

ABSTRACT
The building blocks of intracellular Ca2+ signals evoked by inositol 1,4,5-trisphosphate receptors (IP3Rs) are Ca2+ puffs, transient focal increases in Ca2+ concentration that reflect the opening of small clusters of IP3Rs. We use total internal reflection fluorescence microscopy and automated analyses to detect Ca2+ puffs evoked by photolysis of caged IP3 or activation of endogenous muscarinic receptors with carbachol in human embryonic kidney 293 cells. Ca2+ puffs evoked by carbachol initiated at an estimated 65±7 sites/cell, and the sites remained immobile for many minutes. Photolysis of caged IP3 evoked Ca2+ puffs at a similar number of sites (100±35). Increasing the carbachol concentration increased the frequency of Ca2+ puffs without unmasking additional Ca2+ release sites. By measuring responses to sequential stimulation with carbachol or photolysed caged IP3, we established that the two stimuli evoked Ca2+ puffs at the same sites. We conclude that IP3-evoked Ca2+ puffs initiate at numerous immobile sites and the sites become more likely to fire as the IP3 concentration increases; there is no evidence that endogenous signalling pathways selectively deliver IP3 to specific sites.
KEY WORDS: Ca2+ puff, Endoplasmic reticulum, IP3, IP3 receptor, Muscarinic receptor, Phospholipase C
Summary: Ca2+ puffs are the building blocks for IP3-evoked Ca2+ signals. Ca2+ puffs evoked by caged IP3 or via endogenous signalling pathways initiate at the same fixed intracellular sites.
INTRODUCTION
Most Ca2+ signals in electrically inexcitable cells are initiated by receptors that stimulate phospholipase C (PLC) and, thereby, formation of inositol 1,4,5-trisphosphate (IP3). IP3 then binds to IP3 receptors (IP3Rs) to stimulate Ca2+ release from the endoplasmic reticulum (ER). Vertebrates express three subtypes of IP3R and numerous splice variants but each contributes to formation of tetrameric Ca2+-permeable channels that open when they bind both IP3 and Ca2+ (Foskett et al., 2007; Prole and Taylor, 2016). Higher concentrations of cytosolic free Ca2+ concentration ([Ca2+]c) than those required for IP3R activation inhibit IP3Rs. Within ER membranes, IP3Rs form small clusters (Chalmers et al., 2006; Geyer et al., 2015; Rahman et al., 2009), wherein Ca2+ released by an active IP3R can stimulate the opening of neighbouring IP3Rs that have bound IP3 (Alzayady et al., 2016; Taylor and Konieczny, 2016). Hence, both the distribution of IP3Rs and the prevailing IP3 concentration set the gain on this regenerative Ca2+-induced Ca2+ release mechanism.
Studies conducted in Ian Parker's laboratory have shown that low concentrations of IP3 evoke tiny, local cytosolic Ca2+ signals (‘Ca2+ blips’) that last just a few milliseconds and reflect the opening of single IP3Rs. Higher concentrations of IP3 evoke longer-lasting local Ca2+ signals (‘Ca2+ puffs’) that are typically 5−10 times larger than blips and result from the almost simultaneous openings of a few IP3Rs (Parker et al., 1996). Ca2+ blips and puffs are the building blocks of all IP3-evoked Ca2+ signals (Bootman et al., 1997a). As the global [Ca2+]c or concentration of IP3 increases further, Ca2+ puffs become more frequent until they typically trigger a Ca2+ wave that spreads regeneratively across the cell (Marchant et al., 1999), at frequencies that increase with the concentration of IP3 (Thurley et al., 2014).
Ca2+ puffs evoked by either caged IP3 (ci-IP3) or by IP3 produced after activation of endogenous signalling pathways have been observed in numerous cell types. A common theme is the suggestion that Ca2+ puffs recur at the same small number of subcellular locations and that these sites often initiate global cytosolic Ca2+ signals (Bootman et al., 1997a; Kasai et al., 1993; Marchant and Parker, 2001; Nakamura et al., 2012; Olson et al., 2012; Rooney et al., 1990; Simpson et al., 1997). However, most quantitative analyses of Ca2+ puffs that have used total internal reflection fluorescence microscopy (TIRFM) have also been restricted to a single cell type (SH-SY5Y neuroblastoma cells) responding to photolysis of ci-IP3 (e.g. Smith and Parker, 2009). There is, therefore, a need to both examine other cells and to compare responses to ci-IP3 with those evoked by endogenous signalling pathways. The latter is important because there is evidence that different stimuli that activate endogenous pathways and direct delivery of IP3 may each ignite Ca2+ puffs at the same small number of Ca2+ release sites (Rooney et al., 1990; Thomas et al., 2000), whereas other studies suggest a selective delivery of IP3 to specific IP3Rs by endogenous signalling pathways (Delmas et al., 2002; Olson et al., 2012; Short et al., 2000; Tovey and Taylor, 2013; Tu et al., 1998; Zhao et al., 1990).
Here, we use TIRFM to image human embryonic kidney 293 (HEK293) cells loaded with a fluorescent Ca2+ indicator, and record Ca2+ puffs evoked by the photolysis of ci-IP3, or by IP3 provided by stimulation of endogenous M3 muscarinic acetylcholine receptors with carbachol. By using a custom-written algorithm that allows automated detection of Ca2+ puffs and Ca2+ release sites (Fig. 1, Figs S1 and S2), we show that both stimuli evoke Ca2+ puffs at abundant immobile intracellular sites. We demonstrate that IP3 produced in response to stimulation with carbachol, and i-IP3 produced by photolysis of ci-IP3 are delivered to the same intracellular Ca2+ release sites. Increasing the stimulus intensity causes an increase in the frequency of Ca2+ puffs without recruiting additional Ca2+ release sites.
Fig. 1.

Automated detection and analysis of Ca2+ puffs and sites. (A) Images are acquired and prepared for analysis by correcting for background (BG) fluorescence, identifying cell boundaries and preparing image stacks for each identified cell. Further details in Materials and Methods. (B) PuffFinder is designed to detect Ca2+ puffs. Images are smoothed. The pre-stimulus fluorescence values (F0) are determined for each pixel. F/F0 is then determined for every pixel in every frame. To identify regions where fluorescence changes rapidly, the difference in fluorescence intensity (ΔF) between each image and its immediate successor is determined. PuffFinder then corrects these ΔF values for any creeping increase in F/F0 by subtracting the average ΔF across every frame from every pixel; this provides the ΔΔF stacks used to identify puffs. The mean and s.d. of the ΔΔF values are determined to provide criteria for identifying puffs. Pixels are ranked by ΔΔF value. The pixel with the largest ΔΔF is placed at the centre of a 5×5 pixel matrix, and selection criteria are applied to decide whether the matrix is an area wherein sufficient pixels have large ΔΔF values. The selection criteria and the rationale for choosing them are elaborated in Materials and Methods. The list of matrices ranked by ΣΔΔF is now interrogated to define the boundaries of each puff. This is achieved by returning to the pixel with the largest ΔΔF value and expanding it outwards until ΔΔF of the enclosed pixels falls below a threshold value. The process is then repeated with the next ranked pixel. The locations of puffs are confirmed by visual inspection and their properties defined after fitting a 2D Gaussian function. (C) SiteMapper is used to decide whether Ca2+ puffs originate at the same or different sites. It begins with the puffs identified in PuffMapper and ranks them according to their signal mass. The centroid of the largest puff is identified and if the centroids of neighbouring puffs fall within 0.96 µm of it, they are amalgamated into the same site. The centre of the new site is defined and the assessment of neighbours is repeated. The site is then accepted and its enclosed puffs are excluded from further analysis. The analysis then moves to the next largest unassigned puff, and the process is repeated until all puffs have been assigned to sites to create a map of all sites in the cell. Further details of PuffMapper and SiteMapper are provided in the Materials and Methods.
RESULTS
Extracellular stimuli evoke Ca2+ puffs at many intracellular sites
Our aim was to define the spatial distribution of the local Ca2+ signals evoked by different stimuli and stimulus intensities. This aim effectively restricts the field of view to ∼82 µm×82 µm, within which there are typically about six HEK293 cells and rarely more than three HeLa cells. It was, therefore, important to establish that the submaximal stimuli that evoke local Ca2+ signals stimulate responses in most cells. In populations of HEK293 cells, carbachol evoked Ca2+ signals with a half-maximal effective concentration (EC50) of ∼40 µM (López-Sanjurjo et al., 2013; Tovey and Taylor, 2013). In single HEK293 cells, maximal (1 mM) and submaximal (10 µM) concentrations of carbachol evoked increases of [Ca2+]c in most cells (92±7%, mean±range, n=2 fields; and 90±16%, mean±s.e.m., n=3 fields, respectively).
Ca2+ puffs were very infrequent in unstimulated HEK239 cells, but carbachol (10 µM) evoked a flurry of Ca2+ puffs beginning 3.2±0.2 s after carbachol addition (n=48, 3 challenges of 16 cells) (Fig. 2A, Fig. S1, Movie 1). Within a few seconds most Ca2+ signals then propagated across the cell as a Ca2+ wave, as reported for many other cell types (Bootman et al., 1997b; Charles et al., 1991; Marchant et al., 1999; Rooney et al., 1990; Smith et al., 2009a). By using PuffMapper to detect Ca2+ puffs automatically (see Methods for the criteria used to identify Ca2+ puffs), we confirmed the significant increase in the frequency of Ca2+ puffs in HEK293 cells stimulated with carbachol and in HeLa cells stimulated with histamine (Fig. 2B). In both cell types, stimulation also significantly increased the number of sites at which Ca2+ puffs were observed (Fig. 2C).
Fig. 2.

Extracellular stimuli evoke abundant Ca2+ puffs. (A) Typical TIRFM images from a single Cal520-loaded HEK293 cell, collected at 40-ms intervals, with ΔF shown in pseudocolour at the indicated times before and after addition of carbachol (CCh, 10 µM). Within the montage, images show every 5th frame (i.e. 200 ms between successive frames). Arrows show Ca2+ puffs identified by PuffFinder (see Fig. S1 for additional examples). (B,C) Summary results show the frequency of Ca2+ puffs (Hz/µm2) (B) and the density of sites (sites/µm2) (C) detected before and after stimulation of HEK293 cells with CCh (10 µM) or HeLa cells with histamine (Hist, 5 µM). Events were recorded for the entire period (up to 10 s) after addition of the stimulus until the Ca2+ signal propagated across the cell. Results show the mean and results for individual cells (16 HEK293 cells and 13 HeLa cells). ****P<0.0001, paired Student's t-test, relative to unstimulated cells. (D) Example of a Ca2+ puff evoked by CCh in a HEK293 cell (i). The time course of the fluorescence changes within the boxed area are shown in panels ii and iii. Images were background-subtracted and smoothed prior to analysis. Panel iv shows the fluorescence intensity profile across the line shown within the boxed area. Examples of the time courses (iii) and fluorescence intensity profiles (iv) of additional Ca2+ puffs are shown by red lines. Time courses reflect the average fluorescence intensity of the boxed area (i) over time.
Typical examples of individual carbachol-evoked Ca2+ puffs in HEK293 are shown in Fig. 2D and Fig. S1B, from which it is clear that the puffs identified by the automated analysis are heterogenous, consistent with results from other cells (Wiltgen et al., 2014). Our methods, designed to optimise spatial resolution (see Materials and Methods), do not provide the high temporal resolution of some studies (e.g. Smith and Parker, 2009). Nevertheless, the temporal and spatial profiles of the local Ca2+ signals detected in HeLa and HEK293 cells are consistent with those reported for Ca2+ puffs in other cells (Table 1). However, a striking difference is the substantially larger number of puffs and initiation sites detected in our analyses (Table 1). The densities of Ca2+ initiation sites (number/µm2) detected during a 10-s period of stimulation are similar in HEK293 and HeLa cells (Fig. 2C), but the sites were at least 10-fold more abundant than observed in other studies (Table 1), although none of these studies examined effects of extracellular stimuli on HEK293 cells (Nakamura et al., 2012; Smith and Parker, 2009; Thomas et al., 2000). The unexpectedly large number of Ca2+ puffs is unlikely to be due to the automated analysis mistakenly identifying puffs because the temporal and spatial profiles of the Ca2+ puffs in HeLa and HEK293 cells are similar to those reported previously (Fig. 2D, Fig. S1B, Table 1). The most likely explanation is that the Ca2+ buffering imposed by EGTA in HEK293 and HeLa cells delays the initiation of global Ca2+ signals for longer than in other cells, and so extends the opportunity to identify Ca2+ puffs and sites. Our automated analysis may also have contributed by detecting events that are missed in manual analyses.
Table 1.
Properties of Ca2+ puffs
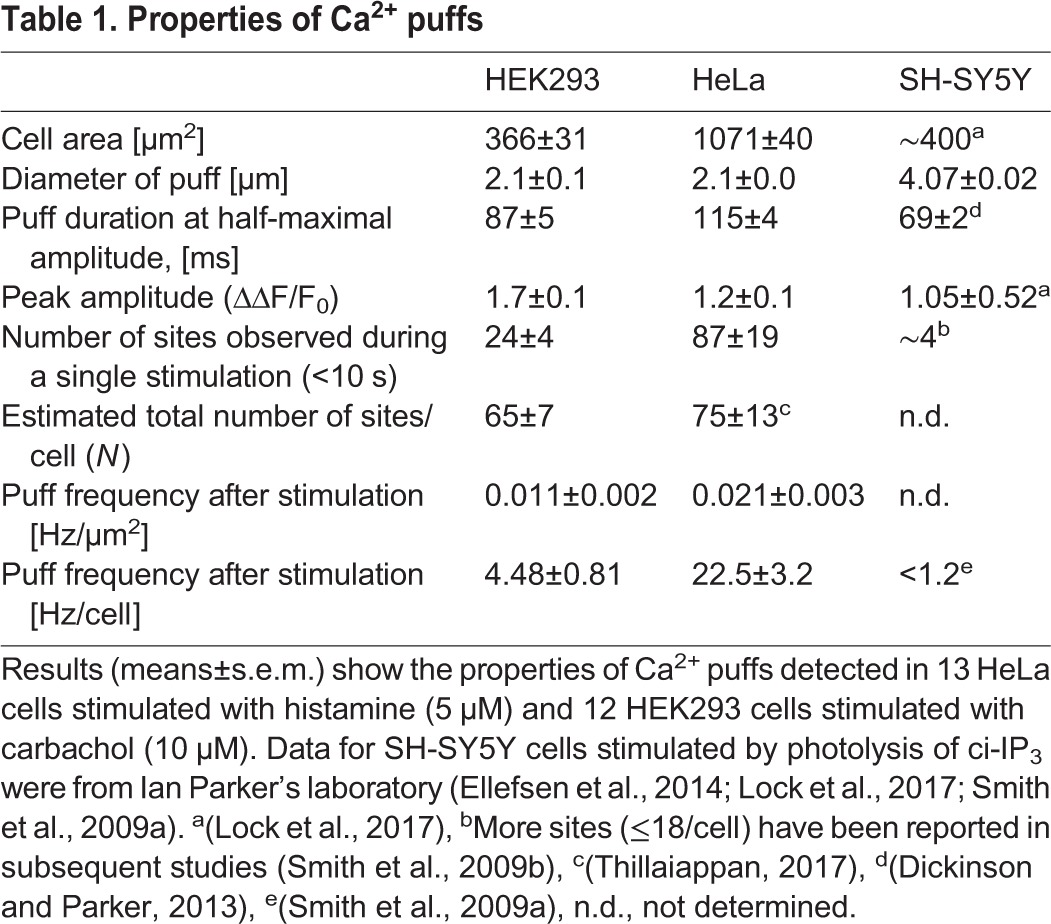
We suggest that Ca2+ puffs are more frequent and initiate at many more sites than hitherto observed. That conclusion is consistent with additional analyses of HeLa cells with endogenously tagged IP3Rs, where we used a different algorithm (xy-spotter) (Steele and Steele, 2014) to identify Ca2+ puffs (Thillaiappan, 2017). Here, too, both histamine and photolysis of ci-IP3 evoked frequent Ca2+ puffs from relatively abundant sites (Thillaiappan, 2017). Subsequent experiments examine the sites at which Ca2+ puffs originate in HEK293 cells.
Repeated stimulation with carbachol identifies many additional Ca2+ release sites
We investigated whether Ca2+ puffs initiate at the same sites during repeated stimulation with carbachol. The optimised protocol, developed to maximise puff frequency and recording intervals while allowing complete recovery between successive stimuli, is shown in Fig. 3A. Basal activity was recorded for 20 s in Ca2+-free HBS, followed by addition of carbachol (10 µM) for 10 s; the carbachol was then removed and replaced with normal HBS. After a 10-min recovery period in normal HBS, the medium was replaced with Ca2+-free HBS (20 s) and the recording and stimulation sequence was repeated (Fig. 3A, Figs S2 and S3). The results demonstrate that three sequential challenges with the same carbachol concentration evoke the same increase in Ca2+ puff activity and with the same frequency distribution of Ca2+ puffs across release sites (Fig. 3B,C). Within a few seconds of stimulation with carbachol at concentrations that evoked substantial increases in the frequency of Ca2+ puffs, the Ca2+ signals usually developed into Ca2+ waves that invaded the entire cell. Hence, although the stimulus regime was consistent, the initiation of global Ca2+ signals usually and unpredictably terminated our ability to resolve Ca2+ puffs before the end of the 10-s incubation period with carbachol. Others have also reported difficulties in recording Ca2+ puffs evoked by extracellular stimuli without rapidly triggering Ca2+ waves (Smith et al., 2009a). In all our analyses of responses to multiple challenges (carbachol or ci-IP3), the shortest interval between detection of the first Ca2+ puff and initiation of a global Ca2+ signal was 2 s. In subsequent quantitative analyses we, therefore, matched the analysis interval for all recordings by analysing puff activity during only the last 2 s before either initiation of the global Ca2+ signal or termination of the carbachol exposure.
Fig. 3.

Carbachol-evoked Ca2+ puffs initiate at abundant immobile sites. (A) The stimulus regime used for repetitive stimulation of Cal520-loaded HEK293 cells with carbachol (CCh, 10 μM). (B) Results show the density of Ca2+ puffs (Hz/μm2) evoked during each pre-incubation (grey in A) and subsequent stimulation period with CCh. Results (mean± s.e.m.) are from 16 cells. Results for ten individual cells are shown in Fig. S3. ***P<0.001, **P<0.01 relative to pre-incubation values, one-way ANOVA with Bonferroni post hoc test. (C) The number of sites at which 1−5 puffs were detected during the last 2 s after CCh addition for each of the three periods of stimulation. Results show all puffs from 16 cells, with the median and interquartile range shown by horizontal and vertical red lines, respectively. (D) For each cell, the new sites detected during each of three sequential stimulations with CCh are expressed as a percentage of all sites visited during that CCh challenge. Results are means±s.e.m., n=16 cells. ***P<0.001 relative to CCh-1, one-way ANOVA with Bonferroni post hoc test. Table S1 shows the data from which this panel was constructed. (E) Analysis used to determine whether Ca2+ puffs more effectively discover new release sites when the interval between measurements is extended. See text for details. (F) Summary results show the number of new sites discovered during the T2 phase of the response to each of the three stimulations with CCh (CCh1−3) relative to the sites identified during the T1 phase of the first CCh challenge (C1T1). Hence, a site is reported as new if it was not detected during C1T1. Results (means±s.e.m., n=16 cells) are reported as percentages of the number of sites discovered during C1T1.
The results so far suggest that Ca2+ puffs initiate at many sites but, even this, may underestimate their number because brief periods of recording may detect too few puffs to reveal the locations of all sites. We would then expect each carbachol challenge to discover new sites, but fewer with each successive stimulus. Fig. 3D confirms this prediction. During the second stimulation with carbachol, for example, only 26±3% (n=16) of the active Ca2+ release sites had also responded during the first challenge (Fig. 3D, Table S1). These results suggest that the second challenge, during which 17±1.8 sites/cell were identified (Table S1), reveal only 26% of the sites capable of initiating Ca2+ puffs. From this, we estimate the total number of Ca2+ release sites in a cell (N) to be 65±7. An alternative approach uses the Poisson distribution to estimate the mean number of puffs/site (µ) and then estimates N from µ (Table S2). This Poisson analysis applied to the first carbachol stimulation suggests that there are 70±13 sites/cell. The two analyses are consistent in suggesting that there are ∼70 carbachol-sensitive Ca2+ release sites/cell. Henceforth, we estimate N from comparisons of responses to the first and second stimuli because it is less prone than the Poisson analysis to errors arising from the small numbers of sites that respond twice during our short analysis intervals (Table S2).
In other cells, it has been concluded that there may be eager and reluctant Ca2+ release sites (Dickinson and Parker, 2013; Rooney et al., 1990; Smith et al., 2009a; Thomas et al., 2000), but our estimates of N assume that each site has a similar probability of initiating a Ca2+ puff. To test this assumption, we first categorised sites as low- or high-activity according to whether they responded once or more than once during the first exposure to carbachol; 78% of sites were categorised as low-activity (Fig. 4A). We then asked how Ca2+ puffs are distributed across the two categories of sites during subsequent responses to carbachol. The results show that the fraction of sites responding with more than one Ca2+ puff during the second and third carbachol challenges was the same for low- and high-activity sites (Fig. 4B). Furthermore, there were no significant differences in the properties of Ca2+ puffs at sites that responded once or several times during each carbachol challenge (Fig. 4C-F). We conclude that, in HEK293 cells, there is no evidence for any systematic difference between sites that respond with multiple Ca2+ puffs and those that respond only once.
Fig. 4.

Most Ca2+ release sites have similar properties. (A,B) Sites at which Ca2+ puffs occurred during the first challenge with carbachol (Fig. 3A) were categorised as ‘low-activity’ if they responded with a single Ca2+ puff and ‘high-activity’ if they responded with several puffs. Summaries show the numbers of each category of site (A) and the percentage of sites evoking more than one Ca2+ puff during subsequent challenges with carbachol according to whether the sites were categorised as high- or low-activity during the first challenge (B). Results are means±s.e.m. from 16 cells. (C-F) Summary data shows puff amplitudes (C), signal mass (D), puff diameters (E) and durations at half-maximal amplitude (F) for puffs evoked at each category of site. There were no significant differences (paired Student's t-test) in the properties of Ca2+ puffs between low- and high-activity sites. Puff amplitude is defined as the maximum ΔΔF value within the puff region. Signal mass is defined as ΣΔΔF for pixels within the puff region that are ≥T2 (see Materials and Methods). Puff diameters are the average of the major and minor elliptical diameters from the 2D Gaussian fit to the puff region. Results are means±s.e.m. from 16 cells responding to carbachol (10 μM).
We obtained matched records from all cells for quantitative analysis by selecting only 2 s from each 10-s recording for analysis, but at the expense of having to extrapolate data to estimate the total number of Ca2+ release sites (N). During the 6 s of stimulation used for analysis of the three sequential carbachol challenges, 41±5 distinct sites/cell (n=16 cells) were directly identified, whereas our extrapolated analyses suggest 65±7 sites/cell. Hence, both the number of directly identified sites and the extrapolated estimate of N suggest that Ca2+ release sites are more abundant than hitherto suggested. We conclude that carbachol evokes Ca2+ release from a large number of distinct sites in HEK293 cells.
Ca2+ release sites are immobile
The large number of sites at which Ca2+ puffs initiate and the frequency with which new sites are discovered during successive stimulations with carbachol (Fig. 3A-D) suggest that there are either many more sites than can be discovered during brief periods of carbachol stimulation or that sites move during the 10-min intervals between stimulations (Fig. 3A). The consistent estimates of N from analyses that compare Ca2+ puffs within (70±13 sites/cell, Table S2) and between (65±7, Table S1) carbachol challenges suggest that movement of sites is probably not a major factor. Nevertheless, to distinguish between the two possibilities we used the method shown in Fig. 3E to compare the rate of discovering new Ca2+ release sites within a short (<10 s) stimulation with carbachol to the rate of discovery after a 10-min or 20-min recovery period. For each cell, all Ca2+ puffs during the first carbachol stimulation were counted before the recording was terminated at either 10 s or by initiation of a Ca2+ wave. This first period of stimulation (C1) was then split to provide two intervals (T1 and T2), each with an equal number of Ca2+ puffs (n). For the same cell, we then examined the second (C2) and third (C3) response to carbachol and identified the interval (T2) during which n Ca2+ puffs occurred (Fig. 3E). This allowed comparison, in each cell and for matched numbers of Ca2+ puffs, of the rates of discovery of new Ca2+ release sites under conditions where the comparison spanned <10 s (C1T1 versus C1T2), 10 min (C1T1 versus C2T2) or 20 min (C1T1 versus C3T2). For the latter, sites have more than 120-times longer to move than during the first comparison (CIT1 versus C1T2). The results demonstrate that the Ca2+ puffs discover new sites at similar rates whether the comparisons span a few seconds or more than 20 min (Fig. 3F).
These results suggest that the sites that respond to carbachol are immobile for many minutes, consistent with previous analyses of SH-SY5Y cells (Smith et al., 2009b) and with recent observations showing that only immobile IP3Rs respond to IP3 (Thillaiappan, 2017). However, we suggest that there are many more of these Ca2+ release sites (65±7 sites/cell) than hitherto supposed (Table 1). Since we record Ca2+ puffs only within the TIRF field (i.e. within ∼62 nm of the basal plasma membrane), we may underestimate the total number of sites in a cell. However, both we for HeLa cells (see Thillaiappan, 2017) and others for SH-SY5Y cells (Smith et al., 2009a) have shown that most IP3-evoked Ca2+ puffs occur within the TIRF field, suggesting that our recordings probably capture most Ca2+ release sites.
Photolysis of ci-IP3 also reveals many Ca2+ release sites
We next examined the distribution of Ca2+ puffs evoked by uniformly delivering the active IP3 analog d-2,3-O-isopropylidene-IP3 (i-IP3) to the cytosol by photolysis of ci-IP3. Photolysis of ci-IP3 evoked a flurry of Ca2+ puffs after a latency of 0.42±0.09 s (Movie 2). We used a stimulus regime similar to that used for sequential stimulation with carbachol, but with the recording period reduced to 8 s (to match the 8 s available for recording after the ∼2 s taken to add carbachol), and with the recovery period extended to 15 min (since i-IP3 is less rapidly metabolised than IP3) (Fig. 5A). It proved impossible to get matched responses to all three rounds of photolysis of ci-IP3 with complete recovery between them (Fig. S4). The incomplete recovery is probably due to i-IP3 being less rapidly degraded than IP3 (Dakin and Li, 2007), and the diminished responses to successive flashes are likely due to depletion of ci-IP3 with each flash. Despite the limitations, the optimised photolysis protocol achieved matched Ca2+ puff activity to two successive UV flashes (Fig. 5B,C, Table S3). We again analysed only the 2 s immediately preceding either initiation of a global Ca2+ signal or termination of the 8-s recording interval and used the rate of discovery of new sites between successive challenges to estimate the total number of Ca2+ release sites/cell (N=100±35, n=10 cells) (Fig. 5D, Table S3). Counting all distinct Ca2+ release sites detected throughout the three 8-s recordings after photolysis of ci-IP3 (mean recording interval 23±0.2 s) identified 63±7 sites/cell (Fig. 5E). There is no significant difference in the estimated numbers of Ca2+ release sites accessible to carbachol (65±7 sites/cell) and to i-IP3 delivered uniformly to the cytosol (100±35). We conclude that both carbachol and photolysis of ci-IP3 evoke Ca2+ release from a large number of immobile sites in HEK293 cells (Fig. 5E).
Fig. 5.

Responses to repeated photolysis of ci-IP3. (A) HEK293 cells loaded with ci-IP3 were exposed to a 40-ms UV flash at the beginning of each of the two stimulation intervals (S1 and S2). (B) Results show the density of Ca2+ puffs (Hz/μm2) evoked during each pre-incubation (grey in A) and after each photolysis of ci-IP3. There was no significant difference between the responses evoked by the first and second flashes. (C) The number of sites at which 1−5 puffs were detected during the last 2 s of the 8-s recording after photolysis of ci-IP3 for each of the two periods of stimulation. Results show all puffs from 10 cells, with the median and interquartile range shown by red lines. (D) For each cell, the new sites detected during each of two sequential UV flashes to photolyse ci-IP3 are expressed as a percentage of all sites visited during that stimulation. Results are means±s.e.m., n=10 cells. Table S3 shows the data from which this panel was constructed. (E) Summary results (mean±s.e.m.) show numbers of Ca2+- release sites identified directly from counting all Ca2+ puffs during all stimulations with carbachol (3×10 s, 16 cells) or photolysis of ci-IP3 (3×8 s, 10 cells) (Observed), or by estimating N as described in Tables S1 and S3.
Increasing the carbachol concentration does not reveal new Ca2+ release sites
To examine the effects of varying stimulus intensity, HEK293 cells were first stimulated for ∼10 s with 10 µM carbachol and then with 30 µM carbachol according to the protocol shown in Fig. 6A. The results demonstrate that the cells fully recovered between the two periods of stimulation, and that increasing the carbachol concentration increased the frequency of Ca2+ puffs and the number of sites detected (Fig. 6B,C). Similar results were obtained with ci-IP3 when the duration of the flash used for photolysis was increased (Fig. 6D-F).
Fig. 6.

Increasing carbachol concentration increases the frequency of Ca2+ puffs, but not the number of sites. (A) Cells were sequentially stimulated with two concentrations of carbachol (CCh). (B,C) Summary results show the frequency of Ca2+ puffs (B) and the number of sites detected (C) before and after stimulation with the indicated CCh concentrations. Events were recorded for the entire period (up to 10 s) after addition of CCh until the Ca2+ signal propagated across the cell. Results (mean±s.e.m.) are from 8 cells. *P<0.05, paired Student's t-test, relative to first stimulus. (D) Cells were stimulated with two sequential UV flashes to photolyse ci-IP3. (E,F) Summary results show the frequency of Ca2+ puffs (E) and the number of sites detected (F) before and after each stimulation. Results (mean±s.e.m.) are from 10 cells. *P<0.05, paired Student's t-test, relative to first stimulus. (G) Relationship between number of Ca2+ puffs/cell and the unique Ca2+ release sites they identify for cells stimulated once or repetitively with CCh (10 µM, black) or by photolysis of ci-IP3 (40 ms, red). The responses to sequential stimulation with 10 µM (blue) and then 30 µM CCh (green) are shown for 8 cells. The dashed line shows the relationship if every Ca2+ puff identified a new Ca2+ release site.
The increase in frequency of Ca2+ puffs with carbachol concentration might be due to an increased probability of every Ca2+ release site responding or, more-intense stimulation might increase the number of sites competent to respond. The first explanation predicts that a fixed number of Ca2+ puffs should discover the same number of Ca2+ release sites whatever the stimulus intensity, whereas the second explanation predicts that Ca2+ puffs evoked by the high carbachol concentration should more effectively discover new Ca2+ release sites. We, therefore, compared the relationship between the number of Ca2+ puffs and the number of unique sites they reveal for cells stimulated with low and high carbachol concentrations. The results demonstrate that the relationship between Ca2+ puffs and the number of Ca2+ release sites they identify is the same for carbachol and photolysis of ci-IP3 and, furthermore, it is the same for low and high concentrations of carbachol (Fig. 6G).
These results demonstrate that a Ca2+ puff has the same probability of discovering a Ca2+ release site, regardless of whether the Ca2+ signals are evoked by carbachol or photolysis of ci-IP3, or are evoked by low or high stimulus intensities. We conclude that the increased frequency of Ca2+ puffs as stimulus intensity is increased results from most sites becoming more likely to respond rather than from recruitment of new Ca2+ release sites.
Carbachol and photolysis of ci-IP3 activate the same Ca2+ release sites
We next considered whether i-IP3 delivered uniformly to the cytosol by photolysis of ci-IP3 or IP3 delivered from endogenous signalling pathways evokes Ca2+ puffs at the same sites. Cells loaded with ci-IP3 were sequentially stimulated with carbachol (10 μM) and a photolysis flash (120 ms) (Fig. 7A). The two stimuli evoked indistinguishable Ca2+ puff activity irrespective of the order in which they were presented (Fig. 7B,C). We then asked whether, with these matched stimulus intensities, the second stimulus from a heterologous sequence of stimuli identified more new Ca2+ release sites than the second stimulus from two sequential carbachol challenges. The results show that the number of new sites identified by the second stimulus was unaffected by the nature of the first stimulus (Fig. 7D,E). The results also suggest that the same Ca2+ release sites are activated by endogenous signalling pathways and by photolysis of ci-IP3.
Fig. 7.

Carbachol and photolysis of ci-IP3 stimulate the same Ca2+ release sites. (A) Cells loaded with ci-IP3 were sequentially stimulated with carbachol (10 μM) and a UV-flash (120 ms), with either stimulus presented first. (B) Density of Ca2+ puffs evoked by the two stimuli presented in either order during the entire 10-s recording interval for each stimulus. The activity recorded before each stimulus is also shown. Results show no significant differences between any of the stimuli. (C) Summary results, from analyses of the entire period of stimulation preceding initiation of a Ca2+ wave show the number of Ca2+ puffs detected during each of the indicated pairs of sequential stimuli. (D) Typical results show centroids of the Ca2+ puffs detected by PuffFinder during stimulation first with carbachol (10 µM, black) and then with ci-IP3 (120 ms, red) (i) or with the same stimuli in reverse order (ii). Sites detected by SiteMapper are shown by blue circles. (E) Summary results show the number of new Ca2+ release sites identified during the second stimulus (S2) that were not detected during the first period of stimulation (S1) for each of the indicated pairs of stimuli. Results (C and E) show means±s.e.m. from 10−18 cells.
DISCUSSION
We used a custom-written algorithm that automatically detects Ca2+ puffs and assigns them to Ca2+ release sites in order to examine the spatial organisation of IP3-evoked Ca2+ signals in human cell lines (Figs 1 and 2, Figs S1 and S2). In both HEK293 and HeLa cells, stimuli that evoke formation of IP3 through endogenous G-protein-coupled receptors (GPCRs) triggered a flurry of Ca2+ puffs whose properties were similar to those of Ca2+ puffs observed in other cells (Table 1, Fig. 2D, Fig. S1B). However, the sites at which these Ca2+ puffs occurred were about 10-fold more abundant than reported previously for other cells (Nakamura et al., 2012; Smith and Parker, 2009; Thomas et al., 2000). In HEK293 cells, for example, we estimate the number of sites to be 65±7 sites/cell (Fig. 5E; Table S1), and our less-comprehensive analysis of HeLa cells suggests at least 87 sites/cell (Table 1). The latter is consistent with our recent, independent analysis of Ca2+ puffs in HeLa cells that express endogenous IP3Rs tagged with EGFP, wherein photolysis of ci-IP3 evoked many Ca2+ puffs at numerous sites (see Thillaiappan, 2017). We conclude that Ca2+ release sites are more abundant than hitherto suggested but, even so, with about eight IP3Rs per Ca2+ release site (see Thillaiappan, 2017) and about 7200 IP3Rs per HEK293 cell (Tovey et al., 2008), it is clear that only a small fraction of IP3Rs (<8%) contribute to the Ca2+ release sites.
Many studies have shown that IP3Rs can move within ER membranes (see references in Geyer et al., 2015); yet the Ca2+ puffs evoked by stimulation of cells with low stimulus intensities are often observed to initiate at immobile sites (Bootman et al., 1997a; Kasai et al., 1993; Marchant and Parker, 2001; Nakamura et al., 2012; Olson et al., 2012; Rooney et al., 1990; Simpson et al., 1997). We confirmed those observations by demonstrating that, in HEK293 cells, Ca2+ puffs initiate at sites that remain immobile for at least 20 min (Fig. 3E,F). Among these immobile Ca2+ release sites, we found no evidence to suggest any systematic difference in their propensity to initiate Ca2+ signals (Fig. 4). We suggest, in keeping with the conclusions of our recent analyses of HeLa cells (Thillaiappan, 2017), that Ca2+ puffs initiate at immobilised IP3R clusters that comprise only a small fraction of the IP3Rs expressed in each cell.
Comparison of the effects of sequential challenges with different carbachol concentrations confirmed that Ca2+ puffs become more frequent as the stimulus intensity increases, with no apparent increase in the number of Ca2+ release sites (Fig. 6). From this, we conclude that more-intense stimulation increases the probability of a site initiating a puff without increasing the underlying number of competent Ca2+ release sites.
By comparing sequential responses of cells to photolysis of ci-IP3 and IP3 delivered to the cell through an endogenous GPCR signalling pathway, we established that each stimulus evoked Ca2+ puffs from the same immobile Ca2+ release sites (Fig. 7). This result suggests that global delivery of i-IP3 and IP3 provided at the plasma membrane by hydrolysis of phosphatidylinositol 4,5-bisphosphate by GPCR-activated PLC reach the same IP3Rs. While our work was in progress, a study of SH-SY5Y cells reached a similar conclusion, namely that the sites where Ca2+ puffs initiate are probably the same, regardless of whether the signals are evoked by photolysis of ci-IP3, or by carbachol or bradykinin activating their respective GPCRs (Lock et al., 2017). These results are unexpected because there is evidence that some GPCRs may selectively deliver IP3 to IP3Rs (Delmas et al., 2002; Olson et al., 2012; Short et al., 2000; Tovey and Taylor, 2013; Tu et al., 1998; Zhao et al., 1990), and both we (Taylor and Konieczny, 2016) and others (Dickinson et al., 2016) have suggested that IP3Rs substantially slow diffusion of IP3 within cells. A possible explanation is that the only IP3Rs that contribute to initiation of Ca2+ puffs are those that lie very close to the plasma membrane (Smith et al., 2009a; Thillaiappan, 2017). In this situation, even if IP3 were locally delivered from GPCR signalling pathways we might fail to distinguish responses evoked by GPCRs from those evoked by uniform photolysis of caged IP3 since the only responsive IP3Rs would be those located close to the plasma membrane.
Our quantitative analyses of the spatial distribution of Ca2+ puffs, the building blocks of all IP3-evoked Ca2+ signals, has established that Ca2+ puffs initiate at relatively abundant immobile sites (∼65 sites/cell) that include only a small fraction of the IP3Rs expressed in a cell. The number of sites appears not to be affected by the intensity of the stimulation, and there is no evidence to suggest that IP3 is selectively delivered to them from endogenous signalling pathways.
MATERIALS AND METHODS
Materials
Cal520-AM was from AAT Bioquest (Sunnyvale, CA). Fluo-4-AM, Dulbecco's Modified Eagle's Medium (DMEM)/Ham's F-12 (50:50) supplemented with GlutaMAX, and TrypLE were from Invitrogen (Paisley, UK). Pluronic acid F-127, dimethyl sulfoxide (DMSO), poly-L-lysine, foetal bovine serum (FBS), histamine and carbamylcholine chloride (carbachol) were from Sigma-Aldrich (Poole, Dorset, UK). Fibronectin and EGTA-AM were from Merck Millipore (Darmstadt, Germany). Caged IP3-PM [ci-IP3-PM, d-2,3-O-isopropylidine-6-O-(2-nitro-4,5-dimethoxy)benzyl-myo-inositol 1,4,5-trisphosphate-hexakis(proprionoxymethyl)ester] was from SiChem GmbH (Bremen, Germany).
Cell culture
We used two cell lines, HEK293 and HeLa cells (ATCC), neither of which express functional ryanodine receptors (RyRs). There is detectable mRNA for RyRs in HeLa cells, but caffeine does not evoke Ca2+ signals (Tovey et al., 2001). HEK293 cells do not express RyRs, and are insensitive to caffeine and ryanodine (López-Sanjurjo et al., 2013; Tong et al., 1999). HEK293 and HeLa cells were maintained in DMEM/Ham's F-12 supplemented with GlutaMAX and 10% FBS in humidified air at 37°C with 5% CO2. The cells were passaged at ∼80% confluence. Short tandem-repeat profiling was used to authenticate HeLa cells (Eurofins, Germany) and HEK293 cells (Public Health England). Regular screening confirmed that all cells were free of mycoplasma infection. Cells were grown on either round glass coverslips for low-resolution measurements (No. 1, 22-mm diameter, Scientific Laboratory Supplies, East Yorkshire, UK) or glass-bottomed dishes for high-resolution measurements (No. 1, 35-mm diameter, MaTek, Ashland, MA). The glass was coated with fibronectin (10 µg/ml) for HeLa cells or poly-l-lysine (10 µg/ml) for HEK293 cells. Coated coverslips in 6-well culture plates were seeded with HEK293 cells (8×105 cells/well) or HeLa cells (9×104 cells/well) and used for imaging after 48 h, when they were still subconfluent.
Imaging of cytosolic Ca2+ signals
To allow recording of local Ca2+ signals, cells were loaded with EGTA to restrict Ca2+ diffusion (Parker and Smith, 2010), a Ca2+ indicator (Cal520) and, in some cases, with ci-IP3. Cells were washed with HEPES-buffered saline (HBS: 135 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 1.5 mM CaCl2, 11.5 mM glucose, 11.6 mM HEPES pH 7.3) and then incubated in darkness with pluronic acid F-127 (0.02% w/v) and either ci-IP3-PM (1 µM) or DMSO (0.02% v/v) at 20°C. After 45 min, Cal520-AM (2 µM) was added and after a further 45 min the medium was replaced with fresh HBS supplemented with EGTA-AM (10 µM). Cells were used for imaging after a further 45 min. Similar loading methods, but without EGTA-AM or ci-IP3-PM, and with Fluo-4 as the Ca2+ indicator, were used for low-resolution measurements of [Ca2+]c.
Local Ca2+ signals were measured using total internal reflection fluorescence microscopy (TIRFM). A glass-bottomed dish containing loaded cells was immobilised on the motorised stage of an Olympus IX83 inverted microscope. Imaging used a 100× TIRF objective [Olympus UApo N; numerical aperture (NA)=1.49] with excitation (488 nm) provided by a diode-pumped solid-state laser (150 mW, iLas Laser System, Cairn) and a band-pass filter (ET-405/488/561/643 quad band filter set, Chroma). The angle of the excitation beam was adjusted to achieve TIRF with a penetration depth of ∼62 nm. Emitted light was captured through a band-pass filter (510−550 nm, Olympus U-MNIBA3 cube) with an Andor iXon 897 EMCCD camera (512×512 pixels, 16 µm×16 µm per pixel). With the 100× objective, each image pixel had dimensions of 160 nm×160 nm. Images were acquired at 25 Hz.
Cells were kept in HBS (2 ml, 20°C) in the intervals between stimulation. The HBS was replaced by Ca2+-free HBS immediately before recordings. Extracellular stimuli were applied in Ca2+-free HBS after careful aspiration of half the medium and its replacement with histamine or carbachol at twice its final concentration in Ca2+-free HBS (1 ml). Medium exchanges took ∼1−2 s. Photolysis of ci-IP3 was achieved using a light-emitting diode (LED, 395 nm, Spectra Lumincor, Beaverton, OR), with the duration of exposure determined by a shutter controlled from within MetaMorph (Molecular Devices, Sunnyvale, CA). A 50:50 mirror in the excitation light path allowed simultaneous excitation with the 488-nm laser (Cal-520) and the 395-nm LED (for photolysis of ci-IP3). Images were captured in Metamorph and saved as tif files for analysis.
Similar methods were used for low-resolution measurements of [Ca2+]c in cells grown on glass coverslips, loaded with Fluo-4, and mounted in a stainless steel chamber (∼1 ml) on the stage of an Olympus IX81 inverted microscope with a 40× objective (Olympus UApo N340, NA=1.35). Fluo-4 was excited (488 nm) by light from a diode-pumped solid-state laser (Olympus, Las/405/50) and a band-pass filter (470−490 nm, Semrock, BrightLine Triple Cube). Emitted light was captured with an Andor iXon 897 EMCCD camera (each image pixel had dimensions of 400 nm×400 nm) after passing through a band-pass filter (510−550 nm, Olympus U-MNIBA3 cube). Images (45-ms capture time) were acquired at 2 Hz.
Automated detection of Ca2+ puffs by using PuffFinder
The need for automated detection of Ca2+ puffs and the unsuitability of the algorithms developed for detection of local Ca2+ signals evoked by RyRs have been discussed by Parker and colleagues (Ellefsen et al., 2014). For our automated analyses, images (in tif, tagged image format) from a complete experiment were compiled into a single stack. The images were corrected for background fluorescence by subtracting from every pixel the average fluorescence intensity from a square (∼40 µm2) region of interest (ROI) outside the cell, determined uniquely for every frame. Cell boundaries were identified by comparison with differential interference contrast images, and fluorescence values within the extracellular regions were set to zero. ROIs corresponding to each cell were then distributed to separate image stacks: one image stack for each cell. A custom-written suite of algorithms (PuffMapper) written in IDL code was used to automatically identify Ca2+ puffs (PuffFinder) from the background-corrected image stacks for each cell and to then allocate Ca2+ puffs to puff sites (SiteMapper) (Fig. 1, Figs S1 and S2).
Fig. 1 summarises the steps used by PuffFinder to identify Ca2+ puffs. The IDL code is available from M.V.K. PuffFinder begins by smoothing the background-corrected images across areas corresponding to 2×2 pixels using the ‘Smooth’ function in IDL. This step aids the initial identification of puffs. The unsmoothed images are retained for subsequent analysis of the Ca2+ puffs once their locations have been identified. The average fluorescence intensity of each pixel (F0) is calculated from 50 pre-stimulation frames. For every subsequent frame, F/F0 is calculated for every pixel (F is the fluorescence intensity at each time). The fluorescence change associated with a Ca2+ puff typically rises very quickly (within ∼40−60 ms) to its peak value (Nakamura et al., 2012; Smith and Parker, 2009). Our recording interval (40 ms) was chosen to maximise the possibility that this rising phase would occur within a single frame and so allow puffs to be detected by searching for rapid frame-to-frame increases in fluorescence intensity. PuffMapper searches for these changes by creating an image stack in which each frame in the F/F0 stack is subtracted from the next frame in the temporal series to create a ΔF stack [ΔF=(Fn+1/F0)−(Fn/F0)]. As stimulation proceeds, Ca2+ puffs become more frequent, the background [Ca2+]c increases and, eventually, a global Ca2+ signal initiates (Movies 1 and 2). To allow detection of Ca2+ puffs against this creeping (and then explosive) increase in F/F0, PuffFinder calculates the average ΔF value across the entire cell for each frame (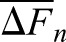 ). This value is negligibly influenced by the appearance of a Ca2+ puff within the frame because Ca2+ puffs are relatively rare and each occupies only a small fraction of the cell area (typically ∼0.2%). The
). This value is negligibly influenced by the appearance of a Ca2+ puff within the frame because Ca2+ puffs are relatively rare and each occupies only a small fraction of the cell area (typically ∼0.2%). The 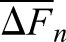 value is subtracted from the ΔF value in each pixel of the same frame (ΔFn) to create a new image stack (
value is subtracted from the ΔF value in each pixel of the same frame (ΔFn) to create a new image stack (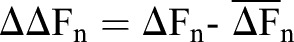 ). In this ΔΔF stack, the average value of all pixels is zero, but those in which the fluorescence increases rapidly above the prevailing global fluorescence intensity have positive values. PuffFinder now scrutinises these ΔΔF values to determine which pixels are likely to be at the centre of a rapid fluorescence increase that extends radially to neighbouring pixels. Since missed events are likely to cause fewer problems in subsequent analyses than mistakenly identified puffs, the algorithm was developed to minimise inclusion of such ‘false-positives’.
). In this ΔΔF stack, the average value of all pixels is zero, but those in which the fluorescence increases rapidly above the prevailing global fluorescence intensity have positive values. PuffFinder now scrutinises these ΔΔF values to determine which pixels are likely to be at the centre of a rapid fluorescence increase that extends radially to neighbouring pixels. Since missed events are likely to cause fewer problems in subsequent analyses than mistakenly identified puffs, the algorithm was developed to minimise inclusion of such ‘false-positives’.
Pixels at the centre of a puff will have the largest ΔΔF values, while surrounding pixels will have lower values. PuffFinder identifies these pixels using two threshold values (T1 and T2). For each pixel, the mean ΔΔF value (µ, invariably close to 0) and the standard deviation (s.d.) are determined over time. The thresholds (determined empirically) are set at T1=µ+(3.4×s.d.), and T2=µ+(2.45×s.d.), to provide two additional frames (T1 and T2). Every frame in an image stack is now compared with the T1 frame to identify all pixels that exceed T1. Since many of these pixels will be part of the same Ca2+ puff, the next step is to identify the pixel that is likely to be at the centre of a puff by finding the pixel with the largest ΔΔF and surrounded by pixels with the largest ΣΔΔF. This is achieved by ranking pixels in each frame in descending order of ΔΔF and by considering the largest first before progressing to the next ranked pixel (excluding those that have already been assigned to a puff). The first pixel is placed at the centre of a 5×5 binary matrix, and each of the 25 pixels is then assigned a binary value (V): V=1 if ΔΔF>T2, and V=0 if ΔΔF≤T2. We chose a 25-pixel area for this analysis because its width (800 nm) would be expected to capture the core of most Ca2+ puffs (diameter 2.1±0.1 μm, Table 1). Matrices are then ranked by their ΣV values (ΣΔΔF for all pixels with ΔΔF>T2, the ‘signal mass’ is used as a tie-breaker for matrices with the same ΣV). The final criterion applied to decide whether a pixel forms part of a puff is to eliminate matrices in which <18 pixels exceed T2 (this was again determined empirically). The output from this analysis is a ranked list for each frame of regions (5×5 pixels) with the largest ΔΔF values.
The next step defines the boundary of each puff, beginning with a 3×3 matrix centred on the initial pixel with the largest ΔΔF value. This is achieved by first expanding each matrix to the left and to the right, incrementally one column at a time, until the number of pixels in the column where ΔΔF>T2 falls to zero. The process is now repeated on this wider matrix by expanding rows incrementally up and then down, until the number of pixels within a row in which ΔΔF>T2 falls below 10%. The matrix delineated by this first round of expansion in 2 dimensions is now re-examined, and expanded outwards (columns then rows) to include only columns or rows in which more than 10% of pixels have ΔΔF>T2. The process is then repeated on the next ranked matrix for which the central pixel has not already been placed within an expanded matrix. The output from this processing is a rectangular map enclosing each puff event. The reliability of PuffFinder was validated by confirming that all automatically identified puffs could be resolved by manual inspection of fluorescence time series.
The ΔΔF values within each rectangular map of a puff are now fitted to a 2-dimensional Gaussian fit using the GAUSS2FIT function in IDL. This provides the radii of the elliptical fit, its rotation angle (from which the area of the puff is calculated), and the coordinates of the centre of the puff with sub-pixel resolution. Comparison of these features across sequential ΔΔF frames provides temporal properties and the maximal amplitude of the puff.
Automated identification of Ca2+ puff sites by using SiteMapper
The second major component of the automated analysis uses SiteMapper to identify the different sites at which Ca2+ puffs occur (Fig. 1C). The algorithm defines a minimal separation between the centroids of all Ca2+ puffs (radius=0.96 µm, i.e. 6 pixels) within a time series to decide whether two puffs occurred at different sites. A similar proximity-based option is provided in the automated analyses described by Parker's group (Ellefsen et al., 2014). The choice of minimal radius is critical for defining the number of sites identified. It was chosen because the average elliptical radii from the 2D Gaussian fits to Ca2+ puffs in HEK293 cells was 1.1±0.1 µm (Table 1) and by empirically comparing the relationship between Ca2+ puffs and sites after selecting different minimal radii.
SiteMapper begins by ranking each Ca2+ puff identified by PuffFinder throughout a time series according to its signal mass. The first site is initially defined as the centroid of the puff with the greatest signal mass. Any neighbouring puffs with centroids that are within 0.96 µm of the first centroid are assigned to this site. The centre of the site is then redefined as the centre of the region enclosing all associated puffs. The process is then repeated to identify any additional puffs that may now reside within 0.96 µm of the new centre of the site. All puffs falling within the site are excluded from further consideration. The process of site identification is then repeated with the next ranked puff, and the site identification process continues until every Ca2+ puff has been assigned to a site. The final output from SiteMapper is a list of all Ca2+ release sites and their coordinates, which can then be used to analyse their distribution and the frequency with which each evokes Ca2+ puffs. SiteMapper was validated by overlaying the elliptical region defining each site with the centroids of the enclosed puffs (Fig. S2), and then visually confirming the number of enclosed puffs and the location of each site centre with the output from SiteMapper.
Statistical analysis
Results are presented as means±s.e.m. unless indicated otherwise. Statistical comparisons used ANOVA and Student's t-tests, with the Bonferroni correction used for multiple comparisons. Significance is denoted by: *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001.
Supplementary Material
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualisation: M.V.K., C.W.T.; Methodology: M.V.K.; Software: M.V.K.; Validation: M.V.K.; Formal analysis: M.V.K., C.W.T.; Investigation: M.V.K.; Writing - original draft: M.V.K., C.W.T.; Writing - review & editing: M.V.K., C.W.T.; Supervision: C.W.T.; Project administration: C.W.T.; Funding acquisition: C.W.T.
Funding
This work was supported by the Wellcome Trust [grant number 101844], by a studentship to M.V.K. from the Cambridge Overseas Trust, and by awards to M.V.K. from St. John's College, Cambridge and the Cambridge Philosophical Society. Deposited in PMC for immediate release.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.208520.supplemental
References
- Alzayady K. J., Wang L., Chandrasekhar R., Wagner L. E. II, Van Petegem F. and Yule D. I. (2016). Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Sci. Signal. 9, ra35 10.1126/scisignal.aad6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootman M. D., Berridge M. J. and Lipp P. (1997a). Cooking with calcium: the recipes for composing global signals from elementary events. Cell 91, 367-373. 10.1016/S0092-8674(00)80420-1 [DOI] [PubMed] [Google Scholar]
- Bootman M., Niggli E., Berridge M. and Lipp P. (1997b). Imaging the hierarchical Ca2+ signalling system in HeLa cells. J. Physiol. 499, 307-314. 10.1113/jphysiol.1997.sp021928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers M., Schell M. J. and Thorn P. (2006). Agonist-evoked inositol trisphosphate receptor (IP3R) clustering is not dependent on changes in the structure of the endoplasmic reticulum. Biochem. J. 394, 57-66. 10.1042/BJ20051130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles A. C., Merrill J. E., Dirksen E. R. and Sanderson M. J. (1991). Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 6, 983-992. 10.1016/0896-6273(91)90238-U [DOI] [PubMed] [Google Scholar]
- Dakin K. and Li W.-H. (2007). Cell membrane permeable esters of d-myo-inositol 1,4,5-trisphosphate. Cell Calcium 42, 291-301. 10.1016/j.ceca.2006.12.003 [DOI] [PubMed] [Google Scholar]
- Delmas P., Wanaverbecq N., Abogadie F. C., Mistry M. and Brown D. A. (2002). Signaling microdomains define the specificity of receptor-mediated InsP3 pathways in neurons. Neuron 34, 209-220. 10.1016/S0896-6273(02)00641-4 [DOI] [PubMed] [Google Scholar]
- Dickinson G. D. and Parker I. (2013). Factors determining the recruitment of inositol trisphosphate receptor channels during calcium puffs. Biophys. J. 105, 2474-2484. 10.1016/j.bpj.2013.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson G. D., Ellefsen K. L., Dawson S. P., Pearson J. E. and Parker I. (2016). Hindered cytoplasmic diffusion of inositol trisphosphate restricts its cellular range of action. Sci. Signal. 9, ra108 10.1126/scisignal.aag1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellefsen K. L., Settle B., Parker I. and Smith I. F. (2014). An algorithm for automated detection, localization and measurement of local calcium signals from camera-based imaging. Cell Calcium 56, 147-156. 10.1016/j.ceca.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett J. K., White C., Cheung K.-H. and Mak D.-O. D. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593-658. 10.1152/physrev.00035.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer M., Huang F., Sun Y., Vogel S. M., Malik A. B., Taylor C. W. and Komarova Y. A. (2015). Microtubule-associated protein EB3 regulates IP3 receptor clustering and Ca2+ signaling in endothelial cells. Cell Rep. 12, 79-89. 10.1016/j.celrep.2015.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H., Li Y. X. and Miyashita Y. (1993). Subcellular distribution of Ca2+ release channels underlying Ca2+ waves and oscillations in exocrine pancreas. Cell 74, 669-677. 10.1016/0092-8674(93)90514-Q [DOI] [PubMed] [Google Scholar]
- Lock J. T., Smith I. F. and Parker I. (2017). Comparison of Ca2+ puffs evoked by extracellular agonists and photoreleased IP3. Cell Calcium 63, 43-47. 10.1016/j.ceca.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Sanjurjo C. I., Tovey S. C., Prole D. L. and Taylor C. W. (2013). Lysosomes shape Ins(1,4,5)P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J. Cell Sci. 126, 289-300. 10.1242/jcs.116103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant J. S. and Parker I. (2001). Role of elementary Ca2+ puffs in generating repetitive Ca2+ oscillations. EMBO J. 20, 65-76. 10.1093/emboj/20.1.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant J., Callamaras N. and Parker I. (1999). Initiation of IP3-mediated Ca2+ waves in Xenopus oocytes. EMBO J. 18, 5285-5299. 10.1093/emboj/18.19.5285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H., Bannai H., Inoue T., Michikawa T., Sano M. and Mikoshiba K. (2012). Cooperative and stochastic calcium releases from multiple calcium puff sites generate calcium microdomains in intact Hela cells. J. Biol. Chem. 287, 24563-24572. 10.1074/jbc.M111.311399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M. L., Sandison M. E., Chalmers S. and McCarron J. G. (2012). Microdomains of muscarinic acetylcholine and Ins(1,4,5)P3 receptors create ‘Ins(1,4,5)P3 junctions’ and sites of Ca2+ wave initiation in smooth muscle. J. Cell Sci. 125, 5315-5328. 10.1242/jcs.105163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker I. and Smith I. F. (2010). Recording single-channel activity of inositol trisphosphate receptors in intact cells with a microscope, not a patch clamp. J. Gen. Physiol. 136, 119-127. 10.1085/jgp.200910390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker I., Choi J. and Yao Y. (1996). Elementary events of InsP3-induced Ca2+ liberation in Xenopus oocytes: hot spots, puffs and blips. Cell Calcium 20, 105-121. 10.1016/S0143-4160(96)90100-1 [DOI] [PubMed] [Google Scholar]
- Prole D. L. and Taylor C. W. (2016). Inositol 1,4,5-trisphosphate receptors and their protein partners as signalling hubs. J. Physiol. 594, 2849-2866. 10.1113/JP271139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman T. U., Skupin A., Falcke M. and Taylor C. W. (2009). Clustering of IP3 receptors by IP3 retunes their regulation by IP3 and Ca2+. Nature 458, 655-659. 10.1038/nature07763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney T. A., Sass E. J. and Thomas A. P. (1990). Agonist-induced cytosolic calcium oscillations originate from a specific locus in single hepatocytes. J. Biol. Chem. 265, 10792-10796. [PubMed] [Google Scholar]
- Short A. D., Winston G. P. and Taylor C. W. (2000). Different receptors use inositol trisphosphate to mobilize Ca2+ from different intracellular pools. Biochem. J. 351, 683-686. 10.1042/bj3510683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson P. B., Mehotra S., Lange G. D. and Russell J. T. (1997). High density distribution of endoplasmic reticulum proteins and mitochondria at specialized Ca2+ release sites in oligodendrocyte processes. J. Biol. Chem. 272, 22654-22661. 10.1074/jbc.272.36.22654 [DOI] [PubMed] [Google Scholar]
- Smith I. F. and Parker I. (2009). Imaging the quantal substructure of single IP3R channel activity during Ca2+ puffs in intact mammalian cells. Proc. Natl. Acad. Sci. USA 106, 6404-6409. 10.1073/pnas.0810799106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith I. F., Wiltgen S. M. and Parker I. (2009a). Localization of puff sites adjacent to the plasma membrane: functional and spatial characterization of Ca2+ signaling in SH-SY5Y cells utilizing membrane-permeant caged IP3. Cell Calcium 45, 65-76. 10.1016/j.ceca.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith I. F., Wiltgen S. M., Shuai J. and Parker I. (2009b). Ca2+ puffs originate from preestablished stable clusters of inositol trisphosphate receptors. Sci. Signal. 2, ra77 10.1126/scisignal.2000466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele E. M. and Steele D. S. (2014). Automated detection and analysis of Ca2+ sparks in x-y image stacks using a thresholding algorithm implemented within the open-source image analysis platform ImageJ. Biophys. J. 106, 566-576. 10.1016/j.bpj.2013.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C. W. and Konieczny V. (2016). IP3 receptors: take four IP3 to open. Sci. Signal. 9, pe1 10.1126/scisignal.aaf6029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thillaiappan N. B. (2017). Dynamics and functional architecture of native inositol 1,4,5-trisphosphate receptors. PhD thesis, University of Cambridge, Cambridge, UK. [Google Scholar]
- Thomas D., Lipp P., Tovey S. C., Berridge M. J., Li W., Tsien R. Y. and Bootman M. D. (2000). Microscopic properties of elementary Ca2+ release sites in non-excitable cells. Curr. Biol. 10, 8-15. 10.1016/S0960-9822(99)00258-4 [DOI] [PubMed] [Google Scholar]
- Thurley K., Tovey S. C., Moenke G., Prince V. L., Meena A., Thomas A. P., Skupin A., Taylor C. W. and Falcke M. (2014). Reliable encoding of stimulus intensities within random sequences of intracellular Ca2+ spikes. Sci. Signal. 7, ra59 10.1126/scisignal.2005237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J., Du G. G., Chen S. R. W. and MacLennan D. H. (1999). Hek-293 cells possess a carbachol- and thapsigargin-sensitive intracellular Ca2+ store that is responsive to stop-flow medium changes and insensitive to caffeine and ryanodine. Biochem. J. 343, 39-44. 10.1042/bj3430039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovey S. C. and Taylor C. W. (2013). Cyclic AMP directs inositol (1,4,5)-trisphosphate-evoked Ca2+ signalling to different intracellular Ca2+ stores. J. Cell Sci. 126, 2305-2313. 10.1242/jcs.126144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovey S. C., De Smet P., Lipp P., Thomas D., Young K. W., Missiaen L., De Smedt H., Parys J., Berridge M. J., Thuring J. et al. (2001). Calcium puffs are generic InsP3-activated elementary calcium signals and are downregulated by prolonged hormonal stimulation to inhibit cellular calcium responses. J. Cell Sci. 114, 3979-3989. [DOI] [PubMed] [Google Scholar]
- Tovey S. C., Dedos S. G., Taylor E. J. A., Church J. E. and Taylor C. W. (2008). Selective coupling of type 6 adenylyl cyclase with type 2 IP3 receptors mediates direct sensitization of IP3 receptors by cAMP. J. Cell Biol. 183, 297-311. 10.1083/jcb.200803172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu J. C., Xiao B., Yuan J. P., Lanahan A. A., Leoffert K., Li M., Linden D. J. and Worley P. F. (1998). Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 21, 717-726. 10.1016/S0896-6273(00)80589-9 [DOI] [PubMed] [Google Scholar]
- Wiltgen S. M., Dickinson G. D., Swaminathan D. and Parker I. (2014). Termination of calcium puffs and coupled closings of inositol trisphosphate receptor channels. Cell Calcium 56, 157-168. 10.1016/j.ceca.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Khademazad M. and Muallem S. (1990). Agonist-mediated Ca2+ release in permeabilized UMR-106-01 cells. Transport properties and generation of inositol 1,4,5-trisphosphate. J. Biol. Chem. 265, 14822-14827. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.