

Abstract
Particular chromatin modifications are associated with different states of gene transcription, yet determining which modifications are causal ‘drivers’ in promoting transcription is incompletely understood. Here, we discuss new developments describing the ordered, mechanistic role of select histone marks occurring during distinct steps in the RNA Polymerase II (Pol II) transcription cycle. In particular, we highlight the interplay between histone marks in specifying the “next step” of transcription. While many studies have described correlative relationships between histone marks and their occupancy at distinct gene regions, we focus on studies that elucidate clear functional consequences of specific histone marks during different stages of transcription. These recent discoveries have refined our current mechanistic understanding of how histone marks promote Pol II transcriptional progression.
Keywords: RNA polymerase II, transcription cycle, chromatin, histone marks, epigenetics
Histone marks & gene transcription
The basic unit of chromatin is the nucleosome, which consists of a histone octamer (two copies each of H3, H4, H2A, and H2B) with 146 base pairs of DNA wrapped around the histones. The N-terminal histone “tails” are unstructured and protruded from the nucleosome [1], and are subject to extensive posttranslational modifications, or histone marks (reviewed in [2]), The potential functional role of histone marks was first postulated by Allfrey in 1964 [3] and subsequently developed into the histone code hypothesis [4]. Histone marks occur as a balance between different groups of enzymes and have diverse functions, many of which are not yet completely understood. In metazoans, the genes encoding histone proteins occur in clusters [5], making direct testing of particular histone modification sites in cells or in vivo extremely technically challenging. Therefore, most studies utilize cell-free systems and/or infer functions of histone modifications through modulating expression or activity of the enzymes that create, remove, or bind these histone marks. The enzymes that deposit histone marks, or “writers,” utilize cofactors that are often metabolites, including ATP, acetyl-CoA, or S-adenosylmethionine (SAM), to write the histone modifications of phosphorylation, acetylation, and methylation, respectively. Other enzymes called “erasers” remove histone modifications. Lastly, “readers” directly bind to modified histones, serving as effector proteins. Readers often do not act alone, but instead are part of larger, multi-subunit protein complexes, that include additional enzymes, scaffolds, etc., to exert downstream functions [4].
Epigenetic reader proteins have particular domains that interact with modified histone residues (reviewed in [6]). Importantly, many reader proteins do not only have one reader domain, but also often have several different domains, suggesting these proteins may be able to recognize combinations of different histone marks at the same time. Therefore, the presence or absence of neighboring marks may impact the affinity of a reader domain binding to a particular residue. Lysine residues on histone proteins are particularly abundant [7], and are more abundant than the other modifiable residues. These residues are subject to many different modifications, the best studied of which are methylated and acetylated lysines. Histone marks on several lysine residues are deposited on particular areas of the genome and associated with distinct states of gene transcription (Figure 1), and these patterns are generally conserved from yeast to humans. Chromatin immunoprecipitation assays first followed by microarrays (ChIP-chip), and now sequencing (ChIP-seq) have yielded extensive information about a variety of histone modifications and localizations in the genome [8,9]. Promoters of transcriptionally active genes are associated with enriched trimethylation on histone H3 lysine 4 (H3K4me3)) and lysine acetylation on histone H3 and H4, while actively transcribing genes tend to have higher levels of H3K36me3 and H3K79me3 in the gene body. Active cis-regulatory enhancer elements are defined by both H3K27ac and high levels of H3K4me1 relative to H3K4me3. Repressed genes have a much higher density of nucleosomes, and can be marked by H3K9 methylation, H3K27me3, and H4K20me3. While the correlation of these modifications with different genomic regions and states of gene expression has been widely described, whether and how they are functional in transcription (i.e. if they are causal or not) is still debated.
Figure 1. Genomic localization of histone H3 modifications associated with active transcription.
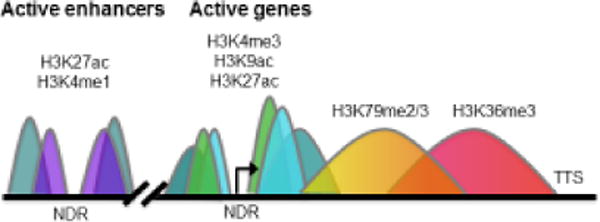
Simplified illustration of histone modification occupancy as defined by ChIP-seq assays is presented. Regions in color represent what a typical ChIP signal would look like at a given eukaryotic gene (teal = H3K27ac, purple = H3K4me1, light blue = H3K9ac, green = H3K4me3, yellow/orange = H3K79me2/3, pink/red = H3K36me3). H3K4me3 and H3K9ac are associated with transcriptionally active gene promoter regions, while H3K36me3 and H3K79me3 are localized to gene bodies of actively transcribing genes. H3K27ac localizes to both active gene promoters and enhancer regions, and H3K4me1 is predominantly enriched at enhancers. In contrast, repressed genes have different methylation marks and lack histone acetylation (not shown). NDR = nucleosome depleted region. Arrow denotes the transcription start site (TSS). TTS = transcription termination site.
Functions of histone marks in the RNA Pol II transcription cycle
Transcription of mRNA is catalyzed by Pol II, and supported by many additional general transcription factors (GTFs), which proceeds in a series of ordered steps known as the transcription cycle [10,11] (Figure 2). While it is estimated that the GTFs and the Mediator complex are composed of about 60 proteins in yeast [12], many more additional factors help regulate transcription [11]. Gene transcriptional programs are driven by transcription factors, which bind directly to specific DNA elements and recruit coregulators to activate or repress transcription. Additional proteins, non-coding RNAs, and cis-regulatory elements are critical for transcriptional regulation and can interact with the GTFs and Pol II, locally at the promoter or genic regions, or at distal regulatory elements such as enhancers. These proteins include coregulators that can function as scaffolds for the assembly of large protein complexes, enzymatic chromatin modifiers, and/or ATP-dependent remodelers [13]. In addition, long non-coding RNAs and enhancer-derived RNAs (eRNA) also can serve critical regulatory functions [14,15]. The chromatin environment is crucial during the process of transcription, serving a plethora of functions, including repressing spurious transcription, facilitating long-range enhancer-promoter interactions, and recruiting regulatory reader protein complexes. Here, we review the role of histone lysine residues associated with gene expression and how select modifications can serve to functionally “drive” different steps of the transcription cycle. The roles of chromatin in co-transcriptional processes such as splicing are comprehensively reviewed elsewhere [16,17].
Figure 2. The RNA Polymerase II (Pol II) transcription cycle.
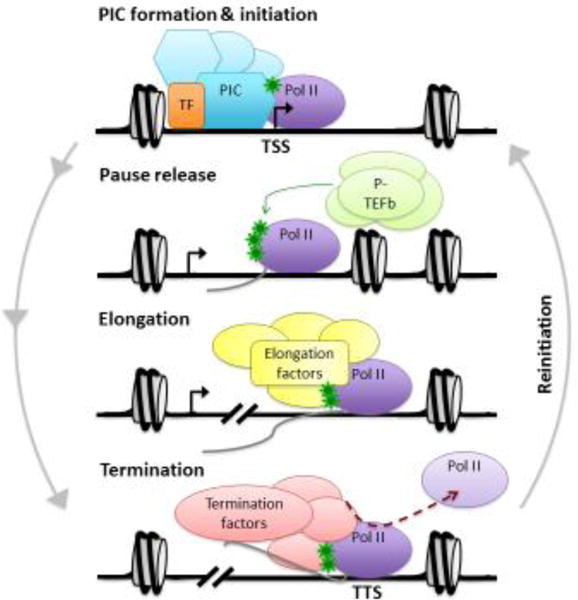
Schematic of the Pol II transcription cycle is presented. The transcription cycle begins with setting up the preinitiation complex (PIC) that includes Pol II and general transcription factors (GTFs), which are localized to core promoter elements around the transcription start site (TSS). Activator binding often drives recruitment of Pol II, designated as TF (transcription factor). On repressed genes, nucleosomes need to be moved or evicted in order to allow access of the GTFs to the promoter prior to PIC formation. Initiation occurs when Pol II creates the first phosphodiester bond and begins incorporating nucleotides into the mRNA chain. Pol II is phosphorylated near the TSS at serine 5 on its C-terminal domain (CTD), indicated by the green stars. After the RNA chain length reaches about 8 nucleotides, Pol II begins to leave the promoter. However, Pol II often pauses and needs to undergo pause release for full promoter clearance. Pause release is accompanied by phosphorylation mediated by P-TEFb targeting the Pol II CTD on serine 2, along with negative elongation factors (not shown). This release allows transcription to proceed to the next step of elongation, in which Pol II proceeds through the gene body and generates the full-length mRNA transcript with the help of elongation factors. The final stage of transcription is termination, in which the mRNA is cleaved and polyadenylated (not shown), and Pol II is released from the DNA template. TTS = transcription termination site. Finally, for the generation of multiple transcripts, the transcription cycle will repeat itself through an additional step of re-initiation.
Formation of the PIC & Transcription Initiation
Recruitment of GTFs and Pol II to promoters and formation of the pre-initiation complex (PIC) is required prior to the first step of transcription initiation, which occurs with opening of the DNA template and incorporation of the first nucleotide to start the mRNA chain. Recruitment of the Pol II GTFs is thought to occur in a stepwise, ordered fashion, and has been extensively reviewed [10]. It is estimated that many genes are regulated at the step of transcription initiation through the rate of recruitment of Pol II, which is driven by transcription factor binding to regulatory elements [18]. Additionally, histone modifications now are used to define transcriptionally active promoters and the best characterized of these modifications is H3K4me3.
Genome-wide studies in a variety of eukaryotic organisms demonstrate that H3K4me3 is enriched near the transcriptional start sites (TSSs) on many gene promoters and is positively associated with gene transcript levels [19]. H3K4me3 is written by a variety of methyltransferases, including the mixed lineage leukemia (MLL) and SET domain containing 1A & B (SETD1A/B) complexes (reviewed in [20,21]). The presence of H3K4me3 at promoters stimulates activator-dependent transcription in cell free assays, demonstrating a causal function [22–24]. H3K4me3 stimulates transcription by recruiting a variety of reader proteins, including TFIID, a key complex of GTFs that includes the TATA-binding protein (TBP) and TBP associated factors (TAFs), and is essential for promoter recognition and PIC formation. This recognition occurs through the direct binding activity of the TBP-associated factor 3 (TAF3) [23,25]. Mutating the plant homeodomain (PHD) domain in TAF3 reduces binding to H3K4me3 in vitro and significantly reduces TAF3 localization to promoter regions. Furthermore, this mutation also reduced in vitro transcription [23] and decreased the activity of a Gal4-ASH2L (a core component of the MLL H3K4 methyltransferase complexes)-dependent reporter construct [25]. The Roeder lab further directly examined the impact of H3K4me3 on PIC formation by performing in vitro ChIP on chromatin-assembled templates. They found that templates bearing H3K4me3 displayed increased occupancy of TFIID and Pol II specifically at the promoter and concluded that H3K4me3 is important for formation and/or stabilization of the PIC [23]. Together, these data suggest that H3K4me3 is integral for PIC assembly and Pol II transcription initiation.
Despite this evidence that H3K4me3 is important for promoting transcription initiation through the recruitment of TFIID, not all cellular genes show dependency on H3K4me3. For example, the loss of H3K4me3 in embryonic stem cells at CpG island-containing promoters did not affect steady state transcription, but the ectopic expression of H3K4me3 at new loci did increase transcriptional output [26]. Additionally, knockdown of core subunits of mammalian H3K4 methyltransferase complexes reduces global levels of H3K4 methylation [27–29], but this loss does not affect the steady state expression of every gene. An interesting example of when H3K4me3 does not correlate with transcription is at “bivalent genes”. In embryonic stem cells, both H3K4me3 and H3K27me3 occur at the same gene, which poises the gene for transcription activation once the repressive H3K27me3 mark is removed in response to differentiation cues [30,31]. Furthermore, TFIID has multiple modes of recruitment to core promoter elements, such as the TATA box and Initiator, that have been extensively characterized (reviewed in [10]), which may be more important in certain chromatin contexts or may serve to compensate for the loss of H3K4me3. Additionally, not all genes are subject to regulation at the level of transcription initiation, as different steps of the transcription cycle can also be rate-limiting and therefore may not impact steady state levels of transcription.
Promoter Clearance & Pol II Pause Release
The transition between transcription initiation and elongation is also a critical regulatory step for the expression of many genes. Studies measuring nascent transcription and Pol II occupancy demonstrate that Pol II is paused in the 5′ region of many genes [32–34], and this pausing is now estimated to impact between 30 – 70% of metazoan genes (reviewed in [35–37]). After initiation, Pol II must translocate from the core promoter region (promoter clearance), which is characterized by further unwinding of the DNA template and stabilization of the transcription bubble upon formation of at least a 6 bp DNA-RNA duplex [12,38,39]. After promoter clearance, Pol II often pauses in the promoter proximal region prior to productive RNA chain extension (elongation). At this point, Pol II will be released once it receives a signal, most often consisting of phosphorylation by positive transcription elongation factor b (P-TEFb). P-TEFb is a complex of cyclin T1 or T2 and cyclin-dependent kinase 9 (CDK9), which phosphorylates serine 2 of the Pol II C-terminal domain (CTD) [40–43]. This phosphorylation event is associated with release of Pol II from a paused state (see below), along with the phosphorylation of negative elongation factors (reviewed in [44]). For examples on the coordination between Pol II CTD phosphorylations and the histone code, see Box 1.
Box 1. Coordination of the Pol II CTD and histone codes.
The highly conserved Pol II CTD (reviewed extensively in [44] and [90]) consists of a series of heptad repeats with a consensus sequence of Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7. The number of repeats varies across species; for example, there are 26 repeats in yeast and 52 in humans. The CTD undergoes extensive posttranslational modifications comprising its own “CTD code”, including phosphorylation on the serines, threonines, and tyrosines, and isomerization of the prolines. Since the heptad sequence is repeated numerously, there is extensive combinatorial complexity of the different modifications. These modifications serve to direct recruitment of proteins involved in the transcription cycle and co-transcriptional processes, such as mRNA capping and splicing [91]. Furthermore, particular modifications are associated with distinct regions of genes and states of transcription, similar to histone modifications. For example, in human cells serine 5 phosphorylation (Ser5phos) peaks at the 5′ end of genes, and displays reduced signal further downstream. In contrast, signals for serine 2 phosphorylation (Ser2phos) are observed starting near the TSS, but peak much further downstream towards the TTS. As these CTD modifications are associated with distinct genic regions, they are also associated with particular histone marks.
Many epigenetic modifying enzymes interact with the Pol II CTD [90]. For example, in budding yeast the SAGA HAT complex has been reported to facilitate not only transcription activation but also RNA export through binding of its subunit Sus1 to the phosphorylated CTD [92]. Here we focus on the best-characterized direct interactions, which occur during transcription elongation. For example, the histone methyltransferase Set2 in S. cerevisiae binds to both Ser5phos and Ser2phos [93], providing a possible explanation for how H3K36me3 is deposited in a co-transcriptional manner. Furthermore, the Rpd3S HDAC complex that reads H3K36me3 also interacts with Ser5phos [94]. Likewise, DOT1L binds to both Ser5phos and Ser2phos [83], linking the CTD code to H3K79 methylation and transcription elongation. However, how and why DOT1L and H3K79me2/3 are possibly exchanged for SET2 and H3K36me3 remains unknown. Recently, the Bentley laboratory reported a connection among the kinetics of Pol II movement, the Pol II CTD, and the histone code. Expressing several different Pol II mutants in mammalian cells that harbor slower elongation rates through chromatin shifted the localization of H3K36me3 more toward the 5′ end of genes, which was accompanied by increased levels of Ser2phos in the same regions. Interestingly, this also altered the occupancy of H3K4me2 and increased divergent anti-sense transcription [95]. These examples illustrate part of the coordination that occurs between the Pol II CTD code and the histone code during transcription elongation, with additional interplay likely occurring throughout the transcription cycle.
Histone lysine acetylation is associated with productive transcription, and several readers of lysine acetylation are positively associated with Pol II pause release in metazoans. Bromodomain protein 4 (BRD4) is a scaffold protein containing two bromodomains, which recognize multiple acetylated residues in close proximity on histones [45]. Importantly, BRD4 can interact with P-TEFb to facilitate Pol II pause release from promoter proximal regions [46,47]. Interestingly, H3K9ac is highly associated with H3K4me3 at active gene promoters, and is selectively promoted on H3K4me3-containing nucleosomes through the reading and writing actions of GCN5/PCAF-containing histone acetyltransferase (HAT) complexes [24,48] (for additional examples of interplay between histone marks, see Box 2). Since H3K4me3 recruits components of the PIC, this interplay raises the question: what is the role of H3K9ac in downstream transcriptional processes? Our recent studies demonstrate a function for this particular histone mark, H3K9ac, in Pol II pause release of a subset of genes in HeLa cells [49]. This action is not through BRD4, but instead through the reading capabilities of the YEATS domain-containing proteins ALL1-fused gene from chromosome 9 protein (AF9), and to a lesser extent, eleven-nineteen leukemia (ENL), to H3K9ac. In this case, AF9 and/or ENL again recruit P-TEFb to acetylated histone residues, but as subunits of the entire super elongation complex (SEC), which functions to release Pol II from a paused state [50]. Notably, we demonstrate that mutating H3K9 to arginine reduces transcription after Pol II pause release in a cell-free system, and loss of H3K9ac leads to an increase in Pol II pausing on select genes in HeLa cells. These data suggest that H3K9ac can mediate Pol II pause release through recruitment of the entire SEC.
Box 2. Interplay between histone tail modifications.
In addition to the interplay described between H3K4me3 and H3K9ac, other histone marks have been demonstrated to impact one another. One well-characterized example is the stimulation of H3K4me3 by ubiquitination on histone H2B (reviewed in [96]). Additionally, there are several examples of histone phosphorylation promoting neighboring lysine acetylation. Recently, the Allis lab demonstrated that phosphorylation on histone H3 serine 28 (H3S28phos) results in increased H3K27ac, mediated by enhanced binding of the HAT p300 to H3S28phos, which is linked with transcriptional activation in stimulated macrophages [97]. Additionally, the HAT GCN5 binds to phosphorylated H3 serine 10 (H3S10phos), resulting in increased acetylation on H3K9 and H3K14 [98,99]. Using the FOSL1 gene as a model to study transcriptional responses to serum stimulation, the Oliviero laboratory demonstrated that the phosphorylation binding protein 14-3-3 also binds to H3S10phos upon serum stimulation. 14-3-3 recruits the HAT MOF, which in turn acetylates histone H4 at lysine 16 and promotes binding of BRD4 along with P-TEFb for subsequent transcription elongation [100]. It therefore seems likely that kinase cascades in response to environmental cues and signaling molecules will alter histone modifications and impact transcriptional responses, such as Pol II pause release via H3K9ac. Furthermore, since p300 can catalyze crotonylation in addition to acetylation [52], it will be interesting to determine if other short chain acyl modifications are stimulated thorough histone phosphorylations.
Furthermore, reader proteins often have multiple domains and have the potential to recognize combinations of histone marks. A prime example of this occurs with the protein bromodomain PHD finger transcription factor (BPTF), which utilizes both a PHD domain to recognize H3K4me3 and a neighboring bromodomain to bind histone H4 lysine 16 acetylation (H4K16ac) on the same nucleosome [101]. This occurrence of multiple epigenetic reader domains located in close proximity to one another is found on many proteins, and moreover, reader proteins often work as part of larger protein complexes, which may contain multiple proteins that have additional epigenetic binding domains. Elucidating the combinatorial binding activities and functions of these reader domains will likely reveal additional functions of histone marks and epigenetic readers in transcription.
In addition to histone lysine acetylation, it is becoming more appreciated that additional acyl short chain modifications are common histone modifications [7] and may also play critical roles in transcription (reviewed in [51]). The Allis and Shi laboratories recently demonstrated that lysine crotonylation is associated with lipopolysaccharide stimulation of Pol II transcription in macrophages [52,53]. Furthermore, p300-dependent crotonylation on H3K18 (H3K18cr) stimulates cell-free transcription, thereby directly demonstrating a causal role. In cells, the level of H3K18cr is sensitive to changes in the cellular crotonyl-CoA pool [52], thus suggesting that different acyl modifications may arise from global cellular changes in metabolites and directly impact transcription. Recently, YEATS domains were characterized as readers of crotonylated histone lysine residues, in addition to acetylated residues. The S. cerevisiae YEATS domain-containing protein Taf14 [54] and the mammalian AF9 and ENL proteins [53] all bind to crotonylated histone lysines. Structural studies suggest that while bromodomains tend to have a shallower binding grove, the YEATS domains can accommodate the additional side-chain length of crotonylation [53], suggesting that the recognition of longer acyl chains on lysines may be selective to YEATS domains. It would therefore be interesting to see if crotonylation or other acyl modifications have any role in recruitment of the SEC and Pol II pause release.
Transcription Elongation
Once Pol II escapes promoter proximal pausing, it undergoes the process of productive elongation. During transcription elongation, Pol II must transcribe the entire length of the gene and contend with nucleosomes in its path and the surrounding chromatin environment. Importantly, there have been many complexes identified that assist Pol II during transcription elongation, including (but not limited to) P-TEFb-containing complexes as mentioned above, the Pol II associated factor 1 (Paf1) complex, TFIIF, facilitator of chromatin transcription (FACT), elongin complexes, and disruptor of telomeric silencing-like (DOT1L)-containing complexes [55–59].
Several lysine methylation events are associated with transcription elongation. H3K36me3 is associated with active transcription and localizes to gene bodies [33,60] and is deposited by Set2 in yeast [61] and SETD2 in humans [62]. Set2 travels with Pol II during transcription and deposits H3K36me3 in a co-transcriptional manner [63,64]. Surprisingly, H3K36me3 is repressive towards transcription, as localization of H3K36me3 to a gene promoter via Set2 tethering decreases transcription [61]. The appearance of H3K36me3 in gene bodies recruits an H3K36me3 reader, which differs between species. For example, in budding yeast the histone deacetylase (HDAC) complex Rpd3S reads H3K36me3 through a PWWP domain in its Eaf3 subunit. H3K36me3 and this subsequent recruitment event in yeast prevents spurious transcription initiation in intragenic regions, including a subset of intragenic antisense transcripts, as Rpd3S is necessary for removing transcription-coupled histone acetylation and restoring nucleosome positioning after transcription elongation [65,66]. Interestingly, a similar mechanism for repressing noncoding RNA transcription initiating from cryptic promoters closer to the 5′ end of yeast genes occurs through the binding another HDAC complex (Set3C) to dimethylated H3 lysine 4 (H3K4me2) via its PHD domain [67].
In contrast, mammalian cells have distinct mechanisms to repress spurious transcription through H3K36me3. The Impey lab demonstrated that a histone H3K4me3 demethylase, KDM5B, co-localizes with H3K36me3 in embryonic stem cells. This co-localization is dependent on the protein MRG15, which is the orthologue of the yeast Eaf3 (the H3K36me3 reader of the Rpd3S complex). Loss of KDM5B localization results in increased intragenic H3K4me3 and increased spurious transcription on a subset of genes in embryonic stem cells [68]. In addition, the de novo DNA methyltransferases DNMT3a and DNMT3b are reported to be readers of H3K36me3. While DNA methylation in promoter regions is associated with transcription repression, gene body DNA methylation is associated with active transcription [69]. The Jeltsch lab first reported that DNMT3a selectively binds to H3K36me3 through its PWWP domain, and that H3K36me3 is necessary for maximal methylation activity of DNMT3a in cell-free assays [70]. Additionally, multiple groups have described that the genome-wide occupancy of DNMT3b in mammalian cells significantly overlaps with H3K36me3 in gene bodies [71–73]. Extensive work by the Oliviero laboratory demonstrated that DNMT3b co-localizes with H3K36me3, and knockdown of either DNMT3b or SETD2 increases intragenic transcription through the creation of new TSSs [73]. Therefore, while H3K36me3 is associated with actively transcribing genes across multiple species, it serves to recruit reader complexes through multiple different mechanisms to prevent transcription arising from unwanted places throughout the gene body.
In addition to recruiting proteins critical for preventing spurious transcription as described above, H3K36me3 also impacts different aspects of transcription. Knockdown of SETD2 and reduction of H3K36me3 decreases the occupancy of the FACT elongation complex on chromatin and FACT-mediated histone displacement during transcription [74]. However, it is unknown at this point whether or not FACT directly interacts with H3K36me3 or SETD2, or if this recruitment is bridged by another reader protein. Additionally, ZMYND11 was discovered to bind to K36me3 specifically on the replication-independent histone variant H3.3 [75,76], which regulates both pre-mRNA processing through intron retention [75] and transcription elongation [76].
Another histone modification associated with transcription elongation is methylation on H3K79. This mark was first demonstrated to be associated with active chromatin on select genes [77,78], and both H3K79me2 and H3K79me3 are associated with active transcription on a global level [60,79,80]. H3K79 methylation is mainly localized to intragenic regions, but in a distinct pattern as compared to H3K36me3: H3K79me2 and H3K79me3 occupancies peak earlier than H3K36me3, and are more enriched towards the 5′ end of genes compared to the 3′ end [60,81]. Methylation on H3K79 is catalyzed by DOT1 in yeast and DOT1L in mammals (reviewed in [82]). In different biochemical purifications, DOT1L is a constituent member of several elongation complexes, including the ENL-associated proteins (EAP) [57], DotCom [59], and AF4-purified complexes [58]. DOT1L binds to the Pol II CTD, and is thought to travel with elongating Pol II and deposit methylation on H3K79 in a co-transcriptional manner [83]. While many studies describe the correlation between H3K79 methylation and transcription, many questions still remain. The mechanistic function of H3K79 methylation is unknown, and readers for H3K79 methylation still need to be identified. The identification of H3K79 methylation readers would likely offer significant insights into direct functional roles of H3K79 methylation in Pol II-mediated transcription.
Transcription Termination
The process of transcription termination is highly regulated and carried out using multiple cellular mechanisms (recently reviewed in [84]). Many events occur during the process of termination, including: polyadenylation of the mRNA transcript after cleavage promoted by AAUAAA elements, slowing down and/or pausing of Pol II, release of the mRNA from Pol II for nuclear export and translation, and eviction of Pol II from the DNA template. The role of histone modifications during this process remains incompletely understood and only recently have particular modifications been found to associate with termination sites [85,86]. Even with the utilization of ChIP-seq studies, particular histone modifications enriched at the transcription termination site (TTS) have not been revealed to the extent at which they define active promoter regions and enhancer elements.
The Proudfoot lab has described one of the few histone marks associated with transcription termination. They discovered that H3K9me2 is deposited near the 3′ end of genes, specifically at genes that show termination-associated Pol II pausing [86]. The pausing of Pol II in the 3′ end of genes often is associated with Pol II CTD serine 2 phosphorylation and proper transcription termination [84]. H3K9me2 is typically associated with heterochromatin formation and can be generated for gene silencing on active genes [87,88]. Interestingly, the deposition of H3K9me2 near TTSs is dependent on R-loop formation and antisense transcription, in which double-stranded RNA is formed, recruiting RNA-induced silencing complex (RISC) components. In addition, the histone methyltransferase G9a is recruited, which deposits H3K9me2 at these sites and in turn recruits the reader protein heterochromatin protein 1 γ (HP1γ) [89]. Importantly, the activity of G9a is necessary for 3′ Pol II pausing and effective termination [86], suggesting H3K9me2 may be a critical mark for transcription termination. It would be interesting to tease apart the mechanism by which HP1γ acts in transcription termination, and if it is similar or distinct from HP1-dependent heterochromatin formation, resulting in transcription repression.
Another histone mark that is associated with transcription termination on DNA damage-response genes is phosphorylation on H3T45. The presence of H3T45phos is necessary for productive transcription termination after exposure to DNA damaging agents, which was revealed by inhibiting the kinase, AKT1, and expressing a mutant histone H3T45A, to decrease levels of H3T45phos [85]. These observations are intriguing, and raise some additional questions. For example, is this phosphorylation event specific to the DNA damage response, or does it occur in response to other cellular stimuli? Additionally, what is the exact mechanistic function of this phosphorylation event: does it recruit a reader protein, impact chromatin structure, etc.? It will be interesting to see what other histone marks contribute to the process of transcription termination.
Concluding Remarks
The observation that histone marks correlate with distinct genomic regions and states of gene expression raises questions regarding their functional role in different steps of the Pol II transcription cycle. Here, we have highlighted studies providing clear roles for H3K4me3, H3K9ac, H3K36me3, and other lysine marks in the context of particular stages of transcription (Figure 3, Key Figure). For discussion on the functional roles of histone marks at enhancers, see Box 3. However, there are many histone marks observed to correlate with transcription for which mechanistic functions are not yet elucidated. Furthermore, with the revelation of additional types of posttranslational modifications, exciting advances remain to be discovered about the role of other histone marks in the transcription cycle.
Figure 3. Readers of select histone marks can drive progression through the transcription cycle.
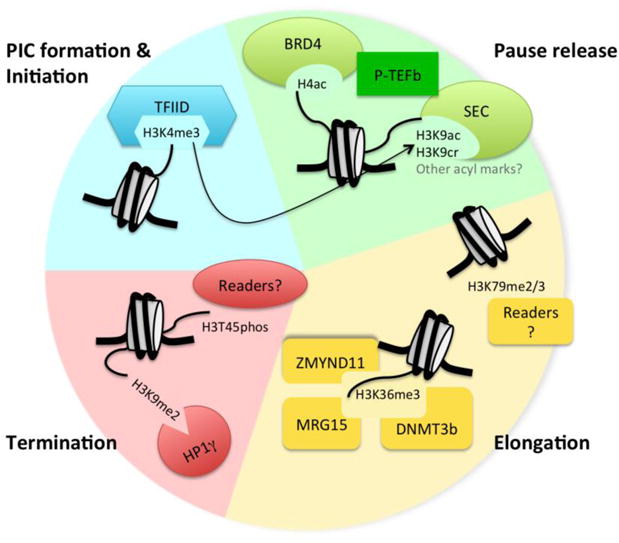
Here the transcription cycle is depicted using different colors: blue = PIC formation and initiation, green = pause release, yellow = elongation, and red = termination. Histone marks discussed in this review are shown, along with some of their readers, which exert particular functions on the transcription cycle. Known interplay between H3K4me3 and H3K9ac is displayed with the arrow.
Box 3. The function of histone marks at enhancers.
Enhancers are designated by some distinct histone marks compared to gene promoters, including high levels of H3K4me1 relative to H3K4me3 (reviewed in [102]). Together with H3K4me1, H3K27ac often denotes active enhancers, and these two modifications have clear interplay with one another. The writers of H3K4me1, MLL3 and MLL4, interact with the writers of H3K27ac, p300 and CBP, and are required for the binding of these HATs to chromatin [103,104]. This suggests that H3K4me1 may precede H3K27ac in a step-wise fashion, and the Roeder lab additionally showed that the presence of the MLL4 complex stimulates p300-mediated H3K27ac in a cell-free system. However, the ability of MLL4 to write H3K4me1 was also dependent on the presence of p300 and acetyl-CoA [104], suggesting that the interplay between H3K4me1 and H3K27ac may be more complex than previously thought.
While H3K4me1 levels are often used to identify enhancer regions along with other chromatin states or modifications, the role of H3K4me1 in enhancer function is unclear. The presence of H3K4me1 is often thought to designate primed enhancers, while increased levels of H3K27ac designates active enhancers [102]. The Wysocka lab recently discovered that while the presence of MLL3 and MLL4 are important for enhancer activity, their enzymatic functions at enhancers is not required, as loss of H3K4me1 through mutating the SET domains in MLL3 and MLL4 does not greatly reduce transcription from either enhancers or promoters [105]. Importantly, this study raises the question of the functional role of H3K4me1 at enhancers.
A potential answer regarding the function of H3K4me1 in enhancer activity has been proposed by the Shi laboratory, which suggests that even though H3K4me1 is a marker of enhancers, this histone mark may actually indicate reduced enhancer activity. When enhancers are activated and transcribe eRNA, there may be a transient signal of H3K4me3 at these enhancers, which needs to be lost in order to repress enhancer function, resulting in H3K4me1 as a “left over” mark. This reduction of enhancer activity is thought to be important to prevent enhancers from becoming overactive, which is associated with an oncogenic state. The Shi laboratory found that the histone H3 K4 lysine demethylase 5C (KDM5C) is recruited to enhancers through receptor for activated C-kinase 7 (RACK7), and then converts H3K4me3 to H3K4me1 after hormone stimulation, in order to reduce enhancer activity [106]. They further demonstrated that knockout of RACK7 reduces KDM5C occupancy at enhancers and results in “overactive enhancers,” and also increases tumorigenicity of breast cancer cells [106]. Taken together, these studies suggest that the potential mechanistic roles of these marks in enhancer function, either in promoting eRNA transcription or communication with the promoter to impact mRNA transcription, still require further study.
Trends Box.
Histone tails are extensively posttranslationally modified (“marked”) by the balance of writers and erasers
Genome-wide localization of histone marks occurs at distinct genic regions (e.g., promoters, enhancers, gene bodies) often correlating with different functional impacts on Pol II transcription
Different histone marks recruit specific readers important for each stage of the Pol II transcription cycle (H3K4me3/TFIID for initiation, H3K9ac/SEC for pause release, H3K36me3/DNMT3a/b for elongation, and H3K9me2/HP1γ for termination)
Interplay between histone modifications can regulate transitions between different stages of the transcription cycle with the best example being H3K4me3 triggering H3K9ac
Outstanding Questions Box.
As Pol II transcriptional regulation is a complex process with many GTFs, sequence-specific DNA binding transcription factors, coregulators, and a plethora of histone marks, what are the particular cellular contexts that rely most on epigenetic mechanisms of transcriptional regulation?
Since donor molecules for epigenetic writers are small metabolites (e.g., ATP, acetyl-CoA, SAM), how does the metabolic state of the cell impact transcriptional regulation?
Might “transcriptional bursting” be influenced by the ‘local’ production of small metabolites utilized in chromatin modifications?
What are the readers for other histone marks associated with transcription elongation (H3K79me2/3) and DNA-damage induced termination (H3T45phos)?
Are other “functions” of Pol II affected in an epigenetic manner, such as: transcription re-initiation, and the intrinsic ability of Pol II to “proofread” or correct misincorporated nucleotides?
Acknowledgments
We thank members of the O’Malley laboratory for their helpful discussions and critical reading of the manuscript. This work was supported by funds from the National Institutes of Health (R01HD008188).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luger K, et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 3.Allfrey VG, et al. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proceedings of the National Academy of Sciences. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 5.Sierra F, et al. Organization of human histone genes. Proceedings of the National Academy of Sciences. 1981;79:1795–1799. doi: 10.1073/pnas.79.6.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Musselman CA, et al. Perceiving the epigenetic landscape through histone readers. Nature Structural & Molecular Biology. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan M, et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pokholok DK, et al. Genome-wide Map of Nucleosome Acetylation and Methylation in Yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 9.Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 10.Roeder RG. The role of general initiation factors in transcription by RNA polymerase II. Trends in Biochemical Sciences. 1996;21:327–335. [PubMed] [Google Scholar]
- 11.Fuda NJ, et al. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature. 2009;461:186–192. doi: 10.1038/nature08449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kornberg RD. The molecular basis of eukaryotic transcription. Proceedings of the National Academy of Sciences. 2007;104:12955–12961. doi: 10.1073/pnas.0704138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lonard DM, O’Malley BW. Expanding functional diversity of the coactivators. Trends in Biochemical Sciences. 2005;30:126–132. doi: 10.1016/j.tibs.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Kugel JF, Goodrich JA. Non-coding RNAs: key regulators of mammalian transcription. Trends in Biochemical Sciences. 2012;37:144–151. doi: 10.1016/j.tibs.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W, et al. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- 16.Naftelberg S, et al. Regulation of Alternative Splicing Through Coupling with Transcription and Chromatin Structure. Annual Review of Biochemistry. 2015;84:165–198. doi: 10.1146/annurev-biochem-060614-034242. [DOI] [PubMed] [Google Scholar]
- 17.Saldi T, et al. Coupling of RNA Polymerase II Transcription Elongation with Pre-mRNA Splicing. Journal of Molecular Biology. 2016;428:2623–2635. doi: 10.1016/j.jmb.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ptashne M, Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- 19.Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruthenburg AJ, et al. Methylation of Lysine 4 on Histone H3: Intricacy of Writing and Reading a Single Epigenetic Mark. Molecular Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 21.Shilatifard A. The COMPASS Family of Histone H3K4 Methylases: Mechanisms of Regulation in Development and Disease Pathogenesis. Annual Review of Biochemistry. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin JJ, et al. Mediator coordinates PIC assembly with recruitment of CHD1. Genes & Development. 2011;25:2198–2209. doi: 10.1101/gad.17554711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lauberth SM, et al. H3K4me3 Interactions with TAF3 Regulate Preinitiation Complex Assembly and Selective Gene Activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foulds CE, et al. Proteomic analysis of coregulators bound to ERα on DNA and nucleosomes reveals coregulator dynamics. Molecular Cell. 2013;51:185–199. doi: 10.1016/j.molcel.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vermeulen M, et al. Selective Anchoring of TFIID to Nucleosomes by Trimethylation of Histone H3 Lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 26.Clouaire T, et al. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes & Development. 2012;26:1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wysocka J, et al. WDR5 Associates with Histone H3 Methylated at K4 and Is Essential for H3 K4 Methylation and Vertebrate Development. Cell. 2005;121:859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 28.Dou Y, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nature Structural & Molecular Biology. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 29.Steward MM, et al. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nature Structural & Molecular Biology. 2006;13:852–854. doi: 10.1038/nsmb1131. [DOI] [PubMed] [Google Scholar]
- 30.Bernstein BE, et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 31.McDonald D, et al. Chromatin signatures of pluripotent cell lines. Nature Cell Biology. 2006;8:532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 32.Kim TH, et al. A high-resolution map of active promoters in the human genome. Nature Cell Biology. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guenther MG, et al. A Chromatin Landmark and Transcription Initiation at Most Promoters in Human Cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Core LJ, et al. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13:720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, et al. Ready, pause, go: regulation of RNA polymerase II pausing and release by cellular signaling pathways. Trends in Biochemical Sciences. 2015;40:516–525. doi: 10.1016/j.tibs.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo J, Price DH. RNA Polymerase II Transcription Elongation Control. Chem Rev. 2013;113:8583–8603. doi: 10.1021/cr400105n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X, et al. Initiation Complex Structure and Promoter Proofreading. Science. 2011;333:633–637. doi: 10.1126/science.1206629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luse DS. Promoter clearance by RNA polymerase II. BBA - Gene Regulatory Mechanisms. 2013;1829:63–68. doi: 10.1016/j.bbagrm.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marshall NF, Price DH. Purification of P-TEFb, a Transcription Factor Required for the Transition into Productive Elongation. Journal of Biological Chemistry. 1995;270:12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 41.Marshall NF, et al. Control of RNA Polymerase II Elongation Potential by a Novel Carboxyl-terminal Domain Kinase. Journal of Biological Chemistry. 1996;271:27176–27183. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- 42.Wada T, et al. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. The EMBO Journal. 1998;17:7395–7403. doi: 10.1093/emboj/17.24.7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamaguchi Y, et al. NELF, a Multisubunit Complex Containing RD, Cooperates with DSIF to Repress RNA Polymerase II Elongation. Cell. 1999;97:41–51. doi: 10.1016/s0092-8674(00)80713-8. [DOI] [PubMed] [Google Scholar]
- 44.Zaborowska J, et al. The pol II CTD: new twists in the tail. Nature Structural & Molecular Biology. 2016;23:771–777. doi: 10.1038/nsmb.3285. [DOI] [PubMed] [Google Scholar]
- 45.Filippakopoulos P, et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jang MK, et al. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Molecular Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 47.Yang Z, et al. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Molecular Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 48.Bian C, et al. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. The EMBO Journal. 2011;30:2829–2842. doi: 10.1038/emboj.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gates LA, et al. Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. Journal of Biological Chemistry. 2017;292:14456–14472. doi: 10.1074/jbc.M117.802074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin C, et al. AFF4, a Component of the ELL/P-TEFb Elongation Complex and a Shared Subunit of MLL Chimeras, Can Link Transcription Elongation to Leukemia. Molecular Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sabari BR, et al. Metabolic regulation of gene expression through histone acylations. Nature Reviews Molecular Cell Biology. 2016;18:90–101. doi: 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sabari BR, et al. Intracellular Crotonyl-CoA Stimulates Transcription through p300-Catalyzed Histone Crotonylation. Molecular Cell. 2015;58:203–215. doi: 10.1016/j.molcel.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y, et al. Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Molecular Cell. 2016;62:181–193. doi: 10.1016/j.molcel.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andrews FH, et al. The Taf14 YEATS domain is a reader of histone crotonylation. Nat Chem Biol. 2016;12:396–398. doi: 10.1038/nchembio.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi X, et al. Cdc73p and Paf1p Are Found in a Novel RNA Polymerase II-Containing Complex Distinct from the Srbp-Containing Holoenzyme. Molecular and Cellular Biology. 1997;17:1160–1169. doi: 10.1128/mcb.17.3.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orphanides G, et al. FACT, a Factor that Facilitates Transcript Elongation through Nucleosomes. Cell. 1998;92:105–116. doi: 10.1016/s0092-8674(00)80903-4. [DOI] [PubMed] [Google Scholar]
- 57.Mueller D, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110:4445–4454. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bitoun E, et al. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Human Molecular Genetics. 2006;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- 59.Mohan M, et al. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom) Genes & Development. 2010;24:574–589. doi: 10.1101/gad.1898410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vakoc CR, et al. Profile of Histone Lysine Methylation across Transcribed Mammalian Chromatin. Molecular and Cellular Biology. 2006;26:9185–9195. doi: 10.1128/MCB.01529-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strahl BD, et al. Set2 Is a Nucleosomal Histone H3-Selective Methyltransferase That Mediates Transcriptional Repression. Molecular and Cellular Biology. 2002;22:1298–1306. doi: 10.1128/mcb.22.5.1298-1306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edmunds JW, et al. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. The EMBO Journal. 2008;27:406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiao T, et al. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes & Development. 2003;17:654–663. doi: 10.1101/gad.1055503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li B, et al. The Set2 Histone Methyltransferase Functions through the Phosphorylated Carboxyl-terminal Domain of RNA Polymerase II. Journal of Biological Chemistry. 2003;278:8897–8903. doi: 10.1074/jbc.M212134200. [DOI] [PubMed] [Google Scholar]
- 65.Carrozza MJ, et al. Histone H3 Methylation by Set2 Directs Deacetylation of Coding Regions by Rpd3S to Suppress Spurious Intragenic Transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 66.Venkatesh S, et al. Selective suppression of antisense transcription by Set2-mediated H3K36 methylation. Nature Communications. 2016;7:1–14. doi: 10.1038/ncomms13610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim T, et al. Set3 HDAC Mediates Effects of Overlapping Noncoding Transcription on Gene Induction Kinetics. Cell. 2012;150:1158–1169. doi: 10.1016/j.cell.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xie L, et al. KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. The EMBO Journal. 2011;30:1473–1484. doi: 10.1038/emboj.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 70.Dhayalan A, et al. The Dnmt3a PWWP Domain Reads Histone 3 Lysine 36 Trimethylation and Guides DNA Methylation. Journal of Biological Chemistry. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baubec T, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015;520:243–247. doi: 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- 72.Morselli M, et al. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. E Life. 2015;4:1–21. doi: 10.7554/eLife.06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neri F, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature. 2017;543:72–77. doi: 10.1038/nature21373. [DOI] [PubMed] [Google Scholar]
- 74.Carvalho S, et al. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Research. 2013;41:2881–2893. doi: 10.1093/nar/gks1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guo R, et al. BS69/ZMYND11 Reads and Connects Histone H3.3 Lysine 36 Trimethylation-Decorated Chromatin to Regulated Pre-mRNA Processing. Molecular Cell. 2014;56:298–310. doi: 10.1016/j.molcel.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wen H, et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508:263–268. doi: 10.1038/nature13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ng HH, et al. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: A potential mechanism for position-effect variegation. Proceedings of the National Academy of Sciences. 2003;100:1820–1825. doi: 10.1073/pnas.0437846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Okada Y, et al. hDOT1L Links Histone Methylation to Leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 79.Schubeler D, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes & Development. 2004;18:1263–1271. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Z, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Steger DJ, et al. DOT1L/KMT4 Recruitment and H3K79 Methylation Are Ubiquitously Coupled with Gene Transcription in Mammalian Cells. Molecular and Cellular Biology. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes & Development. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim SK, et al. Human Histone H3K79 Methyltransferase DOT1L Methyltransferase Binds Actively Transcribing RNA Polymerase II to Regulate Gene Expression. Journal of Biological Chemistry. 2012;287:39698–39709. doi: 10.1074/jbc.M112.384057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Proudfoot NJ. Transcription termination in mammals: Stopping the RNA polymerase II juggernaut. Science. 2016 doi: 10.1126/science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee JH, et al. AKT phosphorylates H3-threonine 45 to facilitate termination of gene transcription in response to DNA damage. Nucleic Acids Research. 2015;43:4505–4516. doi: 10.1093/nar/gkv176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Skourti-Stathaki K, et al. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 2014;516:436–439. doi: 10.1038/nature13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tachibana M, et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes & Development. 2005;19:815–826. doi: 10.1101/gad.1284005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vakoc CR, et al. Histone H3 Lysine 9 Methylation and HP1γ Are Associated with Transcription Elongation through Mammalian Chromatin. Molecular Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 89.Bannister AJ, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 90.Srivastava R, Ahn SH. Modifications of RNA polymerase II CTD: Connections to the histone code and cellular function. Biotechnology Advances. 2015;33:856–872. doi: 10.1016/j.biotechadv.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 91.Harlen KM, et al. Comprehensive RNA Polymerase II Interactomes Reveal Distinct and Varied Roles for Each Phospho-CTD Residue. Cell Reports. 2016;15:2147–2158. doi: 10.1016/j.celrep.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pascual-García P, et al. Sus1 is recruited to coding regions and functions during transcription elongation in association with SAGA and TREX2. Genes & Development. 2008;22:2811–2822. doi: 10.1101/gad.483308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kizer KO, et al. A Novel Domain in Set2 Mediates RNA Polymerase II Interaction and Couples Histone H3 K36 Methylation with Transcript Elongation. Molecular and Cellular Biology. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Govind CK, et al. Phosphorylated Pol II CTD Recruits Multiple HDACs, Including Rpd3C(S), for Methylation-Dependent Deacetylation of ORF Nucleosomes. Mol Cell. 2010;39:234–246. doi: 10.1016/j.molcel.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fong N, et al. RNA Pol II Dynamics Modulate Co-transcriptional Chromatin Modification, CTD Phosphorylation, and Transcriptional Direction. Mol Cell. 2017;66:546–557.e3. doi: 10.1016/j.molcel.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang T, et al. The interplay of histone modifications - writers that read. EMBO reports. 2015;16:1467–1481. doi: 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Josefowicz SZ, et al. Chromatin Kinases Act on Transcription Factors and Histone Tails in Regulation of Inducible Transcription. Mol Cell. 2016;64:347–361. doi: 10.1016/j.molcel.2016.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cheung P, et al. Synergistic Coupling of Histone H3 Phosphorylation and Acetylation in Response to Epidermal Growth Factor Stimulation. Molecular Cell. 2000;5:905–915. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- 99.Lo WS, et al. Phosphorylation of Serine 10 in Histone H3 Is Functionally Linked In Vitro and In Vivo to Gcn5-Mediated Acetylation at Lysine 14. Molecular Cell. 2000;5:917–926. doi: 10.1016/s1097-2765(00)80257-9. [DOI] [PubMed] [Google Scholar]
- 100.Zippo A, et al. Histone Crosstalk between H3S10ph and H4K16ac Generates a Histone Code that Mediates Transcription Elongation. Cell. 2009;138:1122–1136. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 101.Ruthenburg AJ, et al. Recognition of a Mononucleosomal Histone Modification Pattern by BPTF via Multivalent Interactions. Cell. 2011;145:692–706. doi: 10.1016/j.cell.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Calo E, Wysocka J. Modification of Enhancer Chromatin: What, How, and Why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lai B, et al. MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Research. 2017;45:6388–6403. doi: 10.1093/nar/gkx234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang SP, et al. A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription. Mol Cell. 2017;67:308–321. doi: 10.1016/j.molcel.2017.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dorighi KM, et al. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol Cell. 2017;66:568–576. doi: 10.1016/j.molcel.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shen H, et al. Suppression of Enhancer Overactivation by a RACK7-Histone Demethylase Complex. Cell. 2016;165:331–342. doi: 10.1016/j.cell.2016.02.064. [DOI] [PMC free article] [PubMed] [Google Scholar]