

Key Points
TGF-β–induced intracellular PAI-1 regulates the balance of HSPCs localization between BM and periphery.
Intracellular PAI-1 inhibits Furin-dependent maturation of MT1-MMP in HSPCs, resulting in the suppression of HSPC motility.
Abstract
Hematopoietic stem and progenitor cells (HSPCs) reside in the supportive stromal niche in bone marrow (BM); when needed, however, they are rapidly mobilized into the circulation, suggesting that HSPCs are intrinsically highly motile but usually stay in the niche. We questioned what determines the motility of HSPCs. Here, we show that transforming growth factor (TGF)-β–induced intracellular plasminogen activator inhibitor (PAI)-1 activation is responsible for keeping HSPCs in the BM niche. We found that the expression of PAI-1, a downstream target of TGF-β signaling, was selectively augmented in niche-residing HSPCs. Functional inhibition of the TGF-β−PAI-1 signal increased MT1-MMP–dependent cellular motility, causing a detachment of HSPCs from the TGF-β–expressing niche cells, such as megakaryocytes. Furthermore, consistently high motility in PAI-1–deficient HSPCs was demonstrated by both a transwell migration assay and reciprocal transplantation experiments, indicating that intracellular, not extracellular, PAI-1 suppresses the motility of HSPCs, thereby causing them to stay in the niche. Mechanistically, intracellular PAI-1 inhibited the proteolytic activity of proprotein convertase Furin, diminishing MT1-MMP activity. This reduced expression of MT1-MMP in turn affected the expression levels of several adhesion/deadhesion molecules for determination of HSPC localization, such as CD44, VLA-4, and CXCR4, which then promoted the retention of HSPCs in the niche. Our findings open up a new field for the study of intracellular proteolysis as a regulatory mechanism of stem cell fate, which has the potential to improve clinical HSPC mobilization and transplantation protocols.
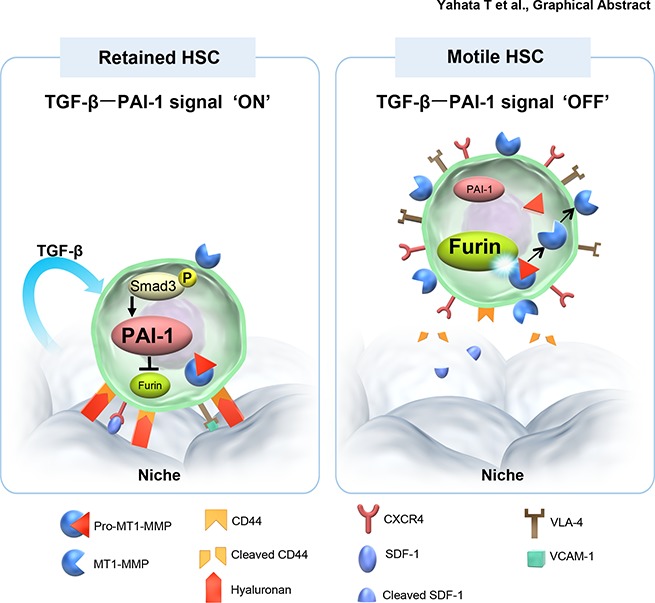
Visual Abstract
Introduction
Hematopoietic stem and progenitor cells (HSPCs) reside in a specialized bone marrow (BM) microenvironment called the niche.1-3 Interactions between HSPCs and the niche govern the survival and differentiation of HSPCs; hence, the localization of HSPCs in the BM niche is a key determinant of stem cell activity and ensures the lifelong production of mature blood cells. Conversely, HSPCs can be rapidly mobilized into the peripheral circulation in response to hematological stresses such as infection, bleeding, and chemotherapy.4,5 Even in the steady state, HSPCs frequently exit the BM into the circulation and replenish blood cells to maintain homeostasis. Thus, HSPCs are intrinsically highly motile cells, and their motility is tightly controlled within the niche. However, determinants of the localization of HSPC in the niche remain largely unknown.
A number of niche factors have been shown to maintain HSPC homeostasis, including thrombopoietin, angiopoietin 1/2, and transforming growth factor-β (TGF-β).6 Yamazaki and colleagues have demonstrated that nonmyelinating Schwann cells can convert latent-form TGF-β to its active form, which stimulates Smad-dependent expression of cyclin-dependent kinase inhibitor, p57Kip2, in HSPCs that inhibits their proliferation.7,8 Another major source of TGF-β is megakaryocytes, which physically associate with ∼20% of HSPCs in the BM.9-11 HSPCs in close proximity to megakaryocytes exhibit Smad2/3 activation, and conditional deletion of TGF-β from megakaryocytes results in reduced phosphorylation of Smad2/3 as well as loss of quiescence and increased HSPC proliferation. Although TGF-β has been characterized as a negative regulator of HSPC proliferation,12-14 its role in HSPC motility has not been studied. Meanwhile, it has been reported that plasminogen activator inhibitor (PAI)-1, a well-known downstream molecule of TGF-β signaling15,16 and a major physiologic inhibitor of the fibrolytic system that inhibits the serine protease activity of urokinase and tissue plasminogen activator (tPA) during blood clotting,16 regulates adhesion and migration of many cell types, including hematopoietic cells.17-23 Pharmacological inhibition of PAI-1 enhances neutrophil and macrophage migration through activation of the tPA-mediated fibrinolytic pathway.24,25 Genetic or pharmacologic blockade of PAI-1 activity increases HSPC mobilization in response to granulocyte-colony stimulating factor (G-CSF)26 and promotes HSPC engraftment after BM transplantation.27 These suggest involvement of TGF-β−PAI-1 signaling in the motility and localization of HSPCs in the niche.
HSPCs are retained in the BM by adhesion interactions with different cell membrane- and pericellular extracellular matrix-anchored molecules, including integrin α4β1/vascular cell adhesion molecule-1, lymphocyte function–associated antigen 1/intercellular adhesion molecule-1, CXC chemokine receptor-4/stromal derived factor-1 (CXCR4/SDF-1), and hyaluronic acid receptor CD44.4,5 Several proteases can cleave these interactions, including cell surface enzyme dipeptidyl peptidase IV (CD26); secreted matrix metalloproteases MMP-9 and MMP-13; elastase; and cathepsins G and K.28,29 This cleavage results in the loss of retention and consequent egress of HSPC. The membrane type-1 MT1-MMP (synonym MMP-14) promotes HSPC motility by degrading pericellular extracellular matrix proteins, vascular cell adhesion molecule-1, SDF-1, and CD44, which triggers the increase of VLA-4 and CXCR4 expression. MT1-MMP also activates pro-MMP-2, and upregulation of MT1-MMP in HSPCs by G-CSF treatment can activate pro-MMP-2,30 which can activate other proteases such as MMP-9 or MMP-13. The interplay of these factors, wherein MT1-MMP might represent the starting point of the cascade with its MMP-2-activating potential, suggests a crucial role for MT1-MMP in the egress of HSPCs from the BM. For instance, G-CSF–induced circulating HSPCs in the peripheral blood (PB) express higher levels of MT1-MMP compared with their quiescent counterparts in the BM,31 whereas downregulation of MT1-MMP in HSPCs impairs their migration, indicating a role for this protease in stem cell egress.30 MT1-MMP is activated by Furin, a proprotein convertase that cleaves the propeptide region of pro-MT1-MMP.32-34 Other than modulating extracellular fibrolytic cascades, PAI-1, along with other serpins, functions intracellularly.35,36 Because intracellular PAI-1 (iPAI-1) forms a stable complex with Furin and inhibits its activity,37 we hypothesized that TGF-β induces iPAI-1 expression in HSPCs, preventing the Furin-dependent maturation of MT1-MMP, which ultimately results in the retention of HSPCs in the BM niche.
Methods
Additional methods are presented in the supplemental Methods, available on the Blood Web site.
Mice
C57BL/6J- and PAI-1-deficient (B6.12952-Serpine1tm1Mg/J)38,39 mice were purchased from CLEA Japan (Tokyo) and the Jackson Laboratory (Bar Harbor, ME), respectively. Transgenic mice constitutively expressing a stable variant form of active human PAI-1 gene were bred as previously described.40 Protocols concerning animal experiments were approved by the Animal Care Committee of Tokai University.
Antibodies
Antibodies used in this study are listed with relevant information in supplemental Table 1.
Statistical analyses
All data were pooled from at least 3 independent experiments. Sample size was limited by ethical considerations. No randomization or blinding was used to allocate experimental groups, and no animals were excluded from analysis. All statistical analyses were conducted with GraphPad Prism, version 5.0 (GraphPad Software). Data are expressed as means ± standard deviation (SD) of 3 to 5 independent experiments. Student 2-tailed unpaired t test was used to determine the significance of the difference between the means of 2 groups. A 1-way analysis of variance, followed by Tukey posttests, was used to compare the means among 3 or more independent groups. A normal distribution of the data was tested using the Kolmogorov-Smirnov test. A value of P < .05 was considered significant.
Results
TGF-β signaling regulates PAI-1 and cell trafficking–related molecules’ expression in HSPCs
To determine the roles of TGF-β signaling in the localization of HSPCs within the niche, we evaluated TGF-β signaling activity in steady-state hematopoietic cells. TGF-β ligands bind type I and type II receptors at the cell surface,12-14 leading to phosphorylation of Smad2 and Smad3, which form a complex with Smad4. The resulting ternary complex translocates to the nucleus, where it recruits transcriptional cofactors to control gene expression. In line with a previous report,8 flow cytometric analyses of TGF-β receptors and p-Smad3 in fractionated murine hematopoietic cells demonstrated the highest levels of TGF-β receptor expression and its downstream signal activation in the most primitive cells (Lin−Sca-1+c-Kit+ [LSK] CD34−, or LSK CD48−CD150+),41,42 a majority of which are hematopoietic stem cells (HSCs) localized in the niche (Figure 1A-C; supplemental Figure 1). A gradual increase in TGF-β activity in immature hematopoietic cell fractions correlated with an increase in PAI-1 expression within the same fractions (Figure 1D; supplemental Figure 1), indicating that the TGF-β−PAI-1 axis is activated in HSPCs.
Figure 1.

TGF-β signaling induces PAI-1 expression and controls cell trafficking–related molecules expression in HSPCs. Representative flow cytometric profiles and MFI (n = 6) for TGF-β RI (A), TGF-β RII (B), p-Smad3 (C), and PAI-1 (D) expressions in freshly isolated immature hematopoietic cells. (E) Schema for in vitro experiments. Representative flow cytometric profiles and MFI (n = 6) for PAI-1 (F), MT1-MMP (G and K), CD44 (H), CXCR4 (I), and VLA-4 (J) expressions in WT or PAI-1 KO LSK cells treated with either TGF-β or LY364947 in vitro. Means ± SD are shown in each bar graph. FACS, fluorescence-activated cell sorter; IgG, immunoglobulin G; LSK, Lin−c-kit+Sca-1+; LS−K, Lin−c-kit+Sca-1−; LS−K−, Lin−c-kit−Sca-1−; MFI, mean fluorescence intensity; NS, not significant; RI, receptor I; RII, receptor II.
To identify the mechanisms that govern the dynamic motion of HSPCs within the niche, we investigated the cellular activities of HSPCs in response to TGF-β (Figure 1E). Addition of TGF-β increased the expression of PAI-1 (Figure 1F) in LSK cells, whereas it simultaneously reduced the expression of MT1-MMP (Figure 1G), a protease involved in the mobilization of HSPCs. This in turn increased the expression of CD44 and reduced the expression of CXCR4 and VLA-4 (Figure 1H-J). These changes were all reversed by the addition of a TGF-β inhibitor (LY364947 or SB431542), each of which is a selective inhibitor of TGF-β receptor I, inhibiting phosphorylation of Smad3 by TGF-β receptor I kinase (Figure 1F-J; supplemental Figure 2). A lack of PAI-1 in LSK cells from mice with genetically disrupted PAI-1 gene expression (PAI-1 knockout [KO] mice)38,39 expressed significantly higher levels of MT1-MMP, even when stimulated with TGF-β in vitro (Figure 1K). TGF-β stimulation did not affect the proliferative response of either wild-type (WT) or PAI-1 KO HSPCs (supplemental Table 2). These findings inform the hypothesis that TGF-β governs the motility and localization of HSPCs through Smad3−PAI-1-dependent downregulation of MT1-MMP expression, which leads to suppression of the molecules necessary for HSPC trafficking, such as CXCR4 and VLA-4, and protects adhesion molecules necessary for niche-anchoring, such as CD44, from cleavage.
PAI-1 regulates localization of HSPCs in the BM niche
To test the hypothesis that PAI-1 is involved in the motility and localization of HSPCs, we examined the effect of inhibition of PAI-1 in the motility of HSPCs in vivo using small molecule-specific inhibitors of PAI-1 (TM5275 or TM5509)27,43,44 or PAI-1 KO mice (Figure 2A). Consistent with the initial observation (Figure 1G-H), inhibition of PAI-1 enhanced MT1-MMP expression, which inversely correlated with CD44 expression, in BM LSK CD34− HSCs in vivo (Figure 2B), resulting in a marked mobilization of LSK cells and granulocytes from the BM to the periphery (Figure 2C-D; supplemental Figure 3A; the absolute number of mobilized hematopoietic cells is shown in supplemental Table 2). Consistently, PAI-1 KO mice had significantly elevated baseline levels of circulating LSK cells (Figure 2C-D). The mobilization of LSK cells was canceled by the administration of anti-MT1-MMP neutralizing antibody (Ab) (Figure 2C-D), suggesting that PAI-1 functions upstream of MT1-MMP and controls the HSPC mobilization.
Figure 2.

PAI-1 regulates mobilization of functional multipotent HSPCs. (A) Schema for mobilization experiments. (B) Representative flow cytometric profiles and MFI (n = 6) for MT1-MMP and CD44 expressions in LSK CD34− cells. (C) Representative flow cytometric profiles of circulating LSK cells. (D-E) Percentages of mobilized LSK cells (D) and the number of colony-forming cells (E) in PB (n = 6). (F) Schema for long-term competitive reconstitution experiments. (G) Percentages of donor cells in PB of primary and secondary recipients at 12 weeks after transplantation. Pooled data (n = 5) of 3 independent experiments are shown. (H) Representative flow cytometric profiles of donor-derived multilineage reconstitution in primary recipients. Means ± SD are shown in each bar graph. CFU, colony-forming unit; MNC, mononuclear cell; NS, not significant; Tx, transplantation.
The MT1-MMP–dependently mobilized cells phenotypically identified as primitive hematopoietic cells were functional multipotent long-term HSPCs, as confirmed by colony forming cell assays (Figure 2E; supplemental Figure 3B; supplemental Table 2) and by long-term competitive reconstitution assays (Figure 2F-H). PB cells isolated from the PAI-1 inhibitor–treated mice contained cells with a remarkable capacity to engraft in secondary recipients (Figure 2G). Flow cytometric analyses confirmed multilineage reconstitution as well as the maintenance of HSPCs in BM cells of primary and secondary recipients (Figure 2H; supplemental Figure 3C), further defining the enhanced long-term repopulation ability of circulating cells derived from the PAI-1 inhibitor–treated mice. In contrast, PB cells from the control mice had no ability to engraft to the secondary recipients (Figure 2G), indicating the roles of PAI-1 in regulation of HSPC mobilization. Competitive repopulating units in the donor cells were calculated as 1 HSC in 10 487 ± 2341 nucleated PB cells. Mice have a mean of 58.5 mL of blood per kilogram.45 A mouse weighing 25 g will therefore have a total blood volume of ∼1.46 mL. The absolute number of nucleated cells was 9.4 × 106 ± 0.34 × 106/mL of PB; thus, it was estimated that >1000 long-term HSCs egressed from the BM to the circulation in the PAI-1 inhibitor–treated mice.
PAI-1 blockade enhances detachment of HSPCs from the BM niche
Given the increased mobilization of HSPCs in PAI-1 blockaded mice (Figure 2), we evaluated whether the localization of HSPCs was affected by PAI-1 blockade in HSPCs by immunofluorescence analysis (Figure 3A). BM sections were stained with antibodies against CD150, CD48, and lineage markers (CD3, B220, Mac-1, Gr-1, and Ter119) to identify HSCs and simultaneously stained with antibodies against CD41 and TGF-β to identify TGF-β-expressing megakaryocytes (Figure 3B-C). We evaluated these sections to determine the positional relationships between HSCs and niche cells. Lin−CD48−CD150+ HSCs with PAI-1 blockade were frequently found farther away from cells staining positive for both TGF-β and CD41 (Figure 3C-D), in contrast to vehicle-treated HSCs that were often in contact with, or within close proximity to, TGF-β+CD41+ niche cells (Figure 3B,D). These results indicate that PAI-1 blockade induces the detachment of HSPCs from TGF-β-expressing megakaryocyte niches.
Figure 3.

PAI-1 regulates HSCs localization in the BM niche. (A) Schema for immunofluorescence analysis. (B-C) Representative pictures of the BM cavity of vehicle- or TM5509-treated mice. BM sections were stained with anti-CD150 (blue), anti-CD41 (green), anti-TGF-β (red), and anti-CD48 and -lineage markers (purple) antibodies. Arrowheads indicate TGF-β–expressing megakaryocytes. Arrows indicate Lin−CD48−CD150+ HSCs. Bars represent 100 μm. (D) Percentages of TGF-β–expressing megakaryocytes in close contact to HSCs. More than 100 cells in random fields on a slide were counted for 3 independent experiments. Means ± SD are shown in each bar graph. Meg, megakaryocyte.
Although the lack of PAI-1 can protect organs from fibrosis, changes in collagen deposition or fibrosis have been documented in some organs in PAI-1 KO mice.16 To exclude the possibility that nonhematopoietic cell effects in PAI-1 KO mice could affect the movement of HSCs, collagen deposition in the BM microenvironment was examined. As in WT mice, the type I collagen uniformly distributed throughout the BM cavity of PAI-1 KO mice (supplemental Figure 4), indicating that PAI-1 blockade-inducing detachment of HSPCs from the niche was an HSPC-autonomous manner.
PAI-1 suppresses MT1-MMP–dependent HSPC migration
To confirm that PAI-1 regulates the motility of HSPCs through the action of MT1-MMP, the motion of cells in response to environmental cues, which reflects the innate motility of functional HSPCs, was assessed. We know that upregulation of HSPC motility enhances their bidirectional trafficking across the endothelium and extracellular membrane barriers during BM egress (HSPCs mobilizing from the BM to the periphery) and homing (HSPCs migrating from the periphery to the BM) processes.5,46,47 The homing of circulating HSPCs to the BM and their capability of engrafting into their niches can be seen as a mirror image of HSPC mobilization, although the kinetics are somewhat different. The chemokine SDF-1 is the major attractant implicated in chemotaxis of HSPCs both in vitro and in vivo.48 SDF-1–induced motility of HSPCs in vitro correlates with HSPC migration, homing, and G-CSF–induced mobilization in vivo.31,49 To study MT1-MMP involvement in SDF-1–induced chemotaxis, we examined the directional motility of HSPCs toward an SDF-1 gradient by a transreconstituted basement membrane (Matrigel) migration assay in vitro. Lin−c-Kit+ immature BM cells isolated from PAI-1 KO mice exhibited significantly higher trans-Matrigel migration activity than WT Lin−c-Kit+ cells (Figure 4A). Addition of MT1-MMP neutralizing Ab to the culture abolished the high migration activity of PAI-1 KO cells (Figure 4A). We therefore tested if the observed higher migration activity of PAI-1–deficient HSPCs in vitro agreed with the enhanced migration and homing of HSPCs in vivo. After transplantation into lethally irradiated WT hosts, PAI-1 KO Lin−c-Kit+ cells demonstrated higher migration and homing activity than WT cells (Figure 4B). Because there was no difference in the fluorescent intensity of lipophilic dye PKH26 staining between WT and PAI-1 KO cells that were homed in the recipients’ BM, the increased homing of PAI-1 KO cells was due to the enhanced migration activity of HSPCs, not to the increased proliferation of cells homed to the BM (supplemental Figure 5A-B). As demonstrated in the experiments using PAI-1 inhibitors (Figure 2C-D), the administration of MT1-MMP neutralizing Ab counteracted the observed high motility of PAI-1 KO Lin−c-Kit+ cells (Figure 4B), confirming that PAI-1 and MT1-MMP cooperate in the regulation of HSPC motility. Importantly, PAI-1-deficient HSPCs demonstrated augmented migration activity even in the WT BM environment, in which extracellular PAI-1 was abundant (Figure 4B). Consistent with this, addition of recombinant PAI-1 did not affect the trans-Matrigel migration activity of WT or PAI-1 KO HSPCs (Figure 4A), indicating that iPAI-1, but not extracellular PAI-1, is a critical determinant of HSPC motility.
Figure 4.

iPAI-1 determines the activity of MT1-MMP–dependent HSPC migration. Percentages of migrated Lin−c-kit+ cells in in vitro (A) or in vivo (B) experiments (n = 6 each). rPAI-1, recombinant plasminogen activator inhibitor-1. Means ± SD are shown in each bar graph.
Intracellular, but not extracellular, PAI-1 determines HSPC motility
To rule out the possibility that extracellular PAI-1 influences MT1-MMP expression, and to confirm the direct involvement of iPAI-1 in the regulation of HSPC motility, we conducted long-term BM transplantation experiments using Ly5.1/Ly5.2 congenic mice (Figure 5A). Lin− BM cells from PAI-1 KO (Ly5.2) or WT (Ly5.1) mice were reciprocally transplanted to lethally irradiated WT (KO to WT) or PAI-1 KO (WT to KO) hosts, respectively. Three to 4 months after transplantation, >90% (95.64% ± 2.08%, n = 25) of hematopoietic cells in the recipients’ BM were identified as being of donor origin. This strategy allowed the selective functional assessment of cells defective in iPAI-1 (ie, PAI-1 KO cells), when housed in the WT environment where extracellular PAI-1 was abundant, and vice versa. Transplantations of PAI-1 KO or WT BM Lin− cells into lethally irradiated recipients of the same genetic background were performed as controls, (KO to KO) or (WT to WT). Consistent with our earlier observation (Figure 1K), a significantly higher level of MT1-MMP expression was detected in PAI-1 KO cells, even when they had been engrafted in the WT environment (Figure 5B). A markedly higher number of PAI-1 KO donor-derived circulating LSK cells was detected in the PB of WT recipients (Figure 5C-D), a result that recapitulates our earlier observation on PAI-1 KO mice (Figure 2C-E), indicating intrinsically high motility of iPAI-1-deficient HSPCs. Furthermore, PAI-1 KO donor-derived cells isolated from the BM of both WT and PAI-1 KO primary recipients demonstrated consistently high homing activity in secondary transfer experiments (Figure 5E-F). In contrast, WT or PAI-1 KO donor-derived cells from the BM of PAI-1 KO primary recipients, in which HSPCs had been housed without extracellular PAI-1, were not influenced in their motility (Figure 5C-F), demonstrating that HSPC motility is determined by the activity of iPAI-1 irrespective of extracellular PAI-1 in the BM environment.
Figure 5.

Intracellular, but not extracellular, PAI-1 regulates the motility of HSPCs. (A) Schema for long-term reciprocal transplantation experiments. (B) Representative flow cytometric profiles and MFI (n = 12) for MT1-MMP expression in LSK CD34− cells of primary recipients. (C-D) Percentages of circulating LSK cells (C) and the number of circulating CFCs (D) in PB samples obtained from the primary recipients of reciprocal transplantation experiments (n = 12). (E-F) Percentages of PKH26+ cells (E) and the number of CFCs (F) homed to the BM of secondary recipients (n = 12). (B-F) Means ± SD of 3 independent experiments are shown.
iPAI-1 suppresses HSPC motility by inhibiting Furin-dependent MT1-MMP expression
To identify how iPAI-1 coordinates the activity of MT1-MMP, we focused on the proteolytic regulation of Furin, an intracellular serine protease. Furin proteolytically activates a large number of proprotein substrates, including MT1-MMP, in the trans-Golgi network, where secretory pathway proteins are transported to final destinations.33 Immunofluorescence microscopic analysis revealed colocalization of Furin and iPAI-1 in Golgi marker–positive (golgi-58K+) cytoplasmic regions in LSK CD34− HSCs (Figure 6A). In addition, intimate molecular interactions between Furin and iPAI-1 were confirmed by a highly sensitive Duolink in situ proximity ligation assay (D-PLA) (Figure 6B). Furthermore, in silico docking simulations based on the 3-dimensional structure of PAI-1 and Furin demonstrated that the reactive central loop of PAI-1 that contains a protease recognition site was tightly fitted into the active site of Furin (Figure 6C). Taking these findings together, it is highly conceivable that PAI-1 inhibits the proteolytic activity of Furin by directly binding to its active site, thereby regulating the motility of HSPCs.
Figure 6.

iPAI-1 modulates MT1-MMP expression through the regulation of Furin. (A) Representative immunofluorescence microscopic images of PAI-1, Furin, and trans-Golgi complex in LSK CD34− cells. (B) D-PLA imaging shows the specific interaction between iPAI-1 and Furin in LSK CD34− cells. As a negative control for D-PLA, slides were treated with either the combination of anti-PAI-1 Ab and mouse IgG or anti-Furin Ab and rabbit IgG followed by the D-PLA secondary Abs. No fluorescent foci were detected by this treatment. (C) The docking simulations between Furin (white) and the active form of PAI-1 (yellow) show the tightly bound PAI-1 covering the active site of Furin, which prevents other substrates from approaching the active site (top). Compare with a structure of Furin (white) with a Furin inhibitor58 (green) bound at the active site of Furin (bottom). (D) Representative flow cytometric profiles and MFI (n = 6) for Furin expression in LSKCD34− cells isolated from WT or PAI-1 KO mice. (E) Representative flow cytometric profiles and MFI (n = 6) for Furin expression in LSK cells treated in vitro with either TGF-β or LY364947. (F-H) Representative flow cytometric profiles and MFI (n = 6) for MT1-MMP (F and H) and Furin (G) expression in PAI-1-overexpressed (OE), PAI-1-deleted (ΔPAI-1), or PAI-1/Furin double-deleted (ΔPAI-1/ΔFurin) hematopoietic cell lines. (D-H) Means ± SD are shown. (I-J) Relative messenger RNA (I) and representative flow cytometric profile (J) for MT1-MMP expression in LSK cells treated with LY364947 in the presence of actinomycin D in vitro. Bar graphs represent means ± SD (n = 5) of MT1-MMP expression. DAPI, 4',6-diamidino-2-phenylindole.
Because the net biological activity of Furin is also determined by the amount of its expression,50,51 we asked if iPAI-1 regulates the expression of Furin as well. Flow cytometric analyses demonstrated that PAI-1 KO LSK CD34− HSCs expressed a significantly higher level of Furin than WT HSCs (Figure 6D). In addition to regulating PAI-1 and MT1-MMP (Figure 1F-G), TGF-β−PAI-1 signaling downregulated the expression of Furin in WT LSK cells, which was reversed by the addition of a TGF-β inhibitor (Figure 6E), suggesting that iPAI-1 controls Furin expression. To further clarify the relationships of the 3 molecules, PAI-1, Furin, and MT1-MMP, involved in the motility of HSPCs, we established hematopoietic cell lines expressing constitutively high levels of PAI-1 overexpressed (supplemental Figure 6A-B) and completely deficient in PAI-1 expression (ΔPAI-1) (supplemental Figure 6C-F). In line with the results presented so far, the intensity of iPAI-1 was inversely correlated with the expressions of MT1-MMP and Furin (Figure 6F-G), demonstrating that iPAI-1 negatively regulates both Furin and MT1-MMP. Importantly, upregulation of MT1-MMP expression in ΔPAI-1 cells was nullified when the Furin gene was simultaneously deleted (ΔPAI-1/ΔFurin) (Figure 6H; supplemental Figure 6G-J), showing direct linkage of iPAI-1, Furin, and MT1-MMP. Therefore, the reciprocal relationships of these 3 proteins appear to govern the motility of HSPCs.
To clarify if the regulation of HSPC motility and localization by TGF-β−iPAI-1−Furin proteolytic sequence occurs at the posttranscriptional level, we evaluated whether MT1-MMP protein expression upon treatment with TGF-β inhibitor was dependent on new transcription and protein synthesis. WT LSK cells were treated with TGF-β inhibitor in the presence of the transcription inhibitor actinomycin D. Although actinomycin D impaired transcription of MT1-MMP messenger RNA in TGF-β inhibitor–treated LSK cells (Figure 6I), cell surface expression of MT1-MMP was still inducible by TGF-β inhibition (Figure 6J). We concluded that regulation of HSPC motility by TGF-β signaling mainly occurs at the posttranscriptional level.
TGF-β–induced iPAI-1 is responsible for the retention of HSPCs in the BM niche
Having established how iPAI-1 regulates the MT1-MMP–dependent motility of HSPCs, we assessed whether TGF-β is indeed involved in the retention of HSPCs in the niche. To prove that TGF-β is responsible for this retention, we investigated the effect of TGF-β signal inhibition in vivo (Figure 7A). As we anticipated, reductions in the levels of p-Smad3, iPAI-1, and CD44, and an increase in MT1-MMP expression, were confirmed in LSK CD34− HSCs (Figure 7B-E). At the same time, administration of TGF-β inhibitor in WT mice augmented the release of HSPCs from the BM to periphery (Figure 7F). In contrast, administration of TGF-β inhibitor in mice expressing a stable form of PAI-1 under the control of a TGF-β-insensitive promoter (PAI-1-stab. Tg),40 in which PAI-1 was constitutively expressed in HSCs even if TGF-β signaling was interrupted, did not facilitate HSPC mobilization (Figure 7G), indicating that downregulation of iPAI-1 activity is necessary for the displacement of HSPCs from the niche and the subsequent mobilization into the circulation. In fact, TGF-β inhibitor administration in PAI-1 KO mice, in which high numbers of circulating LSK cells were consistently detected in a steady state (Figures 2C-D), did not induce further mobilization of LSK cells (Figure 7H), indicating that TGF-β initiates the iPAI-1 proteolytic sequence that controls the motility of HSPCs in vivo. Taken together, our results demonstrate that TGF-β is responsible for the retention of HSPCs within the BM niche by activating a signal pathway, where iPAI-1 plays a critical role in regulating the intracellular proteolytic cascade that ultimately controls the motility of HSPCs (Figure 7I).
Figure 7.

TGF-β–induced iPAI-1 activation is responsible for the retention of HSPCs in the BM niche. (A) Schema for in vivo experiments. Representative flow cytometric profiles and MFI (n = 6) for p-Smad3 (B), iPAI-1 (C), MT1-MMP (D), and CD44 (E) expressions in BM LSK CD34− cells isolated from vehicle- or LY364947-treated WT mice. (F-G) Representative flow cytometric profiles and percentages (n = 6) of mobilized LSK cells in PB of vehicle- or LY364947-treated WT (F), stable PAI-1 transgenic mouse (PAI-1 stab. Tg) (G), or PAI-1 KO (H) mice. Means ± SD are shown. (I) A proposed working model schematically representing the main message of this work. In a steady state, TGF-β signaling stays active in niche-residing HSPCs. TGF-β–induced iPAI-1 inhibits Furin-dependent maturation of MT1-MMP, thereby causing HSPCs to remain within the BM niche. Conversely, when the TGF-β–iPAI-1 signal is suppressed, for example, in response to environmental stimuli, Furin-dependent MT1-MMP maturation becomes enhanced. The enhanced expression of MT1-MMP results in MT1-MMP–mediated CD44 cleavage as well as CXCR4 and VLA-4 activation, which in turn stimulates detachment of HSPCs from the niche and active trafficking into the bloodstream. VCAM-1, vascular cell adhesion molecule-1.
Discussion
In this paper, we elucidated a novel function of TGF-β, a well-known negative regulator of cell proliferation,7,8,12-14 in the retention of HSPCs within the BM niche. Although it is well established that the localization of HSPCs in the niche is essential for lifelong maintenance of hematopoiesis,1-3 how and why HSPCs stay in the niche have been rarely discussed. Here, we show that TGF-β initiates a signaling cascade that “instructs” HSPCs to stay in the niche. TGF-β activates iPAI-1 that downregulates the expression and activity of Furin, which in turn inhibits the activity of MT1-MMP in primitive HSPCs, resulting in the suppression of HSPC motility and retention in the niche. In this context, HSPCs remain accessible to niche signals that govern cellular activities. It is surprising that PAI-1, known conventionally as an extracellular fibrinolysis-related serine protease inhibitor (serpin),16,52 functions intracellularly and coordinates a proteolytic sequence, which ultimately determines the motility and activity of stem cells. TGF-β-induced iPAI-1 is mechanistically responsible for keeping HSPCs in the BM niche, where they are maintained in readiness for activation.
The activities of HSPCs, including their motility, are regulated by a complex network of cell-intrinsic and -extrinsic factors. Although HSPCs are localized in the niche in a steady state, they are quickly displaced from the niche and actively produce functional blood cells in response to stress. For example, during hematopoietic recovery after myelosuppression, the sequential activation of fibrinolytic proteases, namely tPA/plasmin/MMP2/9, and subsequent growth factor production stimulate blood cell regeneration,26,53-55 but these changes swiftly return to normal levels to prevent hematopoietic exhaustion. We have previously demonstrated the substantial elevation of extracellular PAI-1 expression within the BM niche in response to hematopoietic stress, such as irradiation and 5-fluorouracil treatment.27 Elevated extracellular PAI-1 inhibited the activation of tPA/plasmin/MMP2/9 sequence as well as the release of hematopoietic growth factors and chemotactic factors, thereby subduing the stress response. Pharmacological inhibition of PAI-1 significantly promoted hematopoietic regeneration after BM transplantation.27 Thus, extracellular PAI-1 negatively regulates excessive stress response and functions to maintain the steady-state environment. On the other hand, as shown in this study, iPAI-1 controls an intracellular proteolytic cascade that regulates the motility of HSPCs and acts as an instruction signal for HSPC retention in the niche. TGF-β-induced iPAI-1 appeared to regulate the balance of the retention and mobilization of the HSPCs. Taken together, our findings suggest that PAI-1 acts as both a cell-intrinsic and a cell-extrinsic regulatory factor of HSPCs, and is an attractive target for studies that will further our mechanistic understanding of the regulatory controls in stem cell physiology.
Although the expression of iPAI-1 is selectively elevated in HSPCs, other members of the class of intracellular serpins are expressed in a variety of blood cells at different stages of differentiation, working to control the activity of endogenous protease to avoid unexpected intracellular proteolysis.36 Using highly sensitive colocalization analyses and in silico docking simulations, we demonstrated that iPAI-1 binds to the active pocket of Furin and inhibits its activity. Furin has been shown to be responsible for the functional maturation of transmembrane proteins, such as growth factor receptors and Notch.33 It is one of the proprotein convertases, many of which are expressed in the trans-Golgi network and are in charge of posttranslational modification processes of many precursor proteins that are involved in a wide range of physiological events. Proprotein convertases recognize and cleave at the consensus sequence, and they share similar substrate specificities.56,57 Thus, other proprotein convertases could also be targets of iPAI-1. iPAI-1 may regulate the activity of proprotein convertases in general and control the production of functional proteins, thereby maintaining the function of stem cells. The intracellular proteolytic regulation of the physical dynamics and cellular activities of stem cells that we have shown in this study provides a novel insight into the mechanistic aspects of the maintenance of HSPC function.
Acknowledgments
The authors thank Hisako Akatsuka and Takehito Sato (Tokai University School of Medicine) for their expert technical support.
This work was supported by a grant-in-aid for Scientific Research (B) and Strategic Research Foundation Grant-aided Project for Private University from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (grants 15553358 and S1201001); Project Research from Tokai University School of Medicine; Ministry of Education (T.Y. and K.A.), Project for Development of Innovative Research on Cancer Therapeutics (P-DIRECT) and Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP), from the Japan Agency for Medical Research and Development (grants 14533192, 12103262, and 14529205) (T.Y., T.D., T.M., and K.A.); and the National Institutes of Health, National Heart, Lung, and Blood Institute (grant 5R01HL051387) (M.E. and D.E.V.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: T.Y., A.A.I., and Y.M. designed research, performed experiments, analyzed data, and cowrote the paper; M.E., A.M.S., N.W., and S.K. performed experiments; T.N., T.D., and N.H. designed and provided PAI-1 inhibitors; and D.E.V., T.M., and K.A. designed research, analyzed data, and cowrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Takashi Yahata, Tokai University School of Medicine, Kanagawa 259-1193, Japan; e-mail: yahata@tokai-u.jp; and Kiyoshi Ando, Tokai University School of Medicine, Kanagawa 259-1193, Japan; e-mail: andok@keyaki.cc.u-tokai.ac.jp.
References
- 1.Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6(2):93-106. [DOI] [PubMed] [Google Scholar]
- 2.Wilson A, Oser GM, Jaworski M, et al. . Dormant and self-renewing hematopoietic stem cells and their niches. Ann N Y Acad Sci. 2007;1106(1):64-75. [DOI] [PubMed] [Google Scholar]
- 3.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lapidot T, Petit I. Current understanding of stem cell mobilization: the roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp Hematol. 2002;30(9):973-981. [DOI] [PubMed] [Google Scholar]
- 5.Lapid K, Glait-Santar C, Gur-Cohen S, et al. . Egress and Mobilization of Hematopoietic Stem and Progenitor Cells: A Dynamic Multi-facet Process. Cambridge, MA: Harvard Stem Cell Institute; 2012. [PubMed] [Google Scholar]
- 6.Zon LI. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature. 2008;453(7193):306-313. [DOI] [PubMed] [Google Scholar]
- 7.Yamazaki S, Iwama A, Takayanagi S, Eto K, Ema H, Nakauchi H. TGF-β as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood. 2009;113(6):1250-1256. [DOI] [PubMed] [Google Scholar]
- 8.Yamazaki S, Ema H, Karlsson G, et al. . Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147(5):1146-1158. [DOI] [PubMed] [Google Scholar]
- 9.Bruns I, Lucas D, Pinho S, et al. . Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20(11):1315-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao M, Perry JM, Marshall H, et al. . Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20(11):1321-1326. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura-Ishizu A, Takubo K, Kobayashi H, Suzuki-Inoue K, Suda T. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow [published correction appears in J Exp Med. 2015;212(13):2323]. J Exp Med. 2015;212(12):2133-2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsson J, Karlsson S. The role of Smad signaling in hematopoiesis. Oncogene. 2005;24(37):5676-5692. [DOI] [PubMed] [Google Scholar]
- 13.Blank U, Karlsson S. The role of Smad signaling in hematopoiesis and translational hematology. Leukemia. 2011;25(9):1379-1388. [DOI] [PubMed] [Google Scholar]
- 14.Blank U, Karlsson S. TGF-β signaling in the control of hematopoietic stem cells. Blood. 2015;125(23):3542-3550. [DOI] [PubMed] [Google Scholar]
- 15.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17(11):3091-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. 2012;227(2):493-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao C, Lawrence DA, Li Y, et al. . Endocytic receptor LRP together with tPA and PAI-1 coordinates Mac-1-dependent macrophage migration. EMBO J. 2006;25(9):1860-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Czekay R-P, Aertgeerts K, Curriden SA, Loskutoff DJ. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J Cell Biol. 2003;160(5):781-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Czekay R-P, Wilkins-Port CE, Higgins SP, et al. . PAI-1: an integrator of cell signaling and migration. Int. J. Cell Biol. 2011;2011:562481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Degryse B, Sier CF, Resnati M, Conese M, Blasi F. PAI-1 inhibits urokinase-induced chemotaxis by internalizing the urokinase receptor. FEBS Lett. 2001;505(2):249-254. [DOI] [PubMed] [Google Scholar]
- 21.Degryse B, Neels JG, Czekay R-P, Aertgeerts K, Kamikubo Y, Loskutoff DJ. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J Biol Chem. 2004;279(21):22595-22604. [DOI] [PubMed] [Google Scholar]
- 22.Ichimura A, Matsumoto S, Suzuki S, et al. . A small molecule inhibitor to plasminogen activator inhibitor 1 inhibits macrophage migration. Arterioscler Thromb Vasc Biol. 2013;33(5):935-942. [DOI] [PubMed] [Google Scholar]
- 23.Stahl A, Mueller BM. Melanoma cell migration on vitronectin: regulation by components of the plasminogen activation system. Int J Cancer. 1997;71(1):116-122. [DOI] [PubMed] [Google Scholar]
- 24.Tashiro Y, Nishida C, Sato-Kusubata K, et al. . Inhibition of PAI-1 induces neutrophil-driven neoangiogenesis and promotes tissue regeneration via production of angiocrine factors in mice. Blood. 2012;119(26):6382-6393. [DOI] [PubMed] [Google Scholar]
- 25.Honjo K, Munakata S, Tashiro Y, et al. . Plasminogen activator inhibitor-1 regulates macrophage-dependent postoperative adhesion by enhancing EGF-HER1 signaling in mice. FASEB J. 2017;31(6):2625-2637. [DOI] [PubMed] [Google Scholar]
- 26.Tjwa M, Janssens S, Carmeliet P. Plasmin therapy enhances mobilization of HPCs after G-CSF. Blood. 2008;112(10):4048-4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ibrahim AA, Yahata T, Onizuka M, et al. . Inhibition of plasminogen activator inhibitor type-1 activity enhances rapid and sustainable hematopoietic regeneration. Stem Cells. 2014;32(4):946-958. [DOI] [PubMed] [Google Scholar]
- 28.Klein G, Schmal O, Aicher WK. Matrix metalloproteinases in stem cell mobilization. Matrix Biol. 2015;44-46:175-183. [DOI] [PubMed] [Google Scholar]
- 29.Zitka O, Kukacka J, Krizkova S, et al. . Matrix metalloproteinases. Curr Med Chem. 2010;17(31):3751-3768. [DOI] [PubMed] [Google Scholar]
- 30.Shirvaikar N, Marquez-Curtis LA, Shaw AR, Turner AR, Janowska-Wieczorek A. MT1-MMP association with membrane lipid rafts facilitates G-CSF–induced hematopoietic stem/progenitor cell mobilization. Exp Hematol. 2010;38(9):823-835. [DOI] [PubMed] [Google Scholar]
- 31.Vagima Y, Avigdor A, Goichberg P, et al. . MT1-MMP and RECK are involved in human CD34+ progenitor cell retention, egress, and mobilization. J Clin Invest. 2009;119(3):492-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato H, Kinoshita T, Takino T, Nakayama K, Seiki M. Activation of a recombinant membrane type 1-matrix metalloproteinase (MT1-MMP) by furin and its interaction with tissue inhibitor of metalloproteinases (TIMP)-2. FEBS Lett. 1996;393(1):101-104. [DOI] [PubMed] [Google Scholar]
- 33.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3(10):753-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remacle AG, Rozanov DV, Fugere M, Day R, Strongin AY. Furin regulates the intracellular activation and the uptake rate of cell surface-associated MT1-MMP. Oncogene. 2006;25(41):5648-5655. [DOI] [PubMed] [Google Scholar]
- 35.Korpula-Mastalerz R, Dubin A. The intracellular serpin family. Acta Biochim Pol. 1996;43(3):419-429. [PubMed] [Google Scholar]
- 36.Morris EC, Carrell RW, Coughlin PB. Intracellular serpins in haemopoietic and peripheral blood cells. Br J Haematol. 2001;115(4):758-766. [DOI] [PubMed] [Google Scholar]
- 37.Bernot D, Stalin J, Stocker P, et al. . Plasminogen activator inhibitor 1 is an intracellular inhibitor of furin proprotein convertase. J Cell Sci. 2011;124(Pt 8):1224-1230. [DOI] [PubMed] [Google Scholar]
- 38.Carmeliet P, Kieckens L, Schoonjans L, et al. . Plasminogen activator inhibitor-1 gene-deficient mice. I. Generation by homologous recombination and characterization. J Clin Invest. 1993;92(6):2746-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carmeliet P, Stassen JM, Schoonjans L, et al. . Plasminogen activator inhibitor-1 gene-deficient mice. II. Effects on hemostasis, thrombosis, and thrombolysis. J Clin Invest. 1993;92(6):2756-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eren M, Gleaves LA, Atkinson JB, King LE, Declerck PJ, Vaughan DE. Reactive site-dependent phenotypic alterations in plasminogen activator inhibitor-1 transgenic mice. J Thromb Haemost. 2007;5(7):1500-1508. [DOI] [PubMed] [Google Scholar]
- 41.Osawa M, Hanada K, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273(5272):242-245. [DOI] [PubMed] [Google Scholar]
- 42.Kiel MJ, Yilmaz ÖH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109-1121. [DOI] [PubMed] [Google Scholar]
- 43.Izuhara Y, Takahashi S, Nangaku M, et al. . Inhibition of plasminogen activator inhibitor-1: its mechanism and effectiveness on coagulation and fibrosis. Arterioscler Thromb Vasc Biol. 2008;28(4):672-677. [DOI] [PubMed] [Google Scholar]
- 44.Miyata T, Kikuchi K, Kiyomoto H, van Ypersele de Strihou C. New era for drug discovery and development in renal disease. Nat Rev Nephrol. 2011;7(8):469-477. [DOI] [PubMed] [Google Scholar]
- 45.Wolfensohn S, Lloyd M, eds. Handbook of Laboratory Animal Management and Welfare. Hoboken, NJ: Blackwell Publishing; 2003. [Google Scholar]
- 46.Gur-Cohen S, Itkin T, Chakrabarty S, et al. . PAR1 signaling regulates the retention and recruitment of EPCR-expressing bone marrow hematopoietic stem cells. Nat Med. 2015;21(11):1307-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saez B, Ferraro F, Yusuf RZ, et al. . Inhibiting stromal cell heparan sulfate synthesis improves stem cell mobilization and enables engraftment without cytotoxic conditioning. Blood. 2014;124(19):2937-2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. 2002;195(9):1145-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peled A, Petit I, Kollet O, et al. . Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283(5403):845-848. [DOI] [PubMed] [Google Scholar]
- 50.Guimont P, Grondin F, Dubois CM. Sox9-dependent transcriptional regulation of the proprotein convertase furin. Am J Physiol Cell Physiol. 2007;293(1):C172-C183. [DOI] [PubMed] [Google Scholar]
- 51.Laprise M-H, Grondin F, Cayer P, McDonald PP, Dubois CM. Furin gene (fur) regulation in differentiating human megakaryoblastic Dami cells: involvement of the proximal GATA recognition motif in the P1 promoter and impact on the maturation of furin substrates. Blood. 2002;100(10):3578-3587. [DOI] [PubMed] [Google Scholar]
- 52.Ha H, Oh EY, Lee HB. The role of plasminogen activator inhibitor 1 in renal and cardiovascular diseases. Nat Rev Nephrol. 2009;5(4):203-211. [DOI] [PubMed] [Google Scholar]
- 53.Gong Y, Fan Y, Hoover-Plow J. Plasminogen regulates stromal cell-derived factor-1/CXCR4-mediated hematopoietic stem cell mobilization by activation of matrix metalloproteinase-9. Arterioscler Thromb Vasc Biol. 2011;31(9):2035-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heissig B, Lund LR, Akiyama H, et al. . The plasminogen fibrinolytic pathway is required for hematopoietic regeneration. Cell Stem Cell. 2007;1(6):658-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tjwa M, Moura R, Moons L, et al. . Fibrinolysis-independent role of plasmin and its activators in the haematopoietic recovery after myeloablation. J Cell Mol Med. 2009;13(11-12):4587-4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Irving JA, Pike RN, Lesk AM, Whisstock JC. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. 2000;10(12):1845-1864. [DOI] [PubMed] [Google Scholar]
- 57.Seidah NG, Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov. 2012;11(5):367-383. [DOI] [PubMed] [Google Scholar]
- 58.Hardes K, Becker GL, Lu Y, et al. . Novel furin inhibitors with potent anti-infectious activity. ChemMedChem. 2015;10(7):1218-1231. [DOI] [PubMed] [Google Scholar]