

Abstract
Craniosynostosis is a prevalent human birth defect characterized by premature fusion of calvarial bones. In this study, we show that tight regulation of endogenous PDGFRα activity is required for normal calvarium development in the mouse and that dysregulated PDGFRα activity causes craniosynostosis. Constitutive activation of PDGFRα leads to expansion of cartilage underlying the coronal sutures, which contribute to suture closure through endochondral ossification, in a process regulated in part by PI3K/AKT signaling. Our results thus identify a novel mechanism underlying calvarial development in craniosynostosis.
KEY WORDS: Craniosynostosis, Endochondral ossification, Receptor tyrosine kinase, Neural crest, Mesoderm
Summary: PDGFRα-PI3K/AKT signaling plays an essential role in the developing calvarial tissues of mouse by regulating endochondral ossification in cartilage anlagen underlying coronal sutures derived from neural crest and mesoderm.
INTRODUCTION
Bone formation is a complex process in which mesenchymal cells become osteoblasts and deposit bone matrix. There are generally two pathways to form bone. The mesenchymal cells may first form a cartilaginous template that is later replaced by osteoblasts that mineralize into bone, as hypertrophic chondrocytes in the growth plate are thought to be an end state of differentiation. This pathway of endochondral ossification occurs in the axial and appendicular skeleton (Kronenberg, 2003; Ornitz, 2005). Recent studies have however challenged this traditional concept of lineage segregation, as hypertrophic chondrocytes in long bones have been shown by lineage tracing to be able to transdifferentiate into osteoblasts (Yang et al., 2014a,b; Zhou et al., 2014). Alternatively, mesenchymal cells can aggregate and directly differentiate into osteoblasts. This pathway of intramembranous ossification is observed in the formation of the flat bones in the skull, collarbones and jaw bones (Hall and Miyake, 2000).
The skull is composed of the neurocranium and the viscerocranium (facial skeleton). The neurocranium plays an important role in encasing and protecting the brain and other sense organs. Both endochondral ossification and intramembranous ossification occur during neurocranium formation. The base of the neurocranium forms through endochondral ossification (McBratney-Owen et al., 2008), whereas the vault (calvarium) is thought to develop exclusively through intramembranous ossification (Hall and Miyake, 2000). The calvarium is composed of paired frontal bones, paired parietal bones, the squamous region of the occipital bone, and the sutures, which are the fibrous tissues that separate and joint the calvarial bones. Lineage-tracing studies show that the frontal bones are derived from the neural crest, the parietal bones originate from the paraxial mesoderm, while the coronal sutures that separate these bones are heterogeneous but derive mostly from mesoderm cells (Deckelbaum et al., 2012; Jiang et al., 2002; Lenton et al., 2005; Yoshida et al., 2008). During mouse development, the progenitors of sutures and calvarial bones arise at around embryonic day (E) 11 in the supraorbital regions. These cells undergo active proliferation and differentiation while migrating towards their dorsal destination. They develop coordinately in close proximity to the adjacent dura, a thick membrane that surrounds the brain, and surrounding mesenchymal cells through tissue-tissue interactions (Deckelbaum et al., 2012; Opperman, 2000). In addition, the sutures also serve as a growth site and stem cell niche to support continuous growth of calvarial bones after birth (Zhao et al., 2015).
Deformities of the calvarial bones or sutures usually lead to premature fusion of calvarial bones, or craniosynostosis, a common human birth defect with an occurrence of 1/2500 (Senarath-Yapa et al., 2012; Wilkie and Morriss-Kay, 2001). Craniosynostosis may occur as part of a syndrome or more frequently as isolated (non-syndromic) anomalies (Cohen, 2000). Among the genetic alterations identified in humans, mutations are prevalent in genes encoding receptor tyrosine kinases (RTKs), their ligands or downstream signaling components (Johnson and Wilkie, 2011). These include fibroblast growth factor receptor 2 and 3 (FGFR2 and FGFR3), ephrin B1 (EFNB1) and KRAS. Of these, mutations in FGFR2 are most frequent and account for 32% of all cases (Johnson and Wilkie, 2011). Such mutations lead to activation of FGFR2 and frequently cause Apert syndrome, which is characterized by bi-coronal synostosis and other defects (Ibrahimi et al., 2005). These studies suggest that RTKs and their common signaling targets may be involved in the normal development of calvaria and in pathogenesis.
Platelet-derived growth factor receptors (PDGFRs) constitute another family of RTKs. In mouse and human, there are two PDGFRs (α and β) and four PDGF ligands (A-D) (Hoch and Soriano, 2003). While both receptors are required for normal development and homeostasis (Andrae et al., 2008), PDGFRα is essential in neural crest cell (NCC) development through its downstream PI3K/AKT and Rac1 signaling pathways (Fantauzzo and Soriano, 2014, 2016; He and Soriano, 2013; Klinghoffer et al., 2002; Soriano, 1997; Tallquist and Soriano, 2003). Of the four ligands, PDGFA and PDGFC are specific endogenous activators of PDGFRα, and inactivating both ligands causes embryonic lethality at mid-gestation and severe craniofacial malformations, phenocopying Pdgfra null mutants (Ding et al., 2004; Soriano, 1997). It has been reported that both the expression level and activity of PDGFRα are significantly increased in Apert osteoblasts (Miraoui et al., 2010), and generalized activation of PDGFRα signaling has been shown to lead to craniosynostosis (Moenning et al., 2009).
In the present study, we show that PDGFRα and its ligand PDGFA are expressed in the developing calvarial tissues of mice. Expression of an activatable form of PDGFRα knocked into its own locus leads to abnormal ossification and to craniosynostosis of the coronal sutures in neonates. Interestingly, we find that enhanced PDGFRα signaling activity causes abnormal expansion and premature differentiation of cartilage anlagen underlying the coronal sutures, and that endochondral ossification contributes directly to calvarial morphogenesis. Lineage tracing reveals that these chondrocytes derive from both neural crest and mesoderm cells, and activating PDGFRα signaling in chondrocytes, neural crest or mesoderm causes distinct calvarial phenotypes. Further analysis reveals that both PI3K and PLCγ signaling activity are elevated in NCCs expressing constitutively active PDGFRα, and that PDGFRα/PI3K signaling is required for the normal development of coronal sutures and frontal bones.
RESULTS
Generalized activation of the PdgfraK allele leads to craniosynostosis and abnormal calvarial bones
To examine the consequence of dysregulated PDGFRα activity in calvarial development, we drove expression of a conditionally activatable D842V Pdgfra allele utilizing its endogenous promoter by crossing Pdgfra+/K knock-in mice (Olson and Soriano, 2009) with the general epiblast deleter line Meox2Cre (Tallquist and Soriano, 2000). Because Pdgfra+/K; Meox2Cre mice exhibit perinatal lethality (Olson and Soriano, 2009), we analyzed skeletal preparations at E18.5.
In wild-type embryos, the frontal bones and the parietal bones remained separated by coronal sutures (Fig. 1A,C). In Pdgfra+/K; Meox2Cre embryos, however, these bones were fused and the coronal sutures were absent (Fig. 1B,D). Alkaline phosphatase (AP) staining of sagittal sections through these embryos highlighted differentiating osteoblasts and revealed that, in wild-type embryos, the frontal and parietal bones are separated by AP-negative coronal sutures (Fig. 1E). In Pdgfra+/K; Meox2Cre embryos, the frontal and parietal bones were fused, as indicated by continuous AP staining across both bones, which also appeared thicker than in the wild-type controls (Fig. 1E,F).
Fig. 1.
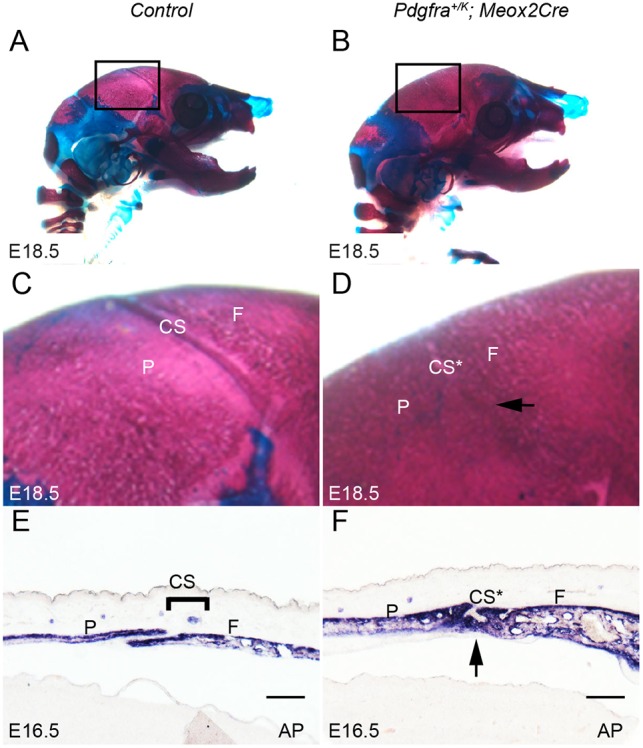
Constitutive PDGFRα activation leads to craniosynostosis of coronal sutures. (A,B) Lateral view of skeletal preparations of control littermate (A) and Pdgfra+/K; Meox2Cre (B) mouse embryos (n=5 for each genotype) at E18.5. Bones are stained with Alizarin Red, and cartilage with Alcian Blue. (C,D) Magnification of the boxed areas in A and B showing closed coronal suture in Pdgfra+/K; Meox2Cre embryo (D, arrow). (E,F) Alkaline phosphatase (AP) staining of sagittal sections from control littermate (E) and Pdgfra+/K; Meox2Cre (F) embryos (n=5 for each genotype) revealed thickened frontal and parietal bones and closed coronal suture (F, arrow) in the mutant. CS, coronal suture; CS*, fused coronal suture; F, frontal bone; P, parietal bone. Scale bars: 50 μm.
These results indicate that PDGFRα activity needs to be tightly regulated for normal calvarial osteogenesis and coronal suture development, and that increased PDGFRα activity leads to craniosynostosis of the coronal suture.
PDGFRα and its ligands are expressed in calvarial rudiments and coronal sutures
Using in situ hybridization and immunohistochemistry, we examined the expression pattern of Pdgfra and its endogenous ligands Pdgfa and Pdgfc. The rudiments of the frontal bones and the parietal bones are composed of differentiating osteoblasts, which we identified using AP and RUNX2 as markers (Fig. 2A,D,H,K). Pdgfa and Pdgfc exhibited differential expression patterns in developing calvarial tissues. Pdgfa mRNA was expressed in a similar domain to that of AP in the rudiments of the frontal and parietal bones at E13.5 and E15.5 (Fig. 2B,E). Pdgfc mRNA was expressed in the ectoderm, but was barely detected in the bone rudiments (Fig. 2C,F). Pdgfra expression, as detected by EGFP expression in PdgfraGFP/+ knock-in embryos, was identified in the mesenchymal cells surrounding RUNX2+ osteoblasts at E13.5 (Fig. 2G-I). At E15.5, Pdgfra expression overlapped with RUNX2 in some osteoblasts as well as in coronal suture cells (Fig. 2J-L).
Fig. 2.

Expression pattern of PDGFRα and its ligands in the developing calvarium. (A,E) AP activity was detected in the osteoprogenitors of the frontal bone and parietal bone on sections through the coronal sutures of E13.5 (A) and E15.5 (D) embryos (n=3 for each stage). (B,E) In situ hybridization showing Pdgfa mRNA in the osteoprogenitors of frontal bone and parietal bone in parallel sections of E13.5 (B) and E15.5 (E) embryonic heads (n=3 for each stage). (C,F) In situ hybridization of Pdgfc in parallel sections of E13.5 (C) and E15.5 (F) embryonic heads (n=3 for each stage). (G-I) In coronal sections of E13.5 and E15.5 embryos, expression of Pdgfra, as tracked by H2B-GFP in PdgfraGFP/+ embryos, was detected in the sutural cells and other mesenchymal cells (G,J) surrounding RUNX2+ osteoprogenitors (H,K) (n=3 for each stage). (I,L) Merged images. At E15.5, Pdgfra expression was identified in the coronal suture, as well as within a portion of endosteal and periosteal osteoprogenitors (yellow cells in L) (n=3). CS, coronal suture; F, frontal bone; P, parietal bone. Scale bars: 50 μm.
Together with the craniosynostosis phenotype observed in Pdgfra+/K; Meox2Cre embryos (Fig. 1), these results are consistent with a role for PDGFA-PDGFRα signaling during calvarial development and coronal suture morphogenesis.
Constitutive activation of PDGFRα leads to ossification of coronal sutures and abnormal development of the underlying cartilage
To trace the onset of the Pdgfra+/K; Meox2Cre craniosynostosis phenotype, we analyzed sections across the coronal sutures at different stages. At E13.5, AP activity became detectable in the differentiating osteoblasts within the frontal and parietal bone rudiments, which were separated by the AP-negative coronal sutures through E15.5 (Fig. 3A,C,E). By contrast, the coronal sutures of Pdgfra+/K; Meox2Cre embryos showed AP activity by E15.5 (Fig. 3B,D,F), suggesting that these sutural cells are transitioning to osteoblasts. In addition, the calvarial rudiments in Pdgfra+/K; Meox2Cre embryos became thicker than in wild-type counterparts at E15.5. The AP-positive domain exhibited sharp edges flanking AP-negative tissues underneath the coronal sutures (Fig. 3F). Alcian Blue (AB) staining of alternate sections indicated that the AP-negative and AB-positive cells were cartilage cells, which were also observed in the control but significantly more so in Pdgfra+/K; Meox2Cre embryos starting at E13.5 (Fig. 3G-L). BrdU labeling results showed proliferation rates for mutant chondrocytes (63%) that trended higher than those for controls (53.6%; Fig. S1, n=9, P<0.01).
Fig. 3.

PDGFRα activation leads to premature closure of coronal sutures and ectopic cartilage formation. (A-F) AP staining on coronal sections of control littermate and Pdgfra+/K; Meox2Cre embryos at E13.5 (A,B), E14.5 (C,D) and E15.5 (E,F) at comparable levels (n=4 for each stage). Arrows point to the boundaries of thickening frontal bones and parietal bones in Pdgfra+/K; Meox2Cre embryo at E15.5. (G-L) Alcian Blue (AB) staining on parallel coronal sections of control littermate and Pdgfra+/K; Meox2Cre embryos at E13.5 (G,H), E14.5 (I,J) and E15.5 (K,L). Sections were counterstained with nuclear Fast Red (NFR). Arrows point to ectopic cartilage formed in Pdgfra+/K; Meox2Cre embryos (H,J). CS, coronal suture; CS*, fused coronal suture; F, frontal bone; P, parietal bone. Scale bars: 50 μm.
To confirm the identity of these cartilage cells, we analyzed the expression of a series of molecular markers. In situ hybridization results showed that Sox9 and Col2a1 were expressed in these cells, indicating that they were chondrocytes and that many more chondrocytes were formed in Pdgfra+/K; Meox2Cre embryos (Fig. 4A-D). Expression of the hypertrophic chondrocyte marker Col10a1 was also detected, suggesting that these chondrocytes underwent differentiation in both wild-type and Pdgfra+/K; Meox2Cre embryos (Fig. 4E,F). Expression of the osteoblast marker Ocn (Bglap) in the Pdgfra+/K; Meox2Cre cartilage indicates that these cells begin to differentiate into osteoblasts (Fig. 4G,H). In line with this result, immunohistochemistry showed that the osteoblast markers RUNX2 and osterix (SP7) were expressed in the chondrocytes of Pdgfra+/K; Meox2Cre, but not wild-type, embryos (Fig. 4I-L), indicating that activation of PDGFRα signaling promotes endochondral ossification. By E18.5, the chondrocytes underlying the parietal bony rudiments were replaced by AP-positive osteoblasts, although some chondrocytes underlying the frontal bone remained (Fig. 4M,N).
Fig. 4.

Increased PDGFRα activity promotes endochondral ossification during calvaria development. (A-H) In situ hybridization shows expression of chondrocyte markers Sox9 (A,B) and Col2a1 (C,D), hypertrophic chondrocyte marker Col10a1 (E,F) and osteoblast marker Ocn (G,H) in coronal sections of littermate control (A,C,E,G) and Pdgfra+/K; Meox2Cre (B,D,F,H) embryos at E15.5 (n=3). Arrows (H) point to weak Ocn expression in cartilage. (I-L) Expression of osteogenic markers RUNX2 (I,J) and osterix (K,L) in coronal sections of littermate control (I,K) and Pdgfra+/K; Meox2Cre (J,L) embryos at E15.5 (n=3). Arrows (J,L) point to differentiating osteoblasts. (M,N) AP staining in coronal sections of littermate control (M) and Pdgfra+/K; Meox2Cre (N) embryos at E18.5 (n=4). Asterisks indicate bones with AP-positive osteoblasts. Sections I-N were stained with AB following immunohistochemistry staining. CS, coronal suture; CS*, fused coronal suture; F, frontal bone; P, parietal bone. Scale bars: 50 μm.
These results indicate that PDGFRα regulates the timing and extent of endochondral ossification during calvarial development, and that constitutive PDGFRα activation promotes chondrocyte formation and subsequent differentiation into osteoblasts.
PDGFRα-directed endochondral ossification contributes to calvarial morphogenesis and coronal suture craniosynostosis
To confirm our observation that endochondral ossification contributes to calvarial morphogenesis in Pdgfra+/K; Meox2Cre embryos, we used Col2a1Cre mice, which express Cre recombinase specifically in chondrocytes (Sakai et al., 2001), to fate map these cells. Lineage tracing with the R26RlacZ reporter line (Soriano, 1999) showed that at E15.5 the lacZ reporter was expressed in both the frontal cartilage and the perichondrium underneath the coronal sutures (Fig. 5A,C). At postnatal day (P) 20, chondrocyte-derivatives were detected in the nasal bones and tissues at the junctions between calvarial bones, including the coronal sutures (Fig. 5E). Compared with littermate controls, Pdgfra+/K; Col2a1Cre embryos exhibited many more chondrocyte-derived cells (Fig. 5B,D) and ectopic chondrocyte-derived tissues adjacent to the coronal sutures at E15.5. At E18.5, skeletal preparations showed enlargement of Pdgfra+/K; Col2a1Cre skulls (Fig. S2). The total length of Pdgfra+/K; Col2a1Cre skulls was 6.9±1.2% (P<0.01) larger than those of the wild type, the mutant frontal bones were 11.7±2.5% (P<0.01) longer, and the mutant parietal bones were 3.1±3% (P>0.05) longer than in the control (n=5). At P20, Pdgfra+/K; Col2a1Cre mice accumulated more chondrocyte derivatives in the nasal bones and calvarial junctions (Fig. 5E,F) and exhibited a domed skull, indicating abnormal calvarial development (Fig. 5G,H). Skeletal preparations revealed abnormal morphology of coronal sutures in Pdgfra+/K; Col2a1Cre mice compared with control littermates (Fig. 5I,J). AP staining of sagittal sections of these skulls revealed that Pdgfra+/K; Col2a1Cre frontal and parietal bones were thinner than in controls (Fig. 5K,L). Further analysis using the R26RtdT reporter (Madisen et al., 2010) for lineage tracing revealed that many cells formerly expressing Col2a1Cre in Pdgfra+/K; Col2a1Cre mice were RUNX2 positive and aligned with calvarial osteoblasts (Fig. 5M,N).
Fig. 5.

Augmentation of PDGFRα activity in the chondrogenic lineage leads to cartilage expansion and calvarial deformity. (A,B) Double staining of AP and with AB on coronal sections of littermate control (A) (n=5) and Pdgfra+/K; R26R+/−; Col2a1Cre (B) embryos (n=3) at E15.5. (C-F) X-gal staining showing derivatives of the chondrogenic lineage on coronal sections of littermate control (C) and Pdgfra+/K; R26R+/−; Col2a1Cre (D) embryos at E15.5 (counterstained with NFR), and in whole-mount heads of P20 littermate control (E) (n=5) and Pdgfra+/K; R26R+/−; Col2a1Cre (F) mice (n=5). Arrows (D) point to chondrocyte derivatives underlying the coronal suture. (G,H) Lateral views of littermate control (G) and Pdgfra+/K; Col2a1Cre (H) mice (n=7 each) at P20. Arrow (H) points to the domed skull of the mutant mouse. (I,J) Dorsal view of skull preparations following removal of the brain of littermate control (I) and Pdgfra+/K; Col2a1Cre (J) mice (n=5 each) at P20. Bones are stained with Alizarin Red, and cartilages with Alcian Blue. Note deformation of the coronal sutures (arrows). (K,L) AP staining on coronal sections of littermate control (K) and Pdgfra+/K; Col2a1Cre (L) mice (n=3 each) at P20. (M,N) Immunofluorescence staining with anti-RUNX2 antibody (green) on sagittal sections of Col2a1Cre; R26R+/tdT (M) and Pdgfra+/K; Col2a1Cre; R26R+/tdT (N) skull (n=3 each) at P20. CS, coronal suture; F, frontal bone; P, parietal bone. Scale bars: 0.5 mm.
These results indicate that ectopic PDGFRα activity in the chondrocyte lineage disrupts normal calvarium development, but does not cause craniosynostosis by P20.
Activating PDGFRα signaling in neural crest or mesoderm leads to distinct phenotypes
The calvarial bones are derived from two different origins: the neural crest and the mesoderm (Chai et al., 2000; Jiang et al., 2000; Yoshida et al., 2008). To examine in which lineage overactive PDGFRα signaling causes craniosynostosis, Pdgfra+/K mice were crossed to the Wnt1Cre-2 (Lewis et al., 2013) or Mesp1Cre (Saga et al., 1999) drivers. Lineage tracing showed that the cartilage underlying the coronal suture was derived from both neural crest and mesodermal cells (Fig. S3). Constitutive activation of PDGFRα in NCCs with Wnt1Cre-2 led to perinatal lethality. Skeletal preparations of E18.5 Pdgfra+/K; Wnt1Cre-2 embryos showed ectopic cartilage formation at the interfrontal, sagittal and coronal sutures (Fig. 6A-D). Lineage tracing revealed that in Pdgfra+/K; Wnt1Cre-2; R26R mice, the neural crest domain expanded posteriorly compared with control littermates (Fig. 6E,F). Constitutive activation of PDGFRα in mesoderm cells with Mesp1Cre, on the other hand, did not cause embryonic lethality. Pdgfra+/K; Mesp1Cre mice survived beyond P20 (n=5) and showed a domed skull (data not shown). Skeletal preparations of Pdgfra+/K; Mesp1Cre mice revealed craniosynostosis of the coronal and lambdoid sutures (Fig. 6G,H). At E16.5, ectopic cartilage was observed posterior to the parietal bone (Fig. 6I,J), and lineage tracing did not detect a displacement of the mesoderm boundary into the anterior skull, although structures resembling vascular plexus appeared increased in the anterior skull (Fig. 6K,L, arrows).
Fig. 6.

Activation of PDGFRα in neural crest cells or in mesoderm gives rise to distinct calvarial phenotypes. (A-D) Lateral (A,B) and dorsal (C,D) views of E18.5 littermate control (A,C) and Pdgfra+/K; Wnt1Cre-2 (B,D) calvaria (n=7 each). Arrows (B,D) point to ectopic cartilage formed at the sutures. Bones are stained with Alizarin Red, and cartilages with Alcian Blue. (E,F) Dorsal views of calvaria of E15.5 littermate control (E) and Pdgfra+/K; Wnt1Cre-2; R26R (F) (n=6 each) following whole-mount X-gal staining. Arrows (F) point to the neural crest expansion at coronal sutures. (G-J) Dorsal views of littermate control (G,I) (n=5 for each stage) and Pdgfra+/K; Mesp1Cre (H,J) (n=5 for each stage) calvaria at P20 (G,H) and E16.5 (I,J). Arrowhead (H) points to fusion at lambdoid suture; arrows (H) point to fusion at coronal sutures. (K,L) Dorsal views of E15.5 calvaria of littermate control (K) and Pdgfra+/K; Mesp1Cre; R26R (L) embryos (n=5 each) following whole-mount X-gal staining. Arrows (L) point to mesoderm staining in vascular plexus-like tissues. F, frontal bone; P, parietal bone. Scale bars: 0.5 mm.
These data demonstrate that activating PDGFRα signaling in two distinct lineages leads to different outcomes during calvarial morphogenesis, and that the phenotype observed in Pdgfra+/K; Meox2Cre embryos could be caused by the synergistic effect of overactive PDGFRα activity in both lineages. In addition, the ectopic cartilage at the suture observed using Wnt1Cre-2 indicates that PDGFRα signaling in NCCs is an important contributor in coronal suture morphogenesis.
PDGFRα-engaged PI3K signaling plays a crucial role in calvaria development and suture morphogenesis
PDGFRα engages the phosphorylation and activation of multiple downstream signaling pathways, including the SRC, ERK1/2 (MAPK3/1), STAT, PI3K/AKT and PLCγ pathways. To identify the mechanisms of PDGFRα regulation in NCCs, we assayed the activity of these signaling pathways in primary cultures prepared from E11.5 frontonasal prominence cells, which are predominantly derived from neural crest.
Western blot analysis revealed that AKT and PLCγ phosphorylation levels are significantly increased in the NCCs of Pdgfra+/K; Wnt1Cre-2 embryos relative to control littermates (Fig. 7A; 1.45±0.14-fold increase in phospho-AKT and 2.98±0.13-fold increase in phospho-PLCγ), as observed previously in E13.5 Pdgfra+/K; Meox2Cre whole-embryo protein lysates (Olson and Soriano, 2009), whereas the activity of SRC, ERK1/2 and STAT3 did not change (Fig. S4). Immunostaining showed increased phospho-AKT and phospho-PLCγ staining in Pdgfra+/K; Meox2Cre embryos compared with control littermates at E13.5 (Fig. 7B-E).
Fig. 7.

PI3K signaling is implicated in the development of frontal cartilage and coronal sutures. (A) Western blot of phospho-AKT, total AKT, phospho-PLCγ and total PLCγ in primary E11.5 littermate control (++) and Pdgfra+/K; Wnt1Cre-2 frontonasal prominence cells. (B-E) Immunostaining of phospho-AKT (B,C) and phospho-PLCγ (D,E) of coronal sections from E13.5 littermate control (B,D) and Pdgfra+/K; Meox2Cre (C,E) embryos (n=3 each). Arrows (C,E) point to chondrocytes showing increased levels of phospho-AKT staining. (F,G) Lateral views of E18.5 littermate control (F) and Ptenfl/fl; Wnt1Cre-2 (G) embryos (n=3 each). (H,I) Double staining of AP and with AB on coronal sections of E14.5 littermate control (H) and Ptenfl/fl; Wnt1Cre-2 (I) embryos (n=4 each). (J,K) Lateral views of E14.5 littermate control (J) and Ptenfl/fl; Meox2Cre (K) embryos (n=4 each). (L,M) Double staining of AP and with AB on coronal sections of E14.5 littermate control (L) and Ptenfl/fl; Meox2Cre (M) embryos (n=3 each). (N,O) Dorsal views of skeletal preparations of E18.5 littermate control (N) and PdgfraPI3K/PI3K (O) embryos (n=6 each). Bones are stained with Alizarin Red, and cartilages with Alcian Blue. Arrowheads indicate frontal bone. (P,Q) Double staining of AP and with AB on coronal sections of E15.5 littermate control (P) and PdgfraPI3K/PI3K (Q) embryos (n=3 each). CS, coronal suture; CS*, fused coronal suture; F, frontal bone; P, parietal bone. Scale bars: 0.5 mm.
PI3K signaling has been established as a crucial mediator of PDGFRα signaling during craniofacial and skeletal development (Fantauzzo and Soriano, 2014, 2016; He and Soriano, 2013; Klinghoffer et al., 2002; Pickett et al., 2008). We therefore asked whether activation of PI3K/AKT signaling in NCCs causes abnormal calvarial development. Our results showed that disruption of the PI3K/AKT inhibitor Pten in NCCs caused enlargement of the head and calvarial bones (Fig. 7F,G) and perinatal lethality (data not shown; n=3). AP and AB staining of Ptenfl/fl; Wnt1Cre-2 embryos at E14.5 revealed enhanced cartilage formation underlying the coronal sutures, and increased AP signal at the coronal sutures and tissues surrounding the cartilage (Fig. 7H,I). Inactivating Pten in the whole embryo led to embryonic lethality at E14.5 accompanied by edema, as also observed in Pdgfra+/K; Meox2Cre embryos (Olson and Soriano, 2009) (Fig. 7J,K). AP staining of the coronal sections showed that the coronal sutures were partially closed and the underlying cartilage was expanded (Fig. 7L,M). By contrast, PdgfraPI3K/PI3K mutant embryos, in which the ability of PDGFRα to activate PI3K signaling is abrogated (Klinghoffer et al., 2002), displayed a wider separation of the frontal and parietal bones at the coronal sutures, and a foramen between the two frontal bones (Fig. 7N,O). AP and AB staining showed hypoplastic cartilage formation in the PdgfraPI3K/PI3K embryos at E15.5 (Fig. 7P,Q).
Taken together, these results indicate that PI3K/AKT signaling plays an essential role in calvarial development and coronal suture morphogenesis.
DISCUSSION
Craniosynostosis is a prevalent human birth defect that arises from genetic or environmental disruptions of normal calvarium development (Wilkie and Morriss-Kay, 2001). Our studies establish PDGFRα and its downstream signaling pathways as crucial regulators of calvarial growth. The calvarial bones are derived from two separate lineages: the neural crest and the mesoderm. Our data showed that increased activity of PDGFRα in both lineages causes the abnormal formation of sutures, as well as expansion and premature ossification of the underlying cartilage. We have identified the origin of these cartilage anlagen, and further demonstrate by marker analysis and lineage tracing that, upon activation of PDGFRα signaling, cartilage contributes to calvaria morphogenesis through endochondral ossification. We found that this process was under the regulation of PDGFRα-PI3K/AKT signaling in the neural crest lineage. Our study also showed that activating PDGFRα signaling in either the neural crest or the mesoderm caused distinct phenotypes, indicating a synergistic role of signaling in both lineages during calvaria development and craniosynostosis.
A role for PDGFRα in craniosynostosis was first suggested by reports showing its increased expression and activity in the osteoblasts from patients with Apert syndrome (Miraoui et al., 2010), as well as premature fusion of the interfrontal and coronal sutures by constitutive activation of PDGFRA in the neural crest (Moenning et al., 2009). Although this particular phenotype is different from that we observe at the coronal suture only, it is likely that this is due a difference in the domain or level of expression of the activated PDGFRα, as the earlier study used constitutive expression from the ROSA26 locus instead of restricted expression from the endogenous Pdgfra locus. Consistent with this interpretation, conditional activation of PDGFRα from these two alleles throughout the epiblast or in NCCs led to different phenotypic outcomes (Kurth et al., 2009; Moenning et al., 2009; Olson and Soriano, 2009). Additional differences in the previous study include the use of a human PDGFRA cDNA, which might signal differently than a mouse cDNA, and the use of the original Wnt1Cre mice (Danielian et al., 1998) that misexpress Wnt1, instead of the Wnt1Cre-2 driver (Lewis et al., 2013). This might confound some results, as ectopic Wnt signaling activity has been shown to affect normal calvarium development and suture morphogenesis (Liu et al., 2007; Maruyama et al., 2010; Mirando et al., 2010). These differences notwithstanding, both our study and the previous report (Moenning et al., 2009) support an important role for PDGFRα signaling in the regulation of cranial sutures and craniosynostosis. We note, however, that PDGFRα activation has not been implicated in human patients with craniosynostosis, possibly because PDGFRα activation leads to very pleiotropic phenotypes (Olson and Soriano, 2009).
RTKs regulate cell fates during development and disease by engaging a number of shared intracellular signaling pathways. Among these, the involvement of the ERK1/2 pathway has been well documented in animal models of craniosynostosis. For example, mice carrying active mutations of Fgfr2 exhibit craniosynostosis correlated with enhanced ERK1/2 signaling activity in the suture cells and osteoprogenitors (Wang et al., 2005; Yin et al., 2008), and inhibiting ERK1/2 phosphorylation or engagement can rescue the craniosynostosis phenotype (Shukla et al., 2007; Eswarakumar et al., 2006). Increased ERK1/2 signaling activity has also been identified in the calvarial bone marrow of Twist1 mutant mice (Connerney et al., 2008). Interestingly, decreased ERK1/2 signaling activity is also observed in Epha4−/− and Twist1+/− craniosynostosis models (Ting et al., 2009). These studies indicate that ERK1/2 signaling must be tightly regulated at multiple stages and in different tissues during calvarium development. Another RTK downstream signaling pathway, PLCγ, is activated with increased PDGFRα activity and has been implicated in osteogenesis of neural crest-derived osteoprogenitors (Moenning et al., 2009; Olson and Soriano, 2009). Our work highlights the role of PI3K/AKT signaling in normal calvarium development and its pathogenesis. In a previous study, we have reported that PDGFAA induces AP activity in mouse embryonic palatal mesenchymal cells via PI3K signaling (Vasudevan et al., 2015). In the present work, we show that PDGFRα-engaged PI3K/AKT signaling regulates development of the coronal sutures and underlying cartilage, and dysregulated PI3K signaling activity causes ossification of suture cells and expansion of chondrocytes. Together, these data reveal an important role for PI3K/AKT signaling in calvarial development and disease.
Bone can be formed by either endochondral ossification or intramembranous ossification, and it is generally believed that calvaria form only through intramembranous ossification. However, cartilage has also been implicated in calvarial ossification. The parietal cartilage has been shown to be remodeled into membranous bone through a non-endochondral ossification pathway regulated by the membrane type 1 matrix metalloproteinase (MT1-MMP; also known as MMP14). In 5-day-old mice, these parietal chondrocytes express the chondrocyte marker Col2a1 but not the hypertrophic chondrocyte marker Col10a1, and finally undergo apoptosis and are replaced by bone cells by day 10 (Holmbeck et al., 2003). In the present study, we show the existence of cartilage anlagen underlying the coronal sutures derived from both the neural crest and mesoderm (Fig. S3). These cells express Col2a1, Sox9 and Col10a1, and exhibit AP activity in E18.5 Pdgfra+/K; Wnt1Cre-2 embryos, indicating that they contribute to the calvaria via the hypertrophic chondrocyte pathway. Interestingly, this pathway does not necessarily follow the traditional endochondral ossification process, since we show by lineage tracing using a chondrocyte-specific Col2a1 Cre driver that descendants of these cartilage cells are incorporated into calvarial osteoblasts and suture cells at P20 (Fig. 5N). This observation is in line with an increasing number of recent studies showing that chondrocytes are not necessarily terminally differentiated, but can instead be transformed into osteoblasts during long-bone development (Enishi et al., 2014; Park et al., 2015; Yang et al., 2014a,b; Zhou et al., 2014). We suggest that this alternate differentiation pathway has also been redeployed in calvarial osteogenesis, at least in part under the regulation of PDGFRα. In addition, identification of Col2a1Cre lineage-traced cells in the coronal suture suggests that chondrocyte progenitors might be multipotent, since the sutures not only function as connective tissues but also provide growth sites and signaling centers of the growing calvaria (Lana-Elola et al., 2007; Morriss-Kay and Wilkie, 2005; Opperman, 2000). A recent study has identified the suture as a mesenchymal stem cell niche for calvarial bone homeostasis and repair (Zhao et al., 2015). Another recent report supports this notion by showing that hypertrophic chondrocytes express pluripotency marker genes and are involved in fracture healing (Hu et al., 2017). In addition, since the perichondrium is the major source of osteoblast precursors, expansion of this tissue primordium might contribute to the enhanced growth of calvarial bones in Pdgfra+/K; Meox2Cre mice.
Compared with the endochondral ossification that occurs in normal posterior frontal suture closure (Moss, 1958; Sahar et al., 2005), dysregulated growth factor signaling pathways have been shown to cause ectopic cartilage formation in the anterior frontal, sagittal and lambdoid sutures. For example, ectopic cartilage formation at the sagittal sutures has been reported in mouse models including Fgfr2S252W (Holmes and Basilico, 2012; Wang et al., 2005) and Axin2−/−; Fgfr1+/− mutants (Maruyama et al., 2010). Inactivating Gli3 in mice causes ectopic cartilage in the lambdoid suture (Rice et al., 2010). Unlike these models, Pdgfra+/K; Wnt1Cre-2 embryos exhibit ectopic cartilage in the coronal sutures (Fig. 6A-D). The prominent cartilage phenotype in Pdgfra+/K; Wnt1Cre-2 could be caused by the persistence and expansion of cartilage that forms transiently within the sutures; alternatively, increased PDGFRα activity might direct mesenchymal stem cell differentiation within the sutures. The fact that we could not phenocopy the ectopic sutural cartilage observed in Pdgfra+/K; Wnt1Cre-2 mice using a chondrocyte Cre driver (Fig. S2) supports this latter possibility. This notion is further supported by transcriptome analyses showing that PDGF signaling may be involved in directing mesenchymal stem cell differentiation towards a chondrogenic lineage (Ng et al., 2008). These observations, together with other reports that examine the effects of PDGFR activation in fibro/adipogenic progenitors or multipotent perivascular cells, underscore the need for tight regulation of PDGFR activity in regulating the homeostatic state of various mesenchymal progenitor cells (Iwayama et al., 2015; Olson and Soriano, 2009, 2011). Together, our findings establish a crucial role for PDGFRα in craniosynostosis, and identify endochondral ossification as a mechanism that can operate during calvarial morphogenesis.
MATERIALS AND METHODS
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee at Icahn School of Medicine at Mount Sinai. The following strains were maintained on a C57BL/6J background: Pdgfratm12Sor (Olson and Soriano, 2009), referred to as Pdgfra+/K in the text; Pdgfratm11(EGFP)Sor (Hamilton et al., 2003), referred to as Pdgfra+/GFP; Gt(ROSA)26Sortm1Sor (Soriano, 1999), referred to as R26RlacZ; and Gt(ROSA)26Sortm9(CAG-tdTomato)Hze (Madisen et al., 2010), referred to as R26RtdT. All other strains were maintained on C57BL/6J; 129SvJaeSor (MGI:3044540) mixed genetic backgrounds: Pdgfratm8Sor (Tallquist and Soriano, 2003), referred to as Pdgfra+/fl; Pdgfratm5Sor (Klinghoffer et al., 2002), referred to as Pdgfra+/PI3K; Meox2tm1(cre)Sor (Tallquist and Soriano, 2000), referred to as Meox2Cre; Tg(Wnt1-cre)2Sor (Lewis et al., 2013), referred to as Wnt1Cre-2; Mesp1tm2(cre)Ysa (Saga et al., 1999), referred to as Mesp1Cre; and Ptentm1Hwu (Groszer et al., 2001), referred to as Pten+/fl.
Skeletal preparations
Staged embryos were sacrificed, dissected in PBS and their skin and internal organs removed. Following incubation in 95% ethanol overnight at room temperature, skeletons were stained with 0.015% Alcian Blue (Sigma, A5268), 0.005% Alizarin Red (Sigma, A5533), 5% glacial acetic acid in 70% ethanol for 72 h at 37°C. The skeletons were then washed briefly in 95% ethanol, cleared with 1% KOH, and subsequently in increasing concentrations of glycerol in KOH until reaching 80% glycerol. For adult mice, the skin was removed using a scalpel, and the cranial base and brain were dissected before staining. Staining and clearing were performed with longer incubation times using the same protocol as for embryos.
X-gal, Alcian Blue and alkaline phosphatase staining
For whole-mount staining, the ectoderm and superficial dermis were carefully peeled off, and embryos were fixed and stained with X-gal (Soriano, 1999). X-gal staining on cryosections was performed using standard procedures. For Alcian Blue staining, sections were quickly dipped in 1% Alcian Blue 8GX (A5268, Sigma) in 0.1 N HCl. For alkaline phosphatase (AP) staining, paraffin sections were deparaffinized in Histoclear (Fisher Scientific, 50-899-90147), rehydrated in decreasing ethanol solutions, and incubated in a solution containing 0.03% nitro-blue tetrazolium chloride (NBT; Roche, 11383213001) and 0.02% 5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt (BCIP; Roche, 11383221001) to detect AP activity.
In situ hybridization and immunostaining
In situ hybridization was performed as previously described (He and Soriano, 2013). Pdgfa and Pdgfc probes were provided by Andras Nagy (Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Toronto), Sox9, Col2a1 and Col10a1 probes originate from YiPing Chen (Tulane University, New Orleans), and the Ocn probe was from Fanxin Long (Washington University, St Louis). Immunostaining was performed according to a standard protocol and antibodies to anti-phospho-AKT (1:25, Cell Signaling Technology 9271), anti-phospho-PLCγ1 (1:400, Novus Biologicals, MAB74542), RUNX2 (1:1000, Cell Signaling Technology, 12556) and osterix (SP7) (1:5000, Abcam, ab94744).
BrdU labeling
BrdU/PBS solution was administered by intraperitoneal injection into pregnant females at 3 mg/100 g body weight. Embryos were dissected in ice-cold PBS 1 h after injection. Embryonic heads were fixed in Carnoy's fixative for 1 h and dehydrated and embedded in paraffin. Immunostaining was performed on 10 μm transverse sections using the BrdU Labeling and Detection Kit II (Roche, 11299964001) following the manufacturer's instructions. Sections were counterstained with nuclear Fast Red (Sigma, N8002) to facilitate quantification. Results from three embryos of each phenotype were collected. Statistical data are presented as mean±s.e.m., and subjected to two-tailed Student's t-tests.
Western blot
Protein lysates were prepared from primary cultures of frontonasal prominences dissected from E11.5 embryos in RIPA buffer. Western blot was carried out using the following primary antibodies at 1:1000: anti-p44/42 MAPK (9102), anti-phospho-p44/42 MAPK (9101), anti-AKT (9272), anti-phospho-AKT (Ser473; 9271), anti-PLCγ (2822) and anti-phospho-PLCγ (2821) from Cell signaling Technology; anti-SRC (44-655G) and anti-phospho-SRC (44-660G) from Life Technologies; and anti-STAT3 (sc-8019) and anti-phospho-STAT3 (sc-8059) from Santa Cruz Biotechnology. Primary antibodies were detected by horseradish peroxidase-conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch Laboratories). Western blot results were quantified using ImageJ from NIH.
Supplementary Material
Acknowledgements
We thank Tianfang Yang, Tony Chen and Matthew Hung for excellent genotyping work; Francesco Ramirez for Col2a1Cre mice and Greg Holmes for the anti-osterix antibody; and our laboratory colleagues and Greg Holmes for constructive comments on the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: F.H., P.S.; Methodology: F.H., P.S.; Formal analysis: F.H., P.S.; Investigation: F.H.; Data curation: F.H., P.S.; Writing - original draft: F.H.; Writing - review & editing: F.H., P.S.; Visualization: F.H., P.S.; Funding acquisition: F.H., P.S.
Funding
This work was supported by NIH/National Institute of Dental and Craniofacial Research grants R01DE022363 to P.S. and R00DE024617 to F.H. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.151068.supplemental
References
- Andrae J., Gallini R. and Betsholtz C. (2008). Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 22, 1276-1312. 10.1101/gad.1653708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y., Jiang X., Ito Y., Bringas P. Jr, Han J., Rowitch D. H., Soriano P., McMahon A. P and Sucov H. M. (2000). Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127, 1671-1679. [DOI] [PubMed] [Google Scholar]
- Cohen M. M. (2000). Craniosynostosis. Diagnosis, Evaluation and Management. New York: Oxford University Press. [Google Scholar]
- Connerney J., Andreeva V., Leshem Y., Mercado M. A., Dowell K., Yang X., Lindner V., Friesel R. E. and Spicer D. B. (2008). Twist1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Dev. Biol. 318, 323-334. 10.1016/j.ydbio.2008.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian P. S., Muccino D., Rowitch D. H., Michael S. K. and McMahon A. P. (1998). Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323-1326. 10.1016/S0960-9822(07)00562-3 [DOI] [PubMed] [Google Scholar]
- Deckelbaum R. A., Holmes G., Zhao Z., Tong C., Basilico C. and Loomis C. A. (2012). Regulation of cranial morphogenesis and cell fate at the neural crest-mesoderm boundary by engrailed 1. Development 139, 1346-1358. 10.1242/dev.076729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H., Wu X., Boström H., Kim I., Wong N., Tsoi B., O'Rourke M., Koh G. Y., Soriano P., Betsholtz C. et al. (2004). A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat. Genet. 36, 1111-1116. 10.1038/ng1415 [DOI] [PubMed] [Google Scholar]
- Enishi T., Yukata K., Takahashi M., Sato R., Sairyo K. and Yasui N. (2014). Hypertrophic chondrocytes in the rabbit growth plate can proliferate and differentiate into osteogenic cells when capillary invasion is interposed by a membrane filter. PLoS ONE 9, e104638 10.1371/journal.pone.0104638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar V. P., Ozcan F., Lew E. D., Bae J. H., Tome F., Booth C. J., Adams D. J., Lax I. and Schlessinger J. (2006). Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proc. Natl. Acad. Sci. USA 103, 18603-18608. 10.1073/pnas.0609157103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantauzzo K. A. and Soriano P. (2014). PI3K-mediated PDGFRalpha signaling regulates survival and proliferation in skeletal development through p53-dependent intracellular pathways. Genes Dev. 28, 1005-1017. 10.1101/gad.238709.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantauzzo K. A. and Soriano P. (2016). PDGFRbeta regulates craniofacial development through homodimers and functional heterodimers with PDGFRalpha. Genes Dev. 30, 2443-2458. 10.1101/gad.288746.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M., Erickson R., Scripture-Adams D. D., Lesche R., Trumpp A., Zack J. A., Kornblum H. I., Liu X. and Wu H. (2001). Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294, 2186-2189. 10.1126/science.1065518 [DOI] [PubMed] [Google Scholar]
- Hall B. K. and Miyake T. (2000). All for one and one for all: condensations and the initiation of skeletal development. BioEssays 22, 138-147. [DOI] [PubMed] [Google Scholar]
- Hamilton T. G., Klinghoffer R. A., Corrin P. D. and Soriano P. (2003). Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol. Cell. Biol. 23, 4013-4025. 10.1128/MCB.23.11.4013-4025.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F. and Soriano P. (2013). A critical role for PDGFRalpha signaling in medial nasal process development. PLoS Genet. 9, e1003851 10.1371/journal.pgen.1003851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch R. V. and Soriano P. (2003). Roles of PDGF in animal development. Development 130, 4769-4784. 10.1242/dev.00721 [DOI] [PubMed] [Google Scholar]
- Holmbeck K., Bianco P., Chrysovergis K., Yamada S. and Birkedal-Hansen H. (2003). MT1-MMP-dependent, apoptotic remodeling of unmineralized cartilage: a critical process in skeletal growth. J. Cell Biol. 163, 661-671. 10.1083/jcb.200307061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes G. and Basilico C. (2012). Mesodermal expression of Fgfr2S252W is necessary and sufficient to induce craniosynostosis in a mouse model of Apert syndrome. Dev. Biol. 368, 283-293. 10.1016/j.ydbio.2012.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D. P., Ferro F., Yang F., Taylor A. J., Chang W., Miclau T., Marcucio R. S. and Bahney C. S. (2017). Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development 144, 221-234. 10.1242/dev.130807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi O. A., Chiu E. S., McCarthy J. G. and Mohammadi M. (2005). Understanding the molecular basis of Apert syndrome. Plast. Reconstr. Surg. 115, 264-270. 10.1097/01.PRS.0000146703.08958.95 [DOI] [PubMed] [Google Scholar]
- Iwayama T., Steele C., Yao L., Dozmorov M. G., Karamichos D., Wren J. D. and Olson L. E. (2015). PDGFRalpha signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev. 29, 1106-1119. 10.1101/gad.260554.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Rowitch D. H., Soriano P., McMahon A. P. and Sucov H. M. (2000). Fate of the mammalian cardiac neural crest. Development 127, 1607-1616. [DOI] [PubMed] [Google Scholar]
- Jiang X., Iseki S., Maxson R. E., Sucov H. M. and Morriss-Kay G. M. (2002). Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 241, 106-116. 10.1006/dbio.2001.0487 [DOI] [PubMed] [Google Scholar]
- Johnson D. and Wilkie A. O. M. (2011). Craniosynostosis. Eur. J. Hum. Genet. 19, 369-376. 10.1038/ejhg.2010.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinghoffer R. A., Hamilton T. G., Hoch R. and Soriano P. (2002). An allelic series at the PDGFalphaR locus indicates unequal contributions of distinct signaling pathways during development. Dev. Cell 2, 103-113. 10.1016/S1534-5807(01)00103-4 [DOI] [PubMed] [Google Scholar]
- Kronenberg H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332-336. 10.1038/nature01657 [DOI] [PubMed] [Google Scholar]
- Kurth P., Moenning A., Jäger R., Beine G. and Schorle H. (2009). An activating mutation in the PDGF receptor alpha results in embryonic lethality caused by malformation of the vascular system. Dev. Dyn. 238, 1064-1072. 10.1002/dvdy.21939 [DOI] [PubMed] [Google Scholar]
- Lana-Elola E., Rice R., Grigoriadis A. E. and Rice D. P. C. (2007). Cell fate specification during calvarial bone and suture development. Dev. Biol. 311, 335-346. 10.1016/j.ydbio.2007.08.028 [DOI] [PubMed] [Google Scholar]
- Lenton K. A., Nacamuli R. P., Wan D. C., Helms J. A. and Longaker M. T. (2005). Cranial suture biology. Curr. Top. Dev. Biol. 66, 287-328. 10.1016/S0070-2153(05)66009-7 [DOI] [PubMed] [Google Scholar]
- Lewis A. E., Vasudevan H. N., O'Neill A. K., Soriano P. and Bush J. O. (2013). The widely used Wnt1-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev. Biol. 379, 229-234. 10.1016/j.ydbio.2013.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Yu H.-M. I. and Hsu W. (2007). Craniosynostosis caused by Axin2 deficiency is mediated through distinct functions of beta-catenin in proliferation and differentiation. Dev. Biol. 301, 298-308. 10.1016/j.ydbio.2006.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R. et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133-140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T., Mirando A. J., Deng C. X. and Hsu W. (2010). The balance of WNT and FGF signaling influences mesenchymal stem cell fate during skeletal development. Sci. Signal. 3, ra40 10.1126/scisignal.2000727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBratney-Owen B., Iseki S., Bamforth S. D., Olsen B. R. and Morriss-Kay G. M. (2008). Development and tissue origins of the mammalian cranial base. Dev. Biol. 322, 121-132. 10.1016/j.ydbio.2008.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirando A. J., Maruyama T., Fu J., Yu H.-M. and Hsu W. (2010). beta-catenin/cyclin D1 mediated development of suture mesenchyme in calvarial morphogenesis. BMC Dev. Biol. 10, 116 10.1186/1471-213X-10-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraoui H., Ringe J., Haupl T. and Marie P. J. (2010). Increased EFG- and PDGFalpha-receptor signaling by mutant FGF-receptor 2 contributes to osteoblast dysfunction in Apert craniosynostosis. Hum. Mol. Genet. 19, 1678-1689. 10.1093/hmg/ddq045 [DOI] [PubMed] [Google Scholar]
- Moenning A., Jager R., Egert A., Kress W., Wardelmann E. and Schorle H. (2009). Sustained platelet-derived growth factor receptor alpha signaling in osteoblasts results in craniosynostosis by overactivating the phospholipase C-gamma pathway. Mol. Cell. Biol. 29, 881-891. 10.1128/MCB.00885-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriss-Kay G. M. and Wilkie A. O. M. (2005). Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J. Anat. 207, 637-653. 10.1111/j.1469-7580.2005.00475.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss M. L. (1958). Fusion of the frontal suture in the rat. Am. J. Anat. 102, 141-165. 10.1002/aja.1001020107 [DOI] [PubMed] [Google Scholar]
- Ng F., Boucher S., Koh S., Sastry K. S. R., Chase L., Lakshmipathy U., Choong C., Yang Z., Vemuri M. C., Rao M. S. et al. (2008). PDGF, TGF-beta, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 112, 295-307. 10.1182/blood-2007-07-103697 [DOI] [PubMed] [Google Scholar]
- Olson L. E. and Soriano P. (2009). Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Dev. Cell 16, 303-313. 10.1016/j.devcel.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson L. E. and Soriano P. (2011). PDGFRbeta signaling regulates mural cell plasticity and inhibits fat development. Dev. Cell 20, 815-826. 10.1016/j.devcel.2011.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opperman L. A. (2000). Cranial sutures as intramembranous bone growth sites. Dev. Dyn. 219, 472-485. [DOI] [PubMed] [Google Scholar]
- Ornitz D. M. (2005). FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 16, 205-213. 10.1016/j.cytogfr.2005.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Gebhardt M., Golovchenko S., Perez-Branguli F., Hattori T., Hartmann C., Zhou X., deCrombrugghe B., Stock M., Schneider H. et al. (2015). Dual pathways to endochondral osteoblasts: a novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol. Open 4, 608-621. 10.1242/bio.201411031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett E. A., Olsen G. S. and Tallquist M. D. (2008). Disruption of PDGFRalpha-initiated PI3K activation and migration of somite derivatives leads to spina bifida. Development 135, 589-598. 10.1242/dev.013763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice D. P. C., Connor E. C., Veltmaat J. M., Lana-Elola E., Veistinen L., Tanimoto Y., Bellusci S. and Rice R. (2010). Gli3Xt-J/Xt-J mice exhibit lambdoid suture craniosynostosis which results from altered osteoprogenitor proliferation and differentiation. Hum. Mol. Genet. 19, 3457-3467. 10.1093/hmg/ddq258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saga Y., Miyagawa-Tomita S., Takagi A., Kitajima S., Miyazaki J. and Inoue T. (1999). MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development 126, 3437-3447. [DOI] [PubMed] [Google Scholar]
- Sahar D. E., Longaker M. T. and Quarto N. (2005). Sox9 neural crest determinant gene controls patterning and closure of the posterior frontal cranial suture. Dev. Biol. 280, 344-361. 10.1016/j.ydbio.2005.01.022 [DOI] [PubMed] [Google Scholar]
- Sakai K., Hiripi L., Glumoff V., Brandau O., Eerola R., Vuorio E., Bösze Z., Fässler R. and Aszódi A. (2001). Stage-and tissue-specific expression of a Col2a1-Cre fusion gene in transgenic mice. Matrix Biol. 19, 761-767. 10.1016/S0945-053X(00)00122-0 [DOI] [PubMed] [Google Scholar]
- Senarath-Yapa K., Chung M. T., McArdle A., Wong V. W., Quarto N., Longaker M. T. and Wan D. C. (2012). Craniosynostosis: molecular pathways and future pharmacologic therapy. Organogenesis 8, 103-113. 10.4161/org.23307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla V., Coumoul X., Wang R.-H., Kim H.-S. and Deng C.-X. (2007). RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat. Genet. 39, 1145-1150. 10.1038/ng2096 [DOI] [PubMed] [Google Scholar]
- Soriano P. (1997). The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 124, 2691-2700. [DOI] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71. 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- Tallquist M. D. and Soriano P. (2000). Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 26, 113-115. [DOI] [PubMed] [Google Scholar]
- Tallquist M. D. and Soriano P. (2003). Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development 130, 507-518. 10.1242/dev.00241 [DOI] [PubMed] [Google Scholar]
- Ting M.-C., Wu N. L., Roybal P. G., Sun J., Liu L., Yen Y. and Maxson R. E. Jr (2009). EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development 136, 855-864. 10.1242/dev.028605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan H. N., Mazot P., He F. and Soriano P. (2015). Receptor tyrosine kinases modulate distinct transcriptional programs by differential usage of intracellular pathways. Elife 4, e07186 10.7554/eLife.07186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Xiao R., Yang F., Karim B. O., Iacovelli A. J., Cai J., Lerner C. P., Richtsmeier J. T., Leszl J. M., Hill C. A. et al. (2005). Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development 132, 3537-3548. 10.1242/dev.01914 [DOI] [PubMed] [Google Scholar]
- Wilkie A. O. M. and Morriss-Kay G. M. (2001). Genetics of craniofacial development and malformation. Nat. Rev. Genet. 2, 458-468. 10.1038/35076601 [DOI] [PubMed] [Google Scholar]
- Yang G., Zhu L., Hou N., Lan Y., Wu X.-M., Zhou B., Teng Y. and Yang X. (2014a). Osteogenic fate of hypertrophic chondrocytes. Cell Res. 24, 1266-1269. 10.1038/cr.2014.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Tsang K. Y., Tang H. C., Chan D. and Cheah K. S. E. (2014b). Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 111, 12097-12102. 10.1073/pnas.1302703111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L., Du X., Li C., Xu X., Chen Z., Su N., Zhao L., Qi H., Li F., Xue J. et al. (2008). A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone 42, 631-643. 10.1016/j.bone.2007.11.019 [DOI] [PubMed] [Google Scholar]
- Yoshida T., Vivatbutsiri P., Morriss-Kay G., Saga Y. and Iseki S. (2008). Cell lineage in mammalian craniofacial mesenchyme. Mech. Dev. 125, 797-808. 10.1016/j.mod.2008.06.007 [DOI] [PubMed] [Google Scholar]
- Zhao H., Feng J., Ho T.-V., Grimes W., Urata M. and Chai Y. (2015). The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nat. Cell Biol. 17, 386-396. 10.1038/ncb3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., von der Mark K., Henry S., Norton W., Adams H. and de Crombrugghe B. (2014). Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 10, e1004820 10.1371/journal.pgen.1004820 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.