

The American Society of Colon and Rectal Surgeons (ASCRS)is dedicated to ensuring high-quality patient care by advancing the science, prevention, and management of disorders and diseases of the colon, rectum, and anus. The Clinical Practice Guidelines Committee is composed of society members who are chosen because they have demonstrated expertise in the specialty of colon and rectal surgery. This committee was created to lead international efforts in defining quality care for conditions related to the colon, rectum, and anus, in addition to the development of Clinical Practice Guidelines based on the best available evidence. These guidelines are inclusive and not prescriptive. Their purpose is to provide information on which decisions can be made, rather than to dictate a specific form of treatment. These guidelines are intended for the use of all practitioners, health care workers, and patients who desire information about the management of the conditions addressed by the topics covered in these guidelines.
It should be recognized that these guidelines should not be deemed inclusive of all proper methods of care or exclusive of methods of care reasonably directed to obtaining the same results. The ultimate judgment regarding the propriety of any specific procedure must be made by the physician in light of all the circumstances presented by the individual patient.
METHODOLOGY
These guidelines are built on the last set of the ASCRS Practice Parameters for the Identification and Testing of Patients at Risk for Dominantly Inherited Colorectal Cancer published in 2003.1 An organized search of MEDLINE (1946 to December Week 1 2016) was performed from 1946 through week 4 of September 2016 (Figure 1). Subject headings for “adenomatous polyposis coli” (4203 results) and “intestinal polyposis” (445 results) were included, using focused search. The results were combined (4629 results) and limited to English language (3981 results), then further limited by study type for potential inclusion in the evidence-based review, using the publication type limits of case reports, clinical trial, comparative study, controlled clinical trial, guideline, meta-analysis, multicenter study, observational study, practice guideline, randomized controlled trial, or systematic reviews (1311 results). A second keyword search was done (not mapped to subject heading) for “familial polyposis” (1143 results), “MYH or MUTYH” (408 results), and “desmoid” (2303 results).These were limited in the same manner as the subject heading search, leaving 1249 results. They were merged with the 1311 prior results, leaving a total of 2343 titles for review. After title review, 361 abstracts were reviewed and selections made for full-text review, with directed search of imbedded references and article citation as needed. A total of 121 full text articles were reviewed. Existing guidelines on this topic, their associated references and cited articles were reviewed for any additional studies that may not have been included.1-5 The final source material used was evaluated for the methodological quality, the evidence base was examined, and a treatment guideline was formulated by the subcommittee for this guideline. A final grade of recommendation was assigned using the Grades of Recommendation, Assessment, Development, and Evaluation (GRADE) system (Table 1).6 When agreement was incomplete regarding the evidence base or treatment guideline, consensus from the committee chair, vice chair, and two assigned reviewers determined the outcome. Members of the ASCRS practice guidelines committee worked in joint production of these guidelines from inception to final publication. Recommendations formulated by the subcommittee were reviewed by the entire Clinical Practice Guidelines Committee. Final recommendations were approved by the ASCRS Clinical Guidelines Committee and ASCRS Executive Committee.
Figure 1.
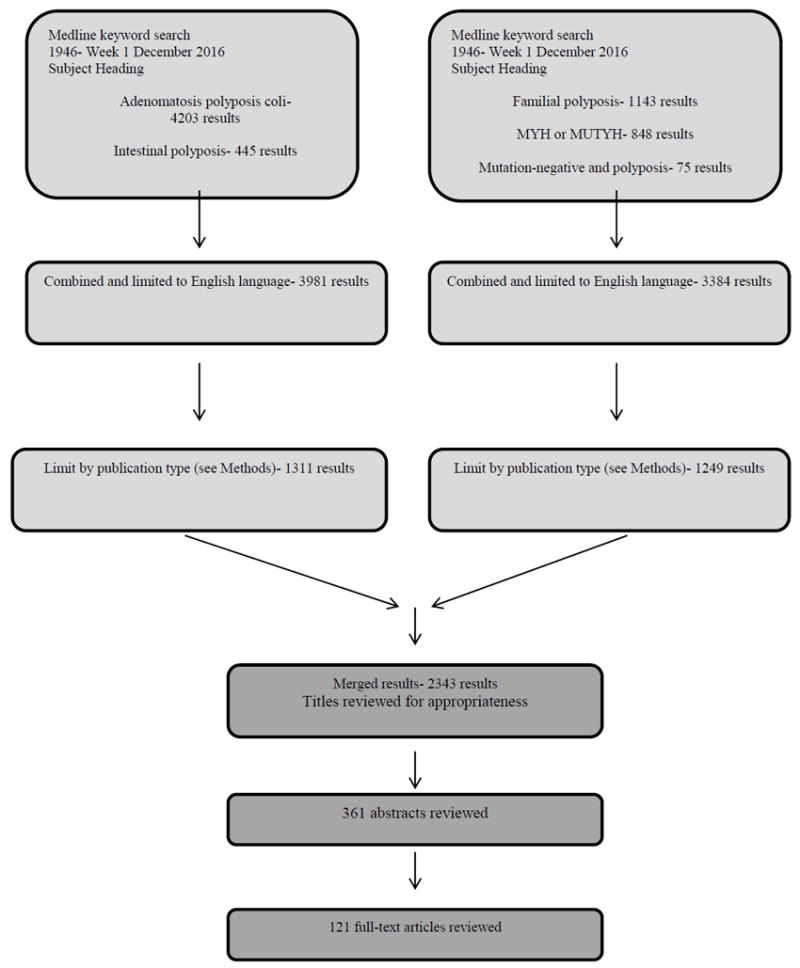
Literature search strategy
Table 1.
The GRADE System - Grading Recommendations
| Description | Benefit vs. Risk and Burdens | Methodologic Quality of Supporting Evidence | Implications | |
|---|---|---|---|---|
| 1A | Strong recommendation, High quality evidence |
Benefits clearly outweigh risk and burdens or vice versa | RCTs without important limitations or overwhelming evidence from observational studies | Strong recommendation, can apply to most patients in most circumstances without reservation |
| 1B | Strong recommendation, Moderate quality evidence |
Benefits clearly outweigh risk and burdens or vice versa | RCTs with important limitations (inconsistent results, methodologic flaws, indirect or imprecise) or exceptionally strong evidence from observational studies | Strong recommendation, can apply to most patients in most circumstances without reservation |
| 1C | Strong recommendation, Low or very low quality evidence |
Benefits clearly outweigh risk and burdens or vice versa | Observational studies or case series | Strong recommendation but may change when higher quality evidence becomes available |
| 2A | Weak recommendation, High quality evidence |
Benefits closely balanced with risks and burdens | RCTs without important limitations or overwhelming evidence from observational studies | Weak recommendation, best action may differ depending on circumstances or patients’ or societal values |
| 2B | Weak recommendations, Moderate quality evidence |
Benefits closely balanced with risks and burdens | RCTs with important limitations (inconsistent results, methodologic flaws, indirect or imprecise) or exceptionally strong evidence from observational studies | Weak recommendation, best action may differ depending on circumstances or patients’ or societal values |
| 2C | Weak recommendation, Low or very low quality evidence |
Uncertainty in the estimates of benefits, risks and burden; benefits, risk and burden may be closely balanced | Observational studies or case series | Very weak recommendations; other alternatives may be equally reasonable |
GRADE = Grades of Recommendation, Assessment, Development, and Evaluation; RCT = randomized controlled trial. Adapted from Guyatt G, Gutermen D, Baumann MH, et al. Grading strength of recommendations and quality of evidence in clinical guidelines: report from an American College of Chest Physicians Task Force. Chest. 2006;129:174-181. Used with permission
STATEMENT OF THE PROBLEM
Colorectal cancer (CRC) is the third most common cancer in men and women in the United States, and the second leading cause of cancer deaths. Approximately 20-30% of colorectal cancer cases are associated with a family history of colorectal polyps or cancer, and approximately 3-5% % of cases are associated with an identifiable inherited colorectal cancer syndrome. Of these inherited syndromes, polyposis syndromes and Lynch Syndrome are the most common. Polyposis syndromes have been recognized for many years, due to a strong phenotype that includes the presence of multiple, even thousands, of polyps of different histologic types.7
Familial adenomatosis polyposis (FAP) is an autosomal dominant syndrome characterized by tens to thousands of colonic adenomas, one or more of which will progress to cancer unless they are diagnosed or treated. There are also higher lifetime risks of several other malignancies (e.g., duodenum, pancreas, thyroid, brain). A germline mutation of the adenomatous polyposis coli (APC) tumor suppressor gene, located on chromosome 5q21, is found in most patients with classical adenomatous polyposis (>100 synchronous adenomas).8, 9 Affected individuals can have a range of disease severity, sometimes predicted by the location of the APC mutation, leading to the designation of “attenuated FAP” (AFAP) in more mildly affected individuals (<100 synchronous adenomas).10, 11 A distinct subset of patients with AFAP have now been recognized to arise from biallelic germline mutations of the base excision repair gene MutY homologue (MYH), in a autosomal recessive syndrome termed MYH-associated polyposis, or MAP.12, 13 Despite recent advances, there are also individuals with clinically evident polyposis, either attenuated adenomatous polyposis or serrated polyposis, in whom a mutation is not found. This clinical practice guideline will cover the identification and management of FAP, AFAP, MAP and polyposis without an identified genotype, and the extraintestinal manifestations included in the adenomatous polyposis syndromes. Hamartomatous polyposis syndromes, such as Peutz-Jeghers Syndrome, Juvenile Polyposis Syndrome, and PTEN Hamartoma Tumor Syndrome have little high-grade evidence to guide treatment and are not reviewed here.
Screening and Genetic Testing for Adenomatous Polyposis Syndromes
1. Polyposis syndromes should typically be considered in patients with greater than 20 lifetime adenomas, patients with a personal history of desmoid tumor or other extracolonic manifestations of FAP, or family members of individuals with known FAP, AFAP, or MAP. Grade of Recommendation: Strong recommendation based on low quality evidence, 1C.
A clinical diagnosis of FAP is generally agreed upon when >100 adenomas are found, and germline testing of the APC gene is recommended for these individuals, as this facilitates screening for the mutation in family members, and may have predictive value for extracolonic manifestations. While most probands with >100 adenomas will have a detectable mutation or deletion in APC, there is a small proportion of cases where no mutation can be found. Recently deletions in APC promoter 1B have been found in such families, and this needs to be requested specifically. In a family with classical FAP but no identifiable mutation in APC, screening and treatment should follow the same principles as those with proven mutations.
For patients with fewer than 100 adenomas, clarifying the diagnosis can be difficult. The recent development of next generation DNA sequencing and multigene panel testing allows these patients to be tested for all the known colorectal cancer genes with a single blood test. This is helpful since many syndromes have been associated with attenuated adenomatous polyposis (AFAP, MAP, Polymerase Proofreading Associated Polyposis, Lynch Syndrome). The clinical question to answer is the threshold of cumulative adenoma numbers at which genetic testing should be sought. While multiple criteria have been proposed, there is no consensus for the diagnosis of AFAP.14-16 The diagnosis of AFAP often requires combining clinical findings, adenoma number (more than 10 or 20 but less than 100), family history, and genetic testing to distinguish the syndrome from MAP, polyposis of unknown etiology, or simply multiple sporadic adenomas.
There are little data to define the cumulative number of polyps that should prompt testing. A cross-sectional study of 8903 individuals who had samples submitted for APC and MYH mutations to Myriad Genetics Laboratories was published in 2012.13 Mutations were found in 82% of individuals with >1000 polyps, 63% of individuals with 100-999 polyps, 17% of individuals with 20-99 polyps, and 9% of individuals with 10-19 polyps. These data show that reliance on genetic testing alone to define these syndromes is not adequate; clinical criteria for the diagnosis of polyposis are required for those in whom no known mutation is detected. Since the risk of finding a genetic abnormality does not rise above 10% until 20 or more adenomas are found, it is reasonable to use a cutoff of 20 cumulative adenomas to prompt genetic counseling and testing. We agree with multiple other existing guidelines that genetic testing should be preceded by genetic counseling, ideally by a certified genetic counselor when available.17-19 Patients in whom no known mutation is found should be treated as having classic or attenuated polyposis, based upon their observed phenotype.
A family history of polyposis is helpful, but not required for making decisions regarding genetic testing. A systematic evidence-based review of the accuracy of a family cancer history was reported by Murff, et al., and established that patient-reported history is accurate and valuable in the evaluation of colon cancer and breast cancer patients.20 The absence of a family history of polyposis or colorectal cancer does not exclude the diagnosis of a polyposis syndrome, as de novo mutations in the APC gene may occur in up to 15% of FAP patients and MAP is recessively inherited.21
Treatment for FAP
1. Treatment should include thorough counseling about the nature of the syndrome, its natural history, its extracolonic manifestations, and the need for compliance with recommendations for management and surveillance. Grade of Recommendation: Strong recommendation based on low quality evidence, 1C.
In untreated patients with classic FAP, colorectal cancer is nearly universal by age 40. There are no randomized or prospective trials of different surveillance strategies. Multiple single-center observational studies and a single systematic review have demonstrated a reduction in CRC incidence and mortality for patients within registry screening programs.22 Affected individuals have a risk of extracolonic manifestations, including gastric, duodenal and small bowel polyps and cancer, adrenal adenomas, thyroid cancer, desmoid tumors, hepatoblastoma and congenital hypertrophy of the retinal pigment epithelium (CHRPE). A recent review of an institutional registry reported a sharp increase in gastric cancer in long term follow up of patients with FAP. Additional research in this area may influence future screening recommendations for gastric cancer.23 Duodenal cancer and desmoid tumors are the most common causes of death in patients with FAP after colorectal cancer, and recent data suggests that gastric cancer incidence is increasing. Surveillance of these organs and management of the post-surgical lower gastrointestinal tract is important. The risk of some of these can be estimated by the position of the codon mutation along the APC gene.24 Based on low-quality evidence, we recommend clinicians caring for polyposis patients should have specialized expertise in the field and, ideally, work within a multidisciplinary team. A recent survey of the membership of the Association of Coloproctology of Great Britain and Ireland showed that even among specialist surgeons, awareness of the nature of polyposis syndromes is low.23 Evidence supporting appropriate diagnosis and treatment, especially for AFAP and MAP, is evolving, and new findings may alter treatment recommendations.
Familial Screening for At-Risk Members
1. At-risk family members of a patient with an identified mutation are screened for the mutation. For children and those who decline genetic testing, endoscopic surveillance is recommended until either genetic testing is performed or a diagnosis is clear based on phenotype. At-risk family members who do not carry the mutation should have the same screening as the average risk population. Grade of Recommendation: Strong recommendation based on moderate quality evidence, 1B.
When a patient is the first in a family to be diagnosed with FAP (proband), genetic testing is indicated. If a mutation is found, then all the at-risk relatives of the proband can be screened for this mutation. This process is much cheaper and quicker than multigene panel sequencing. Most authorities recommend testing children at puberty because cancer is rare before that age. Observational data supports the finding that colorectal cancer has not been reported before age 9 in individuals with FAP. Combining data from five European registries of patients with polyposis, 0.2% developed cancer before age 15 and 1.3% before age 20.6 A survey of 26 registries found only one case of invasive cancer reported before age 17.25 Two features of FAP help to develop rational screening strategies: (1) the rectum is almost always affected in classic FAP; and (2)while polyps form at a much younger age than the general population, there is no clear evidence that progression from polyp to invasive cancer is accelerated.26 Screening for polyps in children of patients with classic FAP can, therefore, begin at age 10 and can be accomplished with flexible sigmoidoscopy. Polyps should be sampled as lymphoid follicles are particularly prominent in children and can be mistaken endoscopically for adenomas. Those without evidence of polyposis should have the exam every two years. There are little data to guide decision-making for children of affected individuals who reach adulthood without a phenotype of polyposis and without a positive genetic diagnosis. It is, however, reasonable for colonoscopy to replace flexible sigmoidoscopy in the later teenage years, (16) and to be done every two years until age 20. If there are still no adenomas, surveillance intervals can be gradually extended. Children who are found to have adenomas on flexible sigmoidoscopy should have a colonoscopy to document the severity of the polyposis. They can generally defer surgical management until they have reached adulthood, based on moderate quality evidence as noted above. Postoperative screening should follow guidance outlined later in this report.
There are incomplete data to strongly support a screening strategy for attenuated FAP. Colorectal cancers occur significantly later with these mutations, and the rectum may be spared. A strategy that focuses on frequent flexible sigmoidoscopy in younger individuals may therefore be inadequate. Two observational studies in AFAP have shown no cancer before the age of 20, and the youngest reported has been at age 24. 15, 27 Observational data and expert opinion support colonoscopy every two years, starting at age 20 until (1) preventive surgery is performed, or (2) genetic testing proves the absence of a known familial mutation. Since screening begins at an older age than with classic FAP, and since rectal sparing is seen with AFAP, colonoscopy instead of flexible sigmoidoscopy is recommended to properly identify affected individuals. Furthermore, besides establishing the clinical diagnosis of polyposis in AFAP, endoscopic clearance of polyps may delay or eliminate the need for preventative surgery. The interval between exams has not been clearly established by available evidence. Existing general post-polypectomy surveillance guidelines may be modified to more frequent exams if endoscopic clearance is the treatment plan, since polyp numbers may increase quickly, and subtle adenomas may be missed.28
Surgical Treatment of FAP and AFAP
1. Proctocolectomy with ileostomy or ileal pouch-anal anastomosis is the treatment of choice for patients with a large number of rectal adenomas, but the optimal timing should be individualized. Grade of Recommendation: Strong recommendation based on moderate quality evidence, 1B.
The aims of treatment of the lower gastrointestinal tract in patients with FAP are to prevent death from cancer and to maximize quality of life. In general, colonoscopic clearance of adenomas in patients with classical FAP cannot guarantee that cancer will be prevented, so removal of the at-risk epithelium is required. The two decisions that need to be made in affected patients to fulfill those aims center around the timing of surgery and the type of operation to be performed.
Multiple factors need to be considered when determining the proper operation and timing of surgery. In addition to more traditional metrics such as medical co-morbidities and nutritional status, the timing and choice of surgical procedure must also take into account the educational, intellectual, and emotional development of the patient and their reliability for follow-up evaluations. The four surgical options are: total colectomy with ileorectal anastomosis (IRA); proctocolectomy with stapled ileal pouch-anal anastomosis (IPAA); proctocolectomy with mucosectomy and hand sewn IPAA; and total proctocolectomy with end ileostomy. There is no randomized trial comparing IRA to proctocolectomy with IPAA. A meta-analysis of twelve nonrandomized studies demonstrated individual merits of each approach.29 Factors that favor total abdominal colectomy and leaving the rectum in place include relative rectal sparing, and the desire to avoid pelvic dissection and possible infertility or sexual dysfunction. Factors that support a proctocolectomy with IPAA include the presence of rectal cancer, large rectal polyp burden (>20 synchronous adenomas, adenoma with high grade dysplasia, large (>30mm) adenomas), or a severe familial phenotype (>1000 synchronous adenomas).
There is a very small risk of adenocarcinoma after an IPAA, with only about two dozen reported cases in the literature to date.30-32 Most of these cases are cancer in the residual rectal or anal transitional zone (ATZ) mucosa. Whether IPAA should be performed with removal of the ATZ by mucosectomy and hand-sewn anastomosis or retaining some ATZ with a stapled anastomosis has been debated. The benefits of a stapled anastomosis include better function and fewer operative complications. The stapled IPAA is also easier to survey, and ATZ adenomas may possibly be treated endoscopically or transanally. The benefit of a handsewn IPAA is a reduced incidence of postoperative ATZ adenomas, but this is achieved at the potential cost of worse function and more complications.33 The largest series analyzing ATZ neoplasia includes 206 patients, with a median follow-up of 10.3 years. The risk of adenoma at the IPAA at ten years was 22% in the mucosectomy group and 51% after stapled IPAA. One patient developed cancer after mucosectomy, and no patient developed cancer after stapled IPAA.34 Pooled data from five registries of 97 patients with at least one year of endoscopic follow-up compared 35 patients with stapled IPAA to 62 patients with mucosectomy.35 The risk of developing a polyp at the IPAA after seven years was 31% after stapled IPAA vs. 10% after mucosectomy. No patient developed an ATZ cancer. A protocol for a Cochrane review on this topic was published in 2014, but the analysis has not been published to date.36 Additional low-quality evidence from multiple studies provides conflicting conclusions about the role of mucosectomy in cancer prevention and functional implications. The evidence available, however, does not support routine mucosectomy if the residual rectal cuff is free of polyps and can be surveyed. Annual endoscopic surveillance of the remaining rectal and ATZ mucosa and ileal pouch must be performed. Total proctocolectomy with end ileostomy can be considered for patients with poor sphincter function, incontinence, distal rectal cancer, cancers requiring radiation, or the desire to avoid the functional limitations of an ileoanal pouch.
While laparoscopic proctocolectomy has some potential advantages over conventional open technique, only one randomized trial has been reported (comparing hand-assisted to open, and including patients with ulcerative colitis and FAP),which showed comparable outcomes.37 Multiple non-randomized studies demonstrating the safety and feasibility of the laparoscopic approach suffer from selection bias and are not adequate to conclude that one technique is superior. A Cochrane review on this topic demonstrated no significant differences in morbidity, recovery, or complications, but reported that cosmesis was better with the laparoscopic approach.38
Total Colectomy with Ileorectal Anastomosis (TAC-IRA)
2. Total colectomy with ileorectal anastomosis (TAC-IRA) can be offered to patients with relative rectal sparing if all rectal adenomas >5mm can be endoscopically removed. Grade of Recommendation: Strong recommendation based on low quality evidence, 1C.
Before restorative proctocolectomy was available, patients typically underwent total colectomy with ileorectal anastomosis and accepted a risk of rectal cancer in order to avoid an ileostomy. Since restorative proctocolectomy has become widely available, the decision whether or not to retain the rectum is made based on functional considerations and on polyposis phenotype that includes rectal sparing.1, 39, 40 Studies of rectal cancer risk in FAP after TAC-IRA should be viewed with caution if they include patients who had their operation before the availability of restorative proctocolectomy. Population-based data from four European centers evaluated 776 patients who had IRA, including 576 prior to the ileoanal pouch era, and 200 after ileoanal pouch became available in these centers.41 The cumulative risk of rectal cancer by Kaplan-Meier analysis was 10% in the pre-pouch era vs. 2% in the pouch era. The most frequently used criteria to offer an ileorectal anastomosis is rectal polyp burden. A cohort study from Church, et al. of 213 FAP patients included 165 patients who had rectal sparing surgery, with 128 of these having fewer than 20 polyps and 37 having greater than 20 polyps. The rectal cancer incidence was 1.6% in the patients with <20 polyps, compared with 10.8% in the patients with >20 polyps.42 A cohort study from the Singapore Polyposis Registry reported that recurrence- and disease-free survival was not different with selective use of ileorectal anastomosis after 98 months of follow-up. It is unlikely that a study will be done comparing functional and oncologic outcomes of IRA vs. IPAA, since most individuals who are eligible for IRA would prefer a rectal sparing procedure. Nevertheless, observational data support clear potential functional benefits of sparing the rectum, including decreased bowel frequency, decreased incontinence, decreased risk of urinary and sexual dysfunction, and the high chance of a single stage operation and avoidance of an ileostomy. Using the current clinical criteria of rectal sparing, most frequently defined as fewer than 20 adenomas, ileorectal anastomosis has support from observational data. Annual surveillance of the rectum is required. Chemoprevention using celecoxib or sulindac can be considered, based on studies showing benefit in management of duodenal adenomas in this population (see below).
MYH-associated Polyposis (MAP)
1. The diagnosis of MAP should be considered in patients presenting with colorectal polyposis (>20 lifetime adenomas). Grade of Recommendation: Strong recommendation based on low quality evidence, 1C.
MAP is an autosomal recessively inherited colorectal polyposis syndrome caused by biallelic germline mutations in the base-excision repair gene MYH, located on chromosome 1.13 As the autosomal recessive inheritance pattern requires that affected individuals have a biallelic mutations, both parents of affected individuals must be at least monoallelic carriers. If so, siblings of affected individuals have a 25% chance of biallelic mutations, and children of affected individuals will be at least heterozygous carriers. It is not clear whether individuals with monoallelic mutations have a higher risk of colorectal neoplasia.43, 44 A multicenter European case-control study of cases matched with population controls identified a standardized incidence rate of colorectal cancer at 2.12.45 Since the reported increases are around the same or lower than the increased risk reported for first-degree relatives of sporadic colorectal cancer, these individuals should be screened in the same way as people with one affected first degree relative.
Population-based cohort studies provide the best estimate for the prevalence of monoallelic MYH mutations, thought to occur in 0.7-1% of the population.44, 46-49 The number of polyps may not correlate with the prevalence of biallelic MYH mutations as well as it does with APC mutations, making it difficult to recommend screening for MAP based on a specific number of polyps. While many reports cite a threshold of 10 polyps as an indication for genetic testing, the National Comprehensive Cancer Network (NCCN) guidelines have moved to a threshold of 20 polyps.2-6, 13, 47, 50 While acknowledging the limited evidence supporting a specific polyp number cut-off, consideration for genetic testing for MAP should be given in most patients with >20 lifetime adenomas. Since most genetic testing today is done with multigene panels, patients who are screened for genetic causes of colonic polyposis syndromes will likely have APC and MYH evaluated at the initial genetic evaluation.
2. Patients with biallelic MYH mutations need yearly colonoscopy and polypectomy, as long as the adenomas can be controlled endoscopically. Siblings or children of an affected individual need to be screened for the family mutations in MYH. Those who have not been tested should undergo colonoscopy every two years, starting at age 20. Grade of Recommendation: Weak recommendation based on moderate quality evidence, 2B.
In general, the colorectal MAP phenotype resembles that of attenuated FAP, but individuals with biallelic mutations may present with an apparently sporadic cancer, as a cancer at a young age, or even mimicking Lynch Syndrome. The average age of colorectal cancer in patients with MAP is 47 years (age range 29–72 years).51-55 Colorectal cancer due to biallelic MYH mutations before the age of 30 is rare, and due to lower polyp numbers, maintaining endoscopic clearance of polyps is possible in some patients. Rectal cancer is uncommon in MAP; a population-based study of 9,268 colorectal cancer patients identified 27 patients with biallelic MYH mutations who had a lower than expected rate of rectal cancer when compared to sporadic cases.56 A registry-based cohort study from the Netherlands demonstrated that 62% of MAP-associated cancers occurred proximal to the splenic flexure.52 Due to an accelerated adenoma-to-carcinoma progression in MAP patients, patients with proven biallelic MYH mutations and siblings who have not been tested should have colonoscopy every year starting at age 20.52 Flexible sigmoidoscopy is not acceptable for screening due to the frequency of proximal colon cancer.
3. Timing and type of surgery in patients with a biallelic MYH mutation depend on the ability to maintain clearance of polyps, the rectal polyp count, and the presence of malignancy. Grade of Recommendation: Weak recommendation based on low quality evidence, 2C.
The lifetime risk of developing colorectal cancer in the setting of biallelic MYH mutation is poorly defined, but a population-based study estimated a 28-fold increased risk of colorectal cancer over the general population with an estimated penetrance (occurrence of colorectal cancer) of 19% by age 50, 43% by age 60, and 80% by age 80.56 There are inadequate data to recommend a specific operation for all patients, and the MAP phenotype varies so much that defining a standard operation is inappropriate. A retrospective review combining two familial cancer registries identified 14 patients. One had a proctocolectomy for severe polyposis, two had proctocolectomies for rectal cancer and polyposis, and 11 had a total colectomy with ileorectal anastomosis. All patients were followed with annual proctoscopy with no loss to follow-up over a median surveillance of 5 years (range 2-23 years), and no patient developed rectal cancer. While polyps were frequently found, with an average of 1.52 adenomas per year per patient, they were all successfully managed with endoscopic resection. Colorectal cancer should be treated in accordance with oncologic principles, with more extensive resections for the purpose of prophylaxis made on a case-by-case basis. Patients with MAP and rectal sparing can be offered colectomy with ileorectal anastomosis.57, 58 Given the typically high polyp burden, long-term endoscopic management of the whole colon is generally not successful, but may be considered in selected cases with a low polyp burden.59
Extraintestinal Manifestations
Screening for Duodenal Neoplasia
1. Screening for duodenal adenomas in individuals with FAP and AFAP should begin with a baseline esophagogastroduodenoscopy beginning at age 20-25, with subsequent exams at intervals based on the endoscopic findings. Grade of Recommendation: Strong recommendation based on moderate quality evidence, 1B.
Three prospective and multiple retrospective studies support screening of the duodenum to detect duodenal polyposis.60-62 A prospective multinational European study of 368 patients with FAP screened with biannual upper endoscopy showed that the cumulative incidence of duodenal adenoma by age 70 was 90%, and the cumulative incidence of duodenal cancer was 4.5%.60 While this is 100-300 times higher than the general population, the low absolute incidence and the prolonged time from adenoma to carcinoma make it hard to design a study that would show a reduction in cancer incidence based on endoscopic screening. Duodenal cancer is rare before the age of 30 and, in the absence of symptoms, screening can begin at age 20-25. The Spigelman classification stratifies the risk of developing cancer based on the polyp number, polyp size, histology, and degree of dysplasia (Table 2).63 While surveillance with selective polypectomy/ampullectomy has been shown to decrease the Spigelman score, the ability of endoscopic polypectomy to prevent cancer has been questioned. A cohort of 114 patients who were prospectively managed and followed for ten years demonstrated that 6/114 (5.2%) developed cancer, but in patients with the most advanced polyps (Spigelman IV), 4/11 (38%) of patients developed cancer This suggests that endoscopic management may only be appropriate for Spigelman I-III disease, and that duodenectomy should be considered for patients with Spigelman IV disease.64 A cohort of FAP patients in an endoscopic surveillance program in Toronto showed that with a prospectively defined endoscopic management strategy, progression to cancer was slow, averaging 15 years after the initial endoscopy, and only occurred in 5/167 (3%) of patients.65 While screening is therefore appropriate for early diagnosis and to slow progression of disease, it remains unclear if endoscopic prevention of all duodenal cancers is possible. The optimal age to start screening is based on consensus opinion, and the appropriate interval for endoscopy should be based on findings at the initial EGD. The strategy reported by Soravia et al. does not use the Spigelman classification but has been evaluated with subsequent reported follow-up.65, 66 Available evidence supports EGD every 5 years after a normal exam, every 2-3 years for Spigelman I, every 1-2 years for Spigelman stage II, and every 6-12 months for Spigelman stage III. Spigelman stage IV patients should be managed by a multidisciplinary team with individualized decision-making regarding ongoing endoscopic surveillance or surgical resection.
Table 2.
Spiegelman Stage
| Duodenal adenomatosis staging system | |||
|---|---|---|---|
| Polyps | 1 Point | 2 Points | 3 Points |
| Number | < 4 | 5-20 | > 20 |
| Size | 0-4 mm | 5-10 mm | > 10 |
| Histology | Tubular | Tubulovilllous | Villious |
| Dysplasia | Mild | Moderate | Severe |
| Spiegelman Stage | Total Points | Frequency of Surveillance | |
| Recommended duodenal surveillance frequency | |||
| 0 | 0 | Every 4 years | |
| I | ≤4 | Every 2-3 years | |
| II | 5-6 | Every 1-3 years | |
| III | 7-8 | Every 6-12 years | |
| IV | 9-12 | Expert surveillance every 3-6 months | |
| Surgical evaluation | |||
| Complete mucosectomy or duodenectomy or Whipple procedure if duodenal papilla is involved | |||
Screening for Thyroid Disease
1. Screening for thyroid disease should be considered in patients with FAP with an annual ultrasound preferred over physical exam alone, especially for women. Grade of Recommendation: Weak recommendation based on low quality evidence, 2C.
Thyroid cancer occurs in 1-2% of the FAP population, compared to 0.2% in the general population, with the majority of cases occurring in women.67-73 There are no prospective studies comparing screening strategies of physical exam or ultrasound. Results of universal screening of 192 FAP patients included in a registry showed 72 (38%) had a thyroid nodule and 5 (2.6%) had thyroid cancer.74 A subsequent study comparing patients with screen-detected cancers to those with incident cancers showed that screening led to detection of smaller tumors with fewer positive lymph nodes.75 In another report, universal screening of 50 patients who underwent ultrasound led to 7 (14%) patients having an FNA and 2 (4%) being diagnosed with papillary thyroid cancer.76 Based on the increased risk of thyroid cancer, screening should be considered and discussed with patients, especially women. Additional data are needed to define the age screening should start and the optimal interval for screening.
Extracolonic Manifestations of MAP
1. Upper gastrointestinal endoscopy is recommended for patients beginning at age 30, with subsequent exams at intervals based on the endoscopic findings. Grade of Recommendation: Weak recommendation based on low quality evidence, 2C.
Extracolonic manifestations of MAP include the risk of duodenal and other intestinal cancers as well as extraintestinal neoplasia. A multicenter registry-based cohort of European centers, including 276 patients from 181 families, identified the prevalence of duodenal polyps at 17% and a lifetime risk of duodenal cancer of 4%. The observed frequency of duodenal adenomas is much lower than that observed in FAP, but greater than the general population. Upper gastrointestinal endoscopy including visualization of the duodenum and ampulla (with visualization improved by using a side-viewing endoscope) is recommended starting at age 30.2, 3 The interval between surveillance exams depends on the number of duodenal adenomas as well as adenoma characteristics including size, histology, and the degree of dysplasia (Spigelman classification) (Table 2).63 The use of the Spigelman criteria is extrapolated from FAP; it was not developed from MAP patients. While there may be an increased incidence of ovarian, bladder, and skin cancer, insufficient data are available to support specific screening for these malignancies.
Surgery for Intra-Abdominal Desmoid Tumors
1. Surgery for intra-abdominal desmoid tumors is generally not recommended and should typically be reserved for small, well-defined tumors when a clear margin can be obtained. Grade of Recommendation: Weak recommendation based on moderate quality evidence, 2B.
Desmoids tumors are histologically benign but potentially locally aggressive growths that affect about 15% of patients with FAP. Abdominal wall desmoids need to be considered separately from mesenteric and intra-abdominal desmoids. In contrast to desmoids found in other populations, FAP-associated desmoids tend to be intra-abdominal, involve the small bowel mesentery, and occur after surgery. The relationship of colectomy and desmoids is a key part of the decision regarding the timing and type of colonic operation to be done.
Church, et al. proposed a desmoid staging system based on symptoms with a subsequent report noting that the staging was predictive of need for treatment and mortality (Table 3).77, 78 Multiple single-center retrospective reports have attempted to define the role of surgery, but the lack of standardization and treatment bias limit the generalizability of the results. Even in selected patients, however, recurrence rates are high, and the overall benefit of resection is not clear.79 Nearly all studies are retrospective and many include both FAP-related desmoid disease and non-FAP related desmoids. A prospective cohort study of 64 FAP patients with intra-abdominal desmoids treated with high-dose selective estrogen receptor modulators (SERMs) and sulindac demonstrated regression in 85% of patients with no progression or treatment failures requiring surgery.80 At this time, insufficient data exist to recommend a specific treatment modality for desmoids, but surgical therapy has a very limited role that should typically be confined to small, well-defined tumors where a clear margin can be obtained.
Table 3.
Desmoid tumor staging system
| Stage | Symptoms | Size | Growth Rate |
|---|---|---|---|
| I | Asymptomatic | < 10 cm maximum diameter | None |
| II | Mild | < 10 cm maximum diameter | None |
| III | Moderate, or with bowel/uretric obstruction | 10 or 20 cm | Slow |
| IV | Severe | > 20 cm | Rapid |
Quality of symptoms are defined as: mild- no restrictions, pain and a sensation of mass, moderate- restrictions, but no hospitalization, pain and a sensation of mass, severe- restrictions and hospitalization, pain and a sensation of mass. (Adapted from Church, et al. Staging intra-abdominal desmoid tumors in familial adenomatous polyposis: a search for a uniform approach to a troubling disease.Dis Colon Rectum, 2005. 48(8): p. 1528-34.)
Chemoprevention of adenomas
1. Individuals with FAP, AFAP, or MAP with any retained rectum or established duodenal adenomas should be considered for chemoprevention with either sulindac or celecoxib after an individualized risk/benefit assessment. Grade of Recommendation: Weak recommendation based on high- quality evidence, 2A.
Most patients are eligible for chemoprevention because proctocolectomy with IPAA or a colectomy with ileorectal anastomosis can retain at-risk rectal mucosa, and the duodenal mucosa remains at risk in all of these patients. It should be noted that although no drug, including those described here, is FDA-approved for the indication of chemoprevention, several drugs have been studied in over a dozen randomized controlled trials and in many observational studies (Table 4).81-96 Of four trials examining the use of sulindac, three reported positive findings.81-84 The negative trial was a primary prevention trial in patients who were phenotypically unaffected but had APC mutations. These findings may not be applicable to the postoperative population of patients with a highly penetrant polyposis phenotype.84 One trial evaluating dual treatment with sulindac and erlotinib for duodenal polyp suppression was stopped early due to superiority of the chemoprevention over placebo, although there was a high rate of grade 1 and 2 adverse events, including an acne-like rash in 87% of treated patients.85 Of seven trials examining the role of selective COX-2 inhibition, six reported positive results.86-92 An international RCT of celecoxib and diflouromethylornithine (DFMO) showed that the addition of DFMO was required to achieve a benefit in reduction of adenoma count when compared to placebo.91 Another trial examined eicosapentaenoic acid with positive results.93 Three studies have examined vitamin C, vitamin E, calcium, or a combination, with mixed but overall negative results.94-96
Table 4.
Randomized controlled trials examining the chemoprevention strategies for polyp suppression.
| Group | Design | N | Patients | Drug | Outcome | Polyp Size | Polyp number | Side Effects |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
|
Sulindac
| ||||||||
| Labayle et al. 199182 | RPCDB | 10 | FAP | Sulindac (300mg/day) | Rectal polyp number | - | Decrease (9/10 people) | None |
|
| ||||||||
| Giardiello et al. 199381 | RPCDB | 22 | FAP | Sulindac (300mg/day) | Colorectal polyp number and size | Decrease (44%) | Decrease (35%) | None |
|
| ||||||||
| Nugent et al. 199383 | RCT | 24 | FAP | Sulindac (400mg/day | Duodenal and rectal polyp number and proliferation | - | Decrease | Indigestion |
|
| ||||||||
| Giardiello et al. 200284 | RPCDB | 41 | FAP (phenotypically negative) | Sulindac (150-300mg/day) | Adenoma development, number, and size | No significant change (p=0.17) | No significant change (p=0.69) | None |
| Samadder et al. 201685 | RPCDB | 92 | FAP | Sulindac (300mg/day) + Erlotinib (75mg/day) | Duodenal polyp number and size | Decrease (by 8.5mm) | Decrease (37.9%) | Rash |
|
| ||||||||
|
COX-2 selective inhibitors
| ||||||||
| Steinbach et al. 200086 | RDBPC | 77 | FAP | Celecoxib (200 or 800mg/day) | Colorectal polyp number and size | Decrease (28%) | Decrease (30.7%) | Diarrhea, abdominal pain |
|
| ||||||||
| Phillips et al. 200292 | RPCDB | 83 | FAP | Celecoxib (200mg/day) V. Celecoxib (800mg/day) | Duodenal polyp burden | - | Decrease with CXB 800mg/day (14.5%) | None |
|
| ||||||||
| Lynch et al. 201087 | Phase I Trial | 18 | FAP (children) | Celecoxib (4-16mg/kg/day) | Colorectal polyp number | - | Decrease (44.2%) | None |
|
| ||||||||
| Lynch et al. 201691 | RCT | 112 | FAP | Celecoxib (800mg/day) V. Celecoxib + DFMO (0.5g/m2/day) | Colorectal polyp number and size | Decrease with CXB + DFMO (40%) | Decrease with CXB+ DFMO (13%) | None |
|
| ||||||||
| Hallak A et al. 200390 | Phase I Trial | 8 | FAP | Rofecoxib (25mg/day) | Colorectal polyp number | - | Decrease (p=0.008) | None |
|
| ||||||||
| Higuchi et al. 200388 | RPCDB | 21 | FAP | Rofecoxib (25mg/day) | Rectal polyp number and size | Decrease (16.2%) | Decrease (6.8%) | Stomatitis, abdominal pain |
|
| ||||||||
| Iwama et al. 200689 | RPCDB | 61 | FAP | Tiracoxib (150-200mg/day) | Colorectal polyp number and size | No change (0%) | No change (0%) | None |
|
| ||||||||
|
Others
| ||||||||
| Bussey et al. 198294 | RDB | 49 | FAP | Ascorbic acid (3g/day) | Rectal polyp number and size | Decrease (trend) | Decrease (p<0.03) | None |
|
| ||||||||
| DeCosse et al. 198995 | RPCDB | 58 | FAP | Vit C (2g/day) + Vit E (400mg/day) +/- Wheat fiber (22.5g/day) | Rectal polyp number and size | No change | No change | None |
|
| ||||||||
| Thomas et al. 199396 | RPCDB | 25 | FAP | Calcium (1500mg/day) | Rectal polyp number and size | No change | No change | None |
|
| ||||||||
| West et al. 201093 | RPCDB | 55 | FAP | EPA (2g/day) | Rectal polyp number and size | Decrease (29.8%) | Decrease (22.4%) | None |
Substantial evidence supports the use of chemoprevention in polyposis patients with duodenal adenomas or at-risk rectal mucosa. Sulindac has a higher rate of gastritis than celecoxib; in patients over age 65, or who have a history of a peptic ulcer, or who require concurrent use of aspirin, corticosteroids or anticoagulants for other medical problems should usually be given with a proton-pump-inhibitor.97, 98 The risk of adverse events or gastrointestinal bleeding, as well as issues with compliance, cost, or patient preference, however, may preclude the use of chemoprevention in all or even many cases. It is important to note that chemoprevention should not replace routine endoscopic surveillance, that the role of chemoprevention for suppression of polyps in the retained rectum is extrapolated from the duodenal adenoma studies, and that compliance with long-term treatment has not been well-studied.
Surveillance and Treatment of Polyposis Without an Identified Gene Mutation
1. Patients with clinical polyposis, but without an identified mutation, should be should be treated and followed based on their phenotype. Grade of Recommendation: Weak recommendation based on low quality evidence, 2C.
Between 20 and 50 percent of patients with oligopolyposis will not have a mutation found in the APC or MYH genes.13, 99-101 Multiple case series have identified alterations that are not included in existing commercial testing that may play a role in polyposis, such as genomic rearrangements involving APC, APC mosaicism, and mutations in the APC promoter.102-104 Other patients may harbor rare or as yet unknown causes of polyposis, such as the recently described polymerase proofreading-associated polyposis.105
Management of mutation-negative polyposis patients has been described in observational studies. Tieu et al. described 27 patients with multiple colorectal adenomas with a phenotype similar to attenuated polyposis with an average of 51 polyps.100 Eighteen patients (67%) underwent colectomy after a mean of 3.1 years after diagnosis due to the concern for cancer or for inability to provide endoscopic clearance. Extracolonic findings in these patients may mirror attenuated polyposis syndromes as EGD identified polyps in 47% of patients. A second observational study of APC-mutation negative polyposis patients, however, showed they were less likely to display extra-colonic manifestations.106 In the absence of a genetic defect, it is reasonable to treat patients according to their phenotype by maintaining endoscopic clearance in patients when possible and proceeding with colectomy or proctocolectomy if required according to the polyp number.
Footnotes
Prepared By:
The Clinical Practice Guidelines Committee of The American Society of Colon and Rectal Surgeons
References
- 1.Church J, Simmang C Standards Task ForceAmerican Society of Colon and Rectal Surgeons; Collaborative Group of the Americas on Inherited Colorectal Cancer and the Standards Committee of The American Society of Colon and Rectal Surgeons. Practice parameters for the treatment of patients with dominantly inherited colorectal cancer (familial adenomatous polyposis and hereditary nonpolyposis colorectal cancer) Dis Colon Rectum. 2003;46:1001–1012. doi: 10.1007/s10350-004-7273-y. [DOI] [PubMed] [Google Scholar]
- 2.Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110:223–262. doi: 10.1038/ajg.2014.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.National Comprehensive Cancer Network. Fort Washington, PA: [October, 14, 2016]. 2016. Genetic/Familial High-Risk Assessment: Colorectal. V2. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp. [Google Scholar]
- 4.Balmaña J, Balaguer F, Cervantes A, Arnold D ESMO Guidelines Working Group. Familial risk-colorectal cancer: ESMO Clinical Practice Guidelines. Ann Oncol. 2013;24(suppl 6):vi73–vi80. doi: 10.1093/annonc/mdt209. [DOI] [PubMed] [Google Scholar]
- 5.Stoffel EM, Mangu PB, Limburg PJ American Society of Clinical Oncology; European Society for Medical Oncology. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology clinical practice guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology clinical practice guidelines. J Oncol Pract. 2015;11:e437–e441. doi: 10.1200/JOP.2015.003665. [DOI] [PubMed] [Google Scholar]
- 6.Guyatt G, Gutterman D, Baumann MH, et al. Grading Strength of Recommendations and Quality of evidence in Clinical Guidelines: Report from an American College of Chest Physicians Task Forect. Chest. 2006;129:174–181. doi: 10.1378/chest.129.1.174. [DOI] [PubMed] [Google Scholar]
- 7.Bussey HJ, Veale AM, Morson BC. Genetics of gastrointestinal polyposis. Gastroenterology. 1978;74:1325–1330. [PubMed] [Google Scholar]
- 8.Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 9.Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat. 1994;3:121–125. doi: 10.1002/humu.1380030206. [DOI] [PubMed] [Google Scholar]
- 10.Giardiello FM, Brensinger JD, Luce MC, et al. Phenotypic expression of disease in families that have mutations in the 5′ region of the adenomatous polyposis coli gene. Ann Intern Med. 1997;126:514–519. doi: 10.7326/0003-4819-126-7-199704010-00003. [DOI] [PubMed] [Google Scholar]
- 11.Lynch HT, Smyrk T, McGinn T, et al. Attenuated familial adenomatous polyposis (AFAP). A phenotypically and genotypically distinctive variant of FAP. Cancer. 1995;76:2427–2433. doi: 10.1002/1097-0142(19951215)76:12<2427::aid-cncr2820761205>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 12.Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 13.Grover S, Kastrinos F, Steyerberg EW, et al. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA. 2012;308:485–492. doi: 10.1001/jama.2012.8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knudsen AL, Bisgaard ML, Bülow S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Fam Cancer. 2003;2:43–55. doi: 10.1023/a:1023286520725. [DOI] [PubMed] [Google Scholar]
- 15.Nielsen M, Hes FJ, Nagengast FM, et al. Germline mutations in APC and MUTYH are responsible for the majority of families with attenuated familial adenomatous polyposis. Clin Genet. 2007;71:427–433. doi: 10.1111/j.1399-0004.2007.00766.x. [DOI] [PubMed] [Google Scholar]
- 16.Knudsen AL, Bülow S, Tomlinson I, Möslein G, Heinimann K, Christensen IJ AFAP Study Group. Attenuated familial adenomatous polyposis: results from an international collaborative study. Colorectal Dis. 2010;12:e243–e249. doi: 10.1111/j.1463-1318.2010.02218.x. [DOI] [PubMed] [Google Scholar]
- 17.Geller G, Botkin JR, Green MJ, et al. Genetic testing for susceptibility to adult-onset cancer. The process and content of informed consent. JAMA. 1997;277:1467–1474. [PubMed] [Google Scholar]
- 18.Riley BD, Culver JO, Skrzynia C, et al. Essential elements of genetic cancer risk assessment, counseling, and testing: updated recommendations of the National Society of Genetic Counselors. J Genet Couns. 2012;21:151–161. doi: 10.1007/s10897-011-9462-x. [DOI] [PubMed] [Google Scholar]
- 19.Statement of the American Society of Clinical Oncology: genetic testing for cancer susceptibility, Adopted on February 20, 1996. J Clin Oncol. 1996;14:1730–1740. doi: 10.1200/JCO.1996.14.5.1730. [DOI] [PubMed] [Google Scholar]
- 20.Murff HJ, Byrne D, Syngal S. Cancer risk assessment: quality and impact of the family history interview. Am J Prev Med. 2004;27:239–245. doi: 10.1016/j.amepre.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 21.Aretz S, Uhlhaas S, Caspari R, et al. Frequency and parental origin of de novo APC mutations in familial adenomatous polyposis. Eur J Hum Genet. 2004;12:52–58. doi: 10.1038/sj.ejhg.5201088. [DOI] [PubMed] [Google Scholar]
- 22.Barrow P, Khan M, Lalloo F, Evans DG, Hill J. Systematic review of the impact of registration and screening on colorectal cancer incidence and mortality in familial adenomatous polyposis and Lynch syndrome. Br J Surg. 2013;100:1719–1731. doi: 10.1002/bjs.9316. [DOI] [PubMed] [Google Scholar]
- 23.Warrier SK, Kalady MF, Kiran RP, Church JM. Results from an American Society of Colon and Rectal Surgeons survey on the management of young-onset colorectal cancer. Tech Coloproctol. 2014;18:265–272. doi: 10.1007/s10151-013-1052-5. [DOI] [PubMed] [Google Scholar]
- 24.Nieuwenhuis MH, Mathus-Vliegen LM, Slors FJ, et al. Genotype-phenotype correlations as a guide in the management of familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2007;5:374–378. doi: 10.1016/j.cgh.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 25.Church JM, McGannon E, Burke C, Clark B. Teenagers with familial adenomatous polyposis: what is their risk for colorectal cancer? Dis Colon Rectum. 2002;45:887–889. doi: 10.1007/s10350-004-6322-x. [DOI] [PubMed] [Google Scholar]
- 26.Muto T, Bussey HJ, Morson BC. The evolution of cancer of the colon and rectum. Cancer. 1975;36:2251–2270. doi: 10.1002/cncr.2820360944. [DOI] [PubMed] [Google Scholar]
- 27.Burt RW, Leppert MF, Slattery ML, et al. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology. 2004;127:444–451. doi: 10.1053/j.gastro.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 28.National Comprehensive Cancer Network. Fort Washington, PA: [October 14, 2016]. 2016. Colorectal Cancer Screening Version 2.2016. https://www.nccn.org/professionals/physician_gls/PDF/colorectal_screening.pdf. [Google Scholar]
- 29.Aziz O, Athanasiou T, Fazio VW, et al. Meta-analysis of observational studies of ileorectal versus ileal pouch-anal anastomosis for familial adenomatous polyposis. Br J Surg. 2006;93:407–417. doi: 10.1002/bjs.5276. [DOI] [PubMed] [Google Scholar]
- 30.Tajika M, Niwa Y, Bhatia V, Tanaka T, Ishihara M, Yamao K. Risk of ileal pouch neoplasms in patients with familial adenomatous polyposis. World J Gastroenterol. 2013;19:6774–6783. doi: 10.3748/wjg.v19.i40.6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith JC, Schäffer MW, Ballard BR, et al. Adenocarcinomas after prophylactic surgery for familial adenomatous polyposis. J Cancer Ther. 2013;4:260–270. doi: 10.4236/jct.2013.41033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boostrom SY, Mathis KL, Pendlimari R, Cima RR, Larson DW, Dozois EJ. Risk of neoplastic change in ileal pouches in familial adenomatous polyposis. J Gastrointest Surg. 2013;17:1804–1808. doi: 10.1007/s11605-013-2319-x. [DOI] [PubMed] [Google Scholar]
- 33.Remzi FH, Church JM, Bast J, et al. Mucosectomy vs. stapled ileal pouch-anal anastomosis in patients with familial adenomatous polyposis: functional outcome and neoplasia control. Dis Colon Rectum. 2001;44:1590–1596. doi: 10.1007/BF02234377. [DOI] [PubMed] [Google Scholar]
- 34.von Roon AC, Will OC, Man RF, et al. Mucosectomy with handsewn anastomosis reduces the risk of adenoma formation in the anorectal segment after restorative proctocolectomy for familial adenomatous polyposis. Ann Surg. 2011;253:314–317. doi: 10.1097/SLA.0b013e318f3f498. [DOI] [PubMed] [Google Scholar]
- 35.van Duijvendijk P, Vasen HF, Bertario L, et al. Cumulative risk of developing polyps or malignancy at the ileal pouch-anal anastomosis in patients with familial adenomatous polyposis. J Gastrointest Surg. 1999;3:325–330. doi: 10.1016/s1091-255x(99)80075-4. [DOI] [PubMed] [Google Scholar]
- 36.Cirocchi RMU, Arezzo A, Trastulli S, et al., editors. Cochrane Database Sys Rev. 2004. Double-stapled anastomosis versus mucosectomy and handsewn anastomosis in ileal pouch-anal anastomosis for ulcerative colitis or familial adenomatous polyposis. CD011089. [Google Scholar]
- 37.Maartense S, Dunker MS, Slors JF, et al. Hand-assisted laparoscopic versus open restorative proctocolectomy with ileal pouch anal anastomosis: a randomized trial. Ann Surg. 2004;240:984–991. doi: 10.1097/01.sla.0000145923.03130.1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahmed Ali U, Keus F, Heikens JT, et al. Open versus laparoscopic (assisted) ileo pouch anal anastomosis for ulcerative colitis and familial adenomatous polyposis. Cochrane Database Syst Rev. 2009;(1) doi: 10.1002/14651858.CD006267.pub2. CD006267. [DOI] [PubMed] [Google Scholar]
- 39.Koskenvuo L, Mustonen H, Renkonen-Sinisalo L, Järvinen HJ, Lepistö A. Comparison of proctocolectomy and ileal pouch-anal anastomosis to colectomy and ileorectal anastomosis in familial adenomatous polyposis. Fam Cancer. 2015;14:221–227. doi: 10.1007/s10689-014-9773-9. [DOI] [PubMed] [Google Scholar]
- 40.Church J, Burke C, McGannon E, Pastean O, Clark B. Risk of rectal cancer in patients after colectomy and ileorectal anastomosis for familial adenomatous polyposis: a function of available surgical options. Dis Colon Rectum. 2003;46:1175–1181. doi: 10.1007/s10350-004-6710-2. [DOI] [PubMed] [Google Scholar]
- 41.Bülow S, Bülow C, Vasen H, Järvinen H, Björk J, Christensen IJ. Colectomy and ileorectal anastomosis is still an option for selected patients with familial adenomatous polyposis. Dis Colon Rectum. 2008;51:1318–1323. doi: 10.1007/s10350-008-9307-3. [DOI] [PubMed] [Google Scholar]
- 42.Church J, Burke C, McGannon E, Pastean O, Clark B. Predicting polyposis severity by proctoscopy: how reliable is it? Dis Colon Rectum. 2001;44:1249–1254. doi: 10.1007/BF02234779. [DOI] [PubMed] [Google Scholar]
- 43.Win AK, Hopper JL, Jenkins MA. Association between monoallelic MUTYH mutation and colorectal cancer risk: a meta-regression analysis. Fam Cancer. 2011;10:1–9. doi: 10.1007/s10689-010-9399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst. 2004;96:1631–1634. doi: 10.1093/jnci/djh288. [DOI] [PubMed] [Google Scholar]
- 45.Jones N, Vogt S, Nielsen M, et al. Increased colorectal cancer incidence in obligate carriers of heterozygous mutations in MUTYH. Gastroenterology. 2009;137:489–494. doi: 10.1053/j.gastro.2009.04.047. [DOI] [PubMed] [Google Scholar]
- 46.Kastrinos F, Syngal S. Inherited colorectal cancer syndromes. Cancer J. 2011;17:405–415. doi: 10.1097/PPO.0b013e318237e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791–799. doi: 10.1056/NEJMoa025283. [DOI] [PubMed] [Google Scholar]
- 48.Fleischmann C, Peto J, Cheadle J, Shah B, Sampson J, Houlston RS. Comprehensive analysis of the contribution of germline MYH variation to early-onset colorectal cancer. Int J Cancer. 2004;109:554–558. doi: 10.1002/ijc.20020. [DOI] [PubMed] [Google Scholar]
- 49.Cleary SP, Cotterchio M, Jenkins MA, et al. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology. 2009;136:1251–1260. doi: 10.1053/j.gastro.2008.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hampel H, Bennett RL, Buchanan A, Pearlman R, Wiesner GL Guideline Development Group, American College of Medical Genetics and Genomics Professional Practice and Guidelines Committee and National Society of Genetic Counselors Practice Guidelines Committee. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70–87. doi: 10.1038/gim.2014.147. [DOI] [PubMed] [Google Scholar]
- 51.Leite JS, Isidro G, Martins M, et al. Is prophylactic colectomy indicated in patients with MYH-associated polyposis? Colorectal Dis. 2005;7:327–331. doi: 10.1111/j.1463-1318.2005.00811.x. [DOI] [PubMed] [Google Scholar]
- 52.Nielsen M, Franken PF, Reinards TH, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP) J Med Genet. 2005;42(9):e54. doi: 10.1136/jmg.2005.033217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nieuwenhuis MH, Vogt S, Jones N, et al. Evidence for accelerated colorectal adenoma--carcinoma progression in MUTYH-associated polyposis? Gut. 2012;61:734–738. doi: 10.1136/gut.2010.229104. [DOI] [PubMed] [Google Scholar]
- 54.Lipton L, Halford SE, Johnson V, et al. Carcinogenesis in MYH-associated polyposis follows a distinct genetic pathway. Cancer Res. 2003;63:7595–7599. [PubMed] [Google Scholar]
- 55.Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39–41. doi: 10.1016/S0140-6736(03)13805-6. [DOI] [PubMed] [Google Scholar]
- 56.Lubbe SJ, Di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol. 2009;27:3975–3980. doi: 10.1200/JCO.2008.21.6853. [DOI] [PubMed] [Google Scholar]
- 57.Valanzano R, Ficari F, Curia MC, et al. Balance between endoscopic and genetic information in the choice of ileorectal anastomosis for familial adenomatous polyposis. J Surg Oncol. 2007;95:28–33. doi: 10.1002/jso.20672. [DOI] [PubMed] [Google Scholar]
- 58.Nascimbeni R, Pucciarelli S, Di Lorenzo D, et al. Rectum-sparing surgery may be appropriate for biallelic MutYH-associated polyposis. Dis Colon Rectum. 2010;53:1670–1675. doi: 10.1007/DCR.0b013e3181ee3d6b. [DOI] [PubMed] [Google Scholar]
- 59.Liang J, Church JM. Rectal cancers in patients with familial adenomatous polyposis. Fam Cancer. 2013;12:749–754. doi: 10.1007/s10689-013-9656-5. [DOI] [PubMed] [Google Scholar]
- 60.Bülow S, Björk J, Christensen IJ, et al. DAF Study Group. Duodenal adenomatosis in familial adenomatous polyposis. Gut. 2004;53:381–386. doi: 10.1136/gut.2003.027771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cordero-Fernández C, Garzón-Benavides M, Pizarro-Moreno A, et al. Gastroduodenal involvement in patients with familial adenomatous polyposis. Prospective study of the nature and evolution of polyps: evaluation of the treatment and surveillance methods applied. Eur J Gastroenterol Hepatol. 2009;21:1161–1167. doi: 10.1097/MEG.0b013e3283297cf2. [DOI] [PubMed] [Google Scholar]
- 62.Saurin JC, Gutknecht C, Napoleon B, et al. Surveillance of duodenal adenomas in familial adenomatous polyposis reveals high cumulative risk of advanced disease. J Clin Oncol. 2004;22:493–498. doi: 10.1200/JCO.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 63.Spigelman AD, Williams CB, Talbot IC, Domizio P, Phillips RK. Upper gastrointestinal cancer in patients with familial adenomatous polyposis. Lancet. 1989;2:783–785. doi: 10.1016/s0140-6736(89)90840-4. [DOI] [PubMed] [Google Scholar]
- 64.Groves CJ, Saunders BP, Spigelman AD, Phillips RK. Duodenal cancer in patients with familial adenomatous polyposis (FAP): results of a 10 year prospective study. Gut. 2002;50:636–641. doi: 10.1136/gut.50.5.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Serrano PE, Grant RC, Berk TC, et al. Progression and Management of Duodenal Neoplasia in Familial Adenomatous Polyposis: A Cohort Study. Ann Surg. 2015;261:1138–1144. doi: 10.1097/SLA.0000000000000734. [DOI] [PubMed] [Google Scholar]
- 66.Soravia C, Berk T, Haber G, Cohen Z, Gallinger S. Management of advanced duodenal polyposis in familial adenomatous polyposis. J Gastrointest Surg. 1997;1:474–478. doi: 10.1016/s1091-255x(97)80136-9. [DOI] [PubMed] [Google Scholar]
- 67.Bülow C, Bülow S. Is screening for thyroid carcinoma indicated in familial adenomatous polyposis? The Leeds Castle Polyposis Group. Int J Colorectal Dis. 1997;12:240–242. doi: 10.1007/s003840050097. [DOI] [PubMed] [Google Scholar]
- 68.Bülow S, Holm NV, Mellemgaard A. Papillary thyroid carcinoma in Danish patients with familial adenomatous polyposis. Int J Colorectal Dis. 1988;3:29–31. doi: 10.1007/BF01649679. [DOI] [PubMed] [Google Scholar]
- 69.van der Linde K, Vasen HF, van Vliet AC. Occurrence of thyroid carcinoma in Dutch patients with familial adenomatous polyposis. An epidemiological study and report of new cases. Eur J Gastroenterol Hepatol. 1998;10:777–781. doi: 10.1097/00042737-199809000-00009. [DOI] [PubMed] [Google Scholar]
- 70.Truta B, Allen BA, Conrad PG, et al. Genotype and phenotype of patients with both familial adenomatous polyposis and thyroid carcinoma. Fam Cancer. 2003;2:95–99. doi: 10.1023/a:1025762706854. [DOI] [PubMed] [Google Scholar]
- 71.Giardiello FM, Offerhaus GJ, Lee DH, et al. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut. 1993;34:1394–1396. doi: 10.1136/gut.34.10.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Plail RO, Bussey HJ, Glazer G, Thomson JP. Adenomatous polyposis: an association with carcinoma of the thyroid. Br J Surg. 1987;74:377–380. doi: 10.1002/bjs.1800740517. [DOI] [PubMed] [Google Scholar]
- 73.Herraiz M, Barbesino G, Faquin W, et al. Prevalence of thyroid cancer in familial adenomatous polyposis syndrome and the role of screening ultrasound examinations. Clin Gastroenterol Hepatol. 2007;5:367–373. doi: 10.1016/j.cgh.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 74.Jarrar AM, Milas M, Mitchell J, et al. Screening for thyroid cancer in patients with familial adenomatous polyposis. Ann Surg. 2011;253:515–521. doi: 10.1097/SLA.0b013e3181fcba8a. [DOI] [PubMed] [Google Scholar]
- 75.Feng X, Milas M, O’Malley M, et al. Characteristics of benign and malignant thyroid disease in familial adenomatous polyposis patients and recommendations for disease surveillance. Thyroid. 2015;25:325–332. doi: 10.1089/thy.2014.0107. [DOI] [PubMed] [Google Scholar]
- 76.Steinhagen E, Hui VW, Levy RA, et al. Results of a prospective thyroid ultrasound screening program in adenomatous polyposis patients. Am J Surg. 2014;208:764–769. doi: 10.1016/j.amjsurg.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 77.Church J, Berk T, Boman BM, et al. Collaborative Group of the Americas on Inherited Colorectal Cancer. Staging intra-abdominal desmoid tumors in familial adenomatous polyposis: a search for a uniform approach to a troubling disease. Dis Colon Rectum. 2005;48:1528–1534. doi: 10.1007/s10350-005-0018-8. [DOI] [PubMed] [Google Scholar]
- 78.Church J, Lynch C, Neary P, LaGuardia L, Elayi E. A desmoid tumor-staging system separates patients with intra-abdominal, familial adenomatous polyposis-associated desmoid disease by behavior and prognosis. Dis Colon Rectum. 2008;51:897–901. doi: 10.1007/s10350-008-9232-5. [DOI] [PubMed] [Google Scholar]
- 79.Berk T, Cohen Z, McLeod RS, Stern HS. Management of mesenteric desmoid tumours in familial adenomatous polyposis. Can J Surg. 1992;35:393–395. [PubMed] [Google Scholar]
- 80.Quast DR, Schneider R, Burdzik E, Hoppe S, Möslein G. Long-term outcome of sporadic and FAP-associated desmoid tumors treated with high-dose selective estrogen receptor modulators and sulindac: a single-center long-term observational study in 134 patients. Fam Cancer. 2016;15:31–40. doi: 10.1007/s10689-015-9830-z. [DOI] [PubMed] [Google Scholar]
- 81.Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328:1313–1316. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 82.Labayle D, Fischer D, Vielh P, et al. Sulindac causes regression of rectal polyps in familial adenomatous polyposis. Gastroenterology. 1991;101:635–639. doi: 10.1016/0016-5085(91)90519-q. [DOI] [PubMed] [Google Scholar]
- 83.Nugent KP, Farmer KC, Spigelman AD, Williams CB, Phillips RK. Randomized controlled trial of the effect of sulindac on duodenal and rectal polyposis and cell proliferation in patients with familial adenomatous polyposis. Br J Surg. 1993;80:1618–1619. doi: 10.1002/bjs.1800801244. [DOI] [PubMed] [Google Scholar]
- 84.Giardiello FM, Yang VW, Hylind LM, et al. Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med. 2002;346:1054–1059. doi: 10.1056/NEJMoa012015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Samadder NJ, Neklason DW, Boucher KM, et al. Effect of Sulindac and Erlotinib vs Placebo on Duodenal Neoplasia in Familial Adenomatous Polyposis: A Randomized Clinical Trial. JAMA. 2016;315:1266–1275. doi: 10.1001/jama.2016.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 87.Lynch PM, Ayers GD, Hawk E, et al. The safety and efficacy of celecoxib in children with familial adenomatous polyposis. Am J Gastroenterol. 2010;105:1437–1443. doi: 10.1038/ajg.2009.758. [DOI] [PubMed] [Google Scholar]
- 88.Higuchi T, Iwama T, Yoshinaga K, Toyooka M, Taketo MM, Sugihara K. A randomized, double-blind, placebo-controlled trial of the effects of rofecoxib, a selective cyclooxygenase-2 inhibitor, on rectal polyps in familial adenomatous polyposis patients. Clin Cancer Res. 2003;9:4756–4760. [PubMed] [Google Scholar]
- 89.Iwama T, Akasu T, Utsunomiya J, Muto T. Does a selective cyclooxygenase-2 inhibitor (tiracoxib) induce clinically sufficient suppression of adenomas in patients with familial adenomatous polyposis? A randomized double-blind placebo-controlled clinical trial. Int J Clin Oncol. 2006;11:133–139. doi: 10.1007/s10147-005-0548-z. [DOI] [PubMed] [Google Scholar]
- 90.Hallak A, Alon-Baron L, Shamir R, et al. Rofecoxib reduces polyp recurrence in familial polyposis. Dig Dis Sci. 2003;48:1998–2002. doi: 10.1023/a:1026130623186. [DOI] [PubMed] [Google Scholar]
- 91.Lynch PM, Burke CA, Phillips R, et al. An international randomised trial of celecoxib versus celecoxib plus difluoromethylornithine in patients with familial adenomatous polyposis. Gut. 2016;65:286–295. doi: 10.1136/gutjnl-2014-307235. [DOI] [PubMed] [Google Scholar]
- 92.Phillips RK, Wallace MH, Lynch PM, et al. FAP Study Group. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut. 2002;50:857–860. doi: 10.1136/gut.50.6.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.West NJ, Clark SK, Phillips RK, et al. Eicosapentaenoic acid reduces rectal polyp number and size in familial adenomatous polyposis. Gut. 2010;59:918–925. doi: 10.1136/gut.2009.200642. [DOI] [PubMed] [Google Scholar]
- 94.Bussey HJ, DeCosse JJ, Deschner EE, et al. A randomized trial of ascorbic acid in polyposis coli. Cancer. 1982;50:1434–1439. doi: 10.1002/1097-0142(19821001)50:7<1434::aid-cncr2820500733>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 95.DeCosse JJ, Miller HH, Lesser ML. Effect of wheat fiber and vitamins C and E on rectal polyps in patients with familial adenomatous polyposis. J Natl Cancer Inst. 1989;81:1290–1297. doi: 10.1093/jnci/81.17.1290. [DOI] [PubMed] [Google Scholar]
- 96.Thomas MG, Thomson JP, Williamson RC. Oral calcium inhibits rectal epithelial proliferation in familial adenomatous polyposis. Br J Surg. 1993;80:499–501. doi: 10.1002/bjs.1800800432. [DOI] [PubMed] [Google Scholar]
- 97.Lanza FL, Chan FK, Quigley EM Practice Parameters Committee of the American College of Gastroenterology. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104:728–738. doi: 10.1038/ajg.2009.115. [DOI] [PubMed] [Google Scholar]
- 98.Rostom A, Muir K, Dube C, et al. Gastrointestinal safety of cyclooxygenase-2 inhibitors: a Cochrane Collaboration systematic review. Clin Gastroenterol Hepatol. 2007;5:818–828 768. doi: 10.1016/j.cgh.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 99.Filipe B, Baltazar C, Albuquerque C, et al. APC or MUTYH mutations account for the majority of clinically well-characterized families with FAP and AFAP phenotype and patients with more than 30 adenomas. Clin Genet. 2009;76:242–255. doi: 10.1111/j.1399-0004.2009.01241.x. [DOI] [PubMed] [Google Scholar]
- 100.Tieu AH, Edelstein D, Axilbund J, et al. Clinical Characteristics of Multiple Colorectal Adenoma Patients Without Germline APC or MYH Mutations. J Clin Gastroenterol. 2016;50:584–588. doi: 10.1097/MCG.0000000000000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Castellsague E, Gonzalez S, Guino E, et al. Allele-specific expression of APC in adenomatous polyposis families. Gastroenterology. 2010;139:439–447. doi: 10.1053/j.gastro.2010.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Heinimann K, Thompson A, Locher A, et al. Nontruncating APC germ-line mutations and mismatch repair deficiency play a minor role in APC mutation-negative polyposis. Cancer Res. 2001;61:7616–7622. [PubMed] [Google Scholar]
- 103.Yamaguchi K, Komura M, Yamaguchi R, et al. Detection of APC mosaicism by next-generation sequencing in an FAP patient. J Hum Genet. 2015;60:227–231. doi: 10.1038/jhg.2015.14. [DOI] [PubMed] [Google Scholar]
- 104.Spier I, Drichel D, Kerick M, et al. Low-level APC mutational mosaicism is the underlying cause in a substantial fraction of unexplained colorectal adenomatous polyposis cases. J Med Genet. 2016;53:172–179. doi: 10.1136/jmedgenet-2015-103468. [DOI] [PubMed] [Google Scholar]
- 105.Bellido F, Pineda M, Aiza G, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2016;18:325–332. doi: 10.1038/gim.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Giarola M, Stagi L, Presciuttini S, et al. Screening for mutations of the APC gene in 66 Italian familial adenomatous polyposis patients: evidence for phenotypic differences in cases with and without identified mutation. Hum Mutat. 1999;13:116–123. doi: 10.1002/(SICI)1098-1004(1999)13:2<116::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]