

Abstract
Background
Previous human and animal studies have demonstrated the ability of exogenously administered basic fibroblast growth factor (FGF2) to act as an antifibrotic agent in the skin. Though the activity of FGF2 as an anti-scarring agent is well-established for fibrotic skin wounds, the mechanisms by which FGF2 exerts these actions are not entirely understood. Canonical FGF2 signaling proceeds in part via FGFR/MAPK pathways in human dermal fibroblasts, and FGF2 has been described to prevent or reverse the fibroblast-to-myofibroblast transition, which is driven by TGFβ signaling and understood to be an important step in the formation of a fibrotic scar in vivo. Thus, we set out to investigate the antagonistic effects of FGF2 on TGFβ signaling as well as the broader effects of MAPK inhibition on the TGFβ-mediated induction of myofibroblast gene expression.
Objective
To better understand the effects of FGF2 signaling pathways on myofibroblastic gene expression and cell phenotypes.
Methods
Human dermal fibroblasts were cultured in vitro in the presence of FGF2, TGFβ, and/or MAPK inhibitors, and the effects of these agents were investigated by molecular biology techniques including qRT-PCR, immunofluorescence, Western blot, and flow cytometry.
Results
FGF2 inhibited TGFβ-mediated fibroblast activation, resulting in more rapidly proliferating, spindle-shaped cells, compared to the more slowly proliferating, flatter TGFβ-treated cells. Treatment with FGF2 also attenuated TGFβ-mediated increase in expression of myofibroblast markers smooth muscle α-actin, calponin, transgelin, connective tissue growth factor, ED-A fibronectin, and collagen I. FGF2-mediated antagonism of the TGFβ-mediated fibroblast-to-myofibroblast transition was reversed by small molecule inhibition of ERK or JNK, and it was potentiated by inhibition of p38. MAPK inhibition was demonstrated to have qualitatively similar effects even in the absence of exogenous FGF2, and small molecule inhibition of p38 MAPK was sufficient to attenuate TGFβ-mediated fibroblast activation.
Conclusions
Inhibition of select MAPK signaling pathways can reverse or potentiate anti-fibrotic FGF2 effects on human dermal fibroblasts, as well as exert their effects independently of exogenous FGF2 supplementation.
Keywords: FGF2, MAPK, TGFβ, Fibrosis, Fibroblast, Myofibroblast
1. Introduction
Fibrosis is an undesirable result of wound healing and subsequent tissue repair that affects myriad tissues in the body. Rather than replacing damaged tissue with functional replacement, the fibrotic response results in deposition of a collagenous scar that preserves barrier function but fails to recapitulate the native function and mechanical properties of the tissue. As various tissue-specific diseases result in fibrotic outcomes, this suggests that common cellular and molecular mechanisms, at least in part, underlie tissue fibrosis across various organs [1]. One of the most important of these mechanisms is the presence and activity of the myofibroblast, an activated fibroblast denoted by the presence of actin stress fibers, contractile capability, and high levels of collagen production and deposition [2]. It has recently been demonstrated that myofibroblasts arise from multiple different cell types in vivo, including fibrocytes, mesenchymal stem cells, and smooth muscle cells [3]. However, one of the most important paradigms in skin fibrosis, in particular, is the activation of resident granulation tissue fibroblasts by transforming growth factor beta (TGFβ) signaling. Mechanical heterogeneity of the granulation tissue leads to the release of TGF-β1 from the provisional extracellular matrix, leading to the differentiation of resident wound fibroblasts into myofibroblasts (termed “fibroblast activation”) [4]. These myofibroblasts, with their contractile potential and high levels of collagen deposition, are important to wound closure, but if they persist aberrantly in the wound past the phase of wound resolution they contribute to formation of a nonfunctional hypertrophic scar [5].
Fibroblast growth factor 2 (FGF2) is one of the most well-studied members of the fibroblast growth factor superfamily, and it has been implicated in cellular processes and paradigms as diverse as mitogenesis, differentiation, proliferative lifespan, survival, oncogenesis, and stem cell self-renewal, among others [6–9]. FGF2 activates its target receptor tyrosine kinases, the FGFRs, on the cell surface in order to activate numerous downstream pathways, including several mitogen activated protein kinase (MAPK) pathways [10]. Importantly, it has been determined that application of exogenous FGF2 has both accelerative and anti-fibrotic effects in various types of skin wounds, reviewed extensively in [11]. This has been demonstrated in humans for acute incisional wounds, avulsions, and burn wounds [12–14], as well as in several animal models [15, 16]. The mechanisms and signaling pathways by which FGF2 inhibits the fibrotic response have been investigated previously but remain incompletely understood. Previously, our lab and others have described anti-fibrotic gene expression paradigms in fibroblasts in response to FGF2 treatment, including downregulation of inflammatory cytokines, upregulation of interstitial collagenases, and inhibition of pro-fibrotic integrin signaling [16–21]. Additionally, FGF2 has been demonstrated under certain circumstances to antagonize TGFβ signaling, including reports in which FGF2 has demonstrated the potential to antagonize TGFβ-mediated myofibroblast phenotypes [15, 16, 18, 22–25]. Thus, since FGF2 is known to act directly on fibroblasts to produce anti-fibrotic and anti-myofibroblastic effects, and since myofibroblasts are one of the most important effectors of the fibrotic response in the skin, we set out to better understand the effects of FGF2 as an antagonist to TGFβ in human dermal fibroblasts, as well as to understand the effects on these same phenotypes of inhibition of specific MAPK pathways known to be activated by FGF2/FGFR signaling.
2. Materials and Methods
2.1 Antibodies
The primary antibodies used were the following: sc-32251 α-SMA, sc-8654-R Histone H3, sc-8783 Collagen I, sc-47778 β-Actin, sc-59826 ED-A Fn, sc-365970 CTGF, sc-136987 Calponin (all from Santa Cruz Biotechnology), and VPA00048KT SM22α (from Bio-Rad). The secondary antibodies used were Alexafluor488-conjugated or Alexafluor568-conjugated (Invitrogen) for immunofluorescence and HRP-conjugated for Western blotting (Bio-Rad).
2.2 Cell culture
CRL-2097 and CRL-2352 human dermal fibroblasts were obtained from ATCC, and CT-1005 human dermal fibroblasts were obtained from the University of Massachusetts Medical School tissue distribution program in Worcester, MA. All fibroblasts were cultured in 1:1 DMEM:Ham’s F12 (Corning) supplemented with 4mM L-glutamine (Mediatech) and 10% Fetal Clone III (Hyclone) at 37°C, 5% O2, 5% CO2, and high humidity. When indicated, cells were treated with 4ng/mL FGF2 (Cell Signaling Technology), 10ng/mL TGF-β1 (Peprotech), 10μM U0126 (MEK1/2 inhibitor; Cell Signaling Technology), 10μM SP600125 (JNK inhibitor; Santa Cruz Biotechnology), and/or 10μM SB202190 (p38 MAPK inhibitor; Santa Cruz Biotechnology). Cells were processed for analysis on day 4.
2.3 Proliferation studies
CRL-2097 human dermal fibroblasts were plated in 60mm tissue culture plastic dishes at 15,000 cells/dish and cultured in the presence or absence of 4ng/mL FGF2 (Cell Signaling Technology) and MAPK inhibitors as listed in 2.2. Media was changed every fourth day and cells were passaged on every seventh day.
2.4 RNA isolation and qRT-PCR
RNA was isolated from snap-frozen cell pellets with the E.Z.N.A. DNA/RNA Isolation Kit (Omega Bio-Tek) according to manufacturer’s instructions. RNA concentration was analyzed using a Nanodrop 2000 spectrophotometer (Thermo Scientific). cDNA was synthesized using SuperScript VILO Master Mix (ThermoFisher) according to manufacturer’s instructions and stored at −20°C. PCR reactions were carried out using established protocols using an AB 7500 (Applied Biosystems) with PowerUp SYBR Master Mix (ThermoFisher), 5ng cDNA per reaction, and 500nM concentration per primer. Primer sequences are listed in Table 1. Fold change was calculated using the ΔΔCt method [26].
Table 1.
Primer sequences used for qRT-PCR
| Gene name | Protein encoded | Forward primer (5′–3′) | Reverse primer (5′–3′) | Amplicon size (bp) |
|---|---|---|---|---|
| ACTA2 | α-SMA | ACTGCCTTGGTGTGTGACAA | 120 | |
| CACCATCACCCCCTGATGTC | ||||
| CNN1 | Calponin | AGGTTAAGAACAAGCTGGCCC | 113 | |
| GAGGCCGTCCATGAAGTTGT | ||||
| TAGLN | SM22α | CACAAGGTGTGTGTAAGGGTG | 132 | |
| GGCTCATGCCATAGGAAGGAC | ||||
| CCN2 | CTGF | GTGCCTGCCATTACAACTGTC | 98 | |
| TCTCACTCTCTGGCTTCATGC | ||||
| ED-A Fn | Fibronectin (ED-A) | CAGTGGAGTATGTGGTTAGTGTC | 119 | |
| GTGACCTGAGTGAACTTCAGG | ||||
| GAPDH | GAPDH | GAGTCCACTGGCGTCTTCAC | 119 | |
| TTCACACCCATGACGAACAT |
2.5 SDS-PAGE and Western blotting
Cells were lysed using Laemmli sample buffer and lysate proteins and separated by SDS-PAGE. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore) using a semi-dry transfer apparatus (GE Healthcare). The membrane was blocked with 5% fat-free dry milk in TBS-T buffer (1xTBS+.1%Tween-20, pH=8.0) and incubated overnight at 4°C in the primary antibody solution at a pre-determined, antibody-specific dilution into 1% fat-free dry milk in TBS-T. The membrane was washed and incubated in a species-specific HRP-conjugated secondary antibody diluted 1:5000 into 1% fat-free dry milk in TBS-T. Signal was visualized using a ChemiDoc XRS system (Bio-Rad) using SuperSignal West Dura Extended Duration Substrate (ThermoFisher).
2.6 Indirect flow cytometry
Cells were harvested with trypsin, washed twice in 1x Dulbecco’s phosphate-buffered saline (DPBS), and fixed for 20 minutes in cold methanol. The cells were then washed in 1x DPBS, followed by a 30 minute incubation in a primary antibody solution (1:200 dilution of α-SMA antibody, Santa Cruz Biotechnology) in PBS+.05% Tween-20 (PBS-T). Cells were then washed again and resuspended in a 1:500 dilution of secondary antibody solution in PBS-T (Alexa Fluor 488-Conjugated goat anti-mouse IgG, Invitrogen). Cell nuclei were counterstained with propidium iodide. Samples were analyzed on an Accuri C6 Flow Cytometer (BD Biosciences). Ten thousand events were collected per sample. Two biological replicates of each treatment condition, including their negative controls, were performed.
2.7 Immunofluorescence
Cells were plated on glass coverslips for 4 days and cultured under various treatment conditions. At the time of analysis, cells were fixed in 4% paraformaldehyde at room temperature, and permeabilized with 0.1% Triton X-100 in 1xPBS, before blocking with 5% BSA in PBS-T. Cells were then incubated overnight at 4°C in primary antibody in PBS-T, washed and incubated in a 1:500 dilution of Alexa Fluor-conjugated secondary antibody or Alexa Fluor-conjugated Phalloidin, (Invitrogen). Cell nuclei were counterstained with Hoechst 33342 and coverslips mounted onto glass slides using Prolong Gold (Life Technologies) and stored at 4°C until imaging. Images were collected using an Axiovert 200M (Zeiss) using identical exposure times and settings between treatments.
3. Results
3.1 FGF2 induces a proliferative response in CRL-2097 human neonatal foreskin fibroblasts
FGF2 has been described by many groups to have myriad, pleiotropic effects on fibroblasts ranging from proliferation to changes in cell morphology. We first verified that exogenous addition of FGF2 would induce proliferation in CRL-2097 human neonatal foreskin fibroblasts, which were chosen for their relatively high basal expression of myofibroblast markers. CRL-2097 fibroblasts grown in the presence of 4ng/mL exogenous FGF2 induced proliferation, indicated by a shift upwards in the growth curve of FGF2-treated fibroblasts compared to fibroblasts grown under control conditions over the course of 21 days in culture (Fig. 1a). These data suggest that CRL-2097 human neonatal foreskin fibroblasts respond to stimulation by FGF2 with a proliferative phenotype, and that this proliferative phenotype can be maintained over an extended period in culture.
Figure 1. Effects of FGF2 on fibroblast proliferation and phenotype.
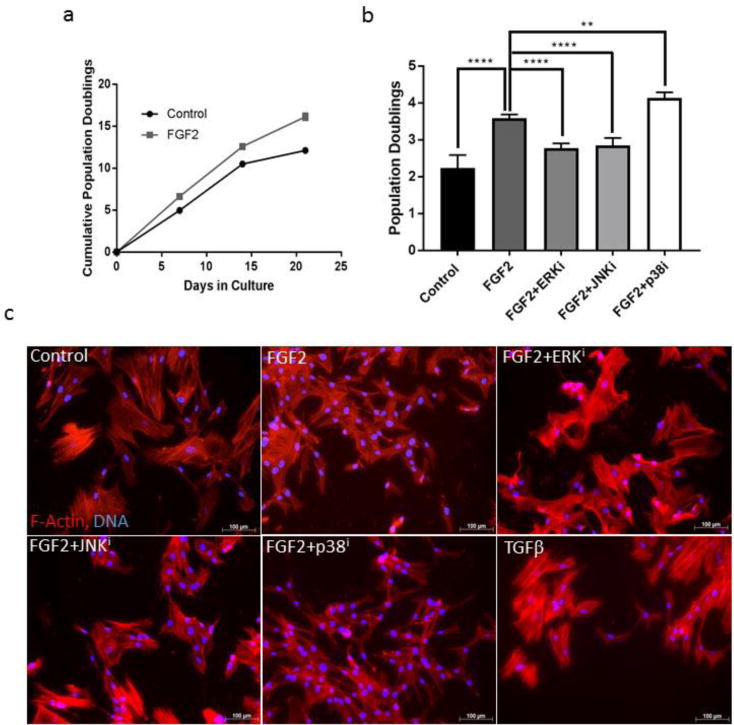
(a) CRL-2097 human neonatal foreskin fibroblasts were grown in the presence or absence of 4ng/mL FGF2. n=2 biological replicates per condition. Error bars=standard deviation. (b) Fibroblasts were cultured for 4 days in the absence (Control) or the presence of FGF2 (FGF2), with or without a specific MAPK inhibitor (U0126 [ERK inhibitor] = ERKi; SP600125 [JNK inhibitor] = JNKi; SB202190 [p38 MAPK inhibitor] = p38i). Statistics were performed by one-way ANOVA, with Tukey’s Multiple Comparisons post-hoc analysis, with each treatment condition being compared to the number of population doublings calculated for the FGF2-treated condition. n=5 biological replicates per condition. Error bars=standard deviation. **<.01, ****p<.0001. (c) Fibroblasts cultured under the same treatment conditions as in (b) were stained for F-actin with Alexa568-conjugated phalloidin and DNA counterstained with Hoechst 33342. Images were purposely overexposed for visualization of cell shape. Scale bar=100μm.
3.2 FGF2 stimulates proliferation and inhibits myofibroblastic morphology, in a manner reversible by specific MAPK inhibition
We next set out to determine whether inhibition of canonical MAPKs was sufficient to inhibit the proliferative effect of FGF2 on fibroblasts in culture. We cultured fibroblasts in the presence or absence of FGF2 and the presence or absence of a specific MAPK inhibitor (U0126, SP600125, or SB202190 to inhibit ERK, JNK, or p38 signaling, respectively) and evaluated the number of population doublings underwent by day 4 in culture (Fig. 1b). Exogenous FGF2 induced proliferation in fibroblasts in a manner that could be reversed by small molecule inhibition of either ERK or JNK, but not by inhibition of p38. Further, addition of the combination of FGF2 and p38 inhibitor to the culture medium induced proliferation to a greater degree than addition of exogenous FGF2 alone, suggesting that these two molecules promote fibroblast proliferation in an additive manner.
We noticed that addition of exogenous FGF2 to the culture medium tended to revert the phenotype of CRL-2097 neonatal foreskin fibroblasts, of which many appear myofibroblastic in nature under normal culture conditions, to a smaller size and more spindle-like shape. Thus, we set out to uncover whether inhibition of specific MAPK pathways was sufficient to reverse this FGF2-mediated change in cell phenotype. Cells were cultured under the same conditions as were used for the proliferation assay, with the inclusion of a TGFβ positive control (which is known to induce and maintain myofibroblastic phenotypes), and then stained with fluorophore-conjugated phalloidin in order to visualize cell size and shape (Fig. 1c). Images were purposely overexposed in order to visualize the shape of cells cultured under all treatment conditions, as FGF2 substantially downregulates both filamentous smooth muscle α-actin (α-SMA) (Fig. 2) and β-actin (Fig. 4b). As expected, treatment with FGF2 reverted CRL-2097 fibroblasts to a thinner, more spindle-like shape with fewer prominent filamentous actin stress fibers. In a similar pattern to the proliferation data, small molecule inhibition of ERK or JNK, but not of p38, each reversed the effects of FGF2 on cell morphology and reverted the cell shape to more closely match that of the TGFβ-treated myofibroblastic control. These data suggest that FGF2-mediated stimulation of proliferation and spindle-like, stress fiber-negative fibroblast phenotypes can be reversed by inhibition of ERK or JNK but not by inhibition of p38.
Figure 2. Effects of FGF2 on TGFβ-mediated expression of α-SMA.
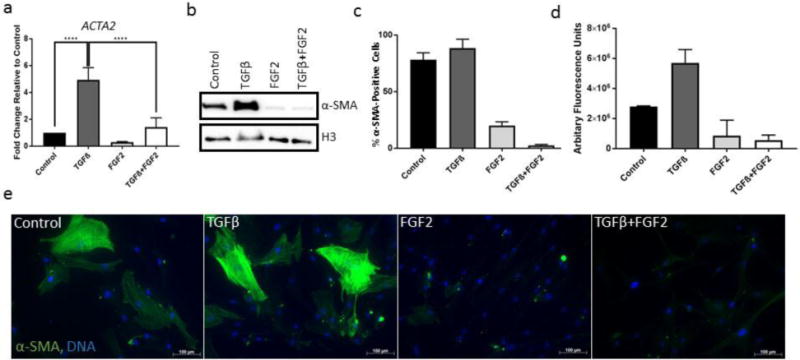
(a) CRL-2097 fibroblasts were cultured in the presence or absence of TGFβ and/or FGF2 until day 4, when they were examined by qRT-PCR for their expression of ACTA2 (α-SMA) relative to loading control GAPDH. n=4 biological replicates per condition. Expression is described as fold change relative to cells cultured under control conditions. Statistics were performed by one-way ANOVA, with Tukey’s Multiple Comparisons post-hoc analysis. ****p<.0001. (b) Fibroblasts cultured under the same conditions for the same length of time were examined by Western blotting for their expression of α-SMA protein. Histone H3 was used as a loading control. (c,d) Fibroblasts cultured under the same conditions for the same length of time were analyzed by flow cytometry. The percentage of α-SMA-positive cells per treatment condition (c), and the average α-SMA fluorescence of all cells (d) were determined. n=2 biological replicates per condition. Error bars=standard deviation. (e) Fibroblasts cultured under the same conditions for the same length of time were fixed and stained by immunofluorescence using an AlexaFluor488-conjugated anti-α-SMA antibody and DNA counterstained with Hoechst 33342. Scale bar=100μm.
Figure 4. Effects of FGF2 on TGFβ-mediated deposition of fibrotic extracellular matrix.
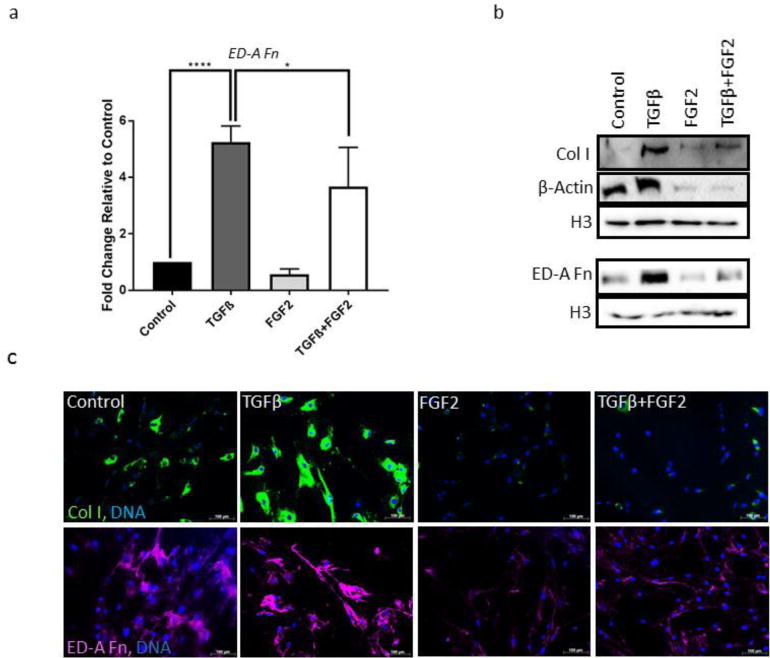
(a) CRL-2097 fibroblasts were cultured in the presence or absence of TGFβ and/or FGF2 until day 4, and expression of ED-A Fn was measured by qRT-PCR. GAPDH was used as a loading control. n=4 biological replicates per condition. Expression is described as fold change relative to cells cultured under control conditions. Statistics were performed by one-way ANOVA, with Tukey’s Multiple Comparisons post-hoc analysis. *p<.05, ****p<.0001. (b) Expression of Col I, β-Actin and ED-A Fn protein was measured by Western blot. Histone H3 was used as a loading control. (c) ECM protein expression was measured by immunofluorescence for type I collagen (Col I; green) and fibronectin (ED-A Fn; pseudo-colored magenta). DNA was counterstained with Hoechst 33342. Scale bar=100μm.
3.3 FGF2 attenuates TGFβ-mediated induction of alpha α actin
The stereotypical marker of the myofibroblast phenotype is the presence of filamentous actin stress fibers enriched in smooth muscle alpha actin. Thus, we wished to determine whether FGF2 was able to inhibit TGFβ-mediated expression of α-SMA in CRL-2097 fibroblasts. Analysis of gene expression on RNA extracted from fibroblasts cultured in the presence or absence of FGF2 and TGFβ demonstrated that FGF2 attenuated TGFβ-mediated induction of ACTA2, the transcript encoding α-SMA (Fig. 2a). Concordantly, Western blot analysis of protein isolated from the same cell lysates demonstrated that FGF2 was able to substantially attenuate TGFβ-mediated expression of α-SMA protein (Fig. 2b). Indirect flow cytometry was performed in order to better determine α-SMA expression at the population level across the different treatment groups. FGF2 treatment substantially reduced both the percentage of α-SMA-positive cells (Fig. 2c) and the amount of α-SMA protein present per cell (Fig. 2d). This observation was further validated by immunofluorescence. Immunofluorescent staining revealed that, while some CRL-2097 fibroblasts expressed detectable baseline levels of α-SMA, treatment with TGFβ induced expression of α-SMA to greater levels, in a manner that could be inhibited substantially by co-treatment with exogenous FGF2 (Fig. 2e). Taken together, these data suggest that FGF2 downregulates α-SMA expression at both the transcript and protein levels.
3.4 FGF2 attenuates TGFβ-mediated expression of cytoplasmic myofibroblast markers across multiple fibroblast cell lines
We next wished to evaluate whether the robust inhibition by FGF2 of TGFβ-mediated α-SMA expression was specific to expression of α-SMA, or whether this effect was applicable to myofibroblast phenotypes more generally. We examined expression of the transcripts CNN1, TAGLN, and CCN2, encoding three other canonical myofibroblast markers (calponin, SM22α, and CTGF, respectively) (Fig. 3a–c). In a manner similar to that which was seen for ACTA2, FGF2 attenuated TGFβ-mediated expression of each of these myofibroblast markers. Similarly, by Western blot analysis, we determined that FGF2 was able to attenuate TGFβ-mediated protein expression of α-SMA, as determined previously, as well as of calponin and SM22α (Fig. 3d). The presence of two immunoreactive CTGF bands likely corresponds to the presence of isoforms both with and without glycosylation modifications, a finding which has been described previously [27]. Both of these bands appear to be induced to a similar degree by TGFβ in a manner able to be partially inhibited by FGF2. In order to verify that FGF2-mediated antagonism of fibroblast activation was not specific to this particular cell line, we performed Western blot analysis to investigate the ability of FGF2 to antagonize TGFβ-mediated expression of myofibroblast proteins in two other primary human dermal fibroblast cell lines (Fig. 3e). In both cases, FGF2 substantially attenuated the ability of exogenous TGFβ to induce expression of myofibroblast markers α-SMA, calponin, and SM22α. Taken together, these data suggest that FGF2 has the ability to antagonize myofibroblast phenotypes and gene expression patterns in a variety of human dermal fibroblast cell lines.
Figure 3. Effects of FGF2 on TGFβ-mediated expression of myofibroblast markers.
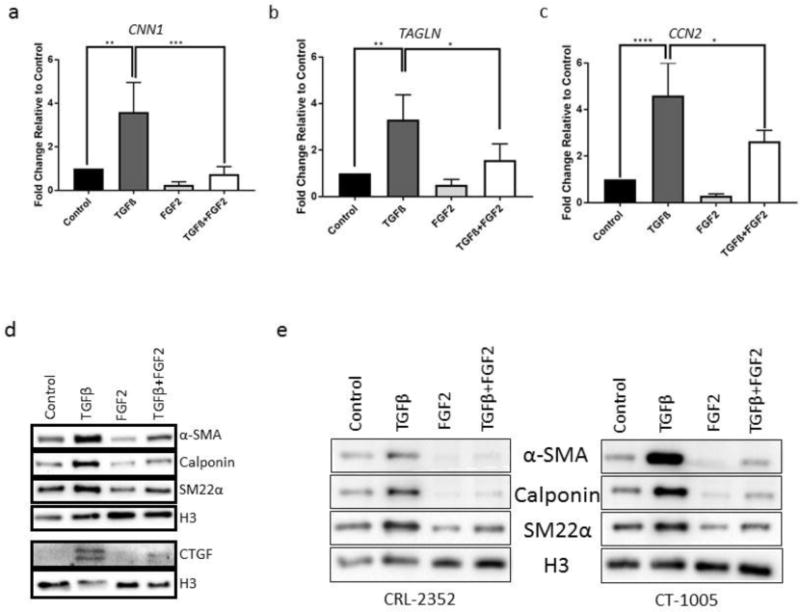
(a–c) CRL-2097 fibroblasts cultured in the presence or absence of TGFβ and/or FGF2 until day 4 and were examined by qRT-PCR for expression of (a) CNN1 (calponin), (b) TAGLN (SM22α), and (c) CCN2 (CTGF). GAPDH was used as a loading control. n=4 biological replicates per condition. Expression is described as fold change relative to cells cultured under control conditions. Statistics were performed by one-way ANOVA, with Tukey’s Multiple Comparisons post-hoc analysis. *p<.05, **p<.01, ***p<.001, ****p<.0001. (d) Western blotting was performed to determine expression of α-SMA, calponin, SM22α, and CTGF protein in CRL-2097s. (e) Western blotting was performed to determine expression of α-SMA, calponin, and SM22α protein in CRL-2352 and CT-1005 primary human dermal fibroblasts. Histone H3 was used as a loading control for all Western blotting.
3.5 FGF2 attenuates TGFβ-mediated expression of cytoskeletal and ECM-associated myofibroblast markers
Aside from the aforementioned markers, we also investigated whether FGF2 was able to attenuate TGFβ-mediated changes in the cytoskeleton and extracellular matrix of fibroblasts. We determined by qRT-PCR (Fig. 4a), Western blot (Fig. 4b), and immunofluorescence (Fig. 4c) that FGF2 attenuates TGFβ-mediated expression of the myofibroblast-specific ED-A splice variant of fibronectin, an extracellular matrix protein that has been causally implicated in the myofibroblast activation and the development of tissue fibrosis [28–31]. We also demonstrated that culture in the presence of exogenous FGF2 was sufficient to attenuate the TGFβ-mediated expression of type I collagen, a major fibrillar collagen deposited in scar tissue. Additionally, we determined by Western blot that FGF2 also is able to attenuate TGFβ-mediated increase in β-actin (Fig. 4b), which is consistent with changes in cellular phenotype observed previously by phalloidin staining (Fig. 1c). Taken together, these data suggest that the ability of FGF2 to attenuate TGFβ-mediated fibroblast activation is generalizable beyond mere downregulation of α-SMA and results in attenuation of several important, functional markers of the myofibroblast phenotype, including the attenuation of deposition of ECM molecules that are causally implicated in fibrotic pathology.
3.6 FGF2-mediated attenuation of myofibroblast phenotypes is antagonized by inhibition of ERK and JNK
Since FGF2 acts canonically via FGFRs and subsequent activation of various MAPK pathways, and since we had previously determined that MAPK inhibition was sufficient to attenuate FGF2-mediated mitogenic activity, we next asked whether inhibition of any of these MAPK pathways could override the antagonism of FGF2 towards expression of some of the myofibroblast markers investigated previously (α-SMA, calponin, and SM22α). For each of these proteins, a qualitatively similar inhibition pattern was seen: FGF2 resulted in a downregulation of expression of each of these markers, which was reversed by inhibition of either ERK or JNK, but not reversed by inhibition of p38 (Fig. 5a). This same pattern was observed by immunofluorescent visualization of α-SMA protein, as well as deposition of collagen I and ED-A fibronectin in cells undergoing the same treatments (Fig. 5b), further validating the MAPK inhibitory effects observed in the Western blots. These data suggest that inhibition of ERK or JNK, individually, is sufficient to antagonize and override the antifibrotic effects of FGF2 on human fibroblasts, but that the inhibition of p38 is not.
Figure 5. Effects of MAPK inhibition on FGF2 antagonism of myofibroblastic phenotypes.
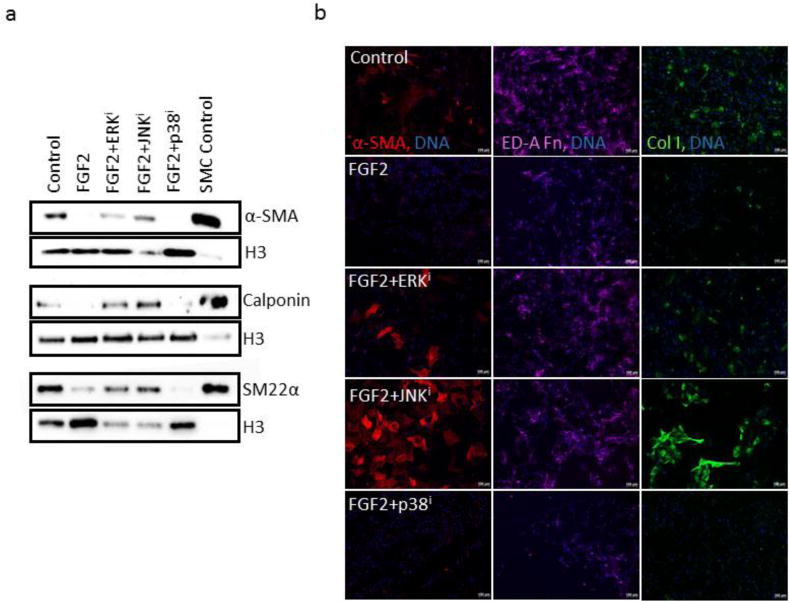
(a) CRL-2097 fibroblasts were cultured in the presence or absence of FGF2 and a specific MAPK inhibitor until day 4, when they were examined for their expression of α-SMA, calponin, and SM22α by Western blot. Histone H3 was used as a loading control. SMC control refers to protein lysate of contractile human vascular smooth muscle cells, which was used as a positive control. (b) Fibroblasts cultured under the same conditions for the same length of time were fixed and stained for immunofluorescence using antibodies against α-SMA (red), collagen I (green), and ED-A fibronectin (pseudo-colored magenta) in order to visualize expression and deposition of these proteins. DNA counterstained with Hoechst 33342. Scale bar=100μm.
3.7 MAPK inhibitors exert their pro-fibrotic or anti-fibrotic effects in the absence of FGF2 signaling
Since FGF2 was able to diminish fibroblast activation in human fibroblasts not only in the presence of exogenous TGFβ but also in its absence, we next investigated whether the effects of MAPK inhibitors on fibroblast activation could act independently of FGF2 signaling as well. Small molecule inhibition of ERK and JNK both notably increased the expression of, and inhibition of p38 decreased the expression of, the canonical myofibroblast marker α-SMA by Western blot, even in the absence of exogenous FGF2 or TGFβ supplementation (Fig. 6a). This finding was verified by immunofluorescence, and qualitatively similar patterns were observed for calponin (Fig. 6b). We also determined by phalloidin staining that fibroblasts cultured in the presence of inhibitors of ERK and JNK both demonstrated a more myofibroblastic morphology, while fibroblasts cultured in the presence of p38 inhibitor appeared more elongated and fibroblastic, and tended to lack visible actin stress fibers (Fig. 6b). These data suggest that MAPK inhibitors have the potential to promote or inhibit fibroblast activation even in the absence of exogenous supplementation with FGF2 or TGFβ.
Figure 6. Effects of MAPK inhibition on myofibroblastic phenotypes.
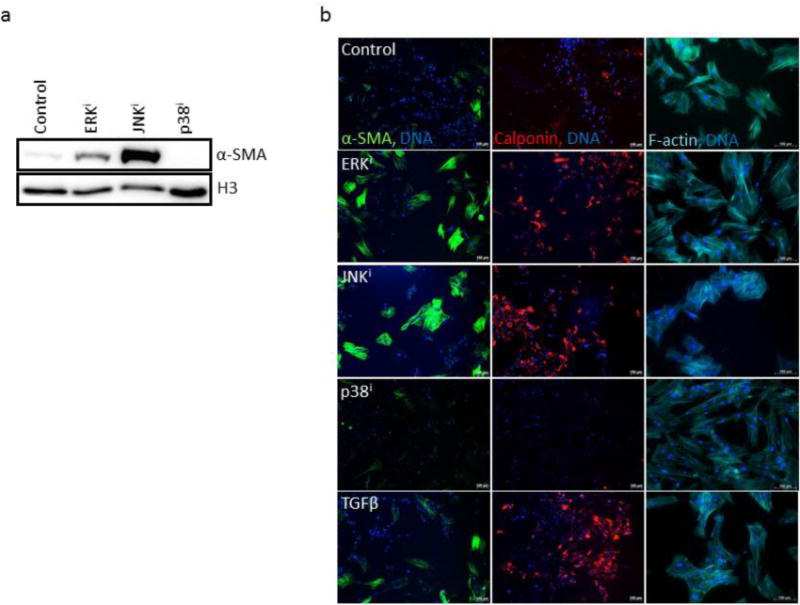
(a) CRL-2097 fibroblasts were cultured in the presence of a specific MAPK inhibitor until day 4, when they were examined for their expression of α-SMA by Western blot. Histone H3 was used as a loading control. (b) Fibroblasts cultured under the same conditions for the same length of time were fixed and stained for immunofluorescence using antibodies against α-SMA (green) or calponin (red), or with fluorophore-conjugated phalloidin (pseudo-colored cyan) for visualization of F-actin. DNA was counterstained with Hoechst 33342. Scale bar=100μm.
3.8 Inhibition of p38 MAPK is sufficient to attenuate TGFβ-mediated fibroblast activation in the absence of exogenous FGF2
Since p38 inhibition appeared to be sufficient to antagonize expression of myofibroblast markers and development of myofibroblast-like morphology in human dermal fibroblasts (Fig. 6), we next examined whether p38 inhibition would also be sufficient to antagonize TGFβ-mediated fibroblast activation in the absence of exogenous FGF2 supplementation. Co-treatment with p38 inhibitor was sufficient to inhibit TGFβ-mediated fibroblast activation, as evidenced by a decrease in expression of α-SMA, calponin, and SM22α by Western blot (Figure 7a). Additionally, this same trend was observed by immunofluorescence, including the ability of p38 inhibition to attenuate TGFβ-mediated transition to a more myofibroblastic morphology by staining with fluorophore-conjugated phalloidin (Figure 7b). Inhibition of p38 signaling also attenuated TGFβ-mediated expression of myofibroblast markers α-SMA and calponin, as well as inhibited TGFβ-mediated deposition of myofibroblastic ECM proteins collagen I and ED-A fibronectin (Figure 7b). Taken together, these data suggest that p38 inhibition is sufficient to antagonize TGFβ-mediated fibroblast activation, as indicated by attenuation of the protein expression of several functional myofibroblast markers.
Figure 7. Effects of p38 inhibition on TGFβ-mediated expression and deposition of myofibroblast-associated proteins.
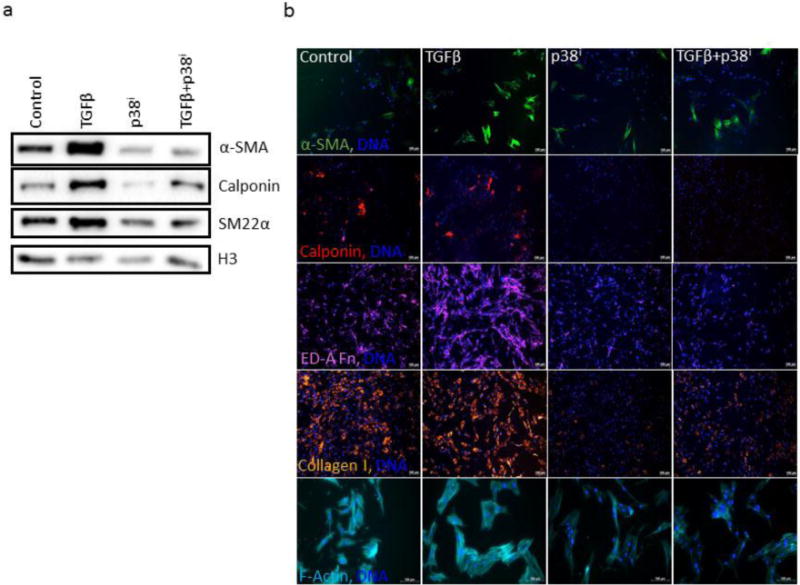
(a) CRL-2097 fibroblasts were cultured in the presence or absence of TGF-β1 and a specific p38 MAPK inhibitor until day 4, when they were examined for expression of α-SMA, calponin, and SM22α by Western blot. Histone H3 was used as a loading control. (b) Fibroblasts cultured under the same conditions for the same length of time were fixed and stained for immunofluorescence for α-SMA (green), calponin (red), collagen I (pseudo-colored orange), ED-A fibronectin (pseudo-colored magenta), and fluorophore-conjugated phalloidin (pseudo-colored cyan) for visualization. DNA was counterstained with Hoechst 33342. Scale bar=100μm.
4. Discussion
The aim of this research was to better understand the effects of FGF2 and specific MAPK pathways and inhibitors on activation of human dermal fibroblasts. Previous research has described a robust ability of FGF2 to contribute to anti-fibrotic effects in dermal wound healing [12–16], but the mechanisms and signaling pathways by which it does so are not entirely understood. Due to the highly pleiotropic nature of FGF2/FGFR signaling [10] as well as the well-described roles of aberrant MAPK signaling in cancer [32–35], it would be beneficial to understand both pathway-specific effects of FGF2 on cell-autonomous phenotypes in fibroblasts, as well as the activity of various MAPK inhibitors on these phenotypes.
We demonstrate that FGF2 contributes to the maintenance of CRL-2097 primary human neonatal foreskin fibroblasts in a highly proliferative, fibroblastic phenotype. Further, we demonstrated that FGF2 has the ability to inhibit TGFβ-induced expression of a whole host of contractile, myofibroblast-associated proteins including α-SMA, calponin, and SM22α in several different human dermal fibroblast lines, as well as the ability to inhibit deposition of fibrosis-associated ECM proteins collagen I and ED-A fibronectin. The observation that FGF2-mediated attenuation of myofibroblastic protein expression is mirrored by transcript expression, as assessed by qRT-PCR, suggests that the mechanisms by which FGF2 attenuates TGFβ-mediated fibroblast activation occur upstream of the level of transcription of these genes, a suggestion supported by similar previous reports [15, 16].
Our data demonstrate that FGF2-mediated inhibition of myofibroblastic markers including α-SMA, calponin, SM22α, type I collagen, and ED-A fibronectin can be overridden by inhibition of ERK or JNK, or potentiated by inhibition of p38. Though our data do not prove definitively that the antifibrotic effects of FGF2 proceed directly via ERK and JNK pathways, as these pathways have numerous activators within a cellular context, they strongly suggest that the antifibrotic effects of FGF2 involve, and perhaps even require, activation of these pathways.
Aside from interaction with FGF2 signaling, we show that inhibition of ERK and JNK signaling promotes fibroblast activation, while inhibition of p38 antagonizes fibroblast activation, even in the absence of exogenous FGF2. It is possible that this pathway inhibition acts on basal MAPK signaling caused by endogenous FGF/FGFR activation, as several FGF family members including FGF2 are known to be secreted by fibroblasts and signal in autocrine or paracrine manners via the FGFRs [36], but there are other possible explanations as well. One plausible explanation is that MAPK inhibitors act on TGFβ signaling. Under certain contexts, TGFβ signaling has been demonstrated to be modulated by activity of specific MAPKs. It has previously been reported that ERK activation is responsible for antagonism of SMAD2 phosphorylation and SMAD2/3 nuclear accumulation by the nonsteroidal anti-inflammatory drug tolfenamic acid, suggesting a mechanism by which ERK activation can antagonize canonical TGFβ signaling [37]. ERK has also been demonstrated to have the ability to phosphorylate serines and threonines in the linker region of SMAD2/3, and mutation of specific ERK-target serine and threonine residues of SMAD3 results in potentiated SMAD3 signaling, suggesting that ERK can inhibit SMAD3 signaling via this mechanism [38]. Previous reports also demonstrate the ability of JNK activation to antagonize TGFβ signaling and TGFβ-mediated SMAD activation [39–41], in support of our findings that pharmacological inhibition of JNK signaling results in increased fibroblast activation.
It has previously been demonstrated that TGFβ can lead to phosphorylation of p38 in a manner distinct and separable from its ability to activate SMAD2/3 [42], and that this p38 activation is able to govern specific TGFβ-mediated phenotypes including apoptosis and epithelial-to-mesenchymal transition in a manner independent of SMAD activation [43]. Thus it is possible that small molecule-mediated inhibition of p38 activity directly inhibits the non-SMAD TGFβ/TGFβR signal transducers that are necessary for, or serve to enhance, fibroblast activation and myofibroblast phenotypes. p38 signaling can modulate the transcriptional activity of TGFβ [43–45], and p38 has also been implicated in regulation of the activity of serum response factor, a major transcription factor involved in the myofibroblastic gene expression program [46, 47].
Whatever the direct mechanism, there is now substantial evidence that regulation of MAPK signaling, including signaling via p38 MAPK, is crucial for certain fibrotic disease states, and for some of the cellular fates underlying tissue fibroses. Inhibition of p38 signaling has been demonstrated to attenuate fibrotic responses in vivo in experimental models of renal fibrosis [48–51], cardiac fibrosis [52, 53], and in myofibroblast activation in vitro [54–56]. Thus, our data demonstrating that p38 has the ability to not only potentiate FGF2-mediated inhibition of myofibroblast phenotypes, but also to inhibit TGFβ-mediated myofibroblast differentiation directly, add the suggestion of p38 inhibition as a potential treatment for dermal fibrosis to a growing body of literature suggesting the potential efficacy of p38 inhibition as antifibrotic therapy in various tissues.
The data presented within this manuscript have shed light on some of the mechanistic details regarding FGF2-mediated inhibition of fibrotic phenotypes in fibroblasts, largely centered around the paradigm of fibroblast activation, a process which FGF2 appears to robustly inhibit. It is our hope that, with a better understanding of the details of gene expression changes in these fibroblasts, along with additional insight into effects of specific MAPK pathways on FGF2-mediated inhibition of fibroblast activation and fibrosis more generally, we will be able to better understand the details of FGF2 in the wound healing process, as well as understand its therapeutic mechanisms and assess whether therapies that act downstream of FGF2/FGFR may be able to attenuate fibroblast activation and fibrosis without the pleiotropic effects that may come with manipulation of signaling at the level of the growth factor or the transmembrane receptor.
Highlights.
FGF2 attenuates TGFβ-mediated myofibroblast activation in human fibroblasts.
ERK, JNK inhibition reverses FGF2-mediated inhibition of myofibroblast phenotypes.
p38 inhibition potentiates FGF2-mediated inhibition of myofibroblast phenotypes.
p38 inhibition sufficient to attenuate myofibroblast activation by TGFβ.
Acknowledgments
We would like to thank Victoria Huntress, M.S. of the WPI imaging core for her training and assistance in fluorescent microscopy.
Funding Sources:
This research was supported by a National Institutes of Health award to Tanja Dominko (grant number R01GM85456) and the National Science Foundation Integrative Graduate Education and Research Traineeship (grant number DGE 1144804) awarded to David Dolivo.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest:
The authors report no conflicts of interest.
References
- 1.Rockey DC, Bell PD, Hill JA. Fibrosis—a common pathway to organ injury and failure. New England Journal of Medicine. 2015;372(12):1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 2.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. The American journal of pathology. 2012;180(4):1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. The American journal of pathology. 2007;170(6):1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinz B. Myofibroblasts. Experimental eye research. 2016;142:56–70. doi: 10.1016/j.exer.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Sarrazy V, Billet F, Micallef L, Coulomb B, Desmoulière A. Mechanisms of pathological scarring: role of myofibroblasts and current developments. Wound Repair and Regeneration. 2011;19(s1):s10–s15. doi: 10.1111/j.1524-475X.2011.00708.x. [DOI] [PubMed] [Google Scholar]
- 6.Fang J, Huang S, Liu H, Crepin M, Xu T, Liu J. Role of FGF-2/FGFR signaling pathway in cancer and its signification in breast cancer. Chinese Science Bulletin. 2003;48(15):1539–1547. [Google Scholar]
- 7.Bailly K, Soulet F, Leroy D, Amalric F. Uncoupling of cell proliferation and differentiation activities of basic fibroblast growth factor. Uncoupling of cell proliferation and differentiation activities of basic fibroblast growth factor. 2000 [PubMed] [Google Scholar]
- 8.Eiselleova L, Matulka K, Kriz V, Kunova M, Schmidtova Z, Neradil J, Tichy B, Dvorakova D, Pospisilova S, Hampl A. A Complex Role for FGF-2 in Self-Renewal, Survival, and Adhesion of Human Embryonic Stem Cells. Stem Cells. 2009;27(8):1847–1857. doi: 10.1002/stem.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dolivo D, Hernandez S, Dominko T. Cellular lifespan and senescence: a complex balance between multiple cellular pathways. BioEssays. 2016;38:S33–S44. doi: 10.1002/bies.201670906. [DOI] [PubMed] [Google Scholar]
- 10.Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdisciplinary Reviews: Developmental Biology. 2015;4(3):215–266. doi: 10.1002/wdev.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nunes QM, Li Y, Sun C, Kinnunen TK, Fernig DG. Fibroblast growth factors as tissue repair and regeneration therapeutics. PeerJ. 2016;4:e1535. doi: 10.7717/peerj.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akita S, Akino K, Imaizumi T, Hirano A. Basic fibroblast growth factor accelerates and improves second-degree burn wound healing. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2008;16(5):635–641. doi: 10.1111/j.1524-475X.2008.00414.x. [DOI] [PubMed] [Google Scholar]
- 13.Ono I, Akasaka Y, Kikuchi R, Sakemoto A, Kamiya T, Yamashita T, Jimbow K. Basic fibroblast growth factor reduces scar formation in acute incisional wounds. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2007;15(5):617–623. doi: 10.1111/j.1524-475X.2007.00293.x. [DOI] [PubMed] [Google Scholar]
- 14.Matsumine H. Treatment of skin avulsion injuries with basic fibroblast growth factor. Plastic and Reconstructive Surgery–Global Open. 2015;3(4):e371. doi: 10.1097/GOX.0000000000000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eto H, Suga H, Aoi N, Kato H, Doi K, Kuno S, Tabata Y, Yoshimura K. Therapeutic potential of fibroblast growth factor-2 for hypertrophic scars: upregulation of MMP-1 and HGF expression. Laboratory investigation; a journal of technical methods and pathology. 2012;92(2):214–223. doi: 10.1038/labinvest.2011.127. [DOI] [PubMed] [Google Scholar]
- 16.Shi HXX, Lin C, Lin BBB, Wang ZGG, Zhang HYY, Wu FZZ, Cheng Y, Xiang LJJ, Guo DJJ, Luo X, Zhang GYY, Fu XBB, Bellusci S, Li XKK, Xiao J. The anti-scar effects of basic fibroblast growth factor on the wound repair in vitro and in vivo. PloS one. 2013;8(4) doi: 10.1371/journal.pone.0059966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kashpur O, LaPointe D, Ambady S, Ryder EF, Dominko T. FGF2-induced effects on transcriptome associated with regeneration competence in adult human fibroblasts. BMC genomics. 2013;14:656. doi: 10.1186/1471-2164-14-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grella A, Kole D, Holmes W, Dominko T. FGF2 Overrides TGFβ1-Driven Integrin ITGA11 Expression in Human Dermal Fibroblasts. Journal of cellular biochemistry. 2016;117(4):1000–1008. doi: 10.1002/jcb.25386. [DOI] [PubMed] [Google Scholar]
- 19.Yasui H, Andoh A, Bamba S, Inatomi O, Ishida H, Fujiyama Y. Role of fibroblast growth factor-2 in the expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human intestinal myofibroblasts. Digestion. 2004;69(1):34–44. doi: 10.1159/000076545. [DOI] [PubMed] [Google Scholar]
- 20.Delrieu I, Jean-Charles F, Bayard F. Inhibition of interleukin-6 promoter activity by the 24 kDa isoform of fibroblast growth factor-2 in HeLa cells. Biochemical …. 1999 [PMC free article] [PubMed] [Google Scholar]
- 21.Newberry EP, Willis D, Latifi T, Boudreaux JM, Towler DA. Fibroblast growth factor receptor signaling activates the human interstitial collagenase promoter via the bipartite Ets-AP1 element. Molecular endocrinology (Baltimore, Md) 1997;11(8):1129–1144. doi: 10.1210/mend.11.8.9958. [DOI] [PubMed] [Google Scholar]
- 22.Ichise T, Yoshida N, Ichise H. FGF2-induced Ras–MAPK signalling maintains lymphatic endothelial cell identity by upregulating endothelial-cell-specific gene expression and suppressing TGFβ …. Journal of cell science. 2014 doi: 10.1242/jcs.137836. [DOI] [PubMed] [Google Scholar]
- 23.Ito T, Sawada R, Fujiwara Y, Seyama Y. FGF-2 suppresses cellular senescence of human mesenchymal stem cells by down-regulation of TGF-β2, … and biophysical research …. 2007 doi: 10.1016/j.bbrc.2007.05.067. [DOI] [PubMed] [Google Scholar]
- 24.Correia AC, Moonen JRA, Brinker MG, Krenning G. FGF2 inhibits endothelial–mesenchymal transition through microRNA-20a-mediated repression of canonical TGF-β signaling. J Cell Sci. 2016;129(3):569–579. doi: 10.1242/jcs.176248. [DOI] [PubMed] [Google Scholar]
- 25.Chen PY, Qin L, Li G, Tellides G, Simons M. Fibroblast growth factor (FGF) signaling regulates transforming growth factor beta (TGFβ)-dependent smooth muscle cell phenotype modulation. Scientific Reports. 2016;6 doi: 10.1038/srep33407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nature protocols. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 27.Yang DH, Kim HS, Wilson EM, Rosenfeld RG, Oh Y. Identification of glycosylated 38-kDa connective tissue growth factor (IGFBP-related protein 2) and proteolytic fragments in human biological fluids, and up-regulation of IGFBP-rP2 expression by TGF-β in Hs578T human breast cancer cells. The Journal of Clinical Endocrinology & Metabolism. 1998;83(7):2593–2596. doi: 10.1210/jcem.83.7.5097. [DOI] [PubMed] [Google Scholar]
- 28.Kohan M, Muro AF, White ES, Berkman N. EDA-containing cellular fibronectin induces fibroblast differentiation through binding to α4β7 integrin receptor and MAPK/Erk 1/2-dependent signaling. The FASEB Journal. 2010;24(11):4503–4512. doi: 10.1096/fj.10-154435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, Flaherty KR, Martinez FJ, Tsui JL, Sheppard D. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. American journal of respiratory and critical care medicine. 2008;177(6):638–645. doi: 10.1164/rccm.200708-1291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. The Journal of cell biology. 1998;142(3):873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohan M, Muro AF, Bader R, Berkman N. The extra domain A of fibronectin is essential for allergen-induced airway fibrosis and hyperresponsiveness in mice. Journal of Allergy and Clinical Immunology. 2011;127(2):439–446.e5. doi: 10.1016/j.jaci.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 32.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 33.Regad T. Targeting RTK signaling pathways in cancer. Cancers. 2015;7(3):1758–1784. doi: 10.3390/cancers7030860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hynes NE. Tyrosine kinase signalling in breast cancer. Breast Cancer Research. 2000;2(3):154. doi: 10.1186/bcr48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samatar AA, Poulikakos PI. Targeting RAS–ERK signalling in cancer: promises and challenges. Nature reviews Drug discovery. 2014;13(12):928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 36.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nature Reviews Cancer. 2010;10(2):116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, Min KW, Liggett J, Baek SJ. Disruption of the transforming growth factor-β pathway by tolfenamic acid via the ERK MAP kinase pathway. Carcinogenesis. 2013;34(12):2900–2907. doi: 10.1093/carcin/bgt250. [DOI] [PubMed] [Google Scholar]
- 38.Kretzschmar M, Doody J, Timokhina I, Massagué J. A mechanism of repression of TGFβ/Smad signaling by oncogenic Ras. Genes & development. 1999;13(7):804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu S, Kasisomayajula K, Peng J, Bancalari E. Inhibition of JNK enhances TGF-β1-activated Smad2 signaling in mouse embryonic lung. Pediatric research. 2009;65(4):381–386. doi: 10.1203/PDR.0b013e3181991c67. [DOI] [PubMed] [Google Scholar]
- 40.Lin Y, Zhang B, Liang H, Lu Y, Ai X, Zhang B, Chen X. JNK inhibitor SP600125 enhances TGF- β- induced apoptosis of RBE human cholangiocarcinoma cells in a Smad- dependent manner. Molecular medicine reports. 2013;8(6):1623–1629. doi: 10.3892/mmr.2013.1711. [DOI] [PubMed] [Google Scholar]
- 41.Verrecchia F, Tacheau C, Wagner EF, Mauviel A. A central role for the JNK pathway in mediating the antagonistic activity of pro-inflammatory cytokines against transforming growth factor-β-driven SMAD3/4-specific gene expression. Journal of Biological Chemistry. 2003;278(3):1585–1593. doi: 10.1074/jbc.M206927200. [DOI] [PubMed] [Google Scholar]
- 42.Galliher AJ, Schiemann WP. Src phosphorylates Tyr284 in TGF-β type II receptor and regulates TGF-β stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer research. 2007;67(8):3752–3758. doi: 10.1158/0008-5472.CAN-06-3851. [DOI] [PubMed] [Google Scholar]
- 43.Yu L, Hébert MC, Zhang YE. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. The EMBO journal. 2002;21(14):3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimoto K, Nakatsuka K, Matsuo N, Yoshioka H. p38 MAPK mediates the expression of type I collagen induced by TGF-β2 in human retinal pigment epithelial cells ARPE-19. Investigative ophthalmology & visual science. 2004;45(7):2431–2437. doi: 10.1167/iovs.03-1276. [DOI] [PubMed] [Google Scholar]
- 45.Hanafusa H, Ninomiya-Tsuji J, Masuyama N, Nishita M, Fujisawa J-i, Shibuya H, Matsumoto K, Nishida E. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-β-induced gene expression. Journal of Biological Chemistry. 1999;274(38):27161–27167. doi: 10.1074/jbc.274.38.27161. [DOI] [PubMed] [Google Scholar]
- 46.Thuerauf DJ, Arnold ND, Zechner D, Hanford DS, DeMartin KM, McDonough PM, Prywes R, Glembotski CC. p38 Mitogen-activated Protein Kinase Mediates the Transcriptional Induction of the Atrial Natriuretic Factor Gene through a Serum Response Element A POTENTIAL ROLE FOR THE TRANSCRIPTION FACTOR ATF6. Journal of Biological Chemistry. 1998;273(32):20636–20643. doi: 10.1074/jbc.273.32.20636. [DOI] [PubMed] [Google Scholar]
- 47.Liang G, Ahlqvist K, Pannem R, Posern G, Massoumi R. Serum response factor controls CYLD expression via MAPK signaling pathway. PloS one. 2011;6(5):e19613. doi: 10.1371/journal.pone.0019613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stambe C, Atkins RC, Tesch GH, Masaki T, Schreiner GF, Nikolic-Paterson DJ. The role of p38α mitogen-activated protein kinase activation in renal fibrosis. Journal of the American Society of Nephrology. 2004;15(2):370–379. doi: 10.1097/01.asn.0000109669.23650.56. [DOI] [PubMed] [Google Scholar]
- 49.Sugiyama N, Kohno M, Yokoyama T. Inhibition of the p38 MAPK pathway ameliorates renal fibrosis in an NPHP2 mouse model. Nephrology Dialysis Transplantation. 2011:gfr550. doi: 10.1093/ndt/gfr550. [DOI] [PubMed] [Google Scholar]
- 50.Nishida M, Okumura Y, Sato H, Hamaoka K. Delayed inhibition of p38 mitogen-activated protein kinase ameliorates renal fibrosis in obstructive nephropathy. Nephrology Dialysis Transplantation. 2008;23(8):2520–2524. doi: 10.1093/ndt/gfn309. [DOI] [PubMed] [Google Scholar]
- 51.Choi SY, Piao ZH, Jin L, Kim JH, Kim GR, Ryu Y, Lin MQ, Kim HS, Kee HJ, Jeong MH. Piceatannol Attenuates Renal Fibrosis Induced by Unilateral Ureteral Obstruction via Downregulation of Histone Deacetylase 4/5 or p38-MAPK Signaling. PloS one. 2016;11(11):e0167340. doi: 10.1371/journal.pone.0167340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.See F, Thomas W, Way K, Tzanidis A, Kompa A, Lewis D, Itescu S, Krum H. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. Journal of the American College of Cardiology. 2004;44(8):1679–1689. doi: 10.1016/j.jacc.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 53.Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K, Davis JM. Fibroblast-Specific Genetic Manipulation of p38 MAPK in vivo Reveals its Central Regulatory Role in Fibrosis. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.116.026238. CIRCULATIONAHA 116.026238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sousa AM, Liu T, Guevara O, Stevens J, Fanburg BL, Gaestel M, Toksoz D, Kayyali US. Smooth muscle α-actin expression and myofibroblast differentiation by TGFβ are dependent upon MK2. Journal of cellular biochemistry. 2007;100(6):1581–1592. doi: 10.1002/jcb.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyer-ter-Vehn T, Gebhardt S, Sebald W, Buttmann M, Grehn F, Schlunck Gn, Knaus P. p38 inhibitors prevent TGF-β–induced myofibroblast transdifferentiation in human Tenon fibroblasts. Investigative ophthalmology & visual science. 2006;47(4):1500–1509. doi: 10.1167/iovs.05-0361. [DOI] [PubMed] [Google Scholar]
- 56.Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki Y. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38(4):879–889. doi: 10.1053/jhep.2003.50384. [DOI] [PubMed] [Google Scholar]