pin2 (0.55 mmol), 1 (0.0275 mmol, 5 mol %), THF (1 mL), 50 °C. Numbers in parentheses are isolated yields after column chromatography.
Unfortunately, fluoroarenes containing bromo and chloro substituents (3m, 3n, and 3o) were incompatible with cobalt-catalyzed C(sp2)–H borlyation (Figure 3). Performing a stoichiometric reaction between 1 and 3n resulted in immediate formation of (4-Me-iPrPNP)CoCl (9).18 Reaction of 3-fluorotoluene (3i) with B2Pin2 at 50 °C (0.55 M in THF) in the presence of 5 mol % of 9 resulted in no product formation after 24 h of heating, establishing formation of 9 as a catalyst deactivation pathway.
Figure 3.
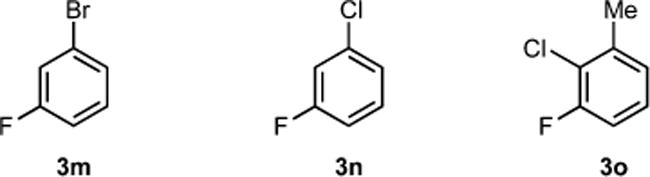
Fluoroarenes incompatible with cobalt-catalyzed C(sp2)–H borylation.
Comparisons with Iridium Catalysts
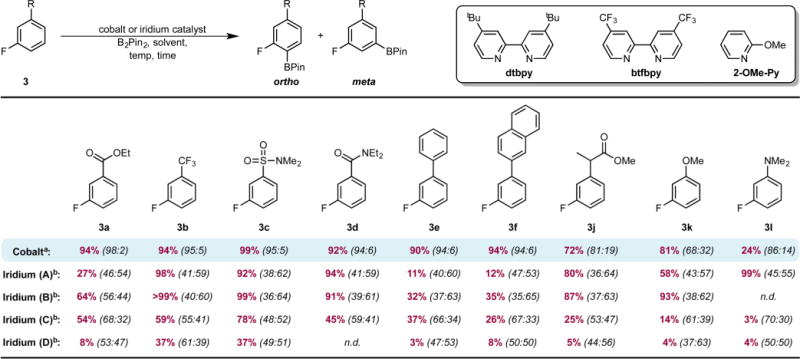
The unique selectivity of the cobalt catalyst was confirmed by direct comparison of the activity and selectivity of 1 to iridium catalysts. A variety of 3-substituted fluoroarenes were selected as substrates given the availability of two sterically accessible but electronically differentiated C(sp2)–H bonds (Table 4). Four different reaction conditions were selected for the iridium cases to ensure fair and representative comparisons between the precious and earth-abundant transition-metal catalysts. Conditions A and B draw on the state-of-the-art and widely used [Ir(COD)OMe]2/dtbpy (dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridine)6c employing the optimized conditions for cobalt (conditions A) and also using conditions reported in the literature (conditions B).26 The other two conditions employed [Ir(COD)OMe]2 in combination with 4,4′-bis-(trifluoromethyl)-2,2′-bipyridine (conditions C) and 2-methoxypyridine (conditions D), ligands claimed to enhance ortho-to-fluorine selectivity in the iridium-catalyzed C–H borylation of 1-chloro-3-fluoro-2-substituted benzenes and 3-fluorotoluene.15 In all cases, iridium catalysis proved significantly less selective for the borylation of 3-substituted fluoroarenes with B2Pin2, as nearly statistical distributions of ortho and meta borylated products were observed. The higher activity of the [Ir(COD)OMe]2/dtbpy catalyst mixture compared to cobalt is highlighted in the borylation of the electron-rich fluoroarene 3l. For conditions A and B, complete conversion of arene was observed in the borylation of 3a, 3j, and 3k with iridium; however, a lower combined NMR yield of the desired monoborylated products was obtained presumably due to competing reactions of the ester (3a, 3j) and the methoxy (3k) groups. The borylation of polyaromatic fluoroarenes (3e27 and 3f) highlights the advantages of the cobalt catalyst. With iridium, complex mixtures of products were observed, a result of competing borylation of the other sterically accessible C–H bonds in the adjacent aryl ring (see Figures S6 and S7, SI). With cobalt, however, the selectivity is high and only borylation of the fluorinated ring was observed, furnishing monoborylated products 4e and 4f in 90% and 94% combined yields, respectively, with 96:4 ortho:meta selectivity in both cases. Using the electron-poor ligand 4,4′-bis-(trifluoromethyl)-2,2′-bipyridine for iridium (conditions C) resulted in an enhancement of the ortho-to-fluorine selectivity for arenes 3a, 3e, 3f, 3k, and 3l relative to the [Ir(COD)-OMe]2/dtbpy mixture; however, the selectivities were still inferior to that observed with cobalt. Finally, the use of 2-methoxypyridine as the ligand for the iridium-catalyzed reaction (conditions D) resulted in poor conversion and nearly statistical distributions of ortho and meta borylated products (Table 4).
Table 4.
Comparison of Cobalt- and Iridium-Catalyzed C–H Borylation of 3-Substituted Fluoroarenesd
|
Using conditions reported in Table 1. Isolated yield after column chromatography.
Reported numbers are combined NMR yield of ortho and meta monoborylated products (crude mixture) determined by 19F NMR spectroscopy using 4-F-toluene as the internal standard. Conditions varied as follows. Iridium (A): arene (0.28 mmol), B2Pin2 (0.28 mmol), [Ir(COD)OMe]2 (2.5 mol %), dtbpy (5 mol %), THF (0.5 mL), 50 °C, 24 h. Iridium (B):26 arene (0.50 mmol), B2Pin2 (0.37 mmol), [Ir(COD)OMe]2 (0.1 mol %), dtbpy (0.2 mol %), THF (1 mL), 50 °C, 24 h. Iridium (C):15 arene (0.34 mmol), B2Pin2 (0.17 mmol), [Ir(COD)OMe]2 (1 mol %), btfbpy (2 mol %), Hünig’s base (1 mL), 60 °C, 12 h. Iridium (D):15 arene (0.17 mmol), B2Pin2 (0.17 mmol), [Ir(COD)OMe]2 (1 mol %), 2-OMe-Py (2 mol %), THF (1 mL), 80 °C, 16 h.
Iridium (E):27 Same as conditions B but using 0.25 mmol of B2Pin2. Combined NMR yield of ortho and meta monoborylated products (crude mixture) determined by 19F NMR spectroscopy using 4-F-toluene as the internal standard.
Regioselectivities were determined by 19F NMR spectroscopy. n.d. = not determined.
Evaluation of the Air-Stable Cobalt Complex 2 for ortho-to-Fluorine C–H Borylation of Fluoroarenes
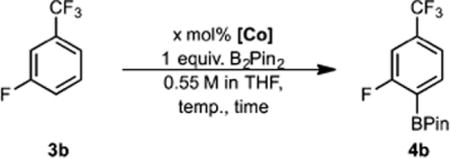
The improved regioselectivity observed with 1 prompted evaluation of the C–H borylation of arene 3b with the air-stable cobalt complex 2 (see Table 5).
Table 5.
C(sp2)–H Borylation of 3b with 2a
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | precatalyst | mol % | time (h) | temp (°C) | % yieldb | o:mc |
| 1 | 1 | 1 | 24 | 50 | 94 | 95:5 |
| 2d,e | 2 | 5 | 1.5 | 50 | >98f | 93:7 |
| 3d,g | 2 | 5 | 1.5 | 50 | >98f | 93:7 |
| 4d | Co(OPiv)2 | 5 | 1.5 | 50 | <5 | N/A |
| 5 | 2 | 1 | 24 | 50 | 46 | 94:6 |
| 6 | 2 | 1 | 24 | 80 | 68 | 92:8 |
| 7h | 2 | 1 | 24 | 80 | 94 | 93:7 |
| 8i | 2 | 1 | 24 | 80 | <5j | N/A |
| 9i,k | 2 | 1 | 24 | 80 | >98j | 92:8 |
Reactions were run with equimolar amounts of 3b and B2Pin2 on a 0.55 mmol scale.
Isolated yield after column chromatography. N/A = Not applicable
Ratio of ortho:meta determined by 19F NMR.
Added 20 mol % of HBPin.
Run in THF-d8.
Percent conversion determined by 19F NMR.
Precatalyst exposed to air for 1 h.
Run on a 5.5 mmol scale.
Precatalyst exposed to air for 14 days.
Percent conversion determined by GC.
Added 4 mol % of HBPin.
Catalytic C–H borylation was accomplished using 2 and B2Pin2 at 50 °C (entry 5), although an approximate 12-h induction period was observed. Addition of HBPin as an activator19a produced comparable activity to 1 (entry 2). Precatalyst 2 was exposed to air for 1 h without any measurable erosion of activity or selectivity (entry 3). At 80 °C, however, comparable activity as well as selectivity with 1 was achieved with 2 (entries 6 and 7) without the need for an external activator. The reaction was successfully scaled to 5.5 mmol using only 1 mol % of 2 to generate the desired o-fluoroboronate ester 4b in 94% isolated yield with 93% regiochemical purity (entry 7). Complete conversion of the arene was observed (92:8 ortho:meta selectivity) when the reaction was carried out at 80 °C using 1 mol % of precatalyst 2 that was exposed to air for 14 days, albeit with the requirement of 4 mol % of HBPin as an activator (entries 8 and 9). Using anhydrous cobalt(II) pivalate as the precatalyst resulted in no reaction, highlighting the necessity of the bis(phosphine)-pyridine ligand for the C–H borylation reaction (entry 4).
The borylation of a variety of fluoroarenes with different functional groups and substitution patterns was explored using the optimized conditions for arene 3b with complex 2 as the precatalyst. The activity and selectivity with 2 proved general among a range of fluorinated arenes, as high isolated yields and ortho-to-fluorine selectivity were observed regardless of the substituent on the arene and its substitution pattern (Table 6). As with 1, exclusive ortho-to-fluorine borylation was observed with polyaromatic substrates 4e and 4f.
Table 6.
Substrate Scope of the ortho-to-Fluorine C(sp2)–H Borylation of Fluoroarenes Catalyzed by the Air-Stable Precatalyst 2g
|
On 5.5 mmol scale.
With 5 mol % of 2.
Percent conversion determined by GC.
Reaction time of 48 h.
With 10 mol % of 2, 72 h.
With 2 equiv of B2Pin2.
Typical reaction conditions: arene (0.55 mmol), B2Pin2 (0.55 mmol), 2 (0.0055 mmol, 1 mol %), THF (1 mL), 80 °C. Reported numbers are isolated yields after column chromatography. Numbers in parentheses correspond to the regioselectivities (ortho:meta:para ratio) determined by 19F NMR spectroscopy.
Application of Cobalt-Catalyzed C(sp2)–H Borylation to the Synthesis of Flurbiprofen
The utility enabled by the increased site selectivity of the cobalt-catalyzed C(sp2)–H functionalization was applied to the total synthesis of the anti-inflammatory drug flurbiprofen (Scheme 5). Fluoroarene 3j was prepared by slightly modifying the procedure reported by Durandetti et al.28 Cobalt-catalyzed ortho-to-fluorine-selective C–H borylation of 3j, followed by Suzuki–Miyaura cross-coupling with phenyl bromide and then ester hydrolysis, afforded flurbiprofen (11) in 33% overall yield from commercially available 10 with 84% regiochemical purity. Thus, our cobalt-catalyzed method enables a four-step synthesis, streamlined from the eight-step route reported previously29 and highlights the utility of regioselective C–H functionalization in synthetic applications.
Scheme 5. Application of ortho-to-Fluorine-Selective C–H Borylation to the Synthesis of Flurbiprofen (11)a.
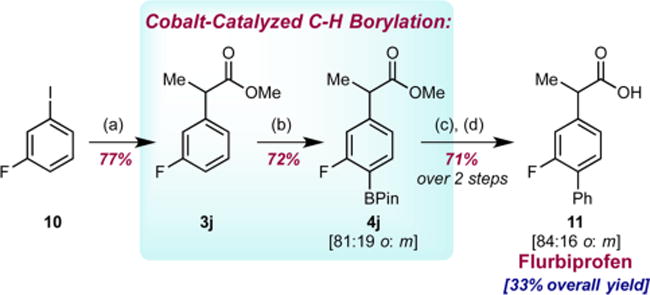
aReagents and conditions: (a) Methyl-2-chloropropionate (5.2 equiv), Mn powder (6 equiv), TFA (30 μL), (bpy)NiBr2 (7 mol %), DMF, 50 °C, 16 h; (b) B2Pin2 (1 equiv), 1 (5 mol %), THF, 50 °C, 48 h; (c) PhBr (1.1 equiv), Pd(dppf)Cl2 (5 mol %), K2CO3 (4 equiv), THF/H2O (20:1), 50 °C, 16 h; (d) NaOH (5 equiv), THF/H2O (1:1), 90 °C, 24 h, then 12 M HCl. Reported numbers are isolated yields after column chromatography. Regioselectivities were determined by 19F NMR spectroscopy. The overall yield of flurbiprofen (11) is corrected for the meta-phenylated regioisomer.
CONCLUSIONS
An efficient, highly ortho-to-fluorine-selective cobalt-catalyzed method for the C(sp2)–H borylation of fluorinated arenes has been developed. An air-stable, pincer-ligated cobalt(II) bis-(pivalate) was synthesized in a single step from the free ligand and appropriate cobalt precursor and was effective for catalytic C(sp2)–H functionalization of electronically diverse substrates, regardless of the substitution pattern on the arene. Common directing groups in iridium-catalyzed C–H functionalization, such as a benzylic dimethylamino substituent or a hydridosilane, did not alter the electronically enhanced site selectivity of the cobalt catalyst, highlighting the complementarity of earth-abundent and precious-metal catalysts. The improved regioselectivity of the cobalt-catalyzed C(sp2)–H borylation was applied to a streamlined synthesis of the anti-inflammatory drug flurbiprofen. These studies represent one of the rare examples of selective C–H functionalization that, in the absence of directing groups, offers new opportunities for reaction development and applications in synthesis.
Supplementary Material
Acknowledgments
J.V.O. acknowledges the 2015 Howard Hughes Medical Institute International Student Research Fellowship and the 2016 Harold W. Dodds Honorific Fellowship (awarded by the Graduate School at Princeton University). M.J.B. thanks the Natural Sciences and Engineering Research Council of Canada for a predoctoral fellowship (PGS-D). We also thank AllyChem for the generous gift of B2Pin2. Financial support was provided by NIH (R01 GM121441).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b13346.
Crystallographic data of 2 in CIF format (CIF)
Complete experimental details, characterization data, NMR spectroscopic data (PDF)
ORCID Paul J. Chirik: 0000-0001-8473-2898
Notes
The authors declare the following competing financial interest(s): J.V.O. and P.J.C. are inventors on U.S. Patent Application 61/913,522 (Filed: December 9, 2014, Published: June 18, 2015).
References
- 1.(a) Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (b) Hagmann WK. J Med Chem. 2008;51:4359. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]; (c) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (d) Ilardi EA, Vitaku E, Njardarson JT. J Med Chem. 2014;57:2832. doi: 10.1021/jm401375q. [DOI] [PubMed] [Google Scholar]; (e) Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 2.(a) Jeschke P. ChemBioChem. 2004;5:570. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]; (b) Jeschke P. Pest Manage Sci. 2010;66:10. doi: 10.1002/ps.1829. [DOI] [PubMed] [Google Scholar]; (c) Fujiwara T, O’Hagan D. J Fluorine Chem. 2014;167:16. [Google Scholar]
- 3.(a) Babudri R, Farinola GM, Naso F, Ragni R. Chem Commun. 2007:1003. doi: 10.1039/b611336b. [DOI] [PubMed] [Google Scholar]; (b) Berger R, Resnati G, Metrangolo P, Weber E, Hulliger J. Chem Soc Rev. 2011;40:3496. doi: 10.1039/c0cs00221f. [DOI] [PubMed] [Google Scholar]
- 4.Hall DG. Boronic Acids. Wiley-VCH; Weinheim, Germany: 2005. [Google Scholar]
- 5.(a) Mkhalid I, Barnard JH, Marder TB, Murphy JM, Hartwig Chem Rev. 2010;110:890. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; (b) Hartwig JF. Chem Soc Rev. 2011;40:1992. doi: 10.1039/c0cs00156b. [DOI] [PubMed] [Google Scholar]; (c) Hartwig JF. Acc Chem Res. 2012;45:864. doi: 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- 6.(a) Cho J–Y, Tse MK, Holmes D, Maleczka RE, Jr, Smith MR., III Science. 2002;295:305. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]; (b) Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J Am Chem Soc. 2002;124:390. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]; (c) Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew Chem, Int Ed. 2002;41:3056. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]; (d) Preshlock SM, Ghaffari B, Maligres PE, Krska SW, Maleczka RE, Smith MR. J Am Chem Soc. 2013;135:7572. doi: 10.1021/ja400295v. [DOI] [PubMed] [Google Scholar]
- 7.Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Hartwig JF. J Am Chem Soc. 2005;127:14263. doi: 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]
- 8.Tamura H, Yamazaki H, Sato H, Sakaki S. J Am Chem Soc. 2003;125:16114. doi: 10.1021/ja0302937. [DOI] [PubMed] [Google Scholar]
- 9.Green AG, Liu P, Merlic CA, Houk KN. J Am Chem Soc. 2014;136:4575. doi: 10.1021/ja411699u. [DOI] [PubMed] [Google Scholar]
- 10.(a) Hall GE, Piccolini R, Roberts JD. J Am Chem Soc. 1955;77:4540. [Google Scholar]; (b) Streitwieser A, Jr, Scannon PJ, Niemeyer HM. J Am Chem Soc. 1972;94:7936. [Google Scholar]
- 11.Palladium-catalyzed C–H arylation has been shown to occur ortho-to-fluorine selectively. For representative examples, see the following:; (a) Campeau LC, Parisien M, Jean A, Fagnou K. J Am Chem Soc. 2006;128:581. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]; (b) Lafrance M, Rowley CN, Woo TK, Fagnou K. J Am Chem Soc. 2006;128:8754. doi: 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]
- 12.Jayasundara CRK, Unold JM, Oppenheimer J, Smith MR, III, Maleczka RE. Org Lett. 2014;16:6072. doi: 10.1021/ol5028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Furukawa T, Tobisu M, Chatani N. J Am Chem Soc. 2015;137:12211. doi: 10.1021/jacs.5b07677. [DOI] [PubMed] [Google Scholar]; (b) Takaya J, Ito S, Nomoto H, Saito N, Kirai N, Iwasawa N. Chem Commun. 2015;51:17662. doi: 10.1039/c5cc07263h. [DOI] [PubMed] [Google Scholar]
- 14.(a) Kalläne SI, Teltewskoi M, Braun T, Braun B. Organometallics. 2015;34:1156. [Google Scholar]; (b) Esteruelas MA, Olivan M, Vélez A. Organometallics. 2015;34:1911. [Google Scholar]
- 15.Smith MR, Maleczka RE, Li H, Jayasundara C, Oppenheimer J, Sabasovs D. US Patent Application 61/874,249
- 16.Chirik PJ, Wieghardt K. Science. 2010;327:794. doi: 10.1126/science.1183281. [DOI] [PubMed] [Google Scholar]
- 17.Obligacion JV, Semproni SP, Chirik PJ. J Am Chem Soc. 2014;136:4133. doi: 10.1021/ja500712z. [DOI] [PubMed] [Google Scholar]
- 18.Obligacion JV, Semproni SP, Pappas I, Chirik PJ. J Am Chem Soc. 2016;138:10645. doi: 10.1021/jacs.6b06144. [DOI] [PubMed] [Google Scholar]
- 19.(a) Scheuermann ML, Johnson EJ, Chirik PJ. Org Lett. 2015;17:2716. doi: 10.1021/acs.orglett.5b01135. [DOI] [PubMed] [Google Scholar]; (b) Palmer WN, Obligacion JV, Pappas I, Chirik PJ. J Am Chem Soc. 2016;138:766. doi: 10.1021/jacs.5b12249. [DOI] [PubMed] [Google Scholar]; (c) Schuster CH, Diao T, Pappas I, Chirik PJ. ACS Catal. 2016;6:2632. [Google Scholar]; (d) Constable EC, Housecroft CE, Jullien V, Neuburger M, Schaffner S. Inorg Chem Commun. 2006;9:504. [Google Scholar]; (e) Léonard NG, Bezdek M, Chirik PJ. Organometallics. 2017;36:142. [Google Scholar]; (f) Pappas I, Treacy S, Chirik PJ. ACS Catal. 2016;6:4105. [Google Scholar]
- 20.Noda D, Tahara A, Sunada Y, Nagashima H. J Am Chem Soc. 2016;138:2480. doi: 10.1021/jacs.5b11311. [DOI] [PubMed] [Google Scholar]
- 21.Aromí G, Batsanov AS, Christian P, Helliwell M, Parkin A, Parsons S, Smith AA, Timco GA, Winpenny REP. Chem -Eur J. 2003;9:5142. doi: 10.1002/chem.200304993. [DOI] [PubMed] [Google Scholar]
- 22.Boebel TA, Hartwig JF. J Am Chem Soc. 2008;130:7534. doi: 10.1021/ja8015878. [DOI] [PubMed] [Google Scholar]
- 23.(a) Ros A, Estepa B, López-Rodriguez R, Álvarez E, Fernández R, Lassaletta JM. Angew Chem, Int Ed. 2011;50:11724. doi: 10.1002/anie.201104544. [DOI] [PubMed] [Google Scholar]; (b) Roering AJ, Hale LVA, Squier PA, Ringgold MA, Wiederspan ER, Clark TB. Org Lett. 2012;14:3558. doi: 10.1021/ol301635x. [DOI] [PubMed] [Google Scholar]
- 24.In a study that compares the regioselectivities of Rh-catalyzed C–H silylation and Ir-catalyzed C–H borylation, the C–H borylation reaction of p-fluorotoluene (7d) with iridium was reported to exclusively undergo borylation ortho to fluorine. See the following:; Cheng C, Hartwig JF. Science. 2014;343:853. doi: 10.1126/science.1248042. [DOI] [PubMed] [Google Scholar]
- 25.The iridium-catalyzed C–H borylation of p-fluorotrifluorome-thoxy benzene (7f) resulted in borylation ortho to fluorine with 97% selectivity. See the following:; Batool F, Parveen S, Emwas AH, Sioud S, Gao X, Munawar MA, Chotana GA. Org Lett. 2015;17:4256. doi: 10.1021/acs.orglett.5b02050. [DOI] [PubMed] [Google Scholar]
- 26.Liskey CW, Liao X, Hartwig JF. J Am Chem Soc. 2010;132:11389. doi: 10.1021/ja104442v. [DOI] [PubMed] [Google Scholar]
- 27.The iridium-catalyzed reaction of 3e using conditions reported in ref 26 was carried out but with only 0.5 equiv of B2Pin2 to ensure that no excess boron reagent is present after consumption of arene (conditions E). A higher combined NMR yield (38%) of the desired monoborylated products was obtained under these conditions; however, polyborylation was still observed.
- 28.Durandetti M, Gosmini C, Périchon J. Tetrahedron. 2007;63:1146. [Google Scholar]
- 29.Quasdorf KW, Riener M, Petrova KV, Garg NK. J Am Chem Soc. 2009;131:17748. doi: 10.1021/ja906477r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.