

Abstract
Mutations in polycystin-1 (PC1) and polycystin-2 (PC2) result in a commonly occurring genetic disorder, called Autosomal Dominant Polycystic Kidney Disease (ADPKD), that is characterized by the formation and development of kidney cysts. Epithelial cells with loss-of-function of PC1 or PC2 show higher rates of proliferation and apoptosis and reduced autophagy. PC1 is a large multifunctional transmembrane protein that serves as a sensor that is usually found in complex with PC2, a calcium (Ca2+)-permeable cation channel. In addition to decreased Ca2+ signaling, several other cell fate-related pathways are de-regulated in ADPKD, including cAMP, MAPK, Wnt, JAK-STAT, Hippo, Src, and mTOR. In this review we discuss how polycystins regulate cell death and survival, highlighting the complexity of molecular cascades that are involved in ADPKD.
Keywords: polycystins, TRPP2, apoptosis, autophagy, ADPKD, calcium signaling
Graphical Abstract
1. Introduction
Loss-of-function of polycystin-1 (PC1) or polycystin-2 (PC2) results in the most common life-threatening renal disease called autosomal dominant polycystic kidney disease (ADPKD), which affects 1 in 400–1,000 people worldwide. PC1 and PC2 are transmembrane proteins that form a heteromeric molecular complex in the plasma membrane and cilia, where PC1 is thought to serve as a sensor and PC2 functions as a calcium (Ca2+) permeant channel [1]. Mutation in PKD1 (encoding PC1) is found in 85% of ADPKD cases, which have typically a more severe phenotype than those with a mutation in PKD2 (encoding PC2), accounting for 15% of cases. The hallmark of ADPKD is the spontaneous generation and continuous growth of kidney cysts. These cysts gradually enlarge to destroy normal renal parenchyma, and lead to end stage renal disease (ESRD) in more than half of patients by the age of 60 [2, 3]. In addition, ADPKD is associated with cysts in other organs, such as liver and pancreas, as well as noncystic manifestations, such as cardiovascular abnormalities, intracranial aneurisms, intestinal diverticuli, and abdominal hernia [4].
The most widely accepted theory for cyst generation in human ADPKD is the “two-hit hypothesis”. Patients with ADPKD are typically heterozygous, with one PKD allele having a germline mutation (first hit), and then the remaining normal PKD allele develops a somatic mutation (second hit) in an extremely small percentage of cells. These doubly mutated cells develop into cysts. This hypothesis is supported by the fact that homozygous mutations in PKD1 or PKD2 are embryonically lethal and renal cysts in ADPKD are clonal in origin [2]. More recently, renal injuries induced by ischemia or nephrotoxic agents have been proposed as a “third hit” in cystogenesis, where there injuries appear to be responsible for promoting cyst initiation and accelerate progression [5].
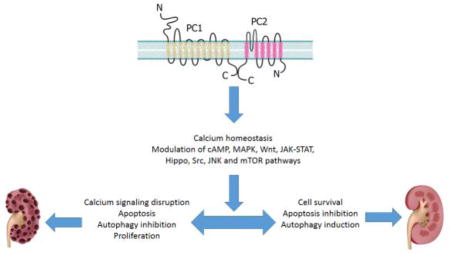
The renal cyst-lining epithelial cells are reported to be converted to a secretory phenotype, instead of functioning as absorptive cells, and these cells have increased rates of proliferation and apoptosis [6, 7]. The cysts gradually increase in size, filling with fluid, dead cells, and necrotic corpus, and are surrounded by interstitial inflammatory cells and matrix deposition [8]. PC1 and PC2 are believed to activate/inhibit a number of key signaling pathways which, in turn, regulate diverse cellular functions including proliferation, tubulogenesis, fluid secretion, cell metabolism, survival, apoptosis, and autophagy. Several signaling pathways have been proposed to be deregulated in ADPKD, including Ca2+, cyclic adenosine monophosphate (cAMP), mitogen-activated protein kinase (MAPK), Wnt, janus kinase (JAK), signal transducer and activator of transcription (STAT), Hippo, Src, and mechanistic target of rapamycin (mTOR) signaling [9] (Fig. 1). Considering the pivotal role of the polycystins in controlling the balance between cell proliferation and death and the consequences for renal cyst development in ADPKD, this review will focus on the newest findings explaining how polycystins regulate cell death and survival.
Fig. 1. Polycystins lead to autosomal dominant polycystic disease.
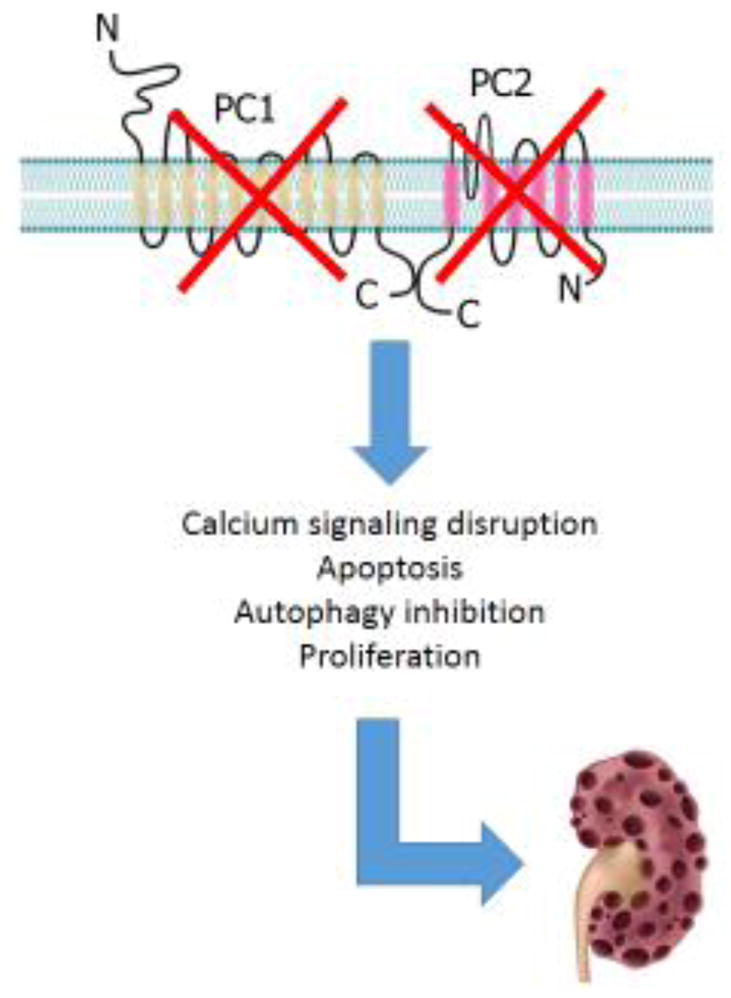
Genetic mutations in PC1 or PC2 affect many signaling cascades, causing progressive cyst growth in the kidney, leading to a dysfunctional polycystic kidney.
2. Polycystins
2.1 Polycystin 1
PC1 is a glycoprotein with a large extracellular N-terminal (3072 aa), 11 transmembrane domains and a cytoplasmic C-terminal region (~200 aa) [10, 11]. The N-terminal domain of PC1, the bulk of the protein, contains several structural motifs that have been reported to be important for interactions with extracellular proteins and carbohydrates [10, 12]. This domain is rapidly cleaved after its synthesis in vivo and remains tethered to the membrane-bound C-terminal domain in a noncovalent fashion, resulting in a high-affinity binding pocket for as-yet unidentified ligands [13]. Regarding the C-terminal cytoplasmic domain of PC1, the most characterized motifs are a coiled-coil sequence that binds specifically to the C-terminus of PC2 [14], and a heterotrimeric G-protein activation sequence that is able to activate heterotrimeric Gi/Go [15]. Interestingly, this coupling of PC1 to Gi/Go proteins is inhibited when PC2 is added to the complex [16]. The C-terminal domain of PC1 also can be cleaved and it is hypothesized that this sequence, once cleaved from the full length PC1, can translocate to the nucleus [17]. When this sequence corresponding to the last 200 amino acids of the C-terminus of PC1 (PC1-P200) is overexpressed, it activates transcription factors after it translocates and accumulates in the nucleus. Again, PC2 expression is able to reduce PC1-P200 translocation, suggesting that the PC2 acts as a buffer regulating the nuclear translocation of the released PC1 C-terminus [17].
Immunohistochemical studies demonstrate that PC1 is developmentally regulated and highly expressed in fetal tissues and polycystic kidney, whereas in adult tissues, PC1 is expressed at low levels in the kidney, predominantly in the distal nephron and collecting duct, as well as in the epithelial ducts of the biliary tree and the pancreas [18–20]. In normal cells, PC1 localizes in the plasma membrane and cilia, whereas an abnormal subcellular localization of PC1 is seen in ADPKD cyst-lining kidney epithelium, where it is largely restricted to the cell cytoplasm [19, 21, 22].
PC1 and PC2 interact through their C-terminal regions. This binding is important for the correct localization and ion channel function of the polycystin complex in the cilia and plasma membrane [23], as well as for PC1 maturation [24]. PC1 and PC2 together can act as an inhibitory complex to cilia-dependent cyst growth–promoting pathway. This hypothesis is supported by observations in double knockout (KO) mice for PC1 or PC2 and structural cilia proteins (kinesin-like protein - Kif3a or intraflagellar transport protein 20 homolog- Ift20) that show a decrease in proliferation rates in cyst-lining cell and in kidney cyst growth when compared to PC1-KO or PC2-KO [25]. The C-terminal domain of PC1 also interacts with the Na,K-ATPase, suggesting that PC1 can modulate renal tubular fluid and electrolyte transport indirectly by increasing Na,K-ATPase activity [26].
In addition to PC2, G-proteins, and the Na,K-ATPase, PC1 also interacts with several focal adhesion proteins (pp125FAK, pp60src, p130Cas, and paxillin), actin-binding proteins (vinculin, talin and α-actinin), and cell adherent junction proteins (E-cadherin, γ-catenin and β-catenin), suggesting that PC1 serves to connect extracellular matrix and the actin cytoskeleton [27, 28]. After the reduction of functional PC1 in kidney epithelial cells of ADPKD patients, the cells change to a partially dedifferentiated state, where E-cadherin is replaced by the mesenchymal N-cadherin in the plasma membrane [29]. PC1 also binds desmosomes during the initial establishment of cell-cell contact [30]. Mislocalization of desmosomal components in apical and basolateral membrane were observed in renal epithelial ADPKD cells, leading to a weak cell-cell interaction [31, 32]. The large variety of proteins that have been identified in complex with PC1 show that the PC1-depletion phenotype is complex and alters several cell events in ADPKD.
2.2 Polycystin 2
PC2, also known as transient receptor potential cation channel, subfamily P, member 2 -TRPP2, is a nonselective cation channel characterized in 1996 [33]. The N-terminal domain of PC2 has sequences for primary cilia localization [34] and oligomerization [35], whereas the C-terminal has an endoplasmatic reticulum (ER)-retention signal [36], an EF-hand motif, and a coiled-coil helix, regions important for channel complex formation and hetero-oligomerization [14, 37, 38], as well as for Ca2+ activation of channel function [39–41]. Recently, the three dimensional structures of truncated and full-length PC2 were solved in open and closed state of the channel, generating information about the channel pore architecture, as well as highlighting the importance of the polycystin domain. This domain is also called the tetragonal opening for polycystins and it is a sequence between transmembrane helices 1 and 2 that can regulate channel function [42–44].
PC2 is diffusely expressed during embryonic and fetal development, persisting in its expression in mature renal tubules, particularly of the distal nephron [45]. PC2 is primarily localized in ER, but it is also found in plasma membrane and cilia [22, 46]. The subcellular localization of PC2 may be dynamic, because the immunostaining of PC2 shows an increased amount of PC2 in the plasma membrane after releasing Ca2+ from ER stores [47].
PC2 interacts with several other proteins in addition to PC1. Among them, PC2 binds cytoskeleton proteins, such as Hax-1, tropomyosin 1, troponin 1, α-actinins, actin-binding and actin-bundling proteins, and filamins [48–53]. PC2 also colocalizes with mDia1, a protein that stabilizes microtubules, the mitotic spindle during cell division [54], and other Ca2+ channels and proteins related to Ca2+ homeostasis, as discussed below.
3. Polycystins and Ca2+ signaling
It is widely accepted that Ca2+ homeostasis is disrupted in ADPKD, either by mutations to PC1 or PC2 [55]. The altered Ca2+ homeostasis can be explained by loss of function of PC2 as a Ca2+ channel, by the lack of regulation of the inositol trisphosphate receptor (InsP3R) and ryanodine receptor (RyR) by PC2, and by the decreased positive modulatory effect of PC1 on PC2 channel activity [23]. The Ca2+ channel function of PC2 has been demonstrated directly in planar lipid bilayers and indirectly in isolated cells [56–58]. PC2 and the PC1/PC2 complex have been shown to function as a Ca2+ channel in cilia [58, 59], a specialized cell compartment where the Ca2+ concentration can change and be regulated independently of alterations in the cytoplasmic Ca2+ [60, 61]. In several studies it was demonstrated that the Ca2+ signaling in the cilia can be initiated by a mechanic stimulus, and these changes in Ca2+ depended on the PC2 channel or PC1/PC2 complex [58, 59, 62–64]. As predicted by these reports, electrophysiological analysis of channel activities measured directly in primary cilium showed that the lack of native PC2 totally abrogates the cation channel activity [65]. There are several recent studies using Ca2+ sensors that are expressed only in the primary cilia [66, 67]. In one report, Ca2+ signals, induced by flow of extracellular fluid, preceded Ca2+ changes in the cytoplasm [66] and these changes required PC2 expression [66]. In another report, there was a complete absence of Ca2+ signal after applying a physiological or a mechanical stimulus [67]. In accordance with the last report, PC2 was not required for the Ca2+ signal in the ciliary membrane [61], and that exogenously expressed polycystins are not sufficient to enhance ciliary mechanosensation [68]. These recent reports suggest that the function of PC1/PC2 in ciliary Ca2+ signaling remains controversial.
Polycystins can also modify Ca2+ release through their interaction with other Ca2+ machinery proteins. PC2 and the InsP3R functionally interact and modulate intracellular Ca2+ signaling by prolonging the half-decay time (t1/2) of InsP3-induced Ca2+ transients [69]. This interaction contributes to the formation of Ca2+ microdomains necessary for initiating Ca2+ -induced Ca2+ release [70]. On the other hand, PC1, when localized in the ER, binds the InsP3R and decreases the InsP3-induced Ca2+ response, suggesting that PC1 competes with PC2 for binding the InsP3R [71].
PC2 also binds and modulates several other Ca2+ channels. The N-terminal and C-terminal cytoplasmic regions of PC2 bind the RyR2 (ryanodine receptor 2) [72], the predominant Ca2+ release channel expressed in cardiac muscle. Interestingly, the C-terminal region of PC2 only binds to the RyR2 in its open state, inhibiting RyR2 channel activity in the presence of Ca2+. This modulatory effect of PC2 explains the increase in the frequency of spontaneous Ca2+ oscillations and decrease in Ca2+ content in sarcoplasmic reticulum stores of cardiomyocytes of PKD2 null mice [72]. The same inhibitory effect of PC2 is observed in the interaction between PC2 and Piezo1, a mechanosensitive ion channel protein. The N-terminal domain of PC2 interacts with Piezo 1 and inhibits the Piezo1-dependent stretch-activated ion channel (SAC) activity in renal tubular epithelial cells [73]. Interestingly, overexpression a PC2 mutant lacking the C-terminal (PC2-740X) produces a large decrease in the amplitude of the Piezo1/SAC currents [73]. In the cilia, PC2 interacts and colocalizes with TRPV4, Transient Receptor Potential cation channel subfamily V member 4, a Ca2+-permeable nonselective cation channel, forming a mechano- and thermosensitive molecular sensor in the cilia [74]. PC2/TRPV4 forms a divalent cation-permeable non-selective ion channel with distinct biophysical, pharmacological, and regulatory profiles when compared to either PC2 or TRPV4 channels. Epidermal growth factor (EGF) can stimulate the PC2/TRPV4 channel through the EGF receptor (EGFR) tyrosine kinase- dependent signaling [75]. PC2 and TRPC1 (transient receptor potential channel 1, a Ca2+-permeable nonselective cation channel) also can assemble to form a channel. PC2/TRPC1 localizes to the primary cilia and plasma membranes and is activated by G-protein-coupled receptors [76].
In addition to forming a Ca2+ channel and regulating Ca2+ fluxes, PC1 and PC2 regulate the ER Ca2+ stores. A fragment of 100 kDa from PC1 C-terminal cleavage binds to the Ca2+-sensor stromal interaction molecule 1 (STIM1) and inhibits the translocation of STIM1 to the cell periphery, which significantly reduces the store operated Ca2+ entry (SOCE) [77]. PC1 expression also increases the interaction between STIM1 and InsP3R, inhibiting Ca2+ release [78]. In PC2-KO cholangiocytes, the interaction of STIM-1 with Orai channel (Ca2+ release-activated Ca2+ channel protein 1) is uncoupled. Cytoplasmic and ER- Ca2+ levels are decreased and SOCE is inhibited, whereas the expression of STIM1 and Orai1 are unchanged [79]. All these functions of PC1 and PC2 related to modulating the Ca2+ machinery activity provide insight into the importance of the polycystins for the Ca2+ signaling. Defects in PC1 or PC2 lead to a decrease in Ca2+ release from ER stores, lowering the cytoplasmic Ca2+ concentration and consequently disrupting Ca2+ homeostasis in ADPKD.
4. Polycystins and cell survival and apoptotic pathway signaling
A link between polycystins and cell survival and cell death has been proposed since 2000. The rates of apoptosis and proliferation are decreased in MDCK cells transfected with full-length human PC1 cDNA (PC1 overexpression – PC1-OE) and these cells preferentially form tubule structures instead of cysts in 3D culture [80]. PC1-OE causes growth arrest in G0/G1, that can be explained by a significant increase in the level of expression of p21, a Cyclin-dependent kinase 2 (Cdk2) inhibitor. The growth inhibitory signals are transmitted by PC1 via direct activation of the JAK-STAT pathway [81]. Conversely, depletion of PC1 causes increases in cyclin A expression and cell proliferation, as well as downregulates p53 expression, a tumor suppressor protein that activates p21 transcription [82, 83]. In agreement with PC1 downregulation results, the overexpression of the PC1 cytoplasmic domain tagged to the plasma membrane showed an increase of cell proliferation through phospholipase C (PLC), protein kinase C alpha (PKCα) and extracellular signal–regulated kinase 1/2 (ERK1/2) activation, thereby upregulating D1 and D3 cyclin, downregulating p21 and p27, and thus inducing cell cycle progression from G0/G1 to the S phase [84]. Together, these results suggest that PC1 acts as a G1 checkpoint, which controls entry into the S phase.
The proposed function of the polycystins as regulators in cell-cycle progression is also supported by the observation that cystic-lining epithelial cells show chromosomal abnormalities. Loss of PC1 produces centrosome amplification and multipolar spindle formation. These events lead to genomic instability, micronucleation, chromatin bridges and aneuploidy [85]. In endothelial primary cells from embryos of PKD1 and PKD2 null mice, it was found that down-regulation of survivin, a protein involved in coordinating proper chromosomal events during mitosis, contributes to these abnormalities, which further results in cell polyploidy [86]. Polycystins also modulate several signaling cascades related to survival, apoptosis and autophagy, as summarized in Figure 2 and discussed below.
Fig. 2. Diagram representing signaling cascades involved in cell survival and cell death that are de-regulated in ADPKD.

Proteins of related to Ca2+ signaling, cyclic adenosine monophosphate (cAMP), mitogen-activated protein kinase (MAPK), Wnt, Janus kinase (JAK) and Signal Transducer and Activator of Transcription (STAT), Hippo, Src, and mechanistic target of rapamycin (mTOR) cascades are represented in the diagram. Ca2+, calcium; PC1, polycystin-1; PC2, polycystin-2; GSK, glycogen synthase kinase; TSC, Tuberous Sclerosis Complex; InsP3R, inositol triphosphate receptor; RYR, ryanodine receptor; TRPV4, Transient Receptor Potential cation channel subfamily V member 4; TRPC1, transient receptor potential channel 1; STIM1, stromal interaction molecule 1; YAP, Yes-associated protein.
4.1 cAMP and MAPK signaling
In vitro and in vivo studies have shown that epithelial cells with mutations in polycystin genes lack a balance between Ca2+ and cAMP (cyclic adenosine monophosphate) signaling [55]. This lack of balance is proposed to be the cause of the change in the phenotype of ADPKD cells from an absorptive and quiescent state to secretory and proliferative state [7]. The secretory phenotype in ADPKD cyst-lining cells can be explained by the activation of protein kinase A (PKA) signaling, a cAMP-dependent protein kinase that leads to the hyperactivation of the chloride channel cystic fibrosis transmembrane conductance regulator (CFTR), and to phosphorylation and translocation of aquaporin 2 (AQP2), a water channel, to the apical plasma membrane [7, 55]. As for the proliferative phenotype, the upregulation of the MAPK pathway partially explains the increase of mitotic rates in ADPKD cyst-lining cells. Overexpression of the PC1 C-terminal in epithelial cells converts the phenotype from one in which cell proliferation is inhibited by cAMP to one in which mitosis is stimulated by cAMP [87, 88]. This change in the phenotype can be attributed to disruption of intracellular Ca2+ mobilization, because epithelial cells treated with Ca2+-lowering reagents (Ca2+ channel blockers or EGTA) or transfected with polycystin-1 C-terminal fragment showed a cAMP-dependent activation of B-Raf and ERK, which are normally inhibited by Akt in a phosphatidylinositol-3-kinases (PI3K)- and Ca2+- dependent manner [88]. The same phenotype switch was also observed in ADPKD renal epithelial cells with germline mutations in PC1 [89]. In accordance with these observations, a reduction of adenylyl cyclase 6 (AC6) expression with the purpose to reduce cAMP formation markedly decreases kidney size and cystogenesis in a PC1 collecting duct-specific knockout mice model, improving renal function, reducing activation of the B-Raf/ERK/MEK pathway, and increasing proliferation and survival [90]. Taken together, the increase in cell proliferation and fluid secretion can be attributed to the intracellular Ca2+ and cAMP changes promoted by polycystin mutations, which trigger the B-Raf/MEK/ERK signaling cascade. These changes contribute to kidney cyst development, as seen in ADPKD.
4.2 Wnt signaling
Wnt signaling is a critical component during embryonic development and also plays an important role in regulating adult tissue homeostasis. Canonical Wnt signaling, a cascade pathway that regulates proliferation, survival and apoptosis through a variety of mechanisms [91], is modulated by PC1 and PC2. Overxpression of the cytoplasmic domain of PC1 tagged to the membrane decreases the glycogen synthase kinase 3 (GSK-3) activity and, consequently, elevates total β-catenin steady-state protein levels, by suppressing β-catenin ubiquitination and degradation. Increasing of transcriptional activity of XWnt8, XDsh, rFz2, and β-catenin, is related to PC1 expression [92]. In addition to the inhibition of GSK-3, the overexpression of C-terminal fragment of PC1 tagged to the membrane also activates Wnt signaling by increasing the degradation of Jade-1, a ubiquitin ligase responsible for β-catenin ubiquitination. ADPKD-associated PC1 mutants failed to regulate Jade-1 [93]. Recently, it was suggested that the PC1/PC2 complex acts either as a direct or indirect receptor to Wnt. Wnt3A and Wnt9B bind to the extracellular domain of PC1 and induce whole-cell currents and Ca2+ influx that depend on PC2. Conversely, pathogenic PKD1 or PKD2 mutations that abrogate PC1/PC2 complex formation suppress activation by the Wnts [94]. However, an inhibitory effect of PC1 C-terminal domain in Wnt signaling has also been described. The last 200 amino acids of the C-terminus of PC1, PC1-P200, binds to β-catenin in the nucleus, inhibiting the ability of β-catenin to activate T-cell factor (TCF) -dependent gene transcription, a major effector of the canonical Wnt signaling pathway [95]. These findings demonstrate that polycystins can directly modulate Wnt signaling, which contributes to cyst formation and development suggesting that the Wnt pathway contains promising targets for treating ADPKD patients.
4.3 JAK/STAT signaling
The JAK-STAT signaling pathway transmits information from extracellular chemical signals to the nucleus resulting in DNA transcription and expression of genes involved in immunity, proliferation, differentiation and apoptosis [96]. The JAK-STAT signaling pathway is also upregulated in ADPKD. Cyst-lining cells in ADPKD exhibit elevated levels of nuclear STAT-1, -3 and -6, and consequently STAT-dependent gene expression. For example, the transcriptional activity of STAT1 is increased in renal tubular epithelial cells overexpressing a 30-kDa peptide sequence of the PC1 C-terminal domain (PC1-P30), via JAK2 phosphorylation [97]. With STAT3, PC1 can regulate its activity by both a direct and an indirect mechanism. Overexpression of the PC1 C-terminal domain tagged to the membrane can directly activate STAT3 in a JAK2-dependent manner, leading to tyrosine phosphorylation and transcriptional activity, whereas the overexpression of the soluble C-terminal domain of PC1 can indirectly activate STAT3 using a mechanism requiring STAT phosphorylation by cytokines or growth factors, resulting in an exaggerated cytokine response [98]. The soluble PC1-P30 C-terminal fragment of PC1 can also activate STAT3 via Src signaling. PC1-P30 interacts with the non-receptor tyrosine kinase Src, resulting in Src-dependent activation of STAT3. Interestingly, this STAT3 activation mechanism is independent of JAK family kinases and amplified by the EGFR or cAMP signaling [99]. STAT6 normally localizes to primary cilia of renal epithelial cells, and translocates to the nucleus after cessation of apical fluid flow. However, in cyst-lining cells from ADPKD, the levels of STAT6, P100 (a STAT6 coactivator), and the soluble PC1 C-terminal domain are constitutively elevated in the nucleus. The soluble PC1 C-terminal fragment interacts with the transcription factor STAT6 and the coactivator P100, and it stimulates STAT6-dependent gene expression [100]. Pharmacological inhibition of STAT6 or backcrossing of STAT6-null mice with PC1-deficient mice leads to a significant inhibition of proliferation and cyst growth, and preservation of renal function [101]. Therefore, the ability of PC1 to regulate STAT proteins may ultimately lead to therapeutic approaches by targeting the aberrantly activated Jak/STAT pathway in ADPKD.
4.4 Hippo signaling
Altered activity of Hippo signaling, a pathway involved in organ size control as a consequence of its regulation of proliferation and survival [102], is described in renal tissues from human ADPKD patients. The transcriptional co-activator Yes-associated protein (YAP), the final effector molecule of the Hippo pathway, is primarily localized in the nucleus of PKD1 knockout cyst-lining cells, and consequently, YAP targets are also up-regulated in cystic cells [103]. The Hippo pathway can also be modulated in ADPKD by the upregulation of cAMP signaling. PKA stimulates Lats1/2, serine/threonine kinases that phosphorylate and inactivate YAP [104]. These initial studies provide some insight into Hippo signaling in ADPKD, yet the pathophysiological implications of a dysregulation of this cascade remain poorly understood.
4.5 Scr signaling
Scr signaling, which is associated with cell proliferation, matrix adhesion, motility, and survival in tumors, is increased in cyst-lining autosomal dominant polycystic kidney disease (ADPKD) epithelial cells in human and mouse ADPKD. In vitro studies on mouse inner medullary collecting duct (mIMCD) cells and human ADPKD cyst-lining epithelial cells showed that a specific inhibitor of pY418-Src inhibits epithelial cell proliferation, decreases adhesion of mIMCD and human ADPKD to extracellular collagen matrix. In vivo, the inhibitor of pY418-Src retards renal cystic phenotype of PDK1 heterozygous mice [105]. Because of the promising preclinical results with Src inhibitor, a phase 2 trial (NCT01233869) is being conducted to test the safety and efficacy of Src inhibition in ADPKD patients [106].
4.6 JNK signaling
Jun N-terminal kinases (JNKs) belong to the superfamily of MAP-kinases involved in the regulation of cell proliferation, differentiation, and apoptosis [107]. The ovexpression of the PC1 C-terminal domain tagged to the membrane leads to JNK signaling activation via Gα and Gβ subunits of heterotrimeric G proteins, and consequently triggers the activation of the transcription factor of activator protein 1 (AP-1), specifically c-Jun and activating transcription factor (ATF2), that modulates a variety of cellular programs including apoptosis [108–110]. This constitutive activation of various AP-1 components occurs in ADPKD [111]. JNK activation, together with an increase in apoptosis, is also observed in PC1-silenced MDCK cells stimulated with thrombin, whereas full length PC1 overexpression shows the opposite effect [112]. Overexpression of PC2 also stimulates the phosphorylation of c-Jun and the induction of AP-1 activity through activation of JNK1 and p38, by PKCε-dependent mechanism. The expression of the C-terminal domain of PC1 tagged to the membrane dramatically augments PC2-mediated AP-1 activity [113]. It is possible that the high rates of apoptosis seen in ADPKD cyst-lining cells is primarily a consequence of JNK cascade activation.
4.7 mTOR signaling
mTOR (mechanistic target of rapamycin) is a serine/threonine kinase, member of the PI3K-related kinase superfamily, and involved in regulating cell cycle progression, translational control, ribosomal biogenesis, and cellular energy responses [114]. The mTOR pathway is inappropriately activated in cyst-lining epithelial cells in human ADPKD patients and mouse models [115]. It was suggested that PC1 modulates mTor signaling by inhibiting Tuberous Sclerosis Complex 2 (TSC2) degradation, a gatekeeper for mTOR activity. This suggestion is based upon the observation that overexpression of full length PC1 downregulates mTOR effectors, S6K1 and 4EBP1 via TSC2-dependent manner [116]. It also was described that the C-terminal domain of PC1 tagged to the membrane directly interacts with and protects TSC2 from Akt phosphorylation at S939, retaining TSC2 at the membrane to inhibit the mTOR pathway. Expression of the C-terminal domain of PC1 also decreases binding of 14-3-3 proteins to TSC2 and increases the interaction between TSC2 and its activating partner TSC1 [117].
Despite of the abnormal mTOR signaling activation in ADPKD, sirolimus, a mTORC1 inhibitor, failed to slow kidney cyst growth in clinical trials. In addition to the lack of tolerance and poor tissue distribution of the drug, the poor performance of sirolimus can be explained by its selective inhibition activity on mTORC1 and its minimal effect on mTORC2 [106]. In a PKD2 knockout mouse model, both mTORC1 and mTORC2 markers (pS6 and pAktSer473, respectively) are increased, suggesting that an inhibitor that targets both molecules is needed. The treatment with mTOR anti-sense oligonucleotides for mTORC1 and mTORC2 significantly decreases S6 and Akt phosphorylation, proliferation and apoptosis of tubular epithelial cells, as well as cyst volume [118]. Therefore, the mTOR pathway inhibition should still be a top contender as a potential therapeutic approach for ADPKD, primarily based upon the new findings that relate cyst development and impairment of autophagy.
5. Polycystins and autophagy
Autophagy, an adaptation mechanism for cellular homeostasis in response to various stress conditions, was recently reported as decreased in ADPKD. The downregulation of autophagy in ADPKD can be partially explained by crosstalk between ciliogenesis and autophagy (for reviews, see [119, 120]). Nutrient deprivation, a well-known autophagy stimulus, was also identified as an activator of primary cilia formation by causing the degradation of oral-facial-digital syndrome 1 (OFD1), a protein related to ciliogenesis [121]. In contrast, the lack of autophagy-related protein (Atg) 5 or Atg3 showed an accumulation of OFD1, leading to fewer and shorter primary cilia [121]. Conversely, disruption of autophagosome synthesis was observed in cells lacking several cilia proteins, such as the intraflagellar transport protein (IFT) 20 and IFT88. Specifically, IFT20 was identified as a common component for both ciliogenesis and starvation-induced autophagosome formation [122]. Together, these studies support the hypothesis that autophagy is downregulated in conditions in which there is a defective ciliogenesis, such as ADPKD.
In addition to a cilia-depended decrease of autophagy, several studies directly related the polycystins to the autophagy. The first report of impaired autophagy in PC1-deficient cells demonstrated that glucose deprivation results in higher apoptotic rates in PKD1-mutant cells instead of autophagy, an event that is normally activated in normal cells to survive the stress stimuli [123]. This inhibition of autophagy-signaling in PC1-deficient cells is partially dependent on the mTOR pathway, one of the main regulators of autophagy. Treatment of PKD1-null cells with rapamycin partially restores autophagy and cell survival under glucose deprivation [123]. In support of these in vitro data describing the lack of autophagy in PC1-deficient cells, it was found that zebrafish mutants for pkd1a, a gene orthologue of mammalian PKD1, and kidney epithelial cells derived from both PKD1-null mice and ADPKD patients have an impaired autophagic flux and mTOR upregulation. The inhibition of autophagy by knocking down Atg5, the core autophagy protein, promotes cystogenesis, whereas pharmacological activation of autophagy, including the use of mTOR-dependent rapamycin or mTOR-independent carbamazepine and minoxidil, markedly attenuated cyst formation and restored kidney function in the zebrafish pkd1a model [124].
The involvement of PC2 in autophagy has also been described. In kidney epithelial cells, autophagy can be induced by fluid flow after activation of the primary cilium [125]. Primary cilium-dependent autophagy is hypothesized to be triggered either by Liver Kinase B1 (LKB1)–AMP-activated protein kinase (AMPK)–mTOR inhibition (noncanonical m-TOR pathway) or by a mechanism dependent on the PC2 channel. In PC2-deficient cells, the activation of autophagy after fluid flow stimuli is impaired, indicating that PC2, most probably in a complex with polycystin 1, stimulates autophagy [125]. These reports show that suppression of autophagy is important in cyst formation and growth and suggests that the induction of autophagy as a potential therapuitc approach for ADPKD.
5. Conclusion
In this review, a large body of evidence was accumulated showing that the polycystins play a key role in cell fate, especially cell differentiation, proliferation, survival and apoptosis, and, more recently, autophagy. In addition to decreases in Ca2+ signaling, several other signaling cascades are directly or indirectly modulated by PC1/PC2, including cAMP, MAPK, Wnt, JAK-STAT, Hippo, Src, and mTOR. This large number of distinct pathways related to the polycystins confirms the complexity of the ADPKD pathophysiology, as well as highlight the challenges to develop pharmacological therapies for this multi-organ disease.
Highlights.
Polycystins form a heteromeric molecular complex important for calcium homeostasis.
Mutations in polycystins cause polycystic kidney disease (ADPKD).
Cyst-lining kidney epithelium show an increase in proliferation and apoptosis and a decrease in autophagy.
Polycystins modulates several signaling cascades related to cell fate.
Acknowledgments
We thank Dr Ivana Kuo and Allison Brill (Yale University) for generously providing many useful suggestions and comments on the manuscript. Grant support is acknowledged: A fellowship from Conselho Nacional de Desenvolvimento Científico e Tecnológico - Brazil (FOL), 5P01DK057751 and P30DK090744 (BEE).
Abbreviation
- PC1
polycystin-1
- PC2
polycystin-2
- ADPKD
autosomal dominant polycystic kidney disease
- PKD1
PC1 gene
- PKD2
PC2 gene
- ESRD
end stage renal disease
- Ca2+
calcium
- cAMP
Cyclic adenosine monophosphate
- MAPK
mitogen-activated protein kinase
- mTOR
mechanistic target of rapamycin
- JAK
Janus kinase
- STAT
Signal Transducer and Activator of Transcription
- Gi and Go
heterotrimeric G protein subunits
- P200
last 200 amino acids of the PC1 C-terminal domain
- Kif3a
kinesin-like protein
- Ift20
intraflagellar transport protein 20 homolog
- KO
knockout
- TRPP2
transient receptor potential cation channel, subfamily P, member 2
- ER
endoplasmatic reticulum
- InsP3R
inositol triphosphate receptor
- t1/2
half-decay time
- InsP3
inositol triphosphate
- RYR2
ryanodine receptor 2
- SAC
stretch-activated ion channel
- TRPV4
Transient Receptor Potential cation channel subfamily V member 4
- EGF
Epidermal growth factor
- EGFR
Epidermal growth factor receptor
- TRPC1
transient receptor potential channel 1
- STIM1
stromal interaction molecule 1
- SOCE
store operated calcium entry
- OE
overexpression
- 3D
tridimensional
- cdk2
Cyclin-dependent kinase 2
- PLC
phospholipase C
- PKCα
protein kinase C alpha
- ERK1/2
extracellular signal–regulated kinase 1/2
- PKA
protein kinase A
- CFTR
chloride channel cystic fibrosis transmembrane conductance regulator
- AQP2
aquaporin 2
- PI3K
phosphatidylinositol-3-kinase
- AC6
adenylyl cyclase 6
- GSK
glycogen synthase kinase
- TCF
T-cell factor
- YAP
Yes-associated protein
- mIMCD
mouse inner medullary collecting duct
- AP-1
activator protein 1
- ATF2
activating transcription factor
- TSC2
Tuberous Sclerosis Complex 2
Footnotes
Competing interests:
The authors state that there are no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Patel A, Honore E. Polycystins and renovascular mechanosensory transduction. Nat Rev Nephrol. 2010;6:530–538. doi: 10.1038/nrneph.2010.97. [DOI] [PubMed] [Google Scholar]
- 2.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet (London, England) 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 3.Ong AC, Devuyst O, Knebelmann B, Walz G. Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet (London, England) 2015;385:1993–2002. doi: 10.1016/S0140-6736(15)60907-2. [DOI] [PubMed] [Google Scholar]
- 4.Luciano RL, Dahl NK. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): considerations for routine screening and management. Nephrol Dial Transplant. 2014;29:247–254. doi: 10.1093/ndt/gft437. [DOI] [PubMed] [Google Scholar]
- 5.Saigusa T, Bell PD. Molecular pathways and therapies in autosomal-dominant polycystic kidney disease. Physiology (Bethesda) 2015;30:195–207. doi: 10.1152/physiol.00032.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simms RJ. Autosomal dominant polycystic kidney disease. Bmj. 2016;352:i679. doi: 10.1136/bmj.i679. [DOI] [PubMed] [Google Scholar]
- 7.Chebib FT, Sussman CR, Wang X, Harris PC, Torres VE. Vasopressin and disruption of calcium signalling in polycystic kidney disease. Nat Rev Nephrol. 2015;11:451–464. doi: 10.1038/nrneph.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grantham JJ, Torres VE. The importance of total kidney volume in evaluating progression of polycystic kidney disease. Nat Rev Nephrol. 2016;12:667–677. doi: 10.1038/nrneph.2016.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seeger-Nukpezah T, Geynisman DM, Nikonova AS, Benzing T, Golemis EA. The hallmarks of cancer: relevance to the pathogenesis of polycystic kidney disease. Nat Rev Nephrol. 2015;11:515–534. doi: 10.1038/nrneph.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–160. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 11.Oatley P, Stewart AP, Sandford R, Edwardson JM. Atomic force microscopy imaging reveals the domain structure of polycystin-1. Biochemistry. 2012;51:2879–2888. doi: 10.1021/bi300134b. [DOI] [PubMed] [Google Scholar]
- 12.Lohning C, Pohlschmidt M, Glucksmann-Kuis MA, Duyk G, Bork P, Schneider MO, Reeders ST, Frischauf AM. Structural motifs of the PKD1 protein. Nephrol Dial Transplant. 1996;11(Suppl 6):2–4. doi: 10.1093/ndt/11.supp6.2. [DOI] [PubMed] [Google Scholar]
- 13.Qian F, Boletta A, Bhunia AK, Xu H, Liu L, Ahrabi AK, Watnick TJ, Zhou F, Germino GG. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc Natl Acad Sci U S A. 2002;99:16981–16986. doi: 10.1073/pnas.252484899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet. 1997;16:179–183. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]
- 15.Parnell SC, Magenheimer BS, Maser RL, Rankin CA, Smine A, Okamoto T, Calvet JP. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem Biophys Res Commun. 1998;251:625–631. doi: 10.1006/bbrc.1998.9514. [DOI] [PubMed] [Google Scholar]
- 16.Delmas P, Nomura H, Li X, Lakkis M, Luo Y, Segal Y, Fernandez-Fernandez JM, Harris P, Frischauf AM, Brown DA, Zhou J. Constitutive activation of G-proteins by polycystin-1 is antagonized by polycystin-2. J Biol Chem. 2002;277:11276–11283. doi: 10.1074/jbc.M110483200. [DOI] [PubMed] [Google Scholar]
- 17.Chauvet V, Tian X, Husson H, Grimm DH, Wang T, Hiesberger T, Igarashi P, Bennett AM, Ibraghimov-Beskrovnaya O, Somlo S, Caplan MJ. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J Clin Invest. 2004;114:1433–1443. doi: 10.1172/JCI21753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward CJ, Turley H, Ong AC, Comley M, Biddolph S, Chetty R, Ratcliffe PJ, Gattner K, Harris PC. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci U S A. 1996;93:1524–1528. doi: 10.1073/pnas.93.4.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ibraghimov-Beskrovnaya O, Dackowski WR, Foggensteiner L, Coleman N, Thiru S, Petry LR, Burn TC, Connors TD, Van Raay T, Bradley J, Qian F, Onuchic LF, Watnick TJ, Piontek K, Hakim RM, Landes GM, Germino GG, Sandford R, Klinger KW. Polycystin: in vitro synthesis, in vivo tissue expression, and subcellular localization identifies a large membrane-associated protein. Proc Natl Acad Sci U S A. 1997;94:6397–6402. doi: 10.1073/pnas.94.12.6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters DJ, van de Wal A, Spruit L, Saris JJ, Breuning MH, Bruijn JA, de Heer E. Cellular localization and tissue distribution of polycystin-1. J Pathol. 1999;188:439–446. doi: 10.1002/(SICI)1096-9896(199908)188:4<439::AID-PATH367>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 21.Palsson R, Sharma CP, Kim K, McLaughlin M, Brown D, Arnaout MA. Characterization and cell distribution of polycystin, the product of autosomal dominant polycystic kidney disease gene 1. Molecular medicine (Cambridge, Mass) 1996;2:702–711. [PMC free article] [PubMed] [Google Scholar]
- 22.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 23.Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 24.Gainullin VG, Hopp K, Ward CJ, Hommerding CJ, Harris PC. Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J Clin Invest. 2015;125:607–620. doi: 10.1172/JCI76972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet. 2013;45:1004–1012. doi: 10.1038/ng.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zatti A, Chauvet V, Rajendran V, Kimura T, Pagel P, Caplan MJ. The C-terminal tail of the polycystin-1 protein interacts with the Na,K-ATPase alpha-subunit. Mol Biol Cell. 2005;16:5087–5093. doi: 10.1091/mbc.E05-03-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huan Y, van Adelsberg J. Polycystin-1, the PKD1 gene product, is in a complex containing E-cadherin and the catenins. J Clin Invest. 1999;104:1459–1468. doi: 10.1172/JCI5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geng L, Burrow CR, Li HP, Wilson PD. Modification of the composition of polycystin-1 multiprotein complexes by calcium and tyrosine phosphorylation. Biochim Biophys Acta. 2000;1535:21–35. doi: 10.1016/s0925-4439(00)00079-x. [DOI] [PubMed] [Google Scholar]
- 29.Roitbak T, Ward CJ, Harris PC, Bacallao R, Ness SA, Wandinger-Ness A. A polycystin-1 multiprotein complex is disrupted in polycystic kidney disease cells. Mol Biol Cell. 2004;15:1334–1346. doi: 10.1091/mbc.E03-05-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheffers MS, van der Bent P, Prins F, Spruit L, Breuning MH, Litvinov SV, de Heer E, Peters DJ. Polycystin-1, the product of the polycystic kidney disease 1 gene, co-localizes with desmosomes in MDCK cells. Hum Mol Genet. 2000;9:2743–2750. doi: 10.1093/hmg/9.18.2743. [DOI] [PubMed] [Google Scholar]
- 31.Silberberg M, Charron AJ, Bacallao R, Wandinger-Ness A. Mispolarization of desmosomal proteins and altered intercellular adhesion in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol. 2005;288:F1153–1163. doi: 10.1152/ajprenal.00008.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russo RJ, Husson H, Joly D, Bukanov NO, Patey N, Knebelmann B, Ibraghimov-Beskrovnaya O. Impaired formation of desmosomal junctions in ADPKD epithelia. Histochemistry and cell biology. 2005;124:487–497. doi: 10.1007/s00418-005-0055-3. [DOI] [PubMed] [Google Scholar]
- 33.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 34.Geng L, Okuhara D, Yu Z, Tian X, Cai Y, Shibazaki S, Somlo S. Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J Cell Sci. 2006;119:1383–1395. doi: 10.1242/jcs.02818. [DOI] [PubMed] [Google Scholar]
- 35.Feng S, Okenka GM, Bai CX, Streets AJ, Newby LJ, DeChant BT, Tsiokas L, Obara T, Ong AC. Identification and functional characterization of an N-terminal oligomerization domain for polycystin-2. J Biol Chem. 2008;283:28471–28479. doi: 10.1074/jbc.M803834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai Y, Maeda Y, Cedzich A, Torres VE, Wu G, Hayashi T, Mochizuki T, Park JH, Witzgall R, Somlo S. Identification and characterization of polycystin-2, the PKD2 gene product. J Biol Chem. 1999;274:28557–28565. doi: 10.1074/jbc.274.40.28557. [DOI] [PubMed] [Google Scholar]
- 37.Giamarchi A, Feng S, Rodat-Despoix L, Xu Y, Bubenshchikova E, Newby LJ, Hao J, Gaudioso C, Crest M, Lupas AN, Honore E, Williamson MP, Obara T, Ong AC, Delmas P. A polycystin-2 (TRPP2) dimerization domain essential for the function of heteromeric polycystin complexes. Embo j. 2010;29:1176–1191. doi: 10.1038/emboj.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc Natl Acad Sci U S A. 1997;94:6965–6970. doi: 10.1073/pnas.94.13.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai Y, Anyatonwu G, Okuhara D, Lee KB, Yu Z, Onoe T, Mei CL, Qian Q, Geng L, Wiztgall R, Ehrlich BE, Somlo S. Calcium dependence of polycystin-2 channel activity is modulated by phosphorylation at Ser812. J Biol Chem. 2004;279:19987–19995. doi: 10.1074/jbc.M312031200. [DOI] [PubMed] [Google Scholar]
- 40.Yang Y, Hodsdon ME, Lolis EJ, Ehrlich BE. Conformational dynamics of Ca2+-dependent responses in the polycystin-2 C-terminal tail. Biochem J. 2016;473:285–296. doi: 10.1042/BJ20151031. [DOI] [PubMed] [Google Scholar]
- 41.Celic AS, Petri ET, Benbow J, Hodsdon ME, Ehrlich BE, Boggon TJ. Calcium-induced conformational changes in C-terminal tail of polycystin-2 are necessary for channel gating. J Biol Chem. 2012;287:17232–17240. doi: 10.1074/jbc.M112.354613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen PS, Yang X, DeCaen PG, Liu X, Bulkley D, Clapham DE, Cao E. The Structure of the Polycystic Kidney Disease Channel PKD2 in Lipid Nanodiscs. Cell. 2016;167:763–773.e711. doi: 10.1016/j.cell.2016.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grieben M, Pike AC, Shintre CA, Venturi E, El-Ajouz S, Tessitore A, Shrestha L, Mukhopadhyay S, Mahajan P, Chalk R, Burgess-Brown NA, Sitsapesan R, Huiskonen JT, Carpenter EP. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2) Nature structural & molecular biology. 2016 doi: 10.1038/nsmb.3343. [DOI] [PubMed] [Google Scholar]
- 44.Wilkes M, Madej MG, Kreuter L, Rhinow D, Heinz V, De Sanctis S, Ruppel S, Richter RM, Joos F, Grieben M, Pike AC, Huiskonen JT, Carpenter EP, Kuhlbrandt W, Witzgall R, Ziegler C. Molecular insights into lipid-assisted Ca2+ regulation of the TRP channel Polycystin-2. Nature structural & molecular biology. 2017 doi: 10.1038/nsmb.3357. [DOI] [PubMed] [Google Scholar]
- 45.Markowitz GS, Cai Y, Li L, Wu G, Ward LC, Somlo S, D’Agati VD. Polycystin-2 expression is developmentally regulated. The American journal of physiology. 1999;277:F17–25. doi: 10.1152/ajprenal.1999.277.1.F17. [DOI] [PubMed] [Google Scholar]
- 46.Kottgen M, Walz G. Subcellular localization and trafficking of polycystins. Pflugers Arch. 2005;451:286–293. doi: 10.1007/s00424-005-1417-3. [DOI] [PubMed] [Google Scholar]
- 47.Miyakawa A, Ibarra C, Malmersjo S, Aperia A, Wiklund P, Uhlen P. Intracellular calcium release modulates polycystin-2 trafficking. BMC Nephrol. 2013;14:34. doi: 10.1186/1471-2369-14-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gallagher AR, Cedzich A, Gretz N, Somlo S, Witzgall R. The polycystic kidney disease protein PKD2 interacts with Hax-1, a protein associated with the actin cytoskeleton. Proc Natl Acad Sci U S A. 2000;97:4017–4022. doi: 10.1073/pnas.97.8.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Q, Dai Y, Guo L, Liu Y, Hao C, Wu G, Basora N, Michalak M, Chen XZ. Polycystin-2 associates with tropomyosin-1, an actin microfilament component. J Mol Biol. 2003;325:949–962. doi: 10.1016/s0022-2836(02)01333-5. [DOI] [PubMed] [Google Scholar]
- 50.Li Q, Shen PY, Wu G, Chen XZ. Polycystin-2 interacts with troponin I, an angiogenesis inhibitor. Biochemistry. 2003;42:450–457. doi: 10.1021/bi0267792. [DOI] [PubMed] [Google Scholar]
- 51.Li Q, Montalbetti N, Shen PY, Dai XQ, Cheeseman CI, Karpinski E, Wu G, Cantiello HF, Chen XZ. Alpha-actinin associates with polycystin-2 and regulates its channel activity. Hum Mol Genet. 2005;14:1587–1603. doi: 10.1093/hmg/ddi167. [DOI] [PubMed] [Google Scholar]
- 52.Wang Q, Dai XQ, Li Q, Wang Z, del Cantero MR, Li S, Shen J, Tu JC, Cantiello H, Chen XZ. Structural interaction and functional regulation of polycystin-2 by filamin. PLoS One. 2012;7:e40448. doi: 10.1371/journal.pone.0040448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Q, Zheng W, Wang Z, Yang J, Hussein S, Tang J, Chen XZ. Filamin-a increases the stability and plasma membrane expression of polycystin-2. PLoS One. 2015;10:e0123018. doi: 10.1371/journal.pone.0123018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rundle DR, Gorbsky G, Tsiokas L. PKD2 interacts and co-localizes with mDia1 to mitotic spindles of dividing cells: role of mDia1 IN PKD2 localization to mitotic spindles. J Biol Chem. 2004;279:29728–29739. doi: 10.1074/jbc.M400544200. [DOI] [PubMed] [Google Scholar]
- 55.Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25:18–32. doi: 10.1681/ASN.2013040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE, Somlo S. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002;4:191–197. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 57.Gonzalez-Perrett S, Kim K, Ibarra C, Damiano AE, Zotta E, Batelli M, Harris PC, Reisin IL, Arnaout MA, Cantiello HF. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc Natl Acad Sci U S A. 2001;98:1182–1187. doi: 10.1073/pnas.98.3.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 59.Yoshiba S, Shiratori H, Kuo IY, Kawasumi A, Shinohara K, Nonaka S, Asai Y, Sasaki G, Belo JA, Sasaki H, Nakai J, Dworniczak B, Ehrlich BE, Pennekamp P, Hamada H. Cilia at the node of mouse embryos sense fluid flow for left-right determination via Pkd2. Science. 2012;338:226–231. doi: 10.1126/science.1222538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE. Primary cilia are specialized calcium signalling organelles. Nature. 2013;504:311–314. doi: 10.1038/nature12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeCaen PG, Delling M, Vien TN, Clapham DE. Direct recording and molecular identification of the calcium channel of primary cilia. Nature. 2013;504:315–318. doi: 10.1038/nature12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–1171. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.AbouAlaiwi WA, Takahashi M, Mell BR, Jones TJ, Ratnam S, Kolb RJ, Nauli SM. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res. 2009;104:860–869. doi: 10.1161/CIRCRESAHA.108.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jin X, Mohieldin AM, Muntean BS, Green JA, Shah JV, Mykytyn K, Nauli SM. Cilioplasm is a cellular compartment for calcium signaling in response to mechanical and chemical stimuli. Cell Mol Life Sci. 2014;71:2165–2178. doi: 10.1007/s00018-013-1483-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kleene SJ, Kleene NK. The native TRPP2-dependent channel of murine renal primary cilia. Am J Physiol Renal Physiol. 2017;312:F96–f108. doi: 10.1152/ajprenal.00272.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yuan S, Zhao L, Brueckner M, Sun Z. Intraciliary calcium oscillations initiate vertebrate left-right asymmetry. Current biology: CB. 2015;25:556–567. doi: 10.1016/j.cub.2014.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delling M, Indzhykulian AA, Liu X, Li Y, Xie T, Corey DP, Clapham DE. Primary cilia are not calcium-responsive mechanosensors. Nature. 2016;531:656–660. doi: 10.1038/nature17426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stavola LK, Prætorius H, Caplan MJ. Expression of Polycystins in LLC-PK1 Cells Does Not Increase Flow-Activated Calcium Fluxes. The FASEB Journal. 2016;30:1219–1211. [Google Scholar]
- 69.Li Y, Wright JM, Qian F, Germino GG, Guggino WB. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J Biol Chem. 2005;280:41298–41306. doi: 10.1074/jbc.M510082200. [DOI] [PubMed] [Google Scholar]
- 70.Sammels E, Devogelaere B, Mekahli D, Bultynck G, Missiaen L, Parys JB, Cai Y, Somlo S, De Smedt H. Polycystin-2 activation by inositol 1,4,5-trisphosphate-induced Ca2+ release requires its direct association with the inositol 1,4,5-trisphosphate receptor in a signaling microdomain. J Biol Chem. 2010;285:18794–18805. doi: 10.1074/jbc.M109.090662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Y, Santoso NG, Yu S, Woodward OM, Qian F, Guggino WB. Polycystin-1 interacts with inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling with implications for polycystic kidney disease. J Biol Chem. 2009;284:36431–36441. doi: 10.1074/jbc.M109.068916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci U S A. 2007;104:6454–6459. doi: 10.1073/pnas.0610324104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peyronnet R, Martins JR, Duprat F, Demolombe S, Arhatte M, Jodar M, Tauc M, Duranton C, Paulais M, Teulon J, Honore E, Patel A. Piezo1-dependent stretch-activated channels are inhibited by Polycystin-2 in renal tubular epithelial cells. EMBO Rep. 2013;14:1143–1148. doi: 10.1038/embor.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kottgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, Nitschke R, Suzuki M, Kramer-Zucker A, Germino GG, Watnick T, Prenen J, Nilius B, Kuehn EW, Walz G. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol. 2008;182:437–447. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang ZR, Chu WF, Song B, Gooz M, Zhang JN, Yu CJ, Jiang S, Baldys A, Gooz P, Steele S, Owsianik G, Nilius B, Komlosi P, Bell PD. TRPP2 and TRPV4 form an EGF-activated calcium permeable channel at the apical membrane of renal collecting duct cells. PLoS One. 2013;8:e73424. doi: 10.1371/journal.pone.0073424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bai CX, Giamarchi A, Rodat-Despoix L, Padilla F, Downs T, Tsiokas L, Delmas P. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep. 2008;9:472–479. doi: 10.1038/embor.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Woodward OM, Li Y, Yu S, Greenwell P, Wodarczyk C, Boletta A, Guggino WB, Qian F. Identification of a polycystin-1 cleavage product, P100, that regulates store operated Ca entry through interactions with STIM1. PLoS One. 2010;5:e12305. doi: 10.1371/journal.pone.0012305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Santoso NG, Cebotaru L, Guggino WB. Polycystin-1, 2, and STIM1 interact with IP(3)R to modulate ER Ca release through the PI3K/Akt pathway. Cell Physiol Biochem. 2011;27:715–726. doi: 10.1159/000330080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spirli C, Locatelli L, Fiorotto R, Morell CM, Fabris L, Pozzan T, Strazzabosco M. Altered store operated calcium entry increases cyclic 3′,5′-adenosine monophosphate production and extracellular signal-regulated kinases 1 and 2 phosphorylation in polycystin-2-defective cholangiocytes. Hepatology. 2012;55:856–868. doi: 10.1002/hep.24723. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 80.Boletta A, Qian F, Onuchic LF, Bhunia AK, Phakdeekitcharoen B, Hanaoka K, Guggino W, Monaco L, Germino GG. Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Molecular cell. 2000;6:1267–1273. doi: 10.1016/s1097-2765(00)00123-4. [DOI] [PubMed] [Google Scholar]
- 81.Bhunia AK, Piontek K, Boletta A, Liu L, Qian F, Xu PN, Germino FJ, Germino GG. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell. 2002;109:157–168. doi: 10.1016/s0092-8674(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 82.Kim H, Bae Y, Jeong W, Ahn C, Kang S. Depletion of PKD1 by an antisense oligodeoxynucleotide induces premature G1/S-phase transition. European journal of human genetics: EJHG. 2004;12:433–440. doi: 10.1038/sj.ejhg.5201136. [DOI] [PubMed] [Google Scholar]
- 83.Nishio S, Hatano M, Nagata M, Horie S, Koike T, Tokuhisa T, Mochizuki T. Pkd1 regulates immortalized proliferation of renal tubular epithelial cells through p53 induction and JNK activation. J Clin Invest. 2005;115:910–918. doi: 10.1172/JCI22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Manzati E, Aguiari G, Banzi M, Manzati M, Selvatici R, Falzarano S, Maestri I, Pinton P, Rizzuto R, del Senno L. The cytoplasmic C-terminus of polycystin-1 increases cell proliferation in kidney epithelial cells through serum-activated and Ca(2+)-dependent pathway(s) Exp Cell Res. 2005;304:391–406. doi: 10.1016/j.yexcr.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 85.Battini L, Macip S, Fedorova E, Dikman S, Somlo S, Montagna C, Gusella GL. Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum Mol Genet. 2008;17:2819–2833. doi: 10.1093/hmg/ddn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.AbouAlaiwi WA, Ratnam S, Booth RL, Shah JV, Nauli SM. Endothelial cells from humans and mice with polycystic kidney disease are characterized by polyploidy and chromosome segregation defects through survivin down-regulation. Hum Mol Genet. 2011;20:354–367. doi: 10.1093/hmg/ddq470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sutters M, Yamaguchi T, Maser RL, Magenheimer BS, St John PL, Abrahamson DR, Grantham JJ, Calvet JP. Polycystin-1 transforms the cAMP growth-responsive phenotype of M-1 cells. Kidney Int. 2001;60:484–494. doi: 10.1046/j.1523-1755.2001.060002484.x. [DOI] [PubMed] [Google Scholar]
- 88.Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 89.Parker E, Newby LJ, Sharpe CC, Rossetti S, Streets AJ, Harris PC, O’Hare MJ, Ong AC. Hyperproliferation of PKD1 cystic cells is induced by insulin-like growth factor-1 activation of the Ras/Raf signalling system. Kidney Int. 2007;72:157–165. doi: 10.1038/sj.ki.5002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J Am Soc Nephrol. 2014;25:232–237. doi: 10.1681/ASN.2013010077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pećina-Šlaus N. Wnt signal transduction pathway and apoptosis: a review. Cancer Cell International. 2010;10:22. doi: 10.1186/1475-2867-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim E, Arnould T, Sellin LK, Benzing T, Fan MJ, Gruning W, Sokol SY, Drummond I, Walz G. The polycystic kidney disease 1 gene product modulates Wnt signaling. J Biol Chem. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- 93.Foy RL, Chitalia VC, Panchenko MV, Zeng L, Lopez D, Lee JW, Rana SV, Boletta A, Qian F, Tsiokas L, Piontek KB, Germino GG, Zhou MI, Cohen HT. Polycystin-1 regulates the stability and ubiquitination of transcription factor Jade-1. Hum Mol Genet. 2012;21:5456–5471. doi: 10.1093/hmg/dds391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim S, Nie H, Nesin V, Tran U, Outeda P, Bai CX, Keeling J, Maskey D, Watnick T, Wessely O, Tsiokas L. The polycystin complex mediates Wnt/Ca(2+) signalling. Nat Cell Biol. 2016;18:752–764. doi: 10.1038/ncb3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lal M, Song X, Pluznick JL, Di Giovanni V, Merrick DM, Rosenblum ND, Chauvet V, Gottardi CJ, Pei Y, Caplan MJ. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum Mol Genet. 2008;17:3105–3117. doi: 10.1093/hmg/ddn208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. Journal of Cell Science. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 97.Wu M, Chen M, Jing Y, Gu J, Mei S, Yao Q, Zhou J, Yang M, Sun L, Wang W, Hu H, Wuthrich RP, Mei C. The C-terminal tail of polycystin-1 regulates complement factor B expression by signal transducer and activator of transcription 1. Am J Physiol Renal Physiol. 2016;310:F1284–1294. doi: 10.1152/ajprenal.00428.2015. [DOI] [PubMed] [Google Scholar]
- 98.Talbot JJ, Shillingford JM, Vasanth S, Doerr N, Mukherjee S, Kinter MT, Watnick T, Weimbs T. Polycystin-1 regulates STAT activity by a dual mechanism. Proc Natl Acad Sci U S A. 2011;108:7985–7990. doi: 10.1073/pnas.1103816108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Talbot JJ, Song X, Wang X, Rinschen MM, Doerr N, LaRiviere WB, Schermer B, Pei YP, Torres VE, Weimbs T. The cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J Am Soc Nephrol. 2014;25:1737–1748. doi: 10.1681/ASN.2013091026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, Kane ME, Obara T, Weimbs T. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Developmental cell. 2006;10:57–69. doi: 10.1016/j.devcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 101.Olsan EE, Mukherjee S, Wulkersdorfer B, Shillingford JM, Giovannone AJ, Todorov G, Song X, Pei Y, Weimbs T. Signal transducer and activator of transcription-6 (STAT6) inhibition suppresses renal cyst growth in polycystic kidney disease. Proc Natl Acad Sci U S A. 2011;108:18067–18072. doi: 10.1073/pnas.1111966108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Harvey KF, Hariharan IK. The hippo pathway. Cold Spring Harb Perspect Biol. 2012;4:a011288. doi: 10.1101/cshperspect.a011288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Happe H, van der Wal AM, Leonhard WN, Kunnen SJ, Breuning MH, de Heer E, Peters DJ. Altered Hippo signalling in polycystic kidney disease. J Pathol. 2011;224:133–142. doi: 10.1002/path.2856. [DOI] [PubMed] [Google Scholar]
- 104.Yu FX, Zhang Y, Park HW, Jewell JL, Chen Q, Deng Y, Pan D, Taylor SS, Lai ZC, Guan KL. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 2013;27:1223–1232. doi: 10.1101/gad.219402.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Elliott J, Zheleznova NN, Wilson PD. c-Src inactivation reduces renal epithelial cell-matrix adhesion, proliferation, and cyst formation. Am J Physiol Cell Physiol. 2011;301:C522–529. doi: 10.1152/ajpcell.00163.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chang MY, Ong AC. New treatments for autosomal dominant polycystic kidney disease. Br J Clin Pharmacol. 2013;76:524–535. doi: 10.1111/bcp.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dhanasekaran DN, Reddy EP. JNK Signaling in Apoptosis. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arnould T, Kim E, Tsiokas L, Jochimsen F, Gruning W, Chang JD, Walz G. The polycystic kidney disease 1 gene product mediates protein kinase C alpha-dependent and c-Jun N-terminal kinase-dependent activation of the transcription factor AP-1. J Biol Chem. 1998;273:6013–6018. doi: 10.1074/jbc.273.11.6013. [DOI] [PubMed] [Google Scholar]
- 109.Le NH, van der Bent P, Huls G, van de Wetering M, Loghman-Adham M, Ong AC, Calvet JP, Clevers H, Breuning MH, van Dam H, Peters DJ. Aberrant polycystin-1 expression results in modification of activator protein-1 activity, whereas Wnt signaling remains unaffected. J Biol Chem. 2004;279:27472–27481. doi: 10.1074/jbc.M312183200. [DOI] [PubMed] [Google Scholar]
- 110.Parnell SC, Magenheimer BS, Maser RL, Zien CA, Frischauf AM, Calvet JP. Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins. J Biol Chem. 2002;277:19566–19572. doi: 10.1074/jbc.M201875200. [DOI] [PubMed] [Google Scholar]
- 111.Le NH, van der Wal A, van der Bent P, Lantinga-van Leeuwen IS, Breuning MH, van Dam H, de Heer E, Peters DJ. Increased activity of activator protein-1 transcription factor components ATF2, c-Jun, and c-Fos in human and mouse autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2005;16:2724–2731. doi: 10.1681/ASN.2004110913. [DOI] [PubMed] [Google Scholar]
- 112.Yu W, Kong T, Beaudry S, Tran M, Negoro H, Yanamadala V, Denker BM. Polycystin-1 protein level determines activity of the Galpha12/JNK apoptosis pathway. J Biol Chem. 2010;285:10243–10251. doi: 10.1074/jbc.M109.070821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Arnould T, Sellin L, Benzing T, Tsiokas L, Cohen HT, Kim E, Walz G. Cellular activation triggered by the autosomal dominant polycystic kidney disease gene product PKD2. Mol Cell Biol. 1999;19:3423–3434. doi: 10.1128/mcb.19.5.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, Weimbs T. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006;103:5466–5471. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Distefano G, Boca M, Rowe I, Wodarczyk C, Ma L, Piontek KB, Germino GG, Pandolfi PP, Boletta A. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol. 2009;29:2359–2371. doi: 10.1128/MCB.01259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dere R, Wilson PD, Sandford RN, Walker CL. Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PLoS One. 2010;5:e9239. doi: 10.1371/journal.pone.0009239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ravichandran K, Zafar I, He Z, Doctor RB, Moldovan R, Mullick AE, Edelstein CL. An mTOR anti-sense oligonucleotide decreases polycystic kidney disease in mice with a targeted mutation in Pkd2. Hum Mol Genet. 2014;23:4919–4931. doi: 10.1093/hmg/ddu208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ravichandran K, Edelstein CL. Polycystic kidney disease: a case of suppressed autophagy? Seminars in nephrology. 2014;34:27–33. doi: 10.1016/j.semnephrol.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 120.Orhon I, Dupont N, Pampliega O, Cuervo AM, Codogno P. Autophagy and regulation of cilia function and assembly. Cell Death Differ. 2015;22:389–397. doi: 10.1038/cdd.2014.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature. 2013;502:254–257. doi: 10.1038/nature12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pampliega O, Orhon I, Patel B, Sridhar S, Diaz-Carretero A, Beau I, Codogno P, Satir BH, Satir P, Cuervo AM. Functional interaction between autophagy and ciliogenesis. Nature. 2013;502:194–200. doi: 10.1038/nature12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, Song XW, Xu H, Mari S, Qian F, Pei Y, Musco G, Boletta A. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. 2013;19:488–493. doi: 10.1038/nm.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhu P, Sieben CJ, Xu X, Harris PC, Lin X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Orhon I, Dupont N, Zaidan M, Boitez V, Burtin M, Schmitt A, Capiod T, Viau A, Beau I, Kuehn EW, Friedlander G, Terzi F, Codogno P. Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat Cell Biol. 2016;18:657–667. doi: 10.1038/ncb3360. [DOI] [PubMed] [Google Scholar]