

Abstract
The prevalence of neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease, is currently a major public health concern due to the lack of efficient disease-modifying therapeutic options. Recent evidence suggests that mitochondrial dysfunction and nitrosative/oxidative stress are key common mediators of pathogenesis. In this review, we highlight molecular mechanisms linking nitric oxide (NO)-dependent post-translational modifications, such as cysteine S-nitrosylation and tyrosine nitration, to abnormal mitochondrial metabolism. We further discuss the hypothesis that pathological levels of NO compromise brain energy metabolism via aberrant S-nitrosylation of key enzymes in the tricarboxylic acid cycle and oxidative phosphorylation, contributing to neurodegenerative conditions. A better understanding of these pathophysiological events may provide a potential pathway for designing novel therapeutics to ameliorate neurodegenerative disorders.
Keywords: S-Nitrosylation, Nitration, Tricarboxylic Acid Cycle, Electron Transport Chain, Mitochondrial Metabolites, Neurodegeneration
Hallmarks of neurodegenerative diseases
Neurodegenerative diseases, including Alzheimer’s disease (AD; see Glossary), Parkinson’s disease (PD), Huntington’s disease (HD), and a variety of other conditions, are characterized by neurological symptoms, including progressive cognitive decline, behavior and personality changes, and movement impairment. In most cases, synaptic dysfunction and neuronal injury underlie these clinical conditions. Although the mechanisms of synaptic loss and neuronal damage have remained elusive, recent progress in the understanding of neurodegenerative diseases has revealed mitochondrial dysfunction, accumulation of misfolded proteins (e.g., β-amyloid and tau in AD, and α-synuclein in PD), and neuroinflammation as common features of the pathogenesis; emerging evidence suggests “crossover” in these diseases with the misfolded proteins classically found in one disease, also encountered in the others. Notably, many of these key neuropathological hallmarks result from elevated production of reactive nitrogen and oxygen species (RNS/ROS), including nitric oxide (NO) [1]. Along these lines, we and others have presented pathophysiological evidence that increased levels of NO and subsequent generation of NO-dependent post-translational modifications (PTMs) contribute to mitochondrial dysfunction, leading to deficits in bioenergetics and consequent synaptic damage and neuronal loss [2].
Compared to other cell types, neurons require higher energy to maintain proper function and activity, particularly synaptic activity. Accordingly, neurons are particularly sensitive to deficits in mitochondrial metabolism. Historically, mitochondrial dysfunction was linked to neurodegenerative conditions in the 1980s; patients who used drugs containing MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), a known precursor to MPP+, which inhibits mitochondrial complex I in the electron transport chain (ETC), developed parkinsonism [3]. In this scenario, impaired mitochondria not only manifest diminished respiratory activity but also produce high ROS and RNS, resulting in oxidative and nitrosative stress [4, 5]. Subsequent studies provided evidence that mitochondrial dysfunction is associated with mitochondrial DNA defects, disturbances in Ca2+ homeostasis, and abnormal mitochondrial morphology, thus playing a critical role in a variety of neurodegenerative conditions [6, 7]. In this review, we focus on the pathophysiological contribution of NO signaling to deficits in mitochondrial energy metabolism, and discuss how such molecular events affect the pathogenesis of neurodegenerative diseases. Specifically, we propose the hypothesis that the NO-dependent PTM, S-nitrosylation, contributes to the neurodegenerative process via impairment of mitochondrial metabolism. Moreover, metabolic disorders, such as type 2 diabetes mellitus (T2DM) and its precursor metabolic syndrome (MetS), manifest several similar aberrant S-nitrosylation reactions on proteins, contributing to the convergence of these metabolic and neurodegenerative disorders.
NO Signaling: Protein S-Nitrosylation and Nitration
In the brain, NO produced under physiological conditions promotes cellular pathways, maintaining synaptic function and neuronal survival [8]. In contrast, under neurodegenerative conditions, elevated production of NO triggers nitrosative stress, compromising neuronal activities. A family of NO synthases (NOSs), including neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS), is responsible for generation of NO. A critical cellular event in neurons leading to stimulation of nNOS activity involves N-methyl-D-aspartate (NMDA)-type glutamate receptor-mediated Ca2+ influx (Figure 1). For instance, physiological activation of synaptic NMDA receptors is essential to produce low levels of NO that facilitate normal synaptic function, whereas excessive activation, particularly of extrasynaptic NMDA receptors, triggers pathological production of NO [9]. Moreover, mitochondrial toxins, such as rotenone and MPTP, recapitulate some aspects of sporadic PD, in part via generation of RNS (from nNOS and iNOS) and ROS (from the mitochondrial ETC) [10, 11].
Figure 1. Protein S-Nitrosylation in NO Signaling under Neurodegenerative Conditions.
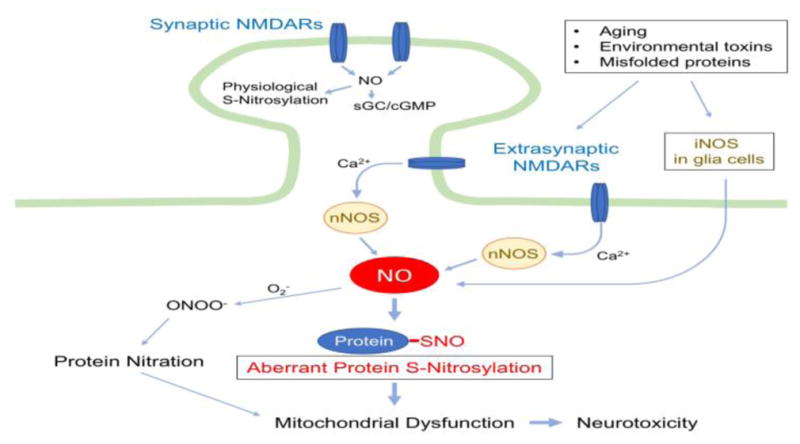
Upon activation by its dual agonists, glutamate and glycine (or D-serine), NMDA receptors (NMDARs) allow entry of Ca2+ into the cytosol of the postsynaptic cell. While synaptic NMDARs mediate synaptic plasticity and neuroprotection, hyperactivation of extrasynaptic NMDARs (eNMDARs) enhances neurotoxicity [94]. Under neurodegenerative conditions, increased Ca2+ influx due predominantly to eNMDAR overactivation stimulates excessive NO production from nNOS. Additionally, NO derived from iNOS in astrocytes augments nitrosative stress. Subsequently, the covalent addition of NO-related species to a reactive cysteine sulfhydryl (or thiolate) group in a target protein leads to the formation of S-nitrosylated (SNO) proteins. For example, pathologically high levels of NO can trigger aberrant S-nitrosylation of mitochondrial proteins, resulting in dysfunction in mitochondrial metabolism and dynamics. NO can also react with superoxide (O2−) to yield peroxynitrite (ONOO−), leading to tyrosine nitration. In contrast, under physiological conditions, synaptic NMDAR activity maintains relatively low levels of NO production, activating soluble guanylyl cyclase (sGC)/cGMP pathways and neuroprotective protein S-nitrosylation-mediated pathways.
Emerging evidence suggests that elevated levels of NO exert their neurodestructive activity in large measure through S-nitrosylation of critical protein thiols (Box 1). Examples of aberrantly S-nitrosylated proteins (SNO-proteins) contributing to neurodegenerative processes include SNO-GAPDH, SNO-Parkin, SNO-PDI, and SNO-Drp1 to name a few (summarized in a recent review [2]). Like other PTMs, S-nitrosylation can either inhibit or activate the activity of the target protein, depending on the exact function of the cysteine involved in the reaction, and thus affect downstream signaling cascades. Another critical mechanism for generating S-nitrosylated proteins is transfer of NO or transnitrosylation from one SNO-protein to another. Such reactions appear to be prodigious; for several proteins, transnitrosylation reactions have been predicted using the Nernst equation, from which the Gibbs free energy generated by the reaction under steady-state conditions can be calculated [14–16]. In many cases NO is a good ‘leaving group’ from S-nitrosothiols, allowing the remaining sulfhydryl to react with ROS to yield sulfenic (S-OH), sulfinic (S-OOH), or sulfonic (S-OOOH) acid derivatives. The latter, more stable oxidative modifications can also participate in neuropathological processes [17].
Box 1. Chemistry of S-Nitrosylation.
Chemically speaking, S-nitrosylation (or R-SNO formation) represents the covalent addition of NO-related species (e.g., nitrosonium cation-like intermediate, [NOx+], where x = 1 or 2) to a cysteine thiol (R-SH) or more properly thiolate anion (R-S−). This reaction scheme requires the presence of a transition metal to accept an electron [2, 12]. The target cysteine residues are typically surrounded by acidic and basic residues (forming a ‘SNO motif’), increasing the susceptibility of the thiol group to SNO modification [13]. However, high levels of NO can interact with a thiol moiety embedded in a partial (incomplete) SNO motif, causing aberrant formation of S-nitrosothiols.
An additional important NO-dependent PTM involves modification of tyrosine residues by peroxynitrite (ONOO−, formed by reaction of NO and O2−). This entails the addition of a nitro group (i.e., -NO2) to the aromatic ring of a tyrosine residue to form 3-nitrotyrosine. Nitration of tyrosine residues occurs in a relatively non-specific manner, and this type of PTM is irreversible, thus commonly serving as a pathological marker of nitrosative/oxidative stress [18]. Here, we examine the hypothesis that NO/nitrosative stress contributes to impairment of mitochondrial function via S-nitrosylation of mitochondrial proteins involved in metabolism. For a discussion of ROS and brain energy metabolism in neurodegeneration, readers are referred to other review articles [6, 19].
Energy metabolism in the brain
The human brain bears the highest energy requirement compared to other organs. Specifically, the brain consumes about 20% of the oxygen and 25% of the glucose taken into the human body, although the brain only constitutes 2% of total body weight. Maintenance of membrane potential and synaptic function appears to contribute to the high energy demands of the brain [20]. In the adult brain, neurons predominantly rely on mitochondrial oxidative metabolism for energy (i.e., ATP) production, whereas glycolysis predominates in glia cells [20]. In addition, mitochondria are critical not only for energy production but also for regulating cell death, calcium homeostasis, and ROS generation [21].
The presence of different metabolic profiles in neurons (mainly oxidative) and astrocytes (mainly glycolytic) is in part due to the cell-type specific activities of several key enzymes in energy metabolism. For instance, under basal conditions, the activity of pyruvate dehydrogenase (PDH) is low in astrocytes, and, therefore, processing of pyruvate, required for entering the tricarboxylic acid (TCA) cycle, is limited [22]. Additionally, during differentiation from neuronal progenitor cells (using predominantly glycolysis) to neurons, expression of hexokinase and lactate dehydrogenase A (LDHA), both of which play critical roles in glycolysis, dramatically declines [23].
The adult brain primarily uses glucose as an energy source; yet, under certain circumstances, such as during development and glucose starvation, ketone bodies are also utilized. To produce ATP from glucose, neurons initially process glucose to pyruvate, mainly via the pentose phosphate pathway (PPP). The PPP also contributes to the regeneration of glutathione (GSH) from its oxidized form (GSSG), thus maintaining the neuronal antioxidant potential against ROS generated during oxidative metabolism [20, 24]. Through the TCA cycle and the ETC in oxidative phosphorylation, neuronal mitochondria catalyze full oxidation of pyruvate to CO2 and water, producing 30–36 ATP (Figure 2). In contrast, the high glycolytic rate of astrocytes produces 2 ATP and pyruvate from a single glucose molecule. In astrocytes, pyruvate is further reduced to lactate by LDH5 (composed of a homo-tetramer of LDHA). The group of Magistretti has proposed the “astrocyte-neuron lactate shuttle (ANLS)” model whereby lactate is released from astrocytes into the extracellular milieu and then shuttled into neurons [22, 24, 25] (Box 2). For instance, a recent study in anesthetized mice revealed the presence of a lactate gradient from astrocytes to neurons under basal conditions, supporting the notion that the ANLS occurs in vivo [28]. Additionally, genetic manipulation studies suggested a physiological role of metabolic neuron-glia coupling in intact mice and drosophila [29–31]. However, the physiological role of the ANLS in brain energetics is still under scrutiny [32]. Future studies are warranted to evaluate actual lactate transport rates in vivo. Collectively, the existing evidence suggests that neurons can probably utilize both glucose and lactate as energy sources, and emerging evidence suggests that neurons prefer lactate over glucose if both are present at similar concentrations [24]. Therefore, the well-orchestrated cooperation of astrocytes and neurons forms the basis of energy metabolism in the adult brain.
Figure 2. Energy Metabolism in the Brain.

Glucose transported into neurons is converted to glucose-6-phosphate (glucose-6P) and processed to pyruvate via glycolysis or the pentose phosphate pathway (PPP). In mature neurons, the PPP is the main route for glucose metabolism. NADPH produced in the PPP regenerates GSH to maintain a proper redox homeostasis in cells. NO-related species affect glucose utilization through S-nitrosylation (SNO) of key enzymes in glycolysis, such as hexokinase (HK), aldolase, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Additional studies are needed to examine if other glycolysis or PPP enzymes are regulated by S-nitrosylation or related redox-mediated events. Glycolysis- or PPP-derived pyruvate enters mitochondria and is fully metabolized through the tricarboxylic acid cycle (TCA) cycle and the electron transport chain (ETC), producing ATP. In astrocytes, high glycolysis activity generates pyruvate; in turn, lactate dehydrogenase 5 (LDH5) converts pyruvate to lactate, which is then shuttled to neurons via the astrocyte-neuron lactate shuttle (ANLS) system. In neurons, the transported lactate is utilized by LDH1, converting lactate back to pyruvate for mitochondrial oxidative metabolism.
Box 2. The Astrocyte-Neuron Lactate Shuttle (ANLS) Model.
In brief, the central points of the ANLS model entail the following steps: i) neuronal activity increases extracellular glutamate, and together with Na+ ions, a large proportion of glutamate is taken up by astrocytes via excitatory amino acid transporters (EAATs); ii) the resulting increase in intra-astrocytic Na+ concentration activates Na+/K+ ATPase, consuming ATP, which in turn increases glucose uptake and glycolysis in astrocytes; iii) glycolysis-derived lactate is then shuttled into neurons as an energy substrate to meet the high energy requirements; iv) simultaneously, in astrocytes, glutamine synthase converts glutamate to glutamine, which is also transported back to neurons for recycling into glutamate. In neurons, LDH1 (composed of a homo-tetramer of LDHB) converts the incorporated lactate to pyruvate for mitochondrial oxidative metabolism [26, 27].
Impairment of mitochondrial metabolism and ATP production during neurodegeneration
Mitochondria represent key organelles for neuronal energy production. Through a combined and interdigitated effort of the TCA cycle and the ETC, mitochondria produce ATP and CO2 from pyruvate. Structurally, the mitochondrion consists of two functionally distinct membranes: the inner and outer membranes. The two membranes create two aqueous compartments known as the intermembrane space and the mitochondrial matrix. The majority of chemical reactions related to the TCA cycle take place in the matrix. The inner membrane folds into cristae, providing additional surface area for the ETC complexes to reside.
After transport of pyruvate from cytosol to mitochondria, the pyruvate dehydrogenase complex produces acetyl-CoA, CO2, and NADH. In addition, β-oxidation of fatty acids represents another important metabolic pathway leading to the production of acetyl-CoA in mitochondria. Acetyl-CoA feeds into the TCA cycle, undergoing a series of enzymatic reactions to generate NADH, FADH2, ATP, and CO2. The enzymes that comprise the TCA cycle include citrate synthase, aconitase, isocitrate dehydrogenase (the rate-limiting step), α-ketoglutarate dehydrogenase (αKGDH), succinyl-CoA synthetase, succinate dehydrogenase, fumarase, and malate dehydrogenase (Figure 3). The reducing equivalents (e.g., NADH) produced from the TCA cycle flow into the ETC for oxidative phosphorylation. The ETC, which contains four multi-subunit protein complexes (I–IV) and electron carriers (ubiquinone and cytochrome C), generates an electrochemical proton gradient across the inner mitochondrial membrane. Importantly, NADH from the TCA cycle interacts with complex I of the ETC to drive transfer of electrons from complex I to complex III and then IV. In addition, a component of complex II in the ETC, succinate dehydrogenase, converts succinate to fumarate in the TCA cycle and actuates the ETC to run from complex II, while bypassing NADH-activated complex I [33]. Thus, the enzyme succinate dehydrogenase represents a key component in both the ETC and TCA cycle and is critical in integrating their coordinated action. Lastly, the proton gradient, resulting from ETC activity, drives ATP production by ATP synthase (complex V), concurrently reducing oxygen to generate water (Figure 3).
Figure 3. Effects of S-Nitrosylation on TCA cycle and ETC Activity.
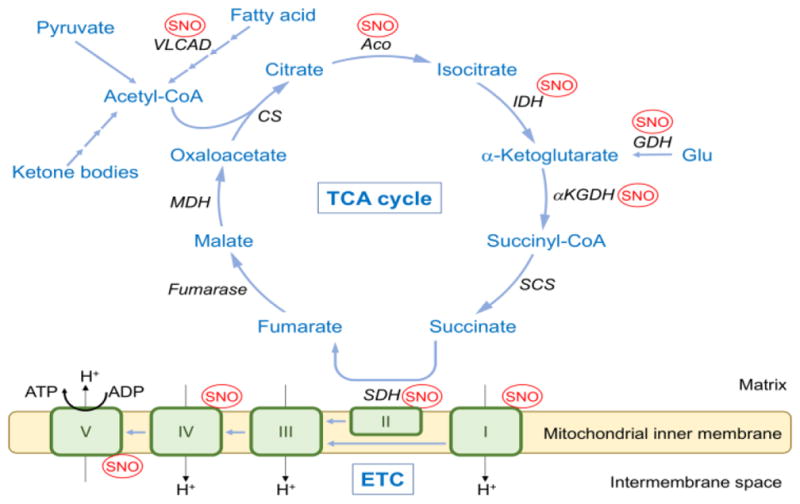
Acetyl-CoA generated from glycolysis, fatty acid oxidation, or ketone bodies enters the TCA cycle to produce NADH, FADH2, ATP, and CO2. Complex I in the ETC accepts an electron from NADH, whereas succinate dehydrogenase (SDH) in the TCA cycle (also serving as complex II in the ETC) provides electrons via generation of FADH2. Electrons flow though the ETC complexes, and these reactions are coupled to the translocation of protons from the mitochondrial matrix to the intermembrane space. The resulting proton electrochemical gradient drives ATP synthesis via ATP synthase (complex V). S-Nitrosylation (SNO) affects the activity of key enzymes in the TCA cycle, ETC, fatty acid oxidation, and glutamate metabolism. Additionally, published MS-based S-nitrosoproteomics has identified citrate synthase (CS), succinyl CoA synthetase (SCS), and malate dehydrogenase (MDH) in the TCA cycle as well as complex III in the ETC as targets of S-nitrosylation, but the effects of this PTM on these enzymes need to be studied further. ACO, aconitase; IDH, isocitrate dehydrogenase; αKGDH, αketoglutarate dehydrogenase; GDH, glutamate dehydrogenase; VLCAD, very long chain acyl-CoA dehydrogenase.
Accumulating evidence suggests that impairment in these mitochondrial processes in neurons leads to synaptic dysfunction and eventually neuronal cell death, and is implicated in the pathophysiology of neurodegenerative diseases [34–36]. For example, the activities of aconitase, isocitrate dehydrogenase, and αKGDH in the TCA cycle are low in AD [37], and defects in mitochondrial function due to environmental insult or mutation in mitochondrial DNA are associated with PD [38–40]. These findings are consistent with the notion that mitochondrial components are compromised under neurodegenerative conditions.
As one example of genetic or environmental insult of mitochondria contributing to PD via aberrant protein S-nitrosylation, our group recently found that dopaminergic neurons derived from a human induced pluripotent stem cells (iPSCs) carrying the A53T mutation in the gene encoding α-synuclein (a known cause of familial PD) manifest decreased mitochondrial respiration compared to isogenic, mutation-corrected controls [41]. This defect was exacerbated by complex I inhibitors, including several pesticides. Further, we showed that the impaired mitochondrial function in these A53T neurons occurred, at least in part, via aberrant S-nitrosylation of the transcription factor MEF2C, resulting in decreased expression of PGC1α, a critical regulator of mitochondrial biogenesis and function. Moreover, we found similar role of SNO-MEF2C in contributing to the neurodegenerative process and disrupting normal adult neurogenesis in AD and cerebral ischemia [42].
In the ensuing paragraphs, we further highlight the role of nitrosative PTMs, represented by protein cysteine S-nitrosylation and tyrosine nitration, in causing mitochondrial dysfunction and thus contributing to neurodegenerative disorders.
TCA cycle enzymes as targets of S-nitrosylation
A series of S-nitrosoproteomic investigations using improved nitrosothiol probes and mass spectrometry (MS) techniques have identified a wide range of mitochondrial SNO-proteins, including those involved in the TCA cycle, ETC complexes, fatty acid oxidation, and molecular chaperones [43–50]. S-Nitrosylated proteins identified heretofore in the TCA cycle include citrate synthase, aconitase, isocitrate dehydrogenase, αKGDH, succinyl-CoA synthetase, succinate dehydrogenase, and malate dehydrogenase (Figure 3) [44–47]. The effects of S-nitrosylation on TCA cycle enzymes are typically inhibitory, since these proteins often contain redox-sensitive cysteine thiol groups at their catalytic or regulatory sites [51]. For instance, NO groups inhibit the activity of aconitase, an enzyme that converts citrate to isocitrate in the first step of the TCA cycle, contributing to an increase in citrate levels [45, 52, 53]. Additionally, endogenously produced NO inhibits NADP+-dependent isocitrate dehydrogenase 1 (IDH1) and IDH2 via S-nitrosylation at Cys297/379 or Cys336/418 in IDH1 and IDH2, respectively [54, 55]. S-Nitrosylation of IDH1/2 results in structural alterations that inhibit its ability to convert isocitrate to α-ketoglutarate. S-Nitrosoproteomics also identified components of mitochondrial IDH3 as SNO-proteins [46]. Importantly, since the activity of IDH represents a key rate-limiting step of the TCA cycle [56], S-nitrosylation-mediated inhibition of IDH would be expected to have a potentially detrimental consequence on mitochondria-mediated metabolism. Concerning the activity of αKGDH, Chouchani et al. demonstrated that an NO donor, MitoSNO decreased enzyme activity via S-nitrosylation [45]; however, Sun et al. reported that another NO donor, S-nitrosoglutathione increased the activity of αKGDH in a dose-dependent manner [44]. To reconcile these apparent discrepancies, future studies are required to examine the effects of endogenously produced S-nitrosylation on αKGDH activity. Although growing evidence points to the fact that several TCA cycle enzymes undergo SNO modification, many questions remain unanswered. For example, the effect of endogenous S-nitrosylation on the regulation of each enzyme as well as on overall TCA cycle flux remain unknown. Moreover, the potential role of SNO-TCA cycle enzymes in the neurodegenerative process requires further study.
Additionally, NO could indirectly regulate the TCA cycle via S-nitrosylation of upstream pathways, such as glycolysis, fatty acid β-oxidation, and glutamate metabolism (Figures 2 and 3). In fact, studies have demonstrated that NO inhibits glycolytic flux [57]. Along these lines, proteomics and other biochemical analyses have found evidence for S-nitrosylation of glycolysis-related proteins, such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and aldolase [47, 58]. For instance, S-nitrosylation inhibits glycolysis by attenuating the activity of GAPDH, which catalyzes the conversion of glyceraldehyde-3-phosphate to 1,3-biphosphoglycerate [58, 59]. S-Nitrosylation of GAPDH occurs at the active site cysteine (Cys149 or 150, depending on the species), thus inhibiting activity [60]. In addition to its role in glycolysis, Solomon Snyder’s group has shown that SNO-GAPDH actively participates in cell death, translocating to the nucleus to stimulate death signaling pathways [61]. S-Nitrosylation may also regulate other redox-sensitive glycolytic enzymes, such as hexokinase [47], as well as possibly pyruvate kinase and phosphofructokinase, the rate limiting step in glycolysis [62].
Furthermore, Doulias et al. discovered that very long-chain acyl-CoA dehydrogenase (VLCAD), which catalyzes the initial, rate-limiting step of mitochondrial β-oxidation of long-chain fatty acids, is S-nitrosylated at Cys 238 [47]. S-Nitrosylation of VLCAD enhances its catalytic activity, possibly via increased substrate binding. Thus, S-nitrosylation of VLCAD may represent a beneficial function of NO, boosting fatty acid metabolism to produce acetyl-CoA for entry into the TCA cycle. Accordingly, VLCAD is S-nitrosylated under basal conditions with low levels of NO, i.e., in the absence of cell stress or disease. Additionally, this same group of Harry Ischiropoulos found that several enzymes involved in glutamate metabolism are S-nitrosylated under basal conditions in mouse brain [48]. One such enzyme, glutamate dehydrogenase (GDH) oxidizes glutamate to α-ketoglutarate, which can feed into the TCA cycle. Since S-nitrosylation of GDH inhibits glutamate oxidation, less α-ketoglutarate may be available to enter the TCA cycle because of this regulation.
NO signaling impairs oxidative phosphorylation – Effect of S-nitrosylation
The ETC is comprised of complexes I to IV plus ATP synthase (complex V), and represents the major site of oxidative phosphorylation in eukaryotic mitochondria. Evidence suggests that S-nitrosylation can inhibit multiple ETC components in the inner mitochondrial membrane (Figure 3) [43, 63]. Among these ETC complexes, inhibitory effects of S-nitrosylation on complex I are the most well characterized. Complex I is an NADH: ubiquinone oxidoreductase, transferring electrons from NADH to ubiquinone to drive proton translocation across the mitochondrial inner membrane. Two catalytically and structurally distinct forms of mammalian complex I exist: the fully competent, “active” form and the disabled, “deactivated” form. Transition from the active form to the deactivated form occurs spontaneously when the supply of enzyme substrates (e.g., NADH and ubiquinone) are low, and it can be slowly reactivated as the substrates become available again. Although, the physiological role of this “active-deactivated” conversion remains unclear, it has been postulated that it is an adaptive metabolic response mechanism to variations in substrate supply [64].
NO can directly inhibit mitochondrial respiration through S-nitrosylation of critical thiol(s) in complex I [64–66]. Interestingly, S-nitrosylation-dependent inhibition of complex I occurs only when its conformational state is in the deactivated form [67]. Although putative S-nitrosylation motifs are present in different subunits of complex I [68], Cys39 of ND3 (a residue exposed to the protein surface only in the “deactivated” form) is primarily responsible for SNO-dependent inhibition [69, 70]. Mechanistically, SNO modification of Cys39 in the ND3 subunit blocks its reactivation, thus decreasing complex I activity. However, the role of SNO-complex I in neurodegenerative conditions remains unknown. Thus, studies of SNO-complex I in disease models are required to determine its potential contribution.
Moreover, proteomics analyses have identified components of complex II and III as targets of S-nitrosylation [45, 46, 50]. These proteins include the succinate dehydrogenase flavoprotein subunit from complex II (or TCA cycle as described above) and the cytochrome b-c1 complex subunit 1 from complex III. Future studies are required to address if NO-related species can affect the enzymatic activity of complexes II or III via S-nitrosylation and if this might contribute to neurodegenerative phenomena.
Additionally, during oxidative phosphorylation, S-nitrosylation inhibits the activity of complex IV (cytochrome oxidase) and ATP synthase (complex V). In subunit II of complex IV, S-nitrosylation can occur at Cys 196 and 200, localized within the active center responsible for the conversion of oxygen to H2O, thus leading to inhibition of complex IV activity [71]. Interestingly, ischemic preconditioning of mouse heart results in S-nitrosylation of the α1 subunit of F1-ATP synthase, inhibiting its activity [44]. Additional studies are needed to determine if S-nitrosylation of complexes IV and V contributes to the pathophysiology of neurodegenerative disorders.
NO signaling impairs oxidative phosphorylation – Effect of nitration and other nitrosylation reactions
Tyrosine nitration inhibits complex I in an irreversible manner, although SNO-dependent inhibition is believed to be more important [72, 73]. In cell culture models, nitration of complex I is associated with decreased ATP synthesis during oxidative phosphorylation, enhancing apoptotic and necrotic cell death [72, 74]. Additionally, high concentrations of peroxynitrite inhibit all ETC complexes, including complex V, consistent with the notion that tyrosine nitration takes place in a relatively non-specific manner [73, 75, 76]. Similar to S-nitrosylation, nitration of ETC complexes decreases ATP production, and is thus associated with mitochondrial dysfunction. However, it is not yet known if these tyrosine nitration events mediate RNS-related neurodegenerative processes. Further studies are warranted to determine the pathophysiological role of tyrosine nitration of the ETC in neurodegenerative disorders.
Additionally, NO species inhibit mitochondrial ATP production via direct reaction with heme and copper centers of ETC components. In fact, one of the earliest studies on NO-related species and mitochondria concerns direct binding to complex IV (cytochrome c oxidase) [77]. More recent studies have suggested that NO-related species may undergo at least two different reactions with cytochrome c oxidase [78]. NO rapidly inhibits cytochrome oxidase at nanomolar levels, suggesting that cytochrome oxidase is a potential (patho)physiologically-relevant target of NO [79]. Subsequent experiments demonstrated that NO binds to the same site that oxygen binds to in complex IV, thus competitively blocking oxygen binding to inhibit the enzyme’s activity [80, 81].
In summary, it is likely that under pathological conditions NO-related species inhibit oxidative phosphorylation at multiple sites, and in large measure via protein S-nitrosylation. Future studies are needed to determine which S-nitrosylation reactions in neuronal systems are instrumental in contributing to the pathogenesis of neurodegenerative diseases.
Nitrosative stress impairs mitochondrial dynamics and mitophagy in neurodegeneration – links to metabolic disease
Mitochondria are highly dynamic organelles, continuously undergoing fission and fusion (collectively termed “mitochondrial dynamics”) to rearrange mitochondrial networks. This process plays a critical role in local ATP production in axons and at synapses. Additionally, mitochondrial fission facilitates selective removal of damaged mitochondria through an autophagic mechanism known as mitophagy. Accordingly, impairment in mitochondrial dynamics and mitophagy is associated with deficient mitochondrial metabolism and function [82], and is thereby linked to neurological conditions.
A crucial role for protein S-nitrosylation in mitochondrial dynamics has recently emerged. Specifically, in AD models our group demonstrated a dramatic increase in S-nitrosylation of the mitochondrial fission protein, dynamin-related protein 1 (Drp1), triggering its hyperactivation. This results in excessive mitochondrial fragmentation, bioenergetic compromise, and consequent synaptic loss and neuronal damage (Figure 4) [34]. Drp1 is a member of the dynamin GTPase superfamily and mediates mitochondrial fission via formation of a ring-like structure, composed of Drp1 oligomers/multimers, at the site of mitochondrial fission. S-Nitrosylation of Drp1 at Cys644 enhances its oligomerization and thus GTPase activity. Importantly, mutation of this target cysteine prevented excessive mitochondrial fission and ameliorated synaptic injury engendered by oligomerized Aβ, which stimulates NO production, suggesting that SNO-Drp1 represents a critical step in the pathogenesis of AD.
Figure 4. Mitochondrial Dynamics and S-Nitrosylation.

Mitochondrial fission and fusion events (termed mitochondrial dynamics) are constantly occurring to maintain a proper mitochondrial functional network in neurons. In Alzheimer’s disease (AD), increased NO production due to the presence of Aβ oligomers hyperstimulates the mitochondrial fission activity of Drp1, resulting in excessive mitochondrial fragmentation. The fragmented mitochondria morphologically manifest abnormal cristae and bioenergetic compromise with decreased ATP production. The disruption in mitochondrial metabolism contributes to synaptic damage and thus to the pathogenesis of AD. Damaged and fragmented mitochondria are selectively eliminated via autophagy (i.e., mitophagy). In conjunction with PINK1, parkin participates in the process of mitophagy, contributing to cytoprotection. However, S-nitrosylation of PINK1 and parkin impairs mitophagy.
Importantly, we also found that elevated glucose levels, as found in T2DM/MetS, representing a known risk factor for AD, enhance NO production synergistically with oligomeric Aβ peptide. In the leptin receptor mutant (db/db) mouse model of T2DM as well as in AD, this increase in NO results in S-nitrosylation of Drp1 and consequent hyperactivation of mitochondrial fission machinery, contributing to bioenergetic compromise and synaptic damage [83]. Moreover, S-nitrosylation of Drp1 has been observed in mouse models of HD [84], suggesting that SNO-Drp1-mediated excessive mitochondrial fragmentation may contribute to a variety of neurodegenerative conditions. Along these lines, we and others have found that SNO-Drp1 levels are significantly elevated in postmortem human brain samples from patients diagnosed with AD or HD [34, 85].
In another example of aberrant protein S-nitrosylation in neurodegenerative disorders, formation of S-nitrosylated parkin, an E3 ubiquitin ligase whose genetic mutation has been linked to autosomal recessive juvenile-onset parkinsonism, may also contribute to abnormal mitochondrial dynamics in models of sporadic PD. S-Nitrosylation of parkin affects its enzymatic activity in a biphasic manner, initially increasing parkin E3 ligase activity followed by inactivation [86]. A recent study showed that mitochondrial toxin-induced S-nitrosylation of parkin also resulted in an increase in Drp1 expression and activity [87]. In this paradigm, SNO-parkin stabilized Drp1, not only via inhibition of parkin E3 ligase activity, but also by decreasing parkin binding to Drp1.
Additionally, in conjunction with PINK1, whose gene can also be mutated to cause familial PD, parkin normally maintains mitochondrial quality control by promoting mitophagy flux [88]. In this PINK1/parkin pathway, PINK1 accumulates on damaged mitochondria, selectively recruits parkin from the cytosol, and then phosphorylates parkin to stimulate its ubiquitin E3 ligase activity. This process promotes flux through the mitophagy pathway, eliminating damaged mitochondria. Recent studies, however, demonstrate that exposure of cells to mitochondrial toxins leads to SNO-parkin formation [86, 89, 90]. The initial increase in E3 ligase activity upon S-nitrosylation of parkin upregulates mitophagy; however, subsequent attenuation in SNO-parkin E3 ligase activity interferes with mitophagy [90]. Moreover, a new study from our group shows that S-nitrosylation of PINK1 precedes formation of SNO-parkin during the development of PD. We report that S-nitrosylation of PINK1 impairs mitophagy in dopaminergic neurons derived from human induced pluripotent stem cells and present evidence for the formation of SNO-PINK1 early in the pathogenesis of PD in transgenic mouse models [91]. Taken together, these results are consistent with the notion that multiple, sequential S-nitrosylation reactions can contribute to dysfunctional mitophagy under at various stages of PD.
Concluding Remarks and Future Perspectives
In this review, we have highlighted some of the molecular links between protein S-nitrosylation and mitochondrial dysfunction and their pathological relationship to neurodegenerative and metabolic diseases. We cite evidence supporting the notion that excessive production of NO-related species contributes to mitochondrial bioenergetic failure, and that these events are key mediators of synaptic damage in neurodegenerative diseases. In animal models, researchers have begun to adopt strategies to improve mitochondrial function and thus ameliorate neurodegenerative phenotypes. For instance, administration of nicotinamide adenine dinucleotide (NAD+) to correct NAD+/NADH imbalance due to impairment in oxidative phosphorylation reportedly ameliorated retinal ganglion cell neuronal damage in a mouse model of glaucoma [92]. Additionally, ketone bodies were used to bypass glycolysis and generate acetyl-CoA for the TCA cycle to mitigate the hypometabolism that precedes loss of memory and cognitive decline in AD models [93]. While these therapeutic approaches may be beneficial by improving metabolic imbalance, they are not expected to affect the underlying nitrosative stress that lead to aberrant S-nitrosylation reactions contributing to mitochondrial dysfunction under pathological conditions. Hence, it is uncertain if these particular modalities will offer lasting benefit.
Concerning the mitochondrial impairment engendered by aberrant NO signaling, additional studies to address the “Outstanding Questions” outlined in this review (and summarized below) are warranted. Further characterization of pathological NO signaling contributing to imbalance in mitochondrial metabolism may lead to the development of more efficient and specific therapies to modify disease pathogenesis.
Outstanding Questions.
Which cysteine residues are selectively S-nitrosylated under pathophysiologically relevant conditions in specific proteins? Additional biochemical studies are needed to verify SNO-site(s) identified by MS-based proteomics and to carefully quantify the percentage of protein that is S-nitrosylated under various pathological conditions.
What is the complete S-nitrosoproteome in various neurodegenerative and metabolic disorders? Emerging MS techniques that employ novel and increasingly selective SNO-protein probes will be used to tackle this problem. Identification of specific SNO-proteins in situ with novel fluorescent probes remains a challenge for the field.
What are the functional consequences of SNO-modification on each protein involved in mitochondrial metabolism that is identified by S-nitrosoproteomics? Experiments are needed to analyze the biochemical, molecular and cellular consequences of these redox modifications under conditions that mimic specific disease states.
Does S-nitrosylation-mediated inhibition of mitochondrial metabolism contribute to neuropathological phenotypes in cellular and animal models? Do the quantitative levels or percentage of S-nitrosylated protein found in human brain or other tissues in a disease state match the degree of protein S-nitrosylation in the animal and cell-based models?
Does tyrosine nitration of specific mitochondrial proteins contribute to mitochondrial dysfunction in models of neurodegenerative diseases? Do the quantitative levels of nitrotyrosine on a specific protein (or percentage of protein affected) in human brain or other relevant tissues in a disease state match the degree of tyrosine nitration found in the animal and cell-based models?
Trends.
Neurons are particularly sensitive to dysfunction in mitochondrial metabolism due to their high-energy demands.
Excessive production of nitric oxide (NO)-related species is linked to mitochondrial metabolic defects in neurodegenerative diseases.
Aberrant protein S-nitrosylation (producing SNO-proteins) mediates pathological NO signaling cascades, contributing to mitochondrial metabolic dysfunction.
A wide range of aberrantly S-nitrosylated mitochondrial proteins that may contribute to neurodegenerative diseases has been identified; these SNO-proteins include enzymes involved in the tricarboxylic acid (TCA) cycle, the electron transport chain (ETC), and mitochondrial dynamics.
Aberrant S-nitrosylation of specific proteins may represent a key mediator of both type 2 diabetes mellitus (T2DM)/metabolic syndrome (MetS) and neurodegenerative disorders such as Alzheimer’s disease (AD).
Acknowledgments
This work was supported in part by NIH grants P01 HD29587, DP1 DA041722, R01 NS086890, R01 AG056259, RF1 AG057409, and NINDS Core grant P30 NS076411 (S.A.L.).
Glossary
- Alzheimer’s disease (AD)
An irreversible, progressive neurodegenerative disorder, representing arguably the most common form of dementia. Pathological key features of this disease include extracellular deposits of β-amyloid (forming plaques) and the intraneuronal aggregation of hyperphosphylated tau (forming neurofibrillary tangles).
- Electron transport chain (ETC)
A series of large enzyme complexes that transfer electrons received from NADH (derived from the TCA cycle) to oxygen. These processes generate a proton gradient across the mitochondrial inner membrane, driving ATP synthesis.
- Glycolysis
A cytosolic enzymatic cascade to produce two molecules of ATP from glucose without the consumption of molecular oxygen. The end product is pyruvate.
- Parkinson’s disease (PD)
A chronic progressive neurodegenerative disease, initially manifesting severe motor symptoms. The major initial neuropathological hallmark of PD is pronounced loss of dopaminergic neurons in the midbrain. Surviving neurons often contain intracellular inclusions known as Lewy bodies composed of aggregated α-synuclein and other misfolded proteins. The disease can spread to other parts in the brain, resulting in cognitive dysfunction in addition to motor deficits.
- Reactive nitrogen and oxygen species (RNS/ROS)
Reactive chemical species containing nitrogen, oxygen, or both. Examples include nitric oxide (·NO), superoxide anion (O2·−), and peroxynitrite (ONOO−). These molecules are typically unstable, easily react with biomolecules such as proteins, lipids, and DNAs, and when excessively produced, elicit cell toxicity. Some species are free radicals, manifesting an unpaired, very reactive electron.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nakamura T, et al. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol Dis. 2015;84:99–108. doi: 10.1016/j.nbd.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakamura T, et al. Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langston JW, et al. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 4.Sipos I, et al. Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J Neurochem. 2003;84:112–118. doi: 10.1046/j.1471-4159.2003.01513.x. [DOI] [PubMed] [Google Scholar]
- 5.Rossetti ZL, et al. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and free radicals in vitro. Biochem Pharmacol. 1988;37:4573–4574. doi: 10.1016/0006-2952(88)90674-0. [DOI] [PubMed] [Google Scholar]
- 6.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 7.Reddy PH, Reddy TP. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr Alzheimer Res. 2011;8:393–409. doi: 10.2174/156720511795745401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lipton SA, et al. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 9.Molokanova E, et al. Differential effects of synaptic and extrasynaptic NMDA receptors on Abeta-induced nitric oxide production in cerebrocortical neurons. J Neurosci. 2014;34:5023–5028. doi: 10.1523/JNEUROSCI.2907-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.St-Pierre J, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 11.Cassarino DS, et al. Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson’s disease. Biochim Biophys Acta. 1997;1362:77–86. doi: 10.1016/s0925-4439(97)00070-7. [DOI] [PubMed] [Google Scholar]
- 12.Smith BC, Marletta MA. Mechanisms of S-nitrosothiol formation and selectivity in nitric oxide signaling. Curr Opin Chem Biol. 2012;16:498–506. doi: 10.1016/j.cbpa.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stamler JS, et al. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 14.Choi MS, et al. Transnitrosylation from DJ-1 to PTEN attenuates neuronal cell death in parkinson’s disease models. J Neurosci. 2014;34:15123–15131. doi: 10.1523/JNEUROSCI.4751-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura T, et al. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell. 2010;39:184–195. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qu J, et al. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc Natl Acad Sci USA. 2011;108:14330–14335. doi: 10.1073/pnas.1105172108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu Z, et al. S-Nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 18.Ischiropoulos H. Protein tyrosine nitration--an update. Arch Biochem Biophys. 2009;484:117–121. doi: 10.1016/j.abb.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 19.Yin F, et al. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal. 2014;20:353–371. doi: 10.1089/ars.2012.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magistretti PJ, Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86:883–901. doi: 10.1016/j.neuron.2015.03.035. [DOI] [PubMed] [Google Scholar]
- 21.Orrenius S, et al. Calcium and mitochondria in the regulation of cell death. Biochem Biophys Res Commun. 2015;460:72–81. doi: 10.1016/j.bbrc.2015.01.137. [DOI] [PubMed] [Google Scholar]
- 22.Itoh Y, et al. Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc Natl Acad Sci USA. 2003;100:4879–4884. doi: 10.1073/pnas.0831078100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng X, et al. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife. 2016:5. doi: 10.7554/eLife.13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belanger M, et al. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 25.Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duka T, et al. Synaptosomal lactate dehydrogenase isoenzyme composition is shifted toward aerobic forms in primate brain evolution. Brain Behav Evol. 2014;83:216–230. doi: 10.1159/000358581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashimoto T, et al. Evidence for the mitochondrial lactate oxidation complex in rat neurons: demonstration of an essential component of brain lactate shuttles. PLoS One. 2008;3:e2915. doi: 10.1371/journal.pone.0002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Machler P, et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016;23:94–102. doi: 10.1016/j.cmet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Supplie LM, et al. Respiration-Deficient Astrocytes Survive As Glycolytic Cells In Vivo. J Neurosci. 2017;37:4231–4242. doi: 10.1523/JNEUROSCI.0756-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Volkenhoff A, et al. Glial Glycolysis Is Essential for Neuronal Survival in Drosophila. Cell Metab. 2015;22:437–447. doi: 10.1016/j.cmet.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Funfschilling U, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–521. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dienel GA, Cruz NF. Aerobic glycolysis during brain activation: adrenergic regulation and influence of norepinephrine on astrocytic metabolism. J Neurochem. 2016;138:14–52. doi: 10.1111/jnc.13630. [DOI] [PubMed] [Google Scholar]
- 33.Giorgi-Coll S, et al. Succinate supplementation improves metabolic performance of mixed glial cell cultures with mitochondrial dysfunction. Sci Rep. 2017;7:1003. doi: 10.1038/s41598-017-01149-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho DH, et al. S-Nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oettinghaus B, et al. Synaptic dysfunction, memory deficits and hippocampal atrophy due to ablation of mitochondrial fission in adult forebrain neurons. Cell Death Differ. 2016;23:18–28. doi: 10.1038/cdd.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mongin AA, et al. Selective vulnerability of synaptic signaling and metabolism to nitrosative stress. Antioxid Redox Signal. 2012;17:992–1012. doi: 10.1089/ars.2012.4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bubber P, et al. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- 38.Parker WD, Jr, Parks JK. Mitochondrial ND5 mutations in idiopathic Parkinson’s disease. Biochem Biophys Res Commun. 2005;326:667–669. doi: 10.1016/j.bbrc.2004.11.093. [DOI] [PubMed] [Google Scholar]
- 39.Bender A, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 40.Kraytsberg Y, et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 41.Ryan SD, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell. 2013;155:1351–1364. doi: 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okamoto S, et al. S-Nitrosylation-mediated redox transcriptional switch modulates neurogenesis and neuronal cell death. Cell Rep. 2014;8:217–228. doi: 10.1016/j.celrep.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piantadosi CA. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim Biophys Acta. 2012;1820:712–721. doi: 10.1016/j.bbagen.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun J, et al. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 45.Chouchani ET, et al. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem J. 2010;430:49–59. doi: 10.1042/BJ20100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bak DW, et al. Identifying Functional Cysteine Residues in the Mitochondria. ACS Chem Biol. 2017;12:947–957. doi: 10.1021/acschembio.6b01074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doulias PT, et al. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci Signal. 2013;6:rs1. doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raju K, et al. Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci Signal. 2015;8:ra68. doi: 10.1126/scisignal.aaa4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seneviratne U, et al. S-nitrosation of proteins relevant to Alzheimer’s disease during early stages of neurodegeneration. Proc Natl Acad Sci USA. 2016;113:4152–4157. doi: 10.1073/pnas.1521318113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kohr MJ, et al. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res. 2011;108:418–426. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fatania HR, et al. Chemical modification of rat liver cytosolic NADP(+)-linked isocitrate dehydrogenase by N-ethylmaleimide. Evidence for essential sulphydryl groups. FEBS Lett. 1993;322:245–248. doi: 10.1016/0014-5793(93)81579-o. [DOI] [PubMed] [Google Scholar]
- 52.Gupta KJ, et al. Inhibition of aconitase by nitric oxide leads to induction of the alternative oxidase and to a shift of metabolism towards biosynthesis of amino acids. J Exp Bot. 2012;63:1773–1784. doi: 10.1093/jxb/ers053. [DOI] [PubMed] [Google Scholar]
- 53.Tortora V, et al. Mitochondrial aconitase reaction with nitric oxide, S-nitrosoglutathione, and peroxynitrite: mechanisms and relative contributions to aconitase inactivation. Free Radic Biol Med. 2007;42:1075–1088. doi: 10.1016/j.freeradbiomed.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 54.Lee JH, et al. Inactivation of NADP+-dependent isocitrate dehydrogenase by peroxynitrite. Implications for cytotoxicity and alcohol-induced liver injury. J Biol Chem. 2003;278:51360–51371. doi: 10.1074/jbc.M302332200. [DOI] [PubMed] [Google Scholar]
- 55.Yang ES, et al. Inactivation of NADP(+)-dependent isocitrate dehydrogenase by nitric oxide. Free Radic Biol Med. 2002;33:927–937. doi: 10.1016/s0891-5849(02)00981-4. [DOI] [PubMed] [Google Scholar]
- 56.Nimmo GA, Nimmo HG. The regulatory properties of isocitrate dehydrogenase kinase and isocitrate dehydrogenase phosphatase from Escherichia coli ML308 and the roles of these activities in the control of isocitrate dehydrogenase. Eur J Biochem. 1984;141:409–414. doi: 10.1111/j.1432-1033.1984.tb08206.x. [DOI] [PubMed] [Google Scholar]
- 57.Diers AR, et al. Differential regulation of metabolism by nitric oxide and S-nitrosothiols in endothelial cells. Am J Physiol Heart Circ Physiol. 2011;301:H803–812. doi: 10.1152/ajpheart.00210.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Molina y Vedia L, et al. Nitric oxide-induced S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase inhibits enzymatic activity and increases endogenous ADP-ribosylation. J Biol Chem. 1992;267:24929–24932. [PubMed] [Google Scholar]
- 59.Izumi Y, Zorumski CF. Neuroprotective effects of pyruvate following NMDA-mediated excitotoxic insults in hippocampal slices. Neurosci Lett. 2010;478:131–135. doi: 10.1016/j.neulet.2010.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mohr S, et al. Mechanism of covalent modification of glyceraldehyde-3-phosphate dehydrogenase at its active site thiol by nitric oxide, peroxynitrite and related nitrosating agents. FEBS Lett. 1994;348:223–227. doi: 10.1016/0014-5793(94)00596-6. [DOI] [PubMed] [Google Scholar]
- 61.Hara MR, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 62.Murray CI, et al. Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol Cell Proteomics. 2012;11:M111 013441. doi: 10.1074/mcp.M111.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Di Giacomo G, et al. Established Principles and Emerging Concepts on the Interplay between Mitochondrial Physiology and S-(De)nitrosylation: Implications in Cancer and Neurodegeneration. Int J Cell Biol. 2012;2012:361872. doi: 10.1155/2012/361872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galkin A, Moncada S. Modulation of the conformational state of mitochondrial complex I as a target for therapeutic intervention. Interface Focus. 2017;7:20160104. doi: 10.1098/rsfs.2016.0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borutaite V, et al. Reversal of nitric oxide-, peroxynitrite- and S-nitrosothiol-induced inhibition of mitochondrial respiration or complex I activity by light and thiols. Biochim Biophys Acta. 2000;1459:405–412. doi: 10.1016/s0005-2728(00)00178-x. [DOI] [PubMed] [Google Scholar]
- 66.Clementi E, et al. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galkin A, Moncada S. S-Nitrosation of mitochondrial complex I depends on its structural conformation. J Biol Chem. 2007;282:37448–37453. doi: 10.1074/jbc.M707543200. [DOI] [PubMed] [Google Scholar]
- 68.Hsu M, et al. Glutathione depletion resulting in selective mitochondrial complex I inhibition in dopaminergic cells is via an NO-mediated pathway not involving peroxynitrite: implications for Parkinson’s disease. J Neurochem. 2005;92:1091–1103. doi: 10.1111/j.1471-4159.2004.02929.x. [DOI] [PubMed] [Google Scholar]
- 69.Chouchani ET, et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Galkin A, et al. Lack of oxygen deactivates mitochondrial complex I: implications for ischemic injury? J Biol Chem. 2009;284:36055–36061. doi: 10.1074/jbc.M109.054346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang J, et al. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am J Physiol Cell Physiol. 2005;288:C840–849. doi: 10.1152/ajpcell.00325.2004. [DOI] [PubMed] [Google Scholar]
- 72.Yamamoto T, et al. Selective nitration of mitochondrial complex I by peroxynitrite: involvement in mitochondria dysfunction and cell death of dopaminergic SH-SY5Y cells. J Neural Transm (Vienna) 2002;109:1–13. doi: 10.1007/s702-002-8232-1. [DOI] [PubMed] [Google Scholar]
- 73.Murray J, et al. Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. J Biol Chem. 2003;278:37223–37230. doi: 10.1074/jbc.M305694200. [DOI] [PubMed] [Google Scholar]
- 74.Davis CW, et al. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic Biol Med. 2010;48:306–317. doi: 10.1016/j.freeradbiomed.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Castro L, et al. Mitochondrial protein tyrosine nitration. Free Radic Res. 2011;45:37–52. doi: 10.3109/10715762.2010.516254. [DOI] [PubMed] [Google Scholar]
- 76.Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- 77.Wainio WW. Reactions of cytochrome oxidase. J Biol Chem. 1955;212:723–733. [PubMed] [Google Scholar]
- 78.Sarti P, et al. The Chemical Interplay between Nitric Oxide and Mitochondrial Cytochrome c Oxidase: Reactions, Effectors and Pathophysiology. Int J Cell Biol. 2012;2012:571067. doi: 10.1155/2012/571067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 80.Taylor CT, Moncada S. Nitric oxide, cytochrome C oxidase, and the cellular response to hypoxia. Arterioscler Thromb Vasc Biol. 2010;30:643–647. doi: 10.1161/ATVBAHA.108.181628. [DOI] [PubMed] [Google Scholar]
- 81.Moncada S, Erusalimsky JD. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat Rev Mol Cell Biol. 2002;3:214–220. doi: 10.1038/nrm762. [DOI] [PubMed] [Google Scholar]
- 82.Wai T, Langer T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 83.Akhtar MW, et al. Elevated glucose and oligomeric beta-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat Commun. 2016;7:10242. doi: 10.1038/ncomms10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Haun F, et al. S-Nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid Redox Signal. 2013;19:1173–1184. doi: 10.1089/ars.2012.4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang X, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yao D, et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci USA. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Z, et al. The Essential Role of Drp1 and Its Regulation by S-Nitrosylation of Parkin in Dopaminergic Neurodegeneration: Implications for Parkinson’s Disease. Antioxid Redox Signal. 2016;25:609–622. doi: 10.1089/ars.2016.6634. [DOI] [PubMed] [Google Scholar]
- 88.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chung KK, et al. S-Nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 90.Ozawa K, et al. S-Nitrosylation regulates mitochondrial quality control via activation of parkin. Sci Rep. 2013;3:2202. doi: 10.1038/srep02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oh CK, et al. S-Nitrosylation of PINK1 Attenuates PINK1/Parkin-Dependent Mitophagy in hiPSC-Based Parkinson’s Disease Models. Cell Rep. 2017 doi: 10.1016/j.celrep.2017.10.068. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Williams PA, et al. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science. 2017;355:756–760. doi: 10.1126/science.aal0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pawlosky RJ, et al. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J Neurochem. 2017;141:195–207. doi: 10.1111/jnc.13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]