

Abstract
The ATP binding cassette transporter P-glycoprotein (ABCB1 or P-gp) plays a major role in cellular resistance to drugs and drug interactions. Experimental studies support a mechanism with nucleotide-dependent fluctuation between inward-facing and outward-facing conformations, which are coupled to nucleotide hydrolysis. However, detailed insight into drug-dependent modulation of these conformational ensembles is lacking. Different drugs likely occupy partially overlapping but distinct sites and are therefore variably coupled to nucleotide binding and hydrolysis. Many fluorescent drug analogues are used in cell-based transport models; however, their specific interactions with P-gp have not been studied, and this limits interpretation of transport assays in terms of molecular models. Here we monitor binding of the fluorescent probe substrates BODIPY-verapamil, BODIPY-vinblastine, and Flutax-2 at low occupancy to murine P-gp in lipid nanodiscs via fluorescence correlation spectroscopy, in variable nucleotide-bound states. Changes in affinity for the different nucleotide-dependent conformations are probe-dependent. For BODIPY-verapamil and BODIPY-vinblastine, there are 2–10-fold increases in KD in the nucleotide-bound or vanadate-trapped state, compared to that in the nucleotide-free state. In contrast, the affinity of Flutax-2 is unaffected by nucleotide or vanadate trapping. In further contrast to BODIPY-verapamil and BODIPY-vinblastine, Flutax-2 does not cause stimulation of ATP hydrolysis despite the fact that it is transported in vesicle-based transport assays. Whereas the established substrates verapamil, paclitaxel, and vinblastine displace BODIPY-verapamil or BODIPY-vinblastine from their high-affinity sites, the transport substrate Flutax-2 is not displaced by any of these substrates. The results demonstrate a unique binding site for Flutax-2 that allows for transport without stimulation of ATP hydrolysis.
Graphical Abstract
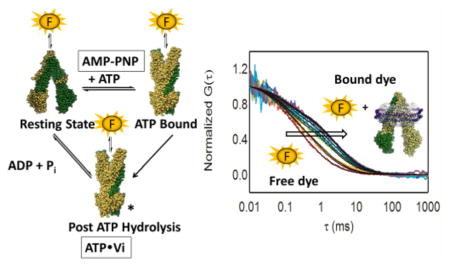
P-Glycoprotein (P-gp or ABCB1) plays a critical role in drug disposition and drug resistance by exporting a wide range of structurally diverse xenobiotics from several cell types.1–3 P-gp is a member of the ABC transporter family and includes a large membrane domain comprised of 12 transmembrane helices (TMHs) connected to N-terminal and C-terminal domains on the cytosolic side of the membrane. These cytosolic domains bind and hydrolyze ATP and are termed nucleotide binding domains (NBDs). On the basis of crystallographic models of mammalian P-gp in the absence of nucleotide,4,5 and results with a bacterial P-gp homologue in the presence of nucleotides,6 it is well-established that binding and hydrolysis of ATP are coordinated to large scale conformational changes7,8 that lead to alternate formation of inward-facing and outward-facing states (Figure 1). The binding and hydrolysis of ATP are coupled to the binding and transport of different drugs in a drug-dependent manner. One possible outward-facing state is stabilized by addition of ATP or ADP with vanadate,9 and the resulting vanadate-trapped state has provided a model for the hydrolytic transition state or the posthydrolysis state, and possibly other outward-facing conformations.
Figure 1.
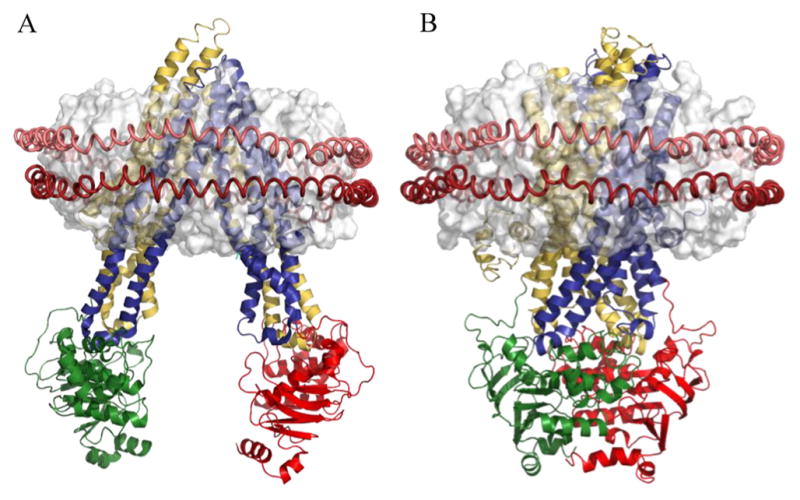
P-gp in inward-facing and outward-facing conformations. (A) Crystal structure of apo murine P-gp (Protein Data Bank entry 4Q9H) and (B) illustrative homology model of human P-gp (see Materials and Methods) manually docked with a discoidal HDL model. Colored segments are transmembrane domains 1 (yellow) and 2 (blue) and nucleotide binding domains 1 (green) and 2 (red). The OPM database10 was used for the spatial arrangement of P-gp with respect to the lipid bilayer (white). The red helical peptides around the lipid bilayer represent the MSP protein of the nanodisc.
However, data suggest that different drugs elicit different conformations and hence differential coupling between drug transport and nucleotide-bound states. In addition, many drugs are known to bind with a stoichiometry of more than one drug per P-gp, possibly as high as four or five drugs per P-gp. It is likely that the two-state nucleotide-dependent scheme (inward-facing vs outward-facing) is oversimplified, and each of these states may include substrate-dependent conformational ensembles that depend on both the identity of the drug bound and the number of drugs bound, as recently emphasized with verapamil.11
Such mechanistic details have been difficult to study and to relate to actual drug transport in vivo. One source of this difficulty results from a lack of biochemical characterization of interactions of the drug or probe substrate with P-gp. For example, many in vitro fluorescent probe substrates are used in cell-based transport assays, but details of their molecular interactions with P-gp are not established, making it difficult to relate transport behavior to molecular mechanism. In effect, rates of probe transport in cell-based assays have not been related to their fundamental biochemical behavior or their specific interactions with P-gp. Interpretation of transport behavior of these fluorescent probe substrates would be facilitated via additional characterization of their interactions with P-gp. Notably, the structures of the fluorescent probes used here deviate significantly from those of their corresponding parent drugs, so it should not be assumed that the drug and corresponding fluorophore bind at the same site.
However, it has been technically difficult to apply many biochemical methods to P-gp in detergent solution or liposomal preparations. The changes in affinity of several substrates for P-gp in different nucleotide-bound states have been nicely demonstrated for detergent-solubilized preparations but contradict analogous nucleotide-dependent changes in affinity of drugs for P-gp in a lipid bilayer.12–14 In general, additional details concerning the nucleotide-dependent interactions of probes and drugs with P-gp are required to fully understand its mechanism, and new methods for studying these interactions may contribute.
We have previously established P-gp in lipid nanodiscs as a platform for its biochemical and biophysical characterization. For example, conformation specific antibodies against human P-gp in nanodiscs differentially recognize the vanadate-trapped nucleotide-bound P-gp versus nucleotide-free and other nucleotide-bound states, as probed by SPR.15 Here we extend the nanodisc platform with a comparison of DMPC and Escherichia coli lipid nanodiscs to monitor the affinity of probe ligands for different nucleotide-bound states of murine P-gp using fluorescence correlation spectroscopy (FCS), which has some advantages over other fluorescence-based approaches. One advantage is the extraordinary sensitivity of FCS that allows for interrogation specifically of low-drug occupancy states. Here we demonstrate that the murine P-gp in nanodiscs exhibits nucleotide-dependent changes in affinity for BODIPY-verapamil (BD-verapamil) and BODIPY-vinblastine (BD-vinblastine), but the affinity of paclitaxel-Oregon Green 488 (Flutax-2) is unaffected by the nucleotide. The chemical structure of each is shown in Figure S1 along with the corresponding unlabeled drug. Most interestingly, Flutax-2 is a transport substrate in murine P-gp vesicles but does not stimulate ATPase activity in either lipid environment. Together, the results confirm that P-gp in lipid nanodiscs has distinct ligand binding sites for commonly used fluorescent probes, at low occupancy, that are differentially coupled to nucleotide binding, and they demonstrate the utility of FCS with P-gp nanodiscs.
MATERIALS AND METHODS
Materials
1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and n-dodecyl β-D-maltoside (DDM) were purchased from Avanti Polar Lipids. BODIPY FL verapamil (also known as Everfluor FL Verapamil) was purchased from Setareh Biotech. Oregon Green 488 Paclitaxel (Flutax-2), BODIPY vinblastine, Alexa Fluor 488 succinimidyl ester, and DiOC16(3) were purchased from ThermoFisher Scientific. Unless stated otherwise, nucleotides and other reagents were from Sigma-Aldrich.
P-gp and MSP1D1 Protein Expression and Purification
Hexahistidine-tagged MSP1D1 was expressed in E. coli and purified via nickel affinity chromatography as described in detail previously.16 The wild-type mouse P-gp mdr1a (or also known as mdr3) was expressed and purified from Pichia pastoris as described previously with some modifications.17 Yeast cells were grown and induced with methanol in a 32 L DCI-Biolafftte fermentor with a 20 L working volume. Frozen, harvested cells were thawed before being resuspended in ice-cold homogenization buffer (100 mM Tris, 250 mM sucrose, 1 mM EDTA, 1 mM EGTA, 100 mM 6-aminohexanoic acid, 2 μg/mL pepstatin A, 2 μg/mL leupeptin, 1 mM PMSF, and 2 mM benzamidine) at a density of 0.5 mg of cells/mL. Cells were lysed with a French press, and P-gp was solubilized in buffer containing 1% (w/v) DDM and protease inhibitors, purified by Ni-NTA and DEAE-cellulose chromatography according to established protocols.
Nanodisc Reconstitution and Purification
P-gp nanodisc formation has been described previously.16 Instead of E. coli total lipid extract, DMPC was used to minimize heterogeneity in lipid composition and to increase the drug-stimulated over basal activity of murine P-gp. Briefly, DMPC lipid films were solubilized in disc forming buffer [20 mM Tris and 100 mM NaCl (pH 7.4)] in the presence of a 6-fold excess of DDM. Purified P-gp, MSP1D1, and lipid were mixed in a 0.1:1:80 molar ratio and incubated for 1 h at room temperature on a nutator. To remove the detergent, prewashed Amberlite XAD2 resin (Sigma-Aldrich) was added at a concentration of 0.6 g/mL for 2 h at room temperature, and P-gp nanodiscs were recovered by passing the sample through a 25 gauge needle. P-gp nanodiscs were separated from empty nanodiscs by size exclusion chromatography and high-performance liquid chromatography (SEC–HPLC) (Superdex 200 10/300 GL column, GE Healthcare). Fractions containing P-gp nanodiscs were collected and concentrated using a 100 kDa MWCO centrifugal filter unit (Millipore). P-gp nanodisc concentrations were estimated by comparing Coomassie-stained P-gp bands with BSA standards of known concentrations using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE).
ATPase Activity Assay
Basal and drug-stimulated ATPase activities of P-gp were measured using a colorimetric assay to monitor the release of inorganic phosphate according to the method described by Chifflet et al.18 P-gp nanodiscs (0.5 μg) were incubated in ATPase buffer [150 mM NH4Cl, 50 mM Tris, 5 mM MgSO4, and 0.02% NaN3 (pH 7.4)] in the presence of 1 mM MgATP for 1 h at 37 °C. Drugs were added from concentrated DMSO stocks, and the final DMSO concentration did not exceed 1% (v/v). The absorbance intensity from the formation of phosphomolybdate was measured at 850 nm using a Tecan Infinite M200 microplate reader.
ATPase activity was measured as a function of drug concentration, and rates were fit to the substrate inhibition equation, as performed in many P-gp studies, which assumes that there are two drug binding sites on P-gp, one that has a high affinity and stimulates ATPase activity and a second one that has a low affinity and inhibits ATPase activity:19
where V is the overall rate of ATP hydrolysis, V0 is the basal rate of ATP hydrolysis in the absence of drug, V1 is the maximal rate of ATP hydrolysis when there is only activation, K1 is the substrate concentration that stimulates half of this maximal increase in ATPase activity, V2 is the rate of ATP hydrolysis at an infinite substrate concentration, and K2 is the substrate concentration that gives a half-maximal decrease in ATPase activity from the value of V1 to the value of V2.
Fluorescence Correlation Spectroscopy (FCS)
FCS measurements were performed on a home-built instrument, consisting of an inverted Zeiss Axio Observer D1 (Carl Zeiss Microscopy, Peabody, MA) microscope equipped with Hydra-Harp 400 detection electronics, a pulsed 485 nm diode laser line, and a Tau-SPAD photon counting detector (PicoQuant GmbH, Berlin, Germany). The laser power was set at 150 μW to maximize the signal-to-noise level and to minimize excitation saturation effects and photobleaching. The laser was passed through a 488 nm/10 nm excitation filter and focused into the sample by a 63×, 1.2 N.A. C-Apochromat water immersion objective. The resulting fluorescence signal of the sample was collected through the same objective and separated from the excitation light by a 488 nm dichroic mirror in combination with a 535 nm/70 nm emission filter (all filters from Chroma Corp., Bellows Falls, VT). Emitted light was focused onto a 50 μm multimode fiber (OZ Optics, Ottawa, ON) and transmitted to the Tau-SPAD detector. Autocorrelation and time-correlated single-photon counting of the fluorescence signal from the detector were performed using the HydraHarp 400 and SymphoTime 64 software (PicoQuant). Binding titrations were performed by adding increasing concentrations of P-gp nanodiscs (receptor) to a fixed nanomolar concentration of the fluorophore (ligand). P-gp and empty nanodiscs were dialyzed in FCS buffer [20 mM Tris, 150 mM NaCl, 2 mM MgSO4, and 4% glycerol (pH 7.4)] overnight and centrifuged at 17500 rpm and 4 °C for 15 min to remove any aggregates before use. A fixed amount of empty nanodiscs was added to minimize adsorption of fluorophores or P-gp to tubes and coverslips. For the data shown, 25 nM BD-verapamil was added to various P-gp nanodisc concentrations in the presence of 1 μM empty nanodiscs. Similarly, 25.7 nM Flutax-2 was added to P-gp nanodiscs in the presence of 0.3 μM empty nanodiscs, and 50 nM BD-vinblastine was added to P-gp nanodiscs in the presence of 0.3 μM empty nanodiscs. The effect of nucleotides on binding of the drug fluorophore to P-gp was characterized by adding 1 mM AMP-PNP, or 1 mM ATP with 240 μM Vi. Orthovanadate stock solutions (100 mM) were prepared from Na3VO4 (Calbiochem) with boiling at pH 10, and substocks were boiled for 5 min and diluted with FCS buffer before each use. Fluorophore concentrations were determined using εM values of 82000 ± 5000 M−1 cm−1 at 506 nm for BODIPY dyes (ThermoFisher) and 49100 ± 1100 M−1 cm−1 at 496 nm for Flutax-2.20
Experiments examining the competition between the fluo-rescent probes and drug substrates for binding were performed in the presence of a fixed concentration of P-gp nanodiscs (300 nM) and dye (as used for binding titrations). Drug stock solutions were created in DMSO, and the final DMSO concentration in FCS samples did not exceed 1% (v/v).
Samples were incubated at room temperature (22.5 ± 0.5 °C) for 90 min before measurement; 1 min measurements were recorded for each sample 5–10 times. Autocorrelation curves with intensity spikes due to aggregation or insolubility were removed. A one-component, free diffusion model was used to describe the autocorrelation function [G(τ)] for single fluorescent molecules:
where N is the average number of molecules in the focal volume, τD is the translational diffusion time of the particles transiting the observation volume, and S (=Z0/r0) is the structural parameter that is the ratio of the axial (Z0) to lateral dimension (r0) of the volume (fixed at 5.0 for our instrumentation). For single-component samples such as free dye alone or dye species fully bound to nanodiscs, the equation presented above was used to obtain τD. The laser point spread function was approximated by a three-dimensional Gaussian model, and 20 nM Alexa Fluor 488 succinimidyl ester was used for calibration and estimating the r02 value (r02 = τD4D, where D is the diffusion coefficient of Alexa Fluor 488 SE, which is reported to be 414 μm2/s).21 Fitting of autocorrelation curves was performed using Igor Pro 6, which uses the Levenberg-Marquardt nonlinear least-squares fitting algorithm. The goodness of fit was derived from the χ2 value or residual plot.
Equilibrium Binding of Fluorophores to P-gp Nano-discs
To analyze a solution containing n non-interacting fluorescent species with n different τDi, the overall autocorrelation function is described as
where S is the structural parameter (Z0/r0) and bi is the relative amplitude of the components that is proportional to the concentration of molecules, assuming the constant brightness of the molecules and the contribution of chemical kinetic processes are negligible.
Here we used the approach of measuring a single, apparent τD, which is a weighted average of all the n components (free dye, empty nanodiscs, and P-gp nanodiscs) in solution to determine the equilibrium dissociation constant, Kd
where τfree, τE, and τP are the known translational diffusion times of the free fluorophore, empty nanodiscs, and P-gp nanodiscs, respectively, and [E], [P], KE, and KP are the concentrations and dissociation constants of empty and P-gp nanodiscs, respectively.
The concentrations of the fluorophore and empty nanodiscs added to the samples were constant and fixed in the equation presented above, and nonspecific binding to the empty nanodiscs was accounted for by determining KE in a separate titration with empty nanodiscs alone. The dissociation constant of binding of the fluorophore to P-gp was obtained by plotting the apparent diffusion time versus different P-gp nanodisc concentrations, and independent data sets collected on different days were globally fitted.
To demonstrate that the FCS binding experiments selectively probe the high-affinity site of P-gp, and that the equation presented above, which assumes a single binding site on P-gp, is appropriate, simulations of the fractional occupancy at the high-and low-affinity sites were performed using the following equation with GraphPad Prism 7, utilizing the parameters recovered from the fits of biphasic ATPase profiles of these probes as well as incorporating a component for nonspecific binding to nanodiscs.
where KD1 and KD2 refer to the dissociation constants of the high- and low-affinity sites of P-gp, respectively, assuming that the KM values for ATP hydrolysis derived from the activity assays are close approximations of the KD values, and [high-affinity sites] = [low-affinity sites] = [P-gp].
The potencies of the drugs in competing for binding of the fluorophore to P-gp nanodiscs were assessed using the following nonlinear regression derived from the Hill equation to determine the IC50 value:
where τmax and τbase refer to the apparent diffusion times of the fluorophore when it is maximally bound to P-gp nanodiscs and when it is unbound, respectively, and n is the Hill coefficient.
Vesicle-Based Transport Assay of Flutax-2
Transport of Flutax-2 was evaluated using inside-out membrane vesicles containing human or murine P-gp (GM0015 or GM0004 from GenoMembrane). The assay made use of an anti-fluorescein antibody [anti-FL IgG (Thermofisher)] that is membrane impermeant to quench the fluorescence of free Flutax-2 in solution but not Flutax-2 transported into vesicles. Fluorescence measurements were performed using a Varian Cary Eclipse spectrophotometer with a temperature-controlled sample compartment. A 75 μL aliquot of membrane vesicles containing 50 μg of protein was preincubated with 133 nM Flutax-2 in transport buffer [20 mM Tris, 150 mM NaCl, and 5 mM MgSO4 (pH 7.4)] at 37 °C for 5 min. To initiate transport, a 10 μL aliquot of ATP or AMP-PNP (control) was added by mixing for 10 s (effective concentration of 4.7 mM). At various time points, a 15 μL aliquot of anti-FL IgG (1 mg/mL stock) was added with 10 s mixing to rapidly quench the fluorescence of Flutax-2 remaining outside of vesicles, and fluorescence readings were collected immediately. The final concentrations of reagents after addition of anti-FL IgG were 100 nM Flutax-2, 4 mM nucleotide, and 1 μM anti-FL Ab. A control experiment with 100 nM Flutax-2 and 1 μM anti-FL Ab demonstrated that up to 90% of fluorescence quenching was achieved within the first few seconds of mixing, in agreement with the results of another study.20 Excitation was set at 495 nm (5 nm slit width), and emission was recorded from 510 to 650 nm (10 nm slit width). Data were collected at 1 nm intervals, and a single measurement was completed within 30 s. Because changes in fluorescence intensity were proportional to the dilution factor, the initial rate of ATP-dependent transport was approximated by taking the difference between the fluorescence intensity in the presence of ATP and AMP-PNP after antibody quenching, scaling it to the fluorescence intensity for 100 nM Flutax-2 in the presence of vesicles, and dividing by the amount of protein used and reaction time (adapted from the GenoMembrane protocol). We recognized that more free dye remaining in AMP-PNP-containing incubations would result in different extents of quenching by the antibody, so the subtraction method may be an underestimate of the rate of transport of Flutax-2.
The initial rate of transport (calculated from the first two or three time points contributing to the linear phase) =
Homology Modeling
For illustrative purposes, we constructed a homology model of P-gp in the outward-facing conformation based on crystal structures of homodimeric bacterial ABC transporter SAV1866. Using the I-TASSER server,22 N-terminal (residues 1–628) and C-terminal (residues 689–1280) portions of the human P-gp sequence were separately modeled with an AMP-PNP-bound crystal structure of SAV1866 (Protein Data Bank entry 2ONJ) specified as the primary template and the inward-facing murine P-gp structure (Protein Data Bank entry 4M1M) excluded as a template. This yielded models for the N- and C-terminal halves of P-gp that were then aligned with the 2ONJ crystal structure to provide an outward-facing model of P-gp (excluding residues 629–688). This model should be considered a rough approximation of the overall conformation of P-gp and possible inter-helical contacts in one of many outward-facing states, rather than as an atomically precise structure.
RESULTS
Reconstitution of Mouse P-gp in DMPC Nanodiscs
Previously published results have demonstrated the utility of incorporating human P-gp into nanodiscs with E. coli mixed lipids.15,16 However, attempts to reconstitute human P-gp into DMPC nanodiscs were unsuccessful. Surprisingly, murine P-gp, which is 87% identical to human P-gp, reconstitutes readily into DMPC nanodiscs using MSP1D1 as a scaffold protein (Figure 2). Size exclusion chromatography and electrophoresis demonstrate that the final P-gp nanodiscs are highly purified, with an MSP1D1 scaffold:P-gp ratio of 1.6:1 for the pooled fractions used (Figure 2). The theoretical ratio is 2:1. A significant advantage of the DMPC nanodiscs is their increased homogeneity and reproducibility compared to those of nanodiscs made with E. coli lipid extracts. P-gp reconstituted in DMPC nanodiscs is more homogeneous than nanodiscs with E. coli lipids based on chromatography, wherein the E. coli nanodisc preparations yield a slightly broader SEC peak (not shown), with slightly more variation between different preparations. We do not know the molecular basis for this. In addition, the basal rate of ATP hydrolysis is higher in the E. coli nanodiscs, as described further below.
Figure 2.

Separation of P-gp-embedded nanodiscs from empty nanodiscs. (A) SEC–HPLC elution profile of the nanodisc reconstitution mixture (blue) and purified P-gp nanodiscs (red). (B) Visualization of eluted fractions of the nanodisc reconstitution mixture from 15 to 28 min, by Coomassie staining of reducing SDS–PAGE. Fractions containing P-gp that co-eluted with histidine tag-cleaved MSP1D1 were pooled together (fractions 17–20) and concentrated using a 100 kDa MWCO spin filter.
ATPase Activity of P-gp Nanodiscs with Probe Substrates
Although fluorescent analogues of probe drugs are used widely in cell-based assays to assess P-gp transport activity, their biochemical characterization is reported in only a few cases, and steady state kinetic parameters and binding constants are particularly sparse for the murine P-gp. Therefore, we determined the concentration dependence of BD-verapamil, BD-vinblastine, and Flutax-2 in standard ATPase assays, in comparison to that of the corresponding unlabeled drugs (Figure 4). For each, the data were fit to a velocity equation that includes P-gp that is singly or doubly bound with substrate, as described in Materials and Methods. Whereas BD-verapamil and BD-vinblastine are known substrates for murine P-gp and human P-gp, biochemical data for Flutax-2 with murine P-gp have not been reported. The recovered parameters are summarized in Table 1. Consistent with findings that show that smaller molecules tend to be more stimulatory and larger molecules tend to bind more tightly with lower levels of ATPase stimulation,23 BD-verapamil and BD-vinblastine exhibit lower KM values for both sites compared to those of their unlabeled analogues. BD-verapamil and BD-vinblastine stimulate ATP hydrolysis ~8- and ~2-fold over basal ATPase activity, respectively. Interestingly, BD-vinblastine is much less stim-ulatory than vinblastine. The KM values for high-affinity sites that stimulate ATP hydrolysis are in the range of 77–330 nM, which are significantly below the corresponding high-affinity KM values for verapamil or vinblastine, suggesting tighter binding of BD-verapamil and BD-vinblastine. Also, both BD-verapamil and BD-vinblastine exhibit clear substrate inhibition with respect to ATPase activity, as observed for many drugs and probe substrates at higher concentrations. In contrast, Flutax-2 does not stimulate ATPase activity and modestly inhibits basal ATPase activity at concentrations approaching the low-affinity KM for paclitaxel.
Figure 4.

Equilibrium binding of fluorescent probes to P-gp nanodiscs under different nucleotide-bound conditions. The top row of three panels shows saturation binding isotherms plotted as the apparent diffusion time of probes as a function of P-gp concentration. The bottom three rows of three panels show the normalized FCS autocorrelation curves of fluorescent probes with increasing nanodisc concentrations (shifts from red to purple from left to right, respectively).
Table 1.
Kinetic Parameters for Substrate Inhibition (two binding sites) of ATPase Activity with Drugs and Fluorescent Analogues
| V1 (nmol of Pi min−1 mg−1) | V2 (nmol of Pi min−1 mg−1) | K1 (μM) | K2 (μM) | |
|---|---|---|---|---|
| DMPC Nanodiscs | ||||
| verapamil | 1004 ± 160 | >0a | 55.7 ± 14.8 | 118.9 ± 38.8 |
| BD-verapamil | 492 ± 10 | 107 ± 13 | 0.08 ± 0.008 | 12.1 ± 1.6 |
| vinblastine | 276 ± 31 | >0a | 2.6 ± 1.0 | 40.1 ± 9.7 |
| BD-vinblastine | 105b | 21 ± 4 | 0.04 ± 0.01 | 0.7 ± 0.1 |
| paclitaxel | 199 ± 23 | 48 ± 6 | 1.3 ± 0.5 | 16.3 ± 24.7c |
| Flutax-2 | 65b | 18 ± 2 | 0.3 ± 0.7d | 13.8 ± 3 |
| E. coli Lipid Nanodiscs | ||||
| BD-verapamil | 627.5 ± 11.2 | 218.4 ± 223.9 | 0.07 ± 0.02 | 113.5 ± 118.4c |
| BD-vinblastine | 465b | 156.5 ± 24.8 | 0.05 ± 0.1d | 1.1 ± 0.4 |
| Flutax-2 | 500b | 33.2 ± 46.8 | 0.2 ± 0.3d | 2.6 ± 0.9 |
The parameter is reaching a constraint set at >0.
The parameter held at a constant value.
Large errors due to only modest inhibition at the highest drug concentration used.
Ambiguous, because of a lack of ATPase stimulation by these ligands (V1 is close to V0). Fits for the DMPC nanodiscs and E. coli nanodiscs with three probes are shown in Figure 4.
For comparison, we also studied the stimulation of ATPase activity for each probe substrate with P-gp in E. coli lipid nanodiscs. The results are summarized in Table 1. Although the basal ATPase activity is higher in the E. coli lipid nanodiscs, the concentration-dependent effects of the three probes on ATPase activity are qualitatively similar in E. coli lipid nanodiscs and DMPC nanodiscs. Flutax-2 exhibits no stimulation in either lipid environment. Neither BD-vinblastine or Flutax-2 stimulated ATPase activity in E. coli lipids. In fact, both BD-vinblastine and Flutax-2 are low-affinity inhibitors that decrease the ATPase activity more in the E. coli lipid than in the DMPC nanodiscs (Figure 3).
Figure 3.

Modulation of ATPase activity of P-gp by drugs and fluorescent analogues. P-gp DMPC nanodiscs (0.5 μg), in the presence of 1 mM ATP, were used for ATPase activity assays with ligand: (A) (○) verapamil and (■) BD-verapamil, (B) (○) vinblastine and (■) BD-vinblastine, and (C) (○) paclitaxel and (■) Flutax-2. (D) Effect of fluorescent probes on the ATPase activity of P-gp reconstituted into E. coli nanodiscs. See ref 16 for details about E. coli lipid nanodisc reconstitution. Each data point represents the mean ± standard deviation of triplicate measurements.
In summary, the three probe ligands behave differently with respect to one another in a lipid-dependent manner, and with respect to their corresponding nonfluorescent analogues, but in particular, Flutax-2 is distinct in its inability to stimulate ATP hydrolysis in either case. We note that the value for K2 in DMPC nanodiscs with paclitaxel includes a high level of error because of insufficient data points at higher concentrations. This was due to the solubility limits of paclitaxel that prohibited the use of higher concentrations. Similarly, there is significant error in the K1 values for the E. coli lipid nanodiscs, because of minimal activation. Although we acknowledge the error, the data still demonstrate substrate inhibition for paclitaxel in DMPC and a behavior distinctly different from that of BD-verapamil in E. coli lipid nanodiscs. We are careful not to overinterpret the recovered parameters, but they clearly indicate that Flutax-2 behaves differently with respect to the other ligands in DMPC nanodiscs and differently with respect to BD-verapamil in E. coli lipid nanodiscs.
Fluorescence Correlation Spectroscopy and Binding of Fluorescent Probe Substrates at Low Occupancies
To obtain more details about the behavior of the fluorescent probes observed in fluorescent transport assays, we measured equilibrium binding affinities for each using FCS. This method is based on a shift in the diffusion times of the free probe when it binds to a P-gp nanodisc that has a much slower rate of diffusion. The P-gp nanodisc system is uniquely suited for FCS. In contrast, P-gp liposomes are not optimal because they provide a a heterogeneous population of particles with different sizes, and hence different diffusion properties, and a large membrane reservoir for nonspecific binding of probe ligands. Also, an important distinction between FCS and other fluorescence-based methods for monitoring binding is the fact that FCS does not require a change in fluorescence intensity or emission wavelength associated with binding. The diffusion constants are independent of the intensity. The experimental design is a reverse titration in which increasing concentrations of P-gp nanodiscs are added to a low, fixed, concentration of dye, and this selectively probes the tightest binding interaction between P-gp and ligand, without significantly populating the low-affinity site(s).
Before monitoring the binding of fluorescent probes to the high-affinity sites of P-gp in nanodiscs, we needed to calibrate the diffusion times of empty nanodiscs and P-gp nanodiscs, to account for any binding of the probe to the lipid bilayer of the nanodisc. This was accomplished by incorporating a trace amount of DiOC16(3) into empty nanodiscs and also into P-gp nanodiscs in a molar ratio of 1:160 (dye:lipid) to determine the diffusion properties of nanodiscs with or without P-gp. The autocorrelation curves fit well to the one-component model, and the average diffusion times of empty and P-gp nanodiscs were 1.71 ± 0.07 ms (n = 5) and 3.02 ± 0.18 ms (n = 12), respectively (Figure S2). These values were used in subsequent experiments in which P-gp nanodiscs were titrated into solutions of BD-verapamil, BD-vinblastine, or Flutax-2.
The addition of P-gp nanodiscs to solutions of each fluorescent probe, in the absence of nucleotide, resulted in concentration-dependent shifts in their diffusion times as shown in Figure 4.
In addition, we monitored the binding of each fluorophore to empty nanodiscs. At the concentrations used, BD-verapamil exhibited negligible binding to the lipid nanodiscs whereas Flutax-2 and BD-vinblastine yielded apparent KD values of 3.6 and 1.7 μM, respectively, which are more than an order of magnitude higher than the KD values recovered for the tight binding site on P-gp in each case. A comparison of binding to P-gp nanodiscs versus empty nanodiscs is shown for each dye in Figure S3. In binding experiments with P-gp nanodiscs, the P-gp nanodisc-induced shift in fluorophore diffusion time was corrected for the membrane partitioning of the dye, as described in Materials and Methods, to obtain an apparent KD specifically for binding to P-gp. The recovered values are listed in Table 2. The fact that the limiting values of τ in the binding isotherms of Figure 4 do not reach the theoretical limit of the bound diffusion time (τ ~ 3 ms) likely arises from some impurity or heterogeneity of the dye in solution, wherein a fraction of fluorescent species that contribute to τ do not bind. Also, it is important to emphasize that with this experimental design, in which P-gp nanodiscs are titrated into a limiting concentration of probe, to a first approximation only the highest-affinity site is occupied throughout the titration. Higher-occupancy states are minimally populated. This is demonstrated with simulated binding isotherms based on the experimentally determined KD values and the experimental conditions in Figure S4.
Table 2.
Nucleotide-Dependent Changes in Affinity for Fluorescent Probe Ligandsa
| nucleotide-bound state | BD-verapamil KD (μM) | BD-vinblastine KD (μM) | Flutax-2 KD (μM) |
|---|---|---|---|
| apo (control) | 0.23 ± 0.08 | 0.042 ± 0.006 | 0.52 ± 0.09 |
| AMP-PNP | 1.16 ± 0.3 | 0.083 ± 0.012 | 0.53 ± 0.09 |
| ATP and Vi | 2.35 ± 0.7 | 0.29 ± 0.04 | 0.49 ± 0.08 |
| empty nanodiscs | NDb | 1.67 ± 0.46 | 3.63 ± 0.32 |
Average values calculated on the basis of global fits of at least two independent experiments.
Not determined because of minimal binding at the nanodisc concentrations used.
The binding affinity of each probe was determined also in the presence of a saturating concentration of AMP-PNP, which is a nonhydrolyzable analogue of ATP, and in the vanadate-trapped state. The vanadate-trapped state is widely used to mimic the posthydrolysis transition state or the transition state.24,25 It is established that binding of AMP-PNP or vanadate trapping leads to a shift in the conformational ensemble toward an outward-facing conformation with NBDs in the proximity of one another, and with rearrangement of the TMHs. These nucleotide-bound states are expected to be predominantly outward-facing, although they are likely different from one another and possibly drug-dependent and isoform-dependent.6,26,27
These results included interesting complexity. We observed that addition of P-gp nanodiscs to BD-verapamil or BD-vinblastine, but not Flutax-2, is accompanied by an increase in fluorescence intensity. Therefore, we measured the brightness per molecule with increasing concentrations of P-gp nanodiscs. Surprisingly, the brightness per molecule did not change with an increasing fraction bound (f). Instead, the data indicate that increasing the concentration of P-gp nanodiscs leads to a change in the apparent Napp. More detail concerning this result, including additional data, is provided in Figure S5a–c. The change in Napp likely results from the recruitment of BD-verapamil or BD-vinblastine from weak nonspecific binding sites on the glass surfaces to the experimental focal volume upon binding. As a result of this complexity, the reported KD values are apparent KD’s and should be considered as relative values.
Regardless of this complexity, the results, with binding parameters summarized in Table 2, clearly demonstrate that BD-verapamil and BD-vinblastine have decreased affinity for their high-affinity site in the vanadate-trapped state, with increases in KD of 10.3- and 6.8-fold, respectively. Interestingly, the results with AMP-PNP are more substrate-dependent upon comparison of BD-verapamil and BD-vinblastine. With BD-verapamil, addition of AMP-PNP induces a 5.1-fold increase in KD, but an only 1.9-fold increase in KD for BD-vinblastine. Furthermore, the results with Flutax-2 suggest a very different behavior. Specifically, the high-affinity binding of Flutax-2 is relatively insensitive to AMP-PNP binding or vanadate trapping. This result is examined in greater detail below.
Competition of the High-Affinity Site for Flutax-2 with Other Probes
It is reported that Flutax-2 is a transport substrate for human P-gp, but no additional characterization has been reported.28 Because Flutax-2 exhibited a high-affinity interaction with the murine P-gp (KD = 519 nM), with no effect on ATPase activity in this concentration range, we hypothesized that Flutax-2 binds at a site distinct from that of drugs that stimulate ATPase activity. Therefore, we performed competition experiments to determine whether it binds competitively with verapamil or vinblastine. For comparison, we also determined whether BD-verapamil and BD-vinblastine bind competitively with these substrates. In separate experiments, each fluorescent probe was poised at a concentration (25–50 nM) that partially saturated the high-affinity site on P-gp, in the presence of excess P-gp nanodiscs (300 nM), and each nonfluorescent drug was titrated into the sample. As the nonfluorescent drug displaces the fluorescent dye, the apparent diffusion time of the dye decreases. This approach revealed whether each dye was competitively displaced by each drug. Results for the titration of verapamil or vinblastine into solutions containing the [P-gp nanodisc·BD-verapamil] or [P-gp nanodisc·Flutax-2] complex are shown in Figure 5 and Figure S5, and the results for each of the dye combinations are summarized in Table 3.
Figure 5.

Comparison of the dose-dependent effect of drugs on fluorescent probes binding to P-gp nanodiscs. Apparent diffusion times of BD-verapamil (○) and Flutax-2 (△) measured as a function of the concentration of unlabeled drug verapamil (left) or vinblastine (right). For this experiment, 25 nM BD-verapamil or 26 nM Flutax-2 was used in the presence of 300 nM P-gp nanodiscs. Error bars represent the standard deviation of five measurements. Verapamil and vinblastine displace BD-verapamil but not Flutax-2. The results of this experiment and competition experiments are summarized in Table 3.
Table 3.
Effects and Potencies of Modulators on Fluorescent Analogues Binding to P-gpa
| verapamil
|
vinblastine
|
paclitaxel
|
||||
|---|---|---|---|---|---|---|
| IC50 (μM) | n | IC50 (μM) | n | IC50 (μM) | n | |
| BD-verapamil | 173 ± 69 | 0.95± 0.3 | 17.5 ± 11 | 0.5± 0.1 | NDb | |
| BD-vinblastine | NDb | 8.8 ± 6 | 0.4± 0.1 | 327 ± 567 | 0.5 ± 0.5 | |
| Flutax-2 | NDb | NDb | NDb | |||
Average values calculated from two independent experiments.
No detectable changes in apparent diffusion time. n refers to the Hill coefficient.
For vinblastine and paclitaxel binding, the data did not fit to a simple 1:1 binding isotherm and a Hill equation was used. Hill coefficients of ~0.5 were recovered for binding of vinblastine and paclitaxel to P-gp complexed with BD-vinblastine, consistent with multiple bindings of these drugs. For binding of verapamil to the complex of BD-verapamil with P-gp, the recovered Hill coefficient was 0.95, suggesting that the binding of multiple verapamil molecules occurs with similar sequential KD values and no cooperativity. Whereas vinblastine and verapamil each displaced BD-verapamil, paclitaxel did not displace it. Furthermore, vinblastine and paclitaxel each displaced BD-vinblastine, but verapamil did not. These results are consistent with previous models that suggest multiple, partially overlapping drug binding sites in P-gp,29,30 although a detailed molecular model for the binding sites was not pursued here. However, the results demonstrate the ability of the FCS approach to monitor overlapping or competitive binding sites. Most striking is the fact that none of the drugs displaced Flutax-2 from its high-affinity site, indicating that it does not overlap substantially with verapamil, vinblastine, or paclitaxel high-affinity sites. Flutax-2 does not compete for the binding sites of vinblastine, verapamil, or paclitaxel.
Transport of Flutax-2
Whereas some probes are known both to be transport substrates for P-gp and to stimulate ATP hydrolysis, there are some that are transported despite the lack of ATPase stimulation.31 Because Flutax-2 exhibited a high affinity for P-gp without having any effect on ATPase activity, we determined whether it is a transport substrate for murine P-gp. The human P-gp has been reported to actively transport Flutax-2, but corresponding experiments with the murine protein have not been reported.28 Murine P-gp vesicles were used as described in Materials and Methods, and we exploited an anti-fluorescein antibody (anti-FL IgG) to monitor the concentration of Flutax-2 remaining outside the vesicles after the addition of ATP or AMPNP. The anti-FL IgG added at various times quenches the fluorescence of free Flutax-2 but not the Flutax-2 that is transported to the inside of the vesicle. As a result, the fluorescence intensity remaining after addition of ATP and anti-FL IgG represents the Flutax-2 that is transported. For comparison, we also measure the transport of Flutax-2 by human P-gp vesicles. The results are shown in Figure 6. Both human and murine P-gp clearly transport Flutax-2 in an ATP-dependent process, with similar initial transport rates of approximately 51 and 42 pmol (mg of total protein)−1 min−1, respectively.
Figure 6.

Transport of Flutax-2 into human or mouse P-gp vesicles. P-gp vesicles (50 μg of protein) were incubated in the presence of 117 nM Flutax-2 and 4.7 mM ATP or AMP-PNP for various times before the remaining free dye was quenched by anti-fluorescein IgG. Overlaid emission spectra of Flutax-2 incubated with human (left) or mouse (right) P-gp vesicles, excited at 495 nm, with fluorescence intensity normalized to the intensity measured after addition of nucleotides (gray, taken to be 100%). Normalized emission spectra measured immediately after addition of anti-fluorescein IgG to incubations with ATP or AMP-PNP are depicted as solid black and dotted lines, respectively. The insets show the normalized emission intensity measured at 526 nm, after being quenched with an antibody, plotted as a function of time.
In light of the combined results that indicate that Flutax-2 is transported (Figure 6) but its binding is unaffected by the nucleotide (Figure 4), we considered whether Flutax-2 stimulated ATPAse in the GenoMembrane system. We performed ATPase assays in both mouse and human P-gp vesicles, and the results are shown in Figure S7. Notably, the GenoMembranes exhibit a basal ATPase activity lower than that of P-gp in nanodiscs. Whereas verapamil exhibited substrate inhibition with a maximal apparent 1.4-fold stimulation of ATPase activity in both human and mouse P-gp vesicles, Flutax-2 did not stimulate ATPase activity in the concentration range studied.
Finally, we note that there is apparent binding of Flutax-2 to the vesicles, distinct from transport, based on the background fluorescence in the samples containing AMP-PNP (Figure 6, insets). To determine whether this resulted from some background P-gp transport that was insensitive to AMPPNP, we also examined the effect of vanadate trapping (Figure S8). Vanadate trapping, like AMP-PNP, completely inhibited the time-dependent increase in fluorescence without decreasing the time-independent background fluorescence.
DISCUSSION
The results described here contribute to three aspects of P-gp research. The first aspect is the demonstration that murine P-gp can be functionally reconstituted into lipid nanodiscs with DMPC. This is interesting because our attempts to reconstitute human P-gp into DMPC nanodiscs have not been successful, despite successful incorporation into nanodiscs made from E. coli lipid extract. It is established that the mouse P-gp and human P-gp have distinct lipid requirements,32 but it is difficult to predict how efficiently various membrane proteins can be incorporated into nanodiscs. Therefore, we considered the possibility that the murine P-gp could be successfully incorporated into nanodiscs with DMPC. The verapamil-stimulated and vinblastine-stimulated ATPase activities of the murine P-gp nanodiscs are nearly identical to previously published results for P-gp in membranes or a detergent solution.33,34 Moreover, larger molecules such as BD-verapamil and BD-vinblastine have an affinity for P-gp higher than that of the unsubstituted drugs verapamil or vinblastine, and this finding further confirms the catalytic and conformational integrity of the P-gp in this lipid environment. As noted elsewhere, incorporation of membrane proteins into lipid nanodiscs allows the application of several biochemical techniques that are difficult or impossible in other membrane platforms.35–37 The ability to reconstitute mouse P-gp into lipid nanodiscs allows for the application of new approaches to its characterization.
The second contribution of this work is the application of FCS to characterize P-gp, which is uniquely permitted by the nanodisc platform. FCS is particularly useful because many in vitro probes of P-gp transport are fluorophores. In fact, many in vitro transport assays utilize fluorescence to monitor uptake into cells, via flow cytometry or fluorescence microscopy. However, understanding the specific interactions of these probes with P-gp requires purified preparations and different methods, and FCS provides some advantages. Neither detergent-solubilized preparations nor proteoliposomes are expected to be optimal with FCS because of heterogeneity in micelle or liposome dimensions. The monodisperse P-gp nanodiscs with a single P-gp per particle, and with both NBDs and cytosolic drug binding sites accessible to solution, are optimal for FCS. FCS provides a previously unexploited approach for characterizing the interactions of commonly used fluorescent transport probes with P-gp and hence better relating biochemical parameters to transport behavior. One potential advantage of FCS is that it does not rely on changes in the emission intensity of ligand fluorescence or P-gp intrinsic fluorescence. Fluorescent probes that undergo no change in emission intensity can be studied by FCS. An additional significant advantage of the FCS is its high sensitivity, which requires minimal amounts of pure protein and allowed for the specific interrogation of low-occupancy states of P-gp in the studies presented here.
The third aspect of P-gp research that is flushed out here concerns the additional detail of its interactions with probe substrates revealed by the combination of FCS with functional activity assays. It is well established that drugs and modulators differentially couple ATP hydrolysis by P-gp to transport and that nucleotides may also alter the affinity of the drug for P-gp. Among the studies that have explored the functional linkage between drug binding sites and NBDs, nucleotide analogues yield variable effects on drug affinity when comparing different drug analogues. For example, “classic” studies indicate that the ATP-vanadate-trapped P-gp has lower affinity for the photo-affinity analogue of prazosin, whereas another study showed that nucleotide binding, even in the absence of ATP hydrolysis, is capable of decreasing drug affinity.12,14 Despite these, and other, elegant studies by a range of investigators, the mechanism by which drug binding is coupled to transport is not completely defined, and there may be drug-dependent mechanisms.
Whereas interactions of BD-verapamil and BD-vinblastine with human P-gp have been studied in various biological systems,38,39 their nucleotide-dependent interactions with murine P-gp have not been described under any conditions to the best of our knowledge. As noted above, the results of interactions of murine P-gp in nanodiscs with verapamil and vinblastine were consistent with previous studies in other lipid or detergent systems, thus validating the utility of the FCS and nanodisc platform. Specifically, AMP-PNP-bound and vanadate-trapped-P-gp demonstrate 5- and 10-fold decreases in affinity for BD-verapamil, respectively, compared to 1.5- and 3.5-fold decreases in affinity for BD-vinblastine, respectively. The results for BD-verapamil and BD-vinblastine support the well-known behavior of P-gp, wherein the equilibrium affinity of drugs may be decreased in the presence of nucleotides, but the magnitude of the decrease in affinity can be drug-dependent and nucleotide-dependent. Addition of AMP-PNP to the complex of BD-vinblastine with human P-gp is sufficient to “release” drug,39 but here the nucleotide causes an increase in KD for BD-vinblastine of only 2-fold.
The human P-gp transports Flutax-2 in cell-based systems, but its binding affinity for any P-gp has not been determined. Interestingly, Flutax-2 is distinct from the other probes or substrates studied here, inasmuch as it causes no detectable stimulation of ATP hydrolysis when bound to its high-affinity site and inhibits only modestly at higher concentrations that approach the low-affinity KM for verapamil. Presumably, the transport of Flutax-2 that we observe requires the well-established conformational switching of Pgp, without formation of the same “NBD dimer” conformation that hydrolyzes ATP at accelerated rates observed with other substrates. The combined results demonstrate that Flutax-2 binds with high affinity to murine P-gp at a site that is distinct from verapamil, paclitaxel, or vinblastine. On the basis of the transport results, two possibilities are evident. The high-affinity Flutax-2 binding site is not coupled to ATP hydrolysis; however, nucleotide-dependent switching between inward-facing and outward-facing conformations still occurs, without an increase in its level over that of basal ATP hydrolysis, and Flutax-2 can dissociate from the outward-facing conformations even though its affinity is not decreased. Alternatively, the transport observed in our assay with vesicles occurs from P-gp complexes with multiple Flutax-2 molecules bound. Possibly, nucleotides do lower the affinity of Flutax-2 at high Flutax-2:P-gp stoichiometries, which are not probed in our FCS experiments, and this would facilitate transport. However, even if this occurs, it does so without stimulation of ATP hydrolysis, based on the lack of an effect of Flutax-2 on ATP hydrolysis even at high concentrations. On the basis of gel electrophoresis (not shown), we estimate that the transport assays contained 150–200 nM P-gp, similar to the level of P-gp expression in vesicles reported by others.40 With 117 nM Flutax-2 in the assays, and dramatically different KD values for the first and second Flutax-2 binding sites, this would be expected to result in minimal P-gp with multiple Flutax-2 molecules bound in the transport assays. Therefore, it is unlikely that the observed transport requires multiple Flutax-2 bindings, and we propose that the first possibility is operative. Apparently, binding to this high-affinity site is not coupled to ATP hydrolysis but allows for transport. Flutax-2 is among the murine P-gp transport substrates that do not stimulate ATP hydrolysis. The combined results are summarized in Figure 7, which emphasizes the similarities and differences between the probes studied here.
Figure 7.
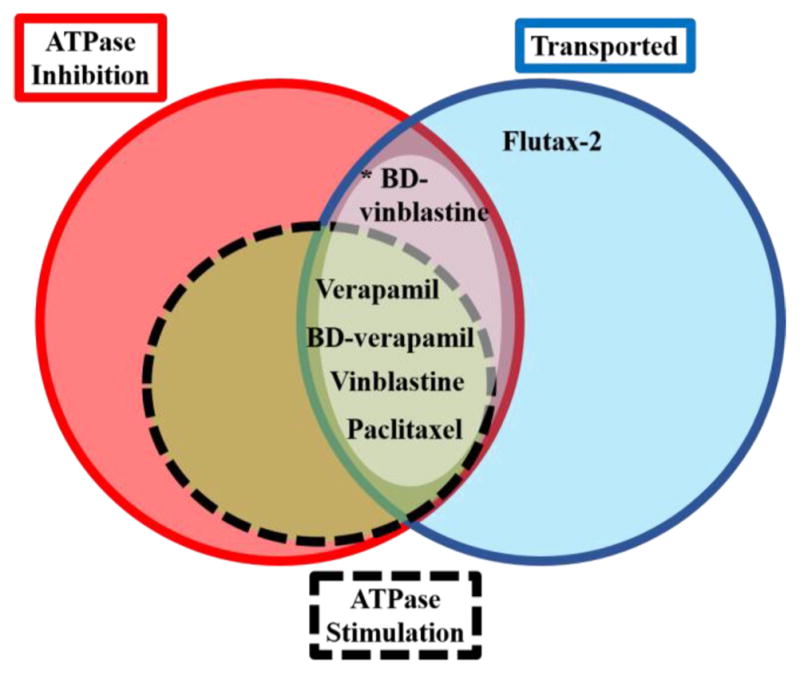
Venn diagram summarizing the interactions of drugs and fluorescent analogues used in this paper with P-glycoprotein in DMPC nanodiscs and GenoMembranes. The dotted circle represents compounds that stimulate ATPase activity and also inhibit activity at high concentrations. Transport of compounds has been demonstrated for both human MDR1 and mouse mdr1a.41–43 The asterisk indicates transport of BD-vinblastine demonstrated for human MDR1.39
Supplementary Material
Acknowledgments
Funding
This work was supported by National Institutes of Health Grants R01GM098457 (W.M.A.) and R01GM121603 (W.M.A.).
ABBREVIATIONS
- ABCB1
ATP binding cassette subfamily B member 1
- AMP-PNP
5′-adenylyl-imidodiphosphate
- BD-verapamil
BODIPY-verapamil
- BD-vinblastine
BODIPY-vinblastine
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- FCS
fluorescence correlation spectroscopy
- mdr1a
multidrug resistance protein 1a
- MSP
membrane scaffold protein
- MWCO
molecular weight cutoff
- OPM
Orientations of Proteins in Membranes database
- SEC–HPLC
size exclusion chromatography–high-performance liquid chromatography
Footnotes
Author Contributions
M.J.L. performed all experiments and analyzed data, with the assistance of A.N. for FCS. A.N. also assisted in the interpretation of data and experimental design for FCS and conducted homology modeling. W.M.A. directed the research.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b01245.
Chemical structures of probes used, FCS calibration data, binding isotherms, including empty nanodiscs, simulated binding isotherms for high- and low-affinity sites for each fluorophore, analysis of the change in Napp upon binding of BD-verapamil and BD-vinblastine to P-gp, ATP hydrolysis data for human and murine P-gp vesicles, and transport data for vanadate-trapped and AMP-PNP-inhibited P-gp (PDF)
References
- 1.Endicott JA, Ling V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem. 1989;58:137–171. doi: 10.1146/annurev.bi.58.070189.001033. [DOI] [PubMed] [Google Scholar]
- 2.Ferry DR, Kerr DJ. P-glycoprotein, a transporter with allosterically coupled drug-acceptor sites as a target for rational drug design. New Mol Targets Cancer Chemother. 1994:177–193. [Google Scholar]
- 3.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter 1. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 4.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawson RJ, Locher KP. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007;581:935–938. doi: 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 7.Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat Struct Mol Biol. 2004;11:918–926. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- 8.Hollenstein K, Dawson RJ, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007;17:412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Urbatsch IL, Tyndall GA, Tombline G, Senior AE. P-glycoprotein catalytic mechanism: studies of the ADP-vanadate inhibited state. J Biol Chem. 2003;278:23171–23179. doi: 10.1074/jbc.M301957200. [DOI] [PubMed] [Google Scholar]
- 10.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 11.Ledwitch KV, Gibbs ME, Barnes RW, Roberts AG. Cooperativity between verapamil and ATP bound to the efflux transporter P-glycoprotein. Biochem Pharmacol. 2016;118:96–108. doi: 10.1016/j.bcp.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin C, Higgins CF, Callaghan R. The vinblastine binding site adopts high- and low-affinity conformations during a transport cycle of P-glycoprotein. Biochemistry. 2001;40:15733–15742. doi: 10.1021/bi011211z. [DOI] [PubMed] [Google Scholar]
- 13.Qu Q, Chu JW, Sharom FJ. Transition state P-glycoprotein binds drugs and modulators with unchanged affinity, suggesting a concerted transport mechanism. Biochemistry. 2003;42:1345–1353. doi: 10.1021/bi0267745. [DOI] [PubMed] [Google Scholar]
- 14.Sauna ZE, Ambudkar SV. Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc Natl Acad Sci U S A. 2000;97:2515–2520. doi: 10.1073/pnas.97.6.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ritchie TK, Kwon H, Atkins WM. Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid nanodiscs and inhibition by the antibodies MRK16 and UIC2. J Biol Chem. 2011;286:39489–39496. doi: 10.1074/jbc.M111.284554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, Sligar SG. Chapter 11 -Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 2009;464:211–231. doi: 10.1016/S0076-6879(09)64011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lerner-Marmarosh N, Gimi K, Urbatsch IL, Gros P, Senior AE. Large scale purification of detergent-soluble P-glycoprotein from Pichia pastoris cells and characterization of nucleotide binding properties of wild-type, Walker A, and Walker B mutant proteins. J Biol Chem. 1999;274:34711–34718. doi: 10.1074/jbc.274.49.34711. [DOI] [PubMed] [Google Scholar]
- 18.Chifflet S, Torriglia A, Chiesa R, Tolosa S. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: application to lens ATPases. Anal Biochem. 1988;168:1–4. doi: 10.1016/0003-2697(88)90002-4. [DOI] [PubMed] [Google Scholar]
- 19.Litman T, Zeuthen T, Skovsgaard T, Stein WD. Structure-activity relationships of P-glycoprotein interacting drugs: kinetic characterization of their effects on ATPase activity. Biochim Biophys Acta, Mol Basis Dis. 1997;1361:159–168. doi: 10.1016/s0925-4439(97)00026-4. [DOI] [PubMed] [Google Scholar]
- 20.Diaz JF, Strobe R, Engelborghs Y, Souto AA, Andreu JM. Molecular recognition of taxol by microtubules. Kinetics and thermodynamics of binding of fluorescent taxol derivatives to an exposed site. J Biol Chem. 2000;275:26265–26276. doi: 10.1074/jbc.M003120200. [DOI] [PubMed] [Google Scholar]
- 21.Petrasek Z, Schwille P. Precise measurement of diffusion coefficients using scanning fluorescence correlation spectroscopy. Biophys J. 2008;94:1437–1448. doi: 10.1529/biophysj.107.108811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szewczyk P, Tao H, McGrath AP, Villaluz M, Rees SD, Lee SC, Doshi R, Urbatsch IL, Zhang Q, Chang G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr, Sect D: Biol Crystallogr. 2015;71:732–741. doi: 10.1107/S1399004715000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Senior AE. Reaction chemistry ABC-style. Proc Natl Acad Sci U S A. 2011;108:15015–15016. doi: 10.1073/pnas.1111863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Urbatsch IL, Sankaran B, Weber J, Senior AE. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J Biol Chem. 1995;270:19383–19390. doi: 10.1074/jbc.270.33.19383. [DOI] [PubMed] [Google Scholar]
- 26.Frank GA, Shukla S, Rao P, Borgnia MJ, Bartesaghi A, Merk A, Mobin A, Esser L, Earl LA, Gottesman MM, Xia D, Ambudkar SV, Subramaniam S. Cryo-EM Analysis of the Conformational Landscape of Human P-glycoprotein (ABCB1) During its Catalytic Cycle. Mol Pharmacol. 2016;90:35–41. doi: 10.1124/mol.116.104190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moeller A, Lee SC, Tao H, Speir JA, Chang G, Urbatsch IL, Potter CS, Carragher B, Zhang Q. Distinct conformational spectrum of homologous multidrug ABC transporters. Structure. 2015;23:450–460. doi: 10.1016/j.str.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patwardhan G, Gupta V, Huang J, Gu X, Liu YY. Direct assessment of P-glycoprotein efflux to determine tumor response to chemotherapy. Biochem Pharmacol. 2010;80:72–79. doi: 10.1016/j.bcp.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin C, Berridge G, Higgins CF, Mistry P, Charlton P, Callaghan R. Communication between Multiple Drug Binding Sites on P-glycoprotein. Mol Pharmacol. 2000;58:624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- 30.Pascaud C, Garrigos M, Orlowski S. Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem J. 1998;333:351–358. doi: 10.1042/bj3330351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polli JW, Wring SA, Humphreys JE, Huang L, Morgan JB, Webster LO, Serabjit-Singh CS. Rational Use of in Vitro P-glycoprotein Assays in Drug Discovery. J Pharmacol Exp Ther. 2001;299:620–628. [PubMed] [Google Scholar]
- 32.Sharom FJ. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front Oncol. 2014;4:41. doi: 10.3389/fonc.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu R, Sharom FJ. Site-directed fluorescence labeling of P-glycoprotein on cysteine residues in the nucleotide binding domains. Biochemistry. 1996;35:11865–11873. doi: 10.1021/bi960823u. [DOI] [PubMed] [Google Scholar]
- 34.Taylor JC, Ferry DR, Higgins CF, Callaghan R. The equilibrium and kinetic drug binding properties of the mouse P-gp1a and P-gp1b P-glycoproteins are similar. Br J Cancer. 1999;81:783–789. doi: 10.1038/sj.bjc.6690764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nath A, Atkins WM, Sligar SG. Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- 36.Nath A, Trexler AJ, Koo P, Miranker AD, Atkins WM, Rhoades E. Single-molecule fluorescence spectroscopy using phospholipid bilayer nanodiscs. Methods Enzymol. 2010;472:89–117. doi: 10.1016/S0076-6879(10)72014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trahey M, Li MJ, Kwon H, Woodahl EL, McClary WD, Atkins WM. Applications of Lipid Nanodiscs for the Study of Membrane Proteins by Surface Plasmon Resonance. Current Protocols in Protein Science. 2015;81:29.13.1–29.13.16. doi: 10.1002/0471140864.ps2913s81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22:7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 39.Bársony O, Szalóki G, Türk D, Tarapcsák S, Gutay-Tóth Z, Bacsó Z, Holb IJ, Székvölgyi L, Szabó G, Csanády L, Szakács G, Goda K. A single active catalytic site is sufficient to promote transport in P-glycoprotein. Sci Rep. 2016;6:24810. doi: 10.1038/srep24810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarkadi B, Price EM, Boucher RC, Germann UA, Scarborough GA. Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J Biol Chem. 1992;267:4854–4858. [PubMed] [Google Scholar]
- 41.Candussio L, Crivellato E, Rosati AM, Bartoli Klugmann F, Granzotto M, Giraldi T, Decorti G. Expression and function of P-glycoprotein and absence of multidrug resistance-related protein in rat and beige mouse peritoneal mast cells. Histochem J. 2001;33:259–266. doi: 10.1023/a:1017920922500. [DOI] [PubMed] [Google Scholar]
- 42.Sadiq MW, Uchida Y, Hoshi Y, Tachikawa M, Terasaki T, Hammarlund-Udenaes M. Validation of a P-Glycoprotein (P-gp) Humanized Mouse Model by Integrating Selective Absolute Quantification of Human MDR1, Mouse Mdr1a and Mdr1b Protein Expressions with In Vivo Functional Analysis for Blood-Brain Barrier Transport. PLoS One. 2015;10:e0118638. doi: 10.1371/journal.pone.0118638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schinkel AH, Wagenaar E, Mol CA, van Deemter L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97:2517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.