

Abstract
Germline mutations in the cyclin-dependent kinase inhibitor 2A gene (CDKN2A) are frequently identified among melanoma kindreds, and are associated with increased atypical nevus counts. However, a clear relationship between pathogenic CDKN2A mutation carriage and other nevus phenotypes including counts of common acquired nevi has not yet been established. Using data from GenoMEL, we investigated the relationships between CDKN2A mutation carriage and 2 mm, 5 mm, and atypical nevus counts among blood-related members of melanoma families. Compared to individuals without a pathogenic mutation, those who carried one had an overall higher prevalence of atypical (OR=1.64; 95% CI: 1.18, 2.28) nevi, but not 2 mm nevi (OR=1.06; 95% CI: 0.92, 1.21) or 5 mm nevi (OR=1.26; 95% CI: 0.94, 1.70). Stratification by case status revealed more pronounced positive associations among non-case family members, who were nearly three times (OR=2.91; 95% CI: 1.75, 4.82) as likely to exhibit nevus counts at or above the median in all three nevus categories simultaneously when harboring a pathogenic mutation (vs. not harboring one). Our results are supportive of the hypothesis that unidentified nevogenic genes are co-inherited with CDKN2A and may influence carcinogenesis.
Introduction
Germline mutations in the cyclin-dependent kinase inhibitor 2A (CDKN2A) gene are frequently identified in familial melanoma (Goldstein et al., 2006; Goldstein et al., 2007), with prevalence in families with three or more members diagnosed with melanoma ranging between 20 and 50% (Goldstein and Tucker, 2001; Harland et al., 2014; Kefford et al., 1999). In contrast, these mutations account for only 1–2% of population-based melanoma cases (Harland et al., 2014). Germline mutations in CDKN2A have also been associated with familial atypical multiple mole melanoma (FAMMM) syndrome, an autosomally dominant condition exemplified by a family history of melanoma and high numbers of atypical nevi (Eckerle Mize et al., 2009; Goldstein et al., 2007). However, estimating the prevalence of FAMMM has been difficult due to intra- and inter-family variability in the FAMMM phenotype (Goldstein et al., 2000; Lynch et al., 2002; Rulyak et al., 2003), and a clear relationship between CDKN2A mutation classification and number of atypical nevi has not yet been established (Bishop et al., 2000; de Snoo et al., 2008; Nielsen et al., 2010).
Few studies have examined the relationship between germline CDKN2A mutational status and number of common melanocytic nevi among melanoma families, even though evidence from previous genome-wide association studies (GWAS) suggests that variation near the CDKN2A locus is associated with nevus count (Barrett et al., 2011; Falchi et al., 2009). Here, we evaluate associations between germline CDKN2A pathogenic mutation classification and nevus phenotype among participants in research performed by the GenoMEL consortium (www.genomel.org). A better understanding of CDKN2A’s influence on nevogenesis among blood-related cases and non-cases of melanoma may aid in the search of other risk modifying nevogenic genes. In addition, robust phenotypic indicators of CDKN2A pathogenic mutation carriers, especially among non-case members (i.e. individuals who have not been diagnosed with melanoma) of melanoma families, could influence clinicians’ surveillance and prevention strategies in this high-risk population.
Results
CDKN2A genotype was available for at least one member of 896 (78%) families comprising 3,990 individuals, of whom 1,651 (41%) also submitted to nevus phenotyping (Table 1). All analyses were confined to this final analytic cohort of 1,651 participants. The median values of 2 mm, 5 mm, and atypical nevus counts were similar among those with and without a pathogenic CDKN2A mutation, although we observed a higher degree of variation among pathogenic mutation carriers compared to those without a pathogenic mutation (Figure 1). Total nevus count (i.e. the sum of 2 mm, 5 mm, and atypical nevus counts) was highly correlated (r=0.99) with number of 2 mm nevi. Median 2 mm nevus counts for those with and without a pathogenic mutation were 54 (interquartile range (IQR)=102) and 47 (IQR=87) respectively. For 5 mm nevus counts, those with a pathogenic mutation had a median value of 2 (IQR=5) whereas a median value of 1 (IQR=5) was observed among individuals without a pathogenic mutation. Those with and without a pathogenic mutation had a median value of 0 for atypical nevus counts with an IQR of 2 for pathogenic mutation carriers and 1 for those without a pathogenic mutation.
Table 1.
CDKN2A status in melanoma families and family members participating in the GenoMEL Study by ascertainment center*
| Center | Total # of families | Number of families with ≥1 member who is CDKN2A genotyped | Number of family members with known CDKN2A genotype | Number of family members phenotyped with known CDKN2A genotype |
|---|---|---|---|---|
| Barcelona, ES | 25 | 25 | 116 | 83 |
| Bethesda, US | 49 | 48 | 782 | 468 |
| Cesena, IT | 24 | 24 | 116 | 17 |
| Copenhagen, DK | 18 | 15 | 18 | 0 |
| Genoa, IT | 14 | 14 | 45 | 31 |
| Leeds, GB | 76 | 74 | 282 | 216 |
| Leiden, NL | 61 | 59 | 600 | 240 |
| Ljubljana, SI | 4 | 4 | 11 | 10 |
| Lund, SE | 8 | 8 | 97 | 74 |
| Montevideo, UY | 4 | 4 | 23 | 23 |
| Paris, FR | 181 | 181 | 588 | 161 |
| Philadelphia, US | 36 | 36 | 104 | 47 |
| Porto Alegre, BR | 10 | 5 | 12 | 4 |
| Queensland, AU | 230 | 22 | 172 | 11 |
| Riga, LV | 5 | 5 | 8 | 5 |
| Salt Lake City, US | 1 | 1 | 3 | 3 |
| Santiago, CL | 2 | 2 | 6 | 6 |
| São Paulo, BR | 12 | 7 | 28 | 25 |
| Stockholm, SE | 27 | 25 | 118 | 113 |
| Sydney, AU | 319 | 311 | 820 | 85 |
| Tel Aviv, IL | 28 | 21 | 25 | 25 |
| Valencia, ES | 15 | 5 | 16 | 4 |
| Total | 1,149 | 896 | 3,990 | 1,651 |
Melanoma families are defined by 3 or more members with a verified melanoma or 2 first degree relatives with verified melanomas. Married-in relatives not belonging to a melanoma family lineage are excluded.
Figure 1.
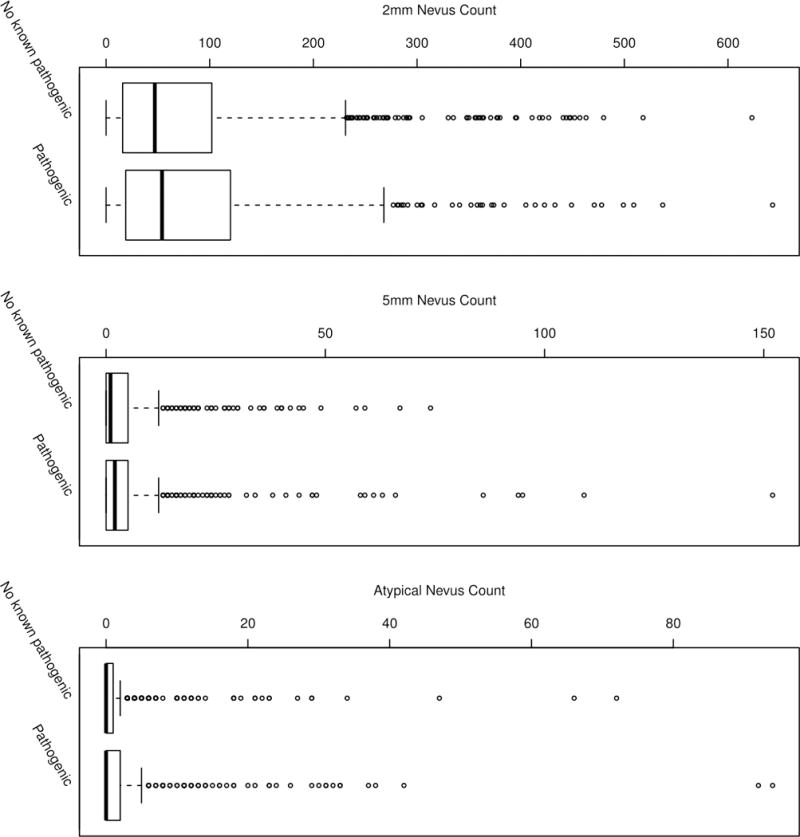
2 mm, 5 mm, and atypical nevus count distributions among GenoMEL melanoma family members across all ascertainment centers according to CDKN2A mutational status. Crude nevus counts are plotted and are not representative of center-specific measures adopted for statistical modeling. Heavy horizonal lines indicate 50th percentile counts, boxes indicate 25th and 75th percentile counts, whiskers indicate 5th and 95th percentile counts, and circles represent values in the top or bottom 5% of counts.
Compared to individuals without a pathogenic CDKN2A mutation, pathogenic mutation carriers had an overall higher prevalence of atypical nevi (OR=1.64; 95% CI: 1.18, 2.28). Moreover, pathogenic mutation carriers were almost twice as likely as those without a pathogenic mutation (OR=1.83; 95% CI: 1.25, 2.67) to exhibit nevus counts at or above the center-specific medians in all three categories of nevi (mole gestalt scores of 3 vs. 0). Pathogenic CDKN2A mutation carriage was not associated with common acquired (2 mm, 5 mm) nevus counts (Table 2). Total nevus count was not associated with carriage of CDKN2A mutations and, as expected, point estimates were nearly identical to those observed for 2 mm nevus counts (data not tabulated).
Table 2.
Associations between nevus phenotypes and CDKN2A mutational status among members of melanoma families
| Nevus Phenotype | Individual CDKN2A mutational status | Overall1 | Cases only (n=757)2 | Non-cases only (n=894)2 | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| OR (95% CI) | p-value | OR (95% CI) | p-value | OR (95% CI) | p-value | p-interaction6 | ||
|
| ||||||||
| 2 mm nevi | No known pathogenic | 1.00 | 0.45 | 1.00 | 0.49 | 1.00 | 0.93 | 0.45 |
| Pathogenic | 1.06 (0.92, 1.21) | 1.06 (0.90, 1.26) | 0.99 (0.83, 1.19) | |||||
|
| ||||||||
| 5 mm nevi | No known pathogenic | 1.00 | 0.18 | 1.00 | 0.31 | 1.00 | 0.27 | 0.95 |
| Pathogenic | 1.26 (0.94, 1.70) | 1.21 (0.87, 1.69) | 1.31 (0.86, 1.99) | |||||
|
| ||||||||
| Atypical nevi | No known pathogenic | 1.00 | 0.02 | 1.00 | 0.16 | 1.00 | 0.01 | 0.27 |
| Pathogenic | 1.64 (1.18, 2.28) | 1.47 (0.92, 2.33) | 1.98 (1.34, 2.90) | |||||
|
| ||||||||
| Mole gestalt (3 vs. 0)3 | No known pathogenic | 1.00 | 0.004 | 1.00 | 0.69 | 1.00 | 0.0001 | 0.002 |
| Pathogenic | 1.83 (1.25, 2.67) | 0.90 (0.53, 1.53) | 2.91 (1.75, 4.82) | |||||
|
| ||||||||
| Mole gestalt (2 vs. 0)4 | No known pathogenic | 1.00 | 0.05 | 1.00 | 0.35 | 1.00 | 0.004 | 0.02 |
| Pathogenic | 1.38 (1.00, 1.91) | 0.79 (0.48, 1.29) | 1.96 (1.26, 3.06) | |||||
|
| ||||||||
| Mole gestalt (1 vs. 0)5 | No known pathogenic | 1.00 | 0.28 | 1.00 | 0.36 | 1.00 | 0.15 | 0.25 |
| Pathogenic | 1.21 (0.86, 1.71) | 0.80 (0.50, 1.29) | 1.42 (0.89, 2.25) | |||||
Adjusted for age at phenotyping, gender, age at phenotyping*gender, melanoma affected status, center, and familial clustering within study center. Married-in relatives not belonging to a melanoma family lineage are excluded. P-values correspond to overall score tests.
Adjusted for age at phenotyping, gender, age at phenotyping*gender, center, and familial clustering within study center; Married-in relatives not belonging to a melanoma family lineage are excluded. P-values correspond to overall score tests.
Mole gestalt is modeled in a GEE model excluding individuals with values of “1” and “2” for mole gestalt to achieve the contrast estimates.
Mole gestalt is modeled in a GEE model excluding individuals with values of “1” and “3” for mole gestalt to achieve the contrast estimates.
Mole gestalt is modeled in a GEE model excluding individuals with values of “2” and “3” for mole gestalt to achieve the contrast estimates.
P-value for the association between the interaction of CDKN2A mutation carriage with case status and nevus phenotype.
Upon stratification by melanoma case status, we observed more pronounced positive associations between CDKN2A pathogenic mutation carriage and nevus counts among the non-case family members. Among non-case participants, those harboring a pathogenic mutation were nearly three times as likely to demonstrate the highest mole gestalt score (3 vs. 0) compared to those without a pathogenic mutation (OR=2.91; 95% CI: 1.75, 4.82) and exhibited approximately twice as many atypical nevi compared to non-cases without a pathogenic mutation (OR=1.98; 95% CI: 1.34, 2.90). In contrast, carriage of a pathogenic mutation among melanoma cases was inversely associated with mole gestalt score (3 vs. 0) compared to those without a pathogenic mutation (OR=0.90; 95% CI: 0.53, 1.53) and showed a modest, but statistically nonsignificant, positive association with number of atypical nevi compared to wildtype carriage (OR=1.47; 95% CI: 0.92, 2.33) (Table 2).
We further explored associations stratified by GenoMEL study centers grouped according to proximity to the equator to assess the relative influence of increasing daylight hours; and one stratified by anatomic site of first melanoma classified by relative duration of UV exposure. Latitude did not demonstrate a statistically significant influence on the association between any CDKN2A mutation carriage and nevus phenotype (p-interaction >0.05 for all nevus phenotype categories), nor could we discern any clear patterns of association according to relative UV exposure of anatomic site of first verified melanoma (Supplemental Tables 2 and 3).
Discussion
Within melanoma families, we observed higher mole gestalt scores among pathogenic CDKN2A mutation carriers compared to those without a pathogenic mutation, indicating that carriers tended to have more nevus laden phenotypes. Estimates within individual nevus phenotype categories (i.e. 2 mm, 5 mm, and atypical nevus counts) indicate pathogenic mutation carriers exhibit greater numbers of atypical nevi compared to non-carriers.
To date, few studies have examined the influence of germline CDKN2A mutation carriage on common acquired nevus counts among melanoma-prone families. A longitudinal study of a large melanoma family from Utah reported increasing nevus counts among carriers of the specific V126D mutation compared to wildtype over a 15 year interval (Florell et al., 2004). However, the impact of the mutation on atypical nevi is unclear as total nevus count was reported. Twin studies identified a quantitative-trait locus (microsatellite marker D9S942) for nevus density in a noncoding region of CDKN2A (Falchi et al., 2006; Zhu et al., 1999; Zhu et al., 2007), which may suggest a broader role of CDKN2A in nevogenesis among the majority of individuals who do not harbor a rare germline mutation. However, an adolescent twin study from the UK found no evidence for D9S942 as a quantitative-trait locus influencing nevus density (Barrett et al., 2003) and a familial-based investigation of a potentially nevogenic variant (A148T) near D9S942 also found no association with common acquired nevus counts (Bertram et al., 2002). Germline mutations in CDKN2A are strongly associated with FAMMM syndrome, and individual members of these families often have abundant numbers of atypical and common nevi (Gruis et al., 1995; Hussussian et al., 1994; Soura et al., 2016). However, not all individuals with CDKN2A mutations present with excessive or even higher nevus counts. Studies of Dutch and Swedish melanoma kindreds have reported low atypical and common nevus counts among CDKN2A mutation carriers (Ipenburg et al., 2016; Nielsen et al., 2010). Similar findings were reported among melanoma families from the UK (Newton Bishop et al., 1994). The range of atypical nevi (0–94) observed in GenoMEL family members with pathogenic CDKN2A mutations further highlights the influence of CDKN2A on phenotypic heterogeneity.
Evaluating individual nevus types among GenoMEL participants suggests that germline pathogenic mutations at CDKN2A are more predictive of number of atypical nevi compared to common acquired nevi (2 mm and 5 mm nevi), a result which is consistent with previous findings (Bishop et al., 2000). These results are also interesting in light of recent research that suggests intermediate lesions, a classification that includes atypical/dysplastic nevi, are likely to exhibit hemizygous loss of CDKN2A, supporting a role for this locus in the development of histological atypia in nevi (Shain et al., 2015). It is important to note that the defining criteria of atypical nevi in the present study were clinical and not pathologically-based; it is possible that very subtle atypical nevi could have been misclassified as 5 mm nevi. Furthermore, although we took a conservative approach when assigning pathogenicity to CDKN2A variants/mutations, it is possible that our designation of some common variants as not pathogenic is not accurate. We based our assessment on evidence of a deleterious effect, and for some of the common variants there is no such evidence to date.
Our observation of distinct differences in associations according to case status is interesting. Non-case members of melanoma-prone families demonstrated relatively strong associations of CDKN2A pathogenic mutation carriage with mole gestalt score and number of atypical nevi, while corresponding associations among case family members tended to be attenuated. Pathogenic germline mutations in CDKN2A and number of nevi are both important risk factors for melanoma. The observed pronounced difference in the relationship between CDKN2A mutational status and nevus phenotype according to case status may be due, in part, to the higher proportion of pathogenic CDKN2A mutations among cases (42%) compared to non-cases (25%). If CDKN2A influences nevogenesis, we might expect to see diminished associations between pathogenic CDKN2A mutation carriage and nevus phenotype among cases compared to non-cases. The higher nevus count distributions we observed among cases compared to non-cases tends to support this hypothesis (Supplemental Figure 1). It is also possible that case members are affected by yet-to-be-discovered nevogenic genes that cosegregate with CDKN2A and either modify CDKN2A’s nevogenic function or influence nevogenesis independently. Another possible explanation is that non-case family members may be more likely to inherit unidentified lower penetrance genes that are important risk modifiers of nevus formation, potentially hinder melanoma initiation, and cosegregate with CDKN2A.
Zhu et al. have speculated that environmental factors affecting spontaneous somatic mutation rates (e.g. UV exposure) in tumor suppressor genes may help to explain nevus count variation between individuals as well as familial correlations in nevus counts (Zhu et al., 2007). However, our analyses by latitude of ascertainment center and anatomic site of melanoma—arguably two proxy measures of UV exposure—did not reveal meaningful nevus phenotype differences across strata. This exploratory analysis did not take into consideration behaviors that influence UV exposure (e.g. sunbathing/tanning, sunscreen usage, apparel).
In summary, our results are consistent with previous studies reporting that CDKN2A plays a role in nevogenesis (Bishop et al., 2000; Cannon-Albright et al., 1994; Florell et al., 2004; Shain et al., 2015; Zhu et al., 1999). In general, pathogenic mutation carriers are significantly more likely to exhibit higher than median nevus counts in all three categories of nevus phenotype simultaneously compared to those without a pathogenic CDKN2A mutations, as evidenced by our mole gestalt score results. Acknowledging the potential nevus phenotype overlap between those with and without a pathogenic CDKN2A mutation (Bishop et al., 2000), we examined associations based on case status among melanoma family members. Associations between CDKN2A pathogenic mutational status and nevus phenotype according to case status contrasted sharply. These differences may be explained if CDKN2A possesses a degree of nevogenic function since case family members exhibited higher nevus counts and were more likely to harbor a pathogenic CDKN2A mutation compared to non-case members, which could result in diminished associations among case members. Our findings are generally supportive of the hypothesis that unidentified nevogenic genes are co-inherited with CDKN2A (Florell et al., 2004).
Materials & Methods
Over the past two decades, GenoMEL has aggregated data from individuals belonging to melanoma families from around the globe. We refer to participants with a melanoma diagnosis at the time of recruitment as cases, whereas family members who had not been diagnosed with melanoma at the time of recruitment are referred to as non-cases. Currently, GenoMEL consists of 29 centers from Australia, Europe, the Middle East, and North and South America.
GenoMEL employed a common protocol for data collection from prospectively enrolled participants, although family identification and recruitment procedures were allowed to differ among study centers. Additionally, centers had a degree of autonomy over the data collection process, which resulted in different contributions across various protocol components. Thus, not all centers completed all portions of the research protocol for each enrolled participant. Regulatory approval was obtained by the institutional review boards of each GenoMEL study center, and written informed consent was obtained for each participant. Individuals who signed informed consent were asked about their personal and familial melanoma history and to submit to a full phenotypic examination by research staff, which included an evaluation of nevus counts by anatomic site. Training was carried out for all staff performing phenotyping on participants in the prospective study in the UK. Consolidation of that training was subsequently carried out in Italy. Several GenoMEL study centers had extant data previously collected from members of melanoma families under local regulatory approval, and where possible this information was harmonized with data arising from participants enrolled in the prospective GenoMEL study.
A melanoma family was defined by the presence of three or more cases of confirmed cutaneous melanoma in the same lineage, or two cases of confirmed cutaneous melanoma in first-degree relatives. Melanoma case family members with a diagnosis of mucosal or ocular melanoma did not contribute to defining a melanoma family and were excluded from analysis. Confirmation of diagnosis was made by: pathology report (75%), physician letter or clinical document verifying melanoma diagnosis (19%), death certificate (2%), or cancer registry data (4%). Individuals who are members of melanoma families by virtue of marriage and not ancestry, or for whom family relationship information was ambiguous or missing, were excluded from this study. Family members who reported a melanoma, but for whom verification of diagnosis was not available, were also excluded from analyses.
Nevi ≥ 2 mm but <5 mm in diameter (hereinafter referred to as 2 mm nevi) were counted on exposed skin, in addition to nevi ≥ 5 mm in diameter (hereinafter referred to as 5 mm nevi) and clinically atypical nevi; sites not examined were the genitalia and female breasts. An atypical nevus was defined as a nevus ≥ 5 mm in diameter and containing a flat component, with at least two of the following characteristics: variable pigmentation, asymmetrical shape or diffuse border. We also derived a summary variable from 2 mm, 5 mm, and atypical nevus counts to describe an individual’s overall nevus phenotypic landscape. Specifically, individuals were assigned a dichotomous score within each category of 2 mm, 5 mm, and atypical nevus count according to the study center-specific median. Individuals with at least the median nevus count were scored as 1, with those exhibiting fewer than the median nevus count scored as 0; each individual then received an aggregate “mole gestalt” summary score between 0 and 3 based on the sum of these three dichotomous scores.
Germline DNA of consenting participants was screened for mutations in CDKN2A (exons 1α, 1β, 2 and 3) as previously described (Harland et al., 2008). Mutation evaluation, predominantly by sequencing or denaturing high performance liquid chromatography followed by sequencing, was conducted at each study center. Previous evaluation has confirmed consistent mutation detection across the consortium (Harland et al., 2008). Sequencing results were collated and mutational status was assigned according to pathogenicity as outlined in Supplemental Table 1. Briefly, pathogenic variants were adjudicated based on demonstrated (i.e. published) impact on the biological function of CDKN2A or bioinformatically inferred deleterious impact on CDKN2A function, and evidence of cosegregation within melanoma families. Variants not meeting any of these criteria were classified as benign (Taylor et al., 2016). Individual participants were classified based on presence of a pathogenic mutation; benign variant carriers and wildtype individuals were combined for analyses and classified as having “no known pathogenic” mutations at CDKN2A. Individuals who carried both a pathogenic mutation and a benign mutation were classified as pathogenic.
We used the generalized estimating equation (GEE) method implemented in SAS v.9.4 (SAS Institute, Cary, NC) to calculate odds ratios and 95% confidence intervals for associations between nevus phenotypes and CDKN2A mutational status. For our nevus count outcomes we used Poisson regression (2 mm nevi) or negative binomial regression when nevus counts were right skewed (5 mm and atypical nevi), whereas a multinomial model was used to evaluate the “mole gestalt” variable. Designating a Type I. error rate of α=0.05, we performed score tests of the null hypothesis that no differences exist between nevus counts within strata of mutational status. Analyses were adjusted for age at phenotyping, sex, the interaction between age and sex, melanoma status and, study center, and we accounted for the non-independence of observations arising from familial clustering within study center using the repeated subject statement of the GENMOD SAS procedure.
We examined associations by latitude by grouping GenoMEL ascertainment centers according to equatorial proximity. Among family members with a diagnosis of melanoma, we also examined associations between CDKN2A mutational status and nevus phenotype by anatomic location of an individual’s first verified melanoma. Anatomic sites were classified as those usually exposed (head, neck, lower arms and scalp-male), intermittently exposed (trunk, back, upper arms, lower legs, and scalp-female), and usually unexposed (buttock, upper legs) to UV radiation.
Supplementary Material
Acknowledgments
This work was performed in participation with members of the following study centers:
Leeds: Paul Affleck, Jennifer H. Barrett, Jane Harrison, Mark M. Iles, Juliette Randerson-Moor, John C. Taylor, Kairen Kukalizch, Susan Leake, Birute Karpavicius, Sue Haynes, Tricia Mack, May Chan, and Yvonne Taylor
Barcelona: Paula Aguilera, Llúcia Alós, Ana Arance, Pedro Arguís, Celia Badenas, Antoni Bennassar, Cristina Carrera, Teresa Castel, Carlos Conill, Daniel Gabriel, Pablo Iglesias, Josep Malvehy, M. Eugenia Moliner, Zighereda Ogbah, Jose Palou, Joan Anton Puig Butille, Ramon Rull, Marcelo Sánchez, Sergi Vidal-Sicart, Antonio Vilalta, and Ramon Vilella
Valencia: Zaida García-Casado, Celia Requena, José Bañuls, Virtudes Soriano, José Antonio López-Guerrero, Manuel Moragón, Vicente Oliver. Samples for CDKN2A analysis were obtained from the Biobank of the Instituto Valenciano de Oncología
NCI at Cesena, Italy: Paola Minghetti, Laura Fontaine, Katie Beebe, and Giorgio Landi
Genoa: Giovanna Bianchi-Scarrà, Lorenza Pastorino, Virginia Andreotti, Claudia Martinuzzi, Bruna Dalmasso, Giulia Ciccarese, Francesco Spagnolo, and Paola Queirolo
Latvia: Kristine Azarjana, Simona Donina, Olita Heisele, Baiba Štreinerte, Aija Ozola and Ludmila Engele
Sydney: Caroline Watts, Gayathri St. George, Robyn Dalziell and Kate McBride who assisted with recruitment of study participants; Leo Raudonikis who assisted with data management; and Chantelle Agha-Hamilton and Svetlana Pianova who assisted with biospecimen management
Uruguay: Virginia Barquet, Javiera Pérez, Miguel Martínez, Jimena Núñez, and Malena Scarone
São Paulo: Dirce Maria Carraro, Alexandre Leon Ribeiro de Ávila, Luciana Facure Moredo, Bianca Costa Soares de Sá, Maria Isabel Waddington Achatz, and João Duprat
Porto Alegre: Renan Rangel Bonamigo and Maria Carolina Widholzer Rey
Leiden: Coby Out-Luiting, Clasine van der Drift and Frans van Nieuwpoort
The French familial melanoma study group: Andry-Benzaquen, B. Bachollet, F. Bérard, P. Berthet, F. Boitier, V. Bonadona, JL. Bonafé, JM. Bonnetblanc, F. Cambazard, O. Caron, F. Caux, J. Chevrant-Breton, A. Chompret (deceased), S. Dalle, L. Demange, O. Dereure, MX. Doré, MS. Doutre, C. Dugast, E. Maubec, L. Faivre, F. Grange, Ph. Humbert, P. Joly, D. Kerob, C. Lasset, MT. Leccia, G. Lenoir, D. Leroux, J. Levang, D. Lipsker, S. Mansard, L. Martin, T. Martin-Denavit, C. Mateus, JL. Michel, P. Morel, L. Olivier-Faivre, JL. Perrot, C. Robert, S. Ronger-Savle, B. Sassolas, P. Souteyrand, D. Stoppa-Lyonnet, L. Thomas, P. Vabres, Lynda Vincent-Fetita, and E. Wierzbicka
Queensland: Nicholas Martin, Grant Montgomery, David Whiteman, Stuart MacGregor and David Duffy
Stockholm: wishes to thank Diana Lindén, R.N. for excellent work collecting and entering data into the study data base and Rainer Tuominen for screening of CDKN2A
Tel Aviv: wishes to acknowledge Yael Laitman
Lund: wishes to thank Anita Zander, R.N. for invaluable help with the data from the Lund Melanoma Study Group and acknowledge Kari Nielsen, Anna Måsbäck, Katja Harbst, Goran Jonsson and Åke Borg
The University of Utah (Salt Lake City): acknowledges the use of the Genetic Counseling Shared Resource supported by National Institutes of Health grant P30CA042014 awarded to the Hunstman Cancer Institute
The University of Pennsylvania: acknowledges the contributions of Patricia Van Belle, Althea Ruffin, Jillian Knorr and Wenting Zhou
Gustave Roussy: wishes to thank Christophe Blondel for technical assistance in CDKN2A genotyping and to acknowledge the work of Gustave Roussy Biobank (BB-0033-00074) in providing DNA resources.
This work was supported by: the European Commission under the 6th and 7th Framework Programme [LSH-CT-2006-018702]; Cancer Research UK Programme Awards (C588/A4994 and C588/ A10589); a Cancer Research UK Project Grant (C8216/A6129); the US National Institutes of Health [R01-CA83115, R01CA65558-01A2 (MTL), 5R25-CA147832-04 (NJT)]; the intramural Research Program of the NIH, National Cancer Institute (NCI), Division of Cancer Epidemiology and Genetics; the National Health and Medical Research Council of Australia [NHMRC 107359, 402761, 633004, 566946, 211172]; the Cancer Council New South Wales (project grant 77/00, 06/10); the Cancer Institute New South Wales [CINSW 05/TPG/1-01, 10/TPG/1-02]; the Cancer Council Victoria and the Cancer Council Queensland (project grant 371); CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior); FAPESP (Fundação para o Amparo da Pesquisa do Estado de São Paulo) – SP, Brazil # 2007/04313-2; the National Health and Medical Research Council of Australia and the NCI (CA88363); the Cancer Research Foundations of Radiumhemmet and the Swedish Cancer Society; the Paulsson Trust, Lund University; grant support from the Swedish Cancer Society and European Research Council Advanced Grant (ERC-2011-294576); grants from Fondo de Investigaciones Sanitarias P.I. 09/01393 and P.I. 12/00840, Spain; the CIBER de Enfermedades Raras of the Instituto de Salud Carlos III, Spain, co-funded by Fondo Europeo de Desarrollo Regional (FEDER), Unión Europea, Una manera de hacer Europa; the AGAUR 2009 SGR 1337 and AGAUR 2014_SGR_603 of the Catalan Government, Spain; Fundació La Marató de TV3, 201331-30, Catalonia, Spain; the Italian Association for Cancer research (AIRC) n. 15460 to PG, Italian Ministry of Health (5 × 1000 funds to IRCCS San Martino-IST, Genoa); the Programme Hospitalier de Recherche Clinique (PHRC-AOM-07-195); grant support from Institut National du Cancer (INCA) was attributed to B B-deP. for coordination of Melanoma Oncogenetics in France; the Comisión Honoraria de Lucha Contra el Cáncer, Montevideo, Uruguay; the work of Nelleke A. Gruis was supported in part by the Dutch Cancer Society (UL 2012-5489); Francisco Cuellar is supported by a scholarship awarded by CONACYT, Mexico (152256/158706); Anne Cust is the recipient of Career Development Fellowships from the NHMRC (1063593) and Cancer Institute NSW (15/CDF/1-14).
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Barrett JH, Gaut R, Wachsmuth R, et al. Linkage and association analysis of nevus density and the region containing the melanoma gene CDKN2A in UK twins. Br J Cancer. 2003;88:1920–4. doi: 10.1038/sj.bjc.6600904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JH, Iles MM, Harland M, et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat Genet. 2011;43:1108–13. doi: 10.1038/ng.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram CG, Gaut RM, Barrett JH, et al. An assessment of the CDKN2A variant Ala148Thr as a nevus/melanoma susceptibility allele. J Invest Dermatol. 2002;119:961–5. doi: 10.1046/j.1523-1747.2002.01825.x. [DOI] [PubMed] [Google Scholar]
- Bishop JA, Wachsmuth RC, Harland M, et al. Genotype/phenotype and penetrance studies in melanoma families with germline CDKN2A mutations. J Invest Dermatol. 2000;114:28–33. doi: 10.1046/j.1523-1747.2000.00823.x. [DOI] [PubMed] [Google Scholar]
- Cannon-Albright LA, Meyer LJ, Goldgar DE, et al. Penetrance and expressivity of the chromosome 9p melanoma susceptibility locus (MLM) Cancer Res. 1994;54:6041–4. [PubMed] [Google Scholar]
- de Snoo FA, Hottenga JJ, Gillanders EM, et al. Genome-wide linkage scan for atypical nevi in p16-Leiden melanoma families. Eur J Hum Genet. 2008;16:1135–41. doi: 10.1038/ejhg.2008.72. [DOI] [PubMed] [Google Scholar]
- Eckerle Mize D, Bishop M, Resse E, et al. Familial Atypical Multiple Mole Melanoma Syndrome. In: Riegert-Johnson DL, Boardman LA, Hefferon T, Roberts M, editors. Cancer Syndromes. Bethesda (MD): 2009. [PubMed] [Google Scholar]
- Falchi M, Bataille V, Hayward NK, et al. Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nat Genet. 2009;41:915–9. doi: 10.1038/ng.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchi M, Spector TD, Perks U, et al. Genome-wide search for nevus density shows linkage to two melanoma loci on chromosome 9 and identifies a new QTL on 5q31 in an adult twin cohort. Hum Mol Genet. 2006;15:2975–9. doi: 10.1093/hmg/ddl227. [DOI] [PubMed] [Google Scholar]
- Florell SR, Meyer LJ, Boucher KM, et al. Longitudinal assessment of the nevus phenotype in a melanoma kindred. J Invest Dermatol. 2004;123:576–82. doi: 10.1111/j.0022-202X.2004.23312.x. [DOI] [PubMed] [Google Scholar]
- Goldstein AM, Chan M, Harland M, et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006;66:9818–28. doi: 10.1158/0008-5472.CAN-06-0494. [DOI] [PubMed] [Google Scholar]
- Goldstein AM, Chan M, Harland M, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. 2007;44:99–106. doi: 10.1136/jmg.2006.043802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AM, Struewing JP, Chidambaram A, et al. Genotype-phenotype relationships in U.S. melanoma-prone families with CDKN2A and CDK4 mutations. J Natl Cancer Inst. 2000;92:1006–10. doi: 10.1093/jnci/92.12.1006. [DOI] [PubMed] [Google Scholar]
- Goldstein AM, Tucker MA. Genetic epidemiology of cutaneous melanoma: a global perspective. Arch Dermatol. 2001;137:1493–6. doi: 10.1001/archderm.137.11.1493. [DOI] [PubMed] [Google Scholar]
- Gruis NA, van der Velden PA, Sandkuijl LA, et al. Homozygotes for CDKN2 (p16) germline mutation in Dutch familial melanoma kindreds. Nat Genet. 1995;10:351–3. doi: 10.1038/ng0795-351. [DOI] [PubMed] [Google Scholar]
- Harland M, Cust AE, Badenas C, et al. Prevalence and predictors of germline CDKN2A mutations for melanoma cases from Australia, Spain and the United Kingdom. Hered Cancer Clin Pract. 2014;12:20. doi: 10.1186/1897-4287-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland M, Goldstein AM, Kukalizch K, et al. A comparison of CDKN2A mutation detection within the Melanoma Genetics Consortium (GenoMEL) Eur J Cancer. 2008;44:1269–74. doi: 10.1016/j.ejca.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8:15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- Ipenburg NA, Gruis NA, Bergman W, et al. The absence of multiple atypical nevi in germline CDKN2A mutations: Comment on “Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndrome”. J Am Acad Dermatol. 2016;75:e157. doi: 10.1016/j.jaad.2016.04.069. [DOI] [PubMed] [Google Scholar]
- Kefford RF, Newton Bishop JA, Bergman W, et al. Counseling and DNA testing for individuals perceived to be genetically predisposed to melanoma: A consensus statement of the Melanoma Genetics Consortium. J Clin Oncol. 1999;17:3245–51. doi: 10.1200/JCO.1999.17.10.3245. [DOI] [PubMed] [Google Scholar]
- Lynch HT, Brand RE, Hogg D, et al. Phenotypic variation in eight extended CDKN2A germline mutation familial atypical multiple mole melanoma-pancreatic carcinoma-prone families: the familial atypical mole melanoma-pancreatic carcinoma syndrome. Cancer. 2002;94:84–96. doi: 10.1002/cncr.10159. [DOI] [PubMed] [Google Scholar]
- Newton Bishop JA, Bataille V, Pinney E, et al. Family studies in melanoma: identification of the atypical mole syndrome (AMS) phenotype. Melanoma Res. 1994;4:199–206. doi: 10.1097/00008390-199408000-00001. [DOI] [PubMed] [Google Scholar]
- Nielsen K, Harbst K, Masback A, et al. Swedish CDKN2A mutation carriers do not present the atypical mole syndrome phenotype. Melanoma Res. 2010;20:266–72. doi: 10.1097/CMR.0b013e3283341339. [DOI] [PubMed] [Google Scholar]
- Rulyak SJ, Brentnall TA, Lynch HT, et al. Characterization of the neoplastic phenotype in the familial atypical multiple-mole melanoma-pancreatic carcinoma syndrome. Cancer. 2003;98:798–804. doi: 10.1002/cncr.11562. [DOI] [PubMed] [Google Scholar]
- Shain AH, Yeh I, Kovalyshyn I, et al. The Genetic Evolution of Melanoma from Precursor Lesions. N Engl J Med. 2015;373:1926–36. doi: 10.1056/NEJMoa1502583. [DOI] [PubMed] [Google Scholar]
- Soura E, Eliades PJ, Shannon K, et al. Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndrome. J Am Acad Dermatol. 2016;74:395–407. doi: 10.1016/j.jaad.2015.08.038. quiz 8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor NJ, Handorf EA, Mitra N, et al. Phenotypic and Histopathological Tumor Characteristics According to CDKN2A Mutation Status among Affected Members of Melanoma Families. J Invest Dermatol. 2016 doi: 10.1016/j.jid.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Duffy DL, Eldridge A, et al. A major quantitative-trait locus for mole density is linked to the familial melanoma gene CDKN2A: a maximum-likelihood combined linkage and association analysis in twins and their sibs. Am J Hum Genet. 1999;65:483–92. doi: 10.1086/302494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Montgomery GW, James MR, et al. A genome-wide scan for naevus count: linkage to CDKN2A and to other chromosome regions. Eur J Hum Genet. 2007;15:94–102. doi: 10.1038/sj.ejhg.5201729. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.