

Abstract
Background and aims
Intrahepatic cholangiocarcinoma (ICC) is an aggressive tumor with a high fatality rate. It was recently found that parathyroid hormone-like hormone (PTHLH) was frequently overexpressed in ICC compared with non-tumor tissue. This study aimed to elucidate the underlying mechanisms of PTHLH in ICC development.
Methods
The CCK-8 assay, colony formation assays, flow cytometry and a xenograft model were used to examine the role of PTHLH in ICC cells proliferation. Immunohistochemistry (IHC) and western blot assays were used to detect target proteins. Luciferase reporter, chromatin immunoprecipitation (ChIP) and DNA pull-down assays were used to verify the transcription regulation of activating transcription factor-2 (ATF2).
Results
PTHLH was significantly upregulated in ICC compared with adjacent and normal tissues. Upregulation of PTHLH indicated a poor pathological differentiation and intrahepatic metastasis. Functional study demonstrated that PTHLH silencing markedly suppressed ICC cells growth, while specific overexpression of PTHLH has the opposite effect. Mechanistically, secreted PTHLH could promote ICC cell growth by activating extracellular signal-related kinase (ERK) and c-Jun N-terminal kinase (JNK) signaling pathways, and subsequently upregulated ATF2 and cyclinD1 expression. Further study found that the promoter activity of PTHLH were negatively regulated by ATF2, indicating that a negative feedback loop exists.
Conclusions
Our findings demonstrated that the ICC-secreted PTHLH plays a characteristic growth-promoting role through activating the canonical ERK/JNK-ATF2-cyclinD1 signaling pathways in ICC development. We identified a negative feedback loop formed by ATF2 and PTHLH. In this study, we explored the therapeutic implication for ICC patients.
Electronic supplementary material
The online version of this article (10.1186/s12967-017-1342-1) contains supplementary material, which is available to authorized users.
Keywords: Parathyroid hormone-like hormone, Activating transcription factor-2, Proliferation, Intrahepatic cholangiocarcinoma
Background
Intrahepatic cholangiocarcinoma arises from epithelial cells lining the bile duct and occurs proximally at the second degree bile ducts within the liver. The condition is commonly associated with cirrhosis, viral hepatitis B and C [1]. ICC displays a feature of rapid progression and a poor outcome, and its global disease incidence has been rapidly increasing [2]. Radical resection and curative liver transplantation are preferred surgical treatments for ICC, however, the patients with vascular and lymph nodes metastases are not eligible for surgical therapy. Although the chemotherapy regimen of gemcitabine and cisplatin and locoregional therapies are additional options for inoperable ICC patients, the 5-year survival rates are very low. An enhanced understanding of the biology pathological progress and the interaction with tumor microenvironment of ICC is needed to improve patient survival.
PTHLH, also referred to parathyroid hormone-related protein (PTHrP), has emerged as an important cytokine with diverse cell functions, including growth, survival, migration, and differentiation [3]. Tumor-derived PTHLH participates in the bone metastatic processes of breast cancer via an intracrine fashion [4]. In addition, PTHLH supports colorectal cancer cell to form distant lung metastatic processes via inducing caspase-independent death in human lung vasculature endothelial cells [5]. A previous report demonstrated that PTHLH produced by proliferating bile duct epithelial cells and may interact with growth factors and hormones to form complex loops that promotes proliferation [6]. Growing evidences indicate that PTHLH-producing cholangiocarcinoma (CHO) patients suffer from humoral hypercalcemia of malignancy [7–9], but litter is known regarding PTHLH’s effect on ICC cells growth. MAP kinase pathways are involved in the process through the PTHLH-induced activation of PTH1R to activate downstream effectors [10–12]. ATF2, as a downstream effector of MAPK in response to cytokines, is phosphorylated on Thr69 and/or Thr71 by either JNK or p38 [13, 14], and is also activated by the ERK1/2 pathway in two step [13]. Several observations support that ATF2 regulates cell cycle progression via controlling the transcriptional output of several key genes, including CyclinD1, CyclinA and RB1 [15–18]. However, no studies have documented a role for the PTHLH-MAPK-ATF2-CyclinD1 signaling axis in the regulation of ICC cells growth. This study aims to elucidate the role and clinical significance of the PTHLH-MAPK-ATF2-CyclinD1 axis in ICC cell cycle progression.
Methods
Patients, tissue samples and microarrays
59 ICC samples and paired non-tumor tissues and 10 normal tissues were obtained from the Department of Hepatobiliary Surgery, NanFang Hospital, Southern Medical University between 2014 and 2016. All patients signed informed consent for therapy and subsequent tissue studies, which were approved by the NanFang Hospital Institutional Review Board. The ICC tissue microarrays, which contained 100 cases, and the extrahepatic cholangiocarcinoma (ECC) tissue microarrays, which contained 27 cases were purchased from Shanghai Outdo Biotech Inc. (Shanghai, China). All tumors were defined as a primary tumor arising from the bile ducts and diagnosed as adenocarcinomas. Tumor stage was defined according to the seventh edition of American Joint Committee on Cancer/International Union against Cancer (AJCC/UICC). All specimens were used for routine pathological processing with comparable clinicopathological features, and complete follow-up data were obtained.
Western blot, real-time PCR analysis, immunohistochemistry and immunofluorescence
RNA and protein lysate extraction, cDNA synthesis, final real-time PCR and western blots were performed according to general protocols. ICC cells were processed for immunofluorescence (IF) using target antibodies with optimized conditions. In addition, human samples and ICC microarrays were subjected to IHC staining to evaluate the expression of relative proteins.
Cell counting kit-8 assay, colony formation assays, cell cycle analysis, cell migration and invasion assay
Cell counting kit-8 assay, colony formation assays, cell cycle analysis, cell migration and invasion assay and Annexin V apoptosis assay were performed according to general protocols and can be found in Additional file 1.
Dual-luciferase reporter gene assay
To determine the effect of ATF2 on PTHLH transcription, RBE cells were transfected with pGL3 as vehicle control, pGL3-PTHLH or pGL3-MUT-PTHLH using Lipofectamine 3000. Firefly and Renilla luciferase activities were measured separately on a fluorescence spectrophotometer (FlOUstar omega, BMG Labtech, Germany) in triplicate according to the manufacturer’s instructions for the dual-luciferase reporter assay kit (Promega). The relative transcriptional activity was normalized by the corresponding vehicle control value.
Chromatin immunoprecipitation (ChIP) assay
Genomic DNA prepared from RBE cells transfected with shControl was crosslinked with 1% formaldehyde and fragmented into 500 ± 100-bp fragments by sonication. Soluble chromatin was then incubated overnight with anti-ATF2 antibodies. Finally, the immunoprecipitated DNA fragments were amplified and quantified using real-time PCR using the following PCR primers specific to the human PTHLH promoter region.
Establishment of a subcutaneous tumor xenograft
RBE cells (shCtrl or shPTHLHx) (1 × 107) were injected subcutaneously into the groins of BALB/c nude mice (6 weeks old, male, n = 5 for each group). Tumor growth was monitored at 2 or 3-day intervals. When the mice were sacrificed after 25 days, tumor weight and size were measured, and the tumor was fixed for additional experimental use.
Statistical analyses
Different statistical analysis methods were used to compare different groups or different categories of data. Extended details regarding materials and methods can be found in Additional file 1.
Results
PTHLH is highly expressed in human CHO tissues specimens and ICC cell lines
To identify the potential role of PTHLH in CHO, we evaluated 59 ICC samples and paired non-tumor tissues from NanFang Hospital. We also screened an additional 10 samples of normal liver tissues for comparison. IHC analyses of ICC tumor regions revealed strong staining of PTHLH compared with that in adjacent regions in the same patients (Fig. 1a, top panel and b). Immunostaining of PTHLP protein was located in the cytoplasm and nucleus of ICC cells. We also observed weak staining in the bile duct of adjacent and normal tissue samples (Fig. 1a, top panel). In addition, we detected the expression of PTH1R, a specific receptor for PTHLH, in membranes of ICC cells (Fig. 1a, middle panel). Cytokeratin19 (CK) staining revealed the presence of adenocarcinoma cells and biliary epithelial cells (Fig. 1a, bottom panel). In addition, to further confirm the expression of PTHLH in CHO, we screened ICC microarrays that contained 100 cases and the ECC microarrays that contained 27 cases (Fig. 1c and Additional file 1: Figure S1). Our results were consistent with the conclusion above that PTHLH was highly expressed in CHO cells. In addition, PTHLH protein expression was examined in ICC cell lines by IF microscopy (Fig. 1e). Microscopy analysis detected cytosolic and nucleus expression of PTHLH in RBE and HCCC-9810 cells, which is consistent with previous observations.
Fig. 1.
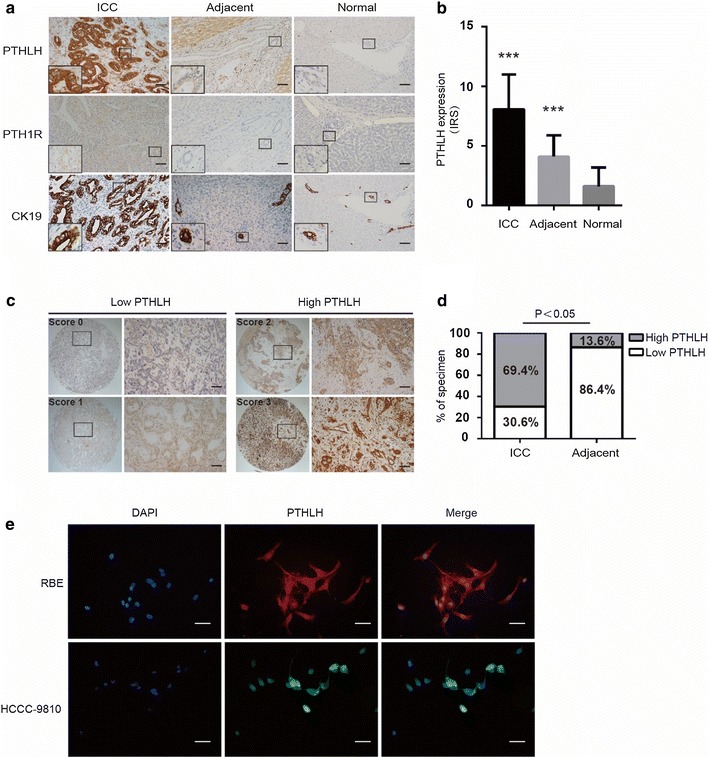
PTHLH is highly expressed in ICC tissues and cells. a Representative images of PTHLH, PTH1R and CK19 protein expression in ICC tumor tissues, paired adjacent tissues and normal tissues as assessed by immunohistochemistry (DAB staining, scale bar, 50 μm). b PTHLH protein expression was significantly increased in primary ICC tumors as compared with their adjacent and normal tissues (***p < 0.01). c, d Scores indicate PTHLH levels in ICC (DAB staining, scale bar, 50 μm). The scores were calculated by intensity and the percentage of stained cells as described in Additional file 1 (DAB staining, scale bar, 50 μm). e Immunofluorescence location. PTHLH was expressed in both nuclear and cytoplasm in ICC cell lines (HCCC-9810 and RBE) by immunofluorescence and fluorescent microscopy (scale bar, 50 μm)
Overexpression PTHLH is positively correlated with poor pathological differentiation in ICC patients
To investigate the clinical significance of PTHLH upregulation in CHO, we further analyzed the relationship between clinicopathological features and PTHLH expression levels in CHO cases. These patients were divided into high- (score, 2–3) or low- (score, 0–1) PTHLH expression groups according to the immunostaining scores (Fig. 1c, d). Scoring was conducted according to the ratio and intensity of positive-staining cells: 0–5% scored 0; 6–35% scored 1; 36–70% scored 2; more than 70% scored 3. The final score was designated as low or high expression group as follows: score 0–1, low expression, score 2–3, high expression. A high expression of PTHLH was positively correlated with poor pathological differentiation (p < 0.05) (Table 1). These findings indicate that PTHLH expression might contribute to ICC progression and be a potential therapeutic target of this disease.
Table 1.
Correlation of the ICC clinicopathological features and TNM staging with PTHLH expression (related to Fig. 1)
| Variable | N | PTHLH expression in ICC (n = 108) (%) |
χ2 | p value | |
|---|---|---|---|---|---|
| High (75) | Low (33) | ||||
| Age (years) | |||||
| ≤50 | 25 | 18 (72.0) | 7 (28.0) | 0.100 | NS |
| >50 | 83 | 57 (68.7) | 26 (31.3) | ||
| Gender | |||||
| Female | 43 | 33 (76.7) | 10 (23.3) | 1.794 | NS |
| Male | 65 | 42 (64.6) | 23 (35.4) | ||
| Pathological differentiation | |||||
| I | 8 | 5 (62.5) | 3 (37.5) | 7.236 | 0.027 |
| II | 53 | 31 (58.5) | 22 (41.5) | ||
| III | 47 | 39 (83.0) | 8 (17.0) | ||
| Tumor size (cm) | |||||
| ≥5 | 71 | 49 (69.0) | 22 (31.0) | 0.018 | NS |
| <5 | 37 | 26 (70.3) | 11 (29.7) | ||
| Intrahepatic metastasis | |||||
| Yes | 44 | 24 (54.5) | 20 (45.5) | 7.768 | 0.005 |
| No | 64 | 51 (79.7) | 13 (20.3) | ||
| Distant metastasis | |||||
| Yes | 5 | 4 (80.0) | 1 (10.0) | 0.275 | NS |
| No | 103 | 71 (68.9) | 32 (31.1) | ||
| TNM | |||||
| I–II | 72 | 24 (33.3) | 48 (66.7) | 1.544 | NS |
| III–IV | 36 | 9 (25.0) | 27 (75.0) | ||
Italic values indicate significance of p value (p < 0.05)
NS not significant between any groups
Note: ICC patients were divided into PTHLH ‘High’ group and ‘Low’ group
Abbreviations: ICC, Intrahepatic cholangiocarcinoma; Differences among variables were assessed by χ2 or Fisher’s exact χ2 test
PTHLH promotes ICC cells growth
The above data suggested that PTHLH may play a critical role in ICC progression. To address whether PTHLH affects cell proliferation, we first investigated endogenous PTHLH levels in tow ICC cell lines. We observed they both have PTHLH endogenous expression (Additional file 1: Figure S2A). We then generated two PTHLH-specific shRNAs to silence the endogenous PTHLH expression in ICC cells. shPTHLHx, which induced the most significant knock-down (KD) effect, was used for vivo study. We stably depleted PTHLH in RBE and HCCC-9810 cells. The relative expression of PTHLH in RBE and HCCC-9810 cells was confirmed by qPCR and western blot (Additional file 1: Figure S2B, C). PTHLH depletion significantly decreased ICC cell proliferation (Fig. 2a and Additional file 1: Figure S3). To evaluate the effects of PTHLH re-expression on tumor growth in vitro, we knock-down endogenous PTHLH and then reintroduced lentivirus-mediated vector (LV-Ctrl) and PTHLH using lentivirus-mediated PTHLHGFP (LV-PTHLHRE) to examine whether the re-expression of PTHLH could rescue the retarded proliferation (Additional file 1: Figure S2D). Compared with the control, incubation with PTHLH-specific shRNA resulted in elongated morphology cells and less confluent cell growth. When exposed to lentivirus-mediated PTHLHGFP, cell growth returned to normal (Additional file 1: Figure S3). Furthermore, we observed that PTHLH secretion was upregulated upon the reintroduction of PTHLH, indicating an autocrine function of PTHLH (Additional file 1: Figure S2E). Similarly, PTHLH re-expression increased LV-PTHLHRE ICC cell proliferation (Fig. 2b and Additional file 1: Figure S3).
Fig. 2.
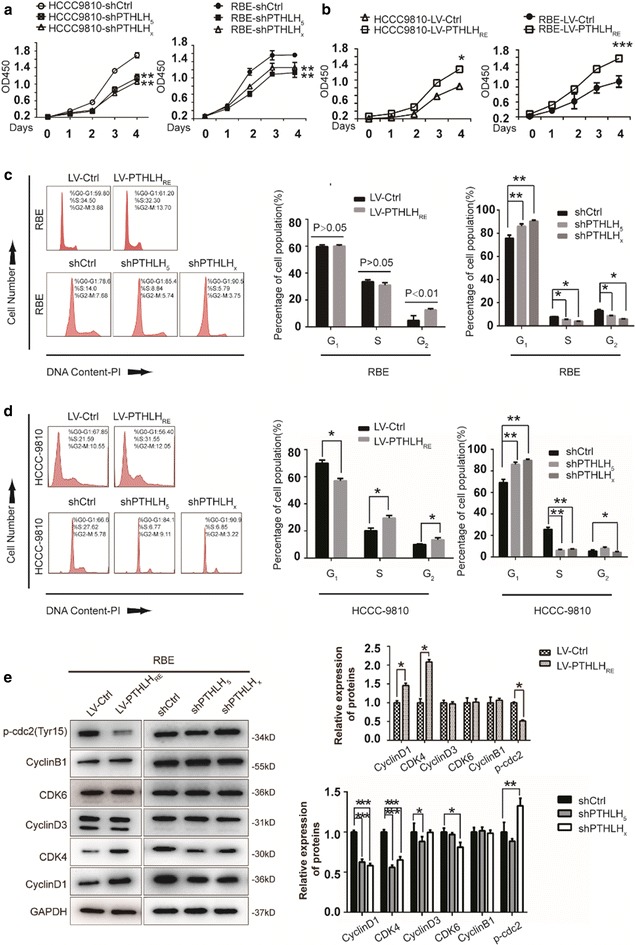
PTHLH promotes ICC cell growth via altering the cell cycle. a Depletion of PTHLH suppresses ICC cell growth (**p < 0.01). b PTHLH re-expression accelerates cell growth in ICC cells. Cell proliferation was examined using the CCK-8 assay in ICC cells with the stable re-expression of PTHLH (*p < 0.05, ***p < 0.001). c The distribution of cell cycle phases in RBE cells. The values are the mean ± SD (*< 0.05, **< 0.01). d The distribution of cell cycle phases in HCCC-9810 cells. Cell population sizes averaged from three independent experiments with standard deviations (*< 0.05, **< 0.01). e Protein expression of CyclinB1, p-cdc2, cell-cycle-related proteins in the G1 phase was determined by western blot (*< 0.05, **< 0.01, ***< 0.001)
PTHLH alters the cell cycle distribution
Cell cycle distribution was analyzed by flow cytometry analysis to determine whether PTHLH enhances cell growth and promotes tumorigenesis via an alteration of the cell cycle. After 12 h of serum starvation for synchronization, the cell population in the G2/M phase is significantly increased upon PTHLH re-expression, whereas the G0/G1 and S phase cell population remained more constant in RBE cells (Fig. 2c top panel). In contrast, the reverse effect was observed when PTHLH was depleted. shPTHLH arrested RBE cells at the G0/G1 phase, and the proportion of cells in the S and G2/M phase decreased (Fig. 2c bottom panel). These results demonstrate that PTHLH re-expression facilitates the S to G2/M phase transition. However, PTHLH deletion blocks the cell cycle by inhibiting the G0/G1 to S phase transition. The similar results were obtained from another ICC cell line, HCCC-9810 (Fig. 2d). To further explore the molecular basis of PTHLH-enhanced tumour development, we investigated the roles of PTHLH on metastasis using in vitro migration and Matrigel invasion assays. The results indicate that PTHLH facilitating RBE cells migration not invasion (Additional file 1: Figure S4A). We also found no significant differences between migration and invasion of HCCC-9810 cells (Additional file 1: Figure S4B). We quantitatively investigated the effect of PTHLH on apoptosis by flow cytometry after staining with Annexin V and 7-amino-actinomycin. The results indicate that ICC cell apoptosis is not regulated by PTHLH (Additional file 1: Figure S4C).
PTHLH regulates the expression of genes controlling the cell cycle
The observed differences in the cell cycle distribution were due to the different expression levels of key cell cycle proteins. We noted an increased accumulation of G2/M-phase cells upon PTHLH re-expression compared with that in LV-Ctrl cells. We observed that p-cdc2 protein levels decreased significantly, whereas CyclinB1 levels remains constant when PTHLH was re-expressed (Fig. 2e, left panel). These results indicate that PTHLH re-expression promotes RBE cells mitosis via downregulating p-cdc2 expression. We also detected G0/G1 phase-related proteins (CDK4/CyclinD1 and CDK6/Cyclin D3). CDK4/CyclinD1 protein levels increased slightly, whereas CDK6 and Cyclin D3 levels remained constant (Fig. 2e, left panel). Given that the cell cycle was altered by PTHLH depletion, we focused our attention on key proteins (CDK4/CyclinD1 and CDK6/Cyclin D3) during the G0/G1 phase. Western blot analysis indicated that CyclinD1 and CDK4 protein levels decreased dramatically when PTHLH expression was deleted in RBE cells (Fig. 2e, right panel). In contrast, Cyclin D3 and CDK6 expression remained constant. These data suggested that PTHLH regulates the expression of cell cycle-related proteins.
Loss of PTHLH expression suppresses tumorigenesis in vivo
To investigate whether PTHLH deletion suppresses tumorigenesis in vivo, PTHLH-KD RBE cells (shPTHLHx) were implanted subcutaneously into the right inguen, and vector cells (shCtrl) were implanted into the left inguen of nude mice (n = 5) (Fig. 3a). Tumor growth was monitored as described in Additional file 1. Consistent with the cell proliferation assay in vitro, tumor growth was significantly decreased in mouse xenografts with shPTHLHx compared with that of shCtrl (Fig. 3b, c). Consistently, the nuclear expression of Ki-67, Cyclin D1 and CDK4 proteins was significantly increased in the shCtrl-RBE tumors compared with that in the shPTHLHx-RBE tumors (Fig. 3e), which is consistent with the in vitro study using western blot (Fig. 2e). These results collectively suggest that PTHLH promotes ICC cell proliferation.
Fig. 3.
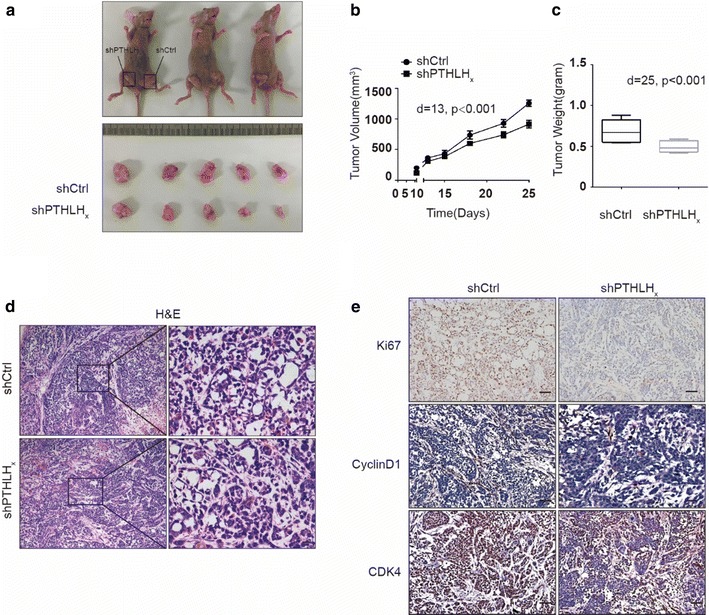
Loss of PTHLH significantly suppressed cell proliferation. a–c PTHLH deletion suppresses tumor growth. Five nude mice were injected subcutaneously with 1 × 107 cells/mouse for each of the indicated stable cell lines. The results are presented as isolated tumors (a), tumor sizes (b), and tumor weights (c). d H&E staining of xenograft tumor from nude mice injected with shCtrl or shPTHLHx cells (scale bar, 20 μm). e Representative images of Ki67, CyclinD1, and CDK4 protein expression in xenograft tumors from nude mice as assessed by immunohistochemistry (DAB staining, scale bar, 50 μm)
PTHLH altered cell cycle genes via activating phosphorylated ATF2 through JNK/ERK1/2 signaling pathways
Accumulating studies highlight PTHLH as a cellular cytokine with actions involved in both cell growth and differentiation [3]. Previous reports supported that the PTHLH can trigger the MAPK signaling cascade by binding with PTH1R, which interacts with the MAPK scaffolding protein β-arrestin2 and G-protein [10–12]. As a downstream effector of MAPK, ATF2 regulates cell cycle progression through the transcriptional control of CyclinD1 (Fig. 4a) [18]. We hypothesized that PTHLH can increase ATF2 transcriptional activity by activating ERK1/2 and JNK signaling cascades. As shown in (Fig. 4b, c), we treated RBE cells with PTHLH (1–34) recombination fragment and assessed the ATF2 expression. PTHLH (1–34) induced a time- and dose-responsive increase of in ATF2 protein expression. In contrast, the effect of PTHLH was attenuated at 100 nM (Fig. 4b, dotted line). These results indicated that PTHLH exhibits dose-dependent biphasic effects on ICC cell dynamics. We also found endogenous PTHLH re-expression upregulated ATF2 protein expression (Additional file 1: Figure S5). In response to PTHLH stimulation, PTHLH/PTH1R signaling triggers JNK and ERK1/2 signaling pathways (Fig. 4d). Therefore, pharmacological approaches were used to confirm that ATF2 transcriptional activity is regulated by PTHLH. When RBE cells were pretreated with an MEK1/2 inhibitor (U0126) or JNK1/2 inhibitor (SP600125) for 1 h followed by PTHLH (1–34) treatment for 4 h, we observed that the MEK1/2 and JNK1/2 inhibitor abrogated the PTHLH-induced phosphorylation of ATF2 (p-ATF2) (Fig. 4e), suggesting that p-ATF2 induction involves the PTHLH-JNK/ERK1/2 signaling cascade. Interestingly, we further found that the MEK inhibitor U0126 and the JNK inhibitor SP600125 inhibited RBE cell growth, arresting cells at the G0/G1 phase (Fig. 4e). These results suggesting that the inhibition of JNK/ERK1/2 attenuated PTHLH-induced ICC growth.
Fig. 4.
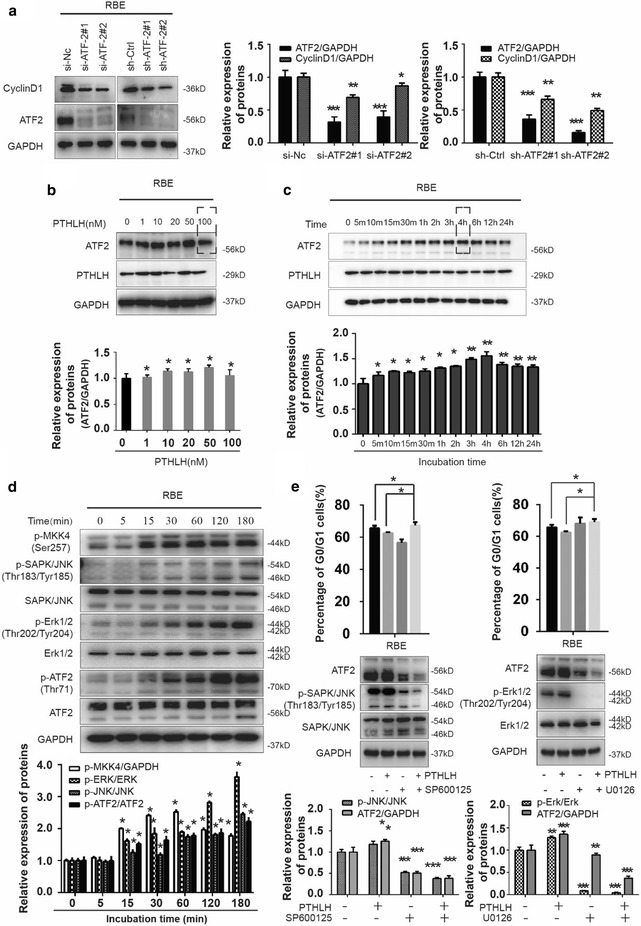
Upstream kinase for ATF2 phosphorylation and expression. a ATF2 regulates the expression of CyclinD1. Western blot indicating the protein level of CyclinD1 in RBE cells where ATF-2 was stably or transiently depleted (*< 0.05, **< 0.01, ***< 0.001). b The cells were treated with 0, 1, 10, 20, 50, and 100 nM PTHLH for 4 h, and western blot was performed for ATF2, PTHLH and GAPDH (*< 0.05). c The cells were treated with 20 nM PTHLH for the indicated times, and western blot was performed for ATF2, PTHLH and GAPDH (*< 0.05, **< 0.01). d Western blot of the expression of ATF2, JNK and ERK1/2 protein and phosphorylation of JNK (Thr183/Thr185), ERK1/2 (T202/Y204), MKK4 (S257) and ATF-2 (Thr71) in the RBE cells (*< 0.05). e Cells were pre-treated with U0126 (10 μM) or SP600125 (20 μM) and then treated with 20 nM PTHLH for 4 h. Cell distribution was determined by flow cytometry (*< 0.05), and the expression of related proteins was determined by western blot (**< 0.01, ***< 0.001)
ATF2 negatively regulate PTHLH expression
ATF2 is a bZIP transcription factors which has an ability to bind to the CRE consensus. According to previous reports, the pthlh gene contains a CRE element within its promoter region. Interestingly, we also found that ATF2 might interact with PTHLH promoter elements in bioinformatics prediction methods (Additional file 1: Figure S5). Next, we mapped the ATF2 response element(s) on the PTHLH promoter. Analysis of the proximal region revealed the presence of ATF2 target sequences at positions − 2210 to − 2243 (Site #3) (Fig. 5a). Further support for the role of ATF2 in the regulation of PTHLH transcription was provided by ChIP analysis. Sheared chromatin was immunoprecipitated with antibodies to ATF-2 (or control IgG) followed by the PCR amplification of PTHLH promoter sequences. Immunoprecipitation of ATF2 enabled the amplification of PTHLH promoter sequences, demonstrating the in vivo binding of ATF2 to the PTHLH promoter. DNA pull-down assays demonstrated ATF2 binding to the PTHLH promoter region (Fig. 5b). Consistent with this finding, a mutation within this site attenuated the basal level of reporter activity and the binding of ATF2 (Fig. 5c), confirming that ATF2 regulates PTHLH transcription via binding its response element at the Site #3. Our studies also indicated that PTHLH expression was regulated by si-ATF2, as confirmed by qPCR and western blot (Fig. 5d). To further confirm the correlation between PTHLH and ATF2 expression in ICC, we detected ATF2 expression using the same samples (Fig. 5e). Further statistical analysis revealed that the ATF2 expression correlated with PTHLH expression in the tissue samples (r = 0.624, p < 0.05), suggesting a potentially complicated regulatory mechanism between PTHLH and ATF2.
Fig. 5.
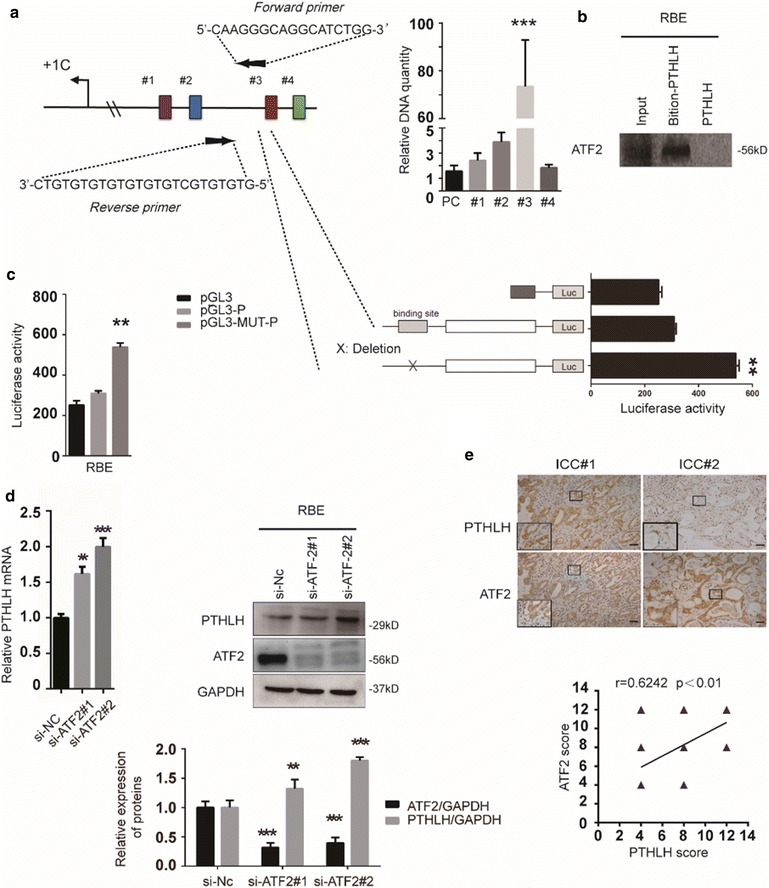
Mapping the ATF2 response element on the PTHLH promoter. a Structure of the PTHLH promoter. Putative ATF2 response elements and fragments of the promoter that were cloned are depicted (colorful boxes, region “#1”–“#4”). Mutation of ATF2 element in the chromatin immunoprecipitation assay. Immunoprecipitated DNA was used as the template in PCR with primers corresponding to the proximal region of the PTHLH promoter (***p < 0.001). PC primers were used as control. b DNA pull-down assay. An equal amount of cell lysate was pulled down with biotinylated PTHLH DNA probe followed by immunoblotting with anti-ATF2 antibody. c Mutation of a PTHLH element at site #3 inhibits reporter activity. PTHLH was mutated, and the relative luciferase activity of the WT and mutant construct was assessed in RBC cells. The results are presented as the mean ± SD. The data were standardized to β-galactosidase (**p < 0.01). d An equal amount of cell lysate from RBE cells transfected with ATF2 small interfering RNA plasmid (si-ATF2), control plasmids (si-NC) or control was subjected to qPCR and immunoblotting with PTHLH or ATF-2 antibody, GAPDH reveals equal loading. e Stains of the same cohorts of ICC sections for analysis of the related expression of PTHLH and ATF2
Discussion
ICC is one of the most lethal epithelial cancers united by poor diagnoses and adverse outcomes. The condition frequently arises in the presence of chronic injury and inflammation. Previous literatures documenting that ICC is commonly associated with cirrhosis, viral hepatitis B and C, and metabolic abnormalities [19–22]. The molecular pathogenesis of ICC proliferation and metastasis, as the main cause of ICC-related mortality, but the mechanisms remain obscure. Our findings demonstrate that PTHLH knockdown in ICC cells suppressed tumor growth, while re-expression of PTHLH has the opposite effect, highlighting the role of PTHLH as a critical oncoprotein in ICC progression.
PTHLH/PTH1R signaling is aberrantly induced or activated in different cancer types and is associated with poor prognosis [5, 23–25]. In the present study, we found that ICC cells produced PTHLH ligands that respond via the expression of cognate receptors PTH1R, resulting in the continuous activation of downstream signaling pathways. Extensive evidence suggest that PTHLH is viewed as a cellular cytokine, particularly in epithelial cancer cells, that exhibits an autocrine or paracrine role in both cell growth and differentiation [26, 27]. In our previous study, we found that overexpression of the PTHLH (LV-PTHLH), which transfects lentivirus-mediated PTHLHGFP without deleting endogenous PTHLH expression in ICC cells, may enter into the non-proliferative cells (data not show). In contrast, LV-PTHLHRE ICC cells promoted cell growth. Moreover, endogenous PTHLH sustains the activation of MAPK signaling pathways, and this effect was more pronounced after the addition of a PTHLH recombination peptide. In contrast, the effect of PTHLH on activation was attenuated at a higher concentration (100 nM) compared with 50 nM (Fig. 5b, dotted portion). These results clearly indicated that PTHLH may function as a tumor cell growth promoter within a certain concentration range. When the range is exceeding, PTHLH becomes saturated and suppresses cells proliferation. In our study, we also found re-expression PTHLH promotes RBE cells migration and specific overexpression PTHLH associates with intrahepatic metastasis in ICC patients. All results indicated a potential ability of facilitating tumor invasiveness. The data would suggest that PTHLH may potentially transform ICC cells into an aggressive form of the disease. And this also indicates that PTHLH influences ICC cells growth and differentiation with a low signal expression.
To investigate the effect of ICC secreted PTHLH on cancer cell growth, we established an in-vitro PTHLH secretion system. In our present work, we found that PTHLH regulated cell growth by altering the cell cycle. Cell cycle dysregulation is a major feature of tumorigenesis, which occurs by shortening the G1 phase or activating CDKs may favor tumor development [28]. Herein, we demonstrate that PTHLH controlled cell cycle progression. Secreted PTHLH protein act on target cells by binding to its specific cell surface receptor: PTH1R. The potential molecular mechanisms could be explained by the finding that the PTHLH protein activates the JNK/ERK1/2-ATF2 axis via interacting with the MAPK scaffolding protein β-arrestin2, or triggering an early G protein-dependent pathway meditated by PKA and PKC [10] leading to cell cycle proliferation. Furthermore, we provided evidence suggesting that PTHLH upregulates ATF2 phosphorylation via activating the ERK1/2 and JNK signaling pathways, which transcriptionally upregulate CyclinD1 expression. When ERK1/2 and JNK are pharmacologically inhibited in ICC cells (i.e., via U0126 and SP600125), which blocked PTHLH-induced activation of ERK1/2 and JNK signal pathways, and transcriptional activity of ATF2 (Fig. 6). Cyclin D1 is frequently deregulated in cancer and is a biomarker of cancer phenotypes and disease progression [29]. Previous findings suggested that the deregulation of CyclinD1 expression and CDK4 activation directly lead to some cancer hallmarks by inducing proliferation [30–32]. We observed that reduced CyclinD1 expression and CDK4 inactivation directly inhibited proliferation, which is consistent with previous reports. Consistently, our present work demonstrated that PTHLH re-expression accelerated the G2 to M phase transition, which is similar to the effects of numerous other oncogenes. Among the genes functioning during the G2 and M phase transition, we observed that the p-cdc2 levels were rapidly reduced upon PTHLH re-expression. Several experimental findings indicate that cdc2 is one of the master regulators of mitosis that controls the centrosome cycle in complex with A- or B-type cyclins [33]. Previous reports have suggested that cdc2 activity upon mitosis entry depends on p-cdc2 levels [34]. However, the reduction of cdc2 activity primarily drives the exit from mitosis [33]. Our present study indicates that PTHLH promote mitosis in ICC cells via downregulating p-cdc2 expression. Interestingly, we observed a paradoxical phenomenon that PTHLH overexpression without knocking-down endogenous secretion arrests ICC cells in the G1 phase and decreases CyclinD1 expression (data not show). We hypothesized that ostensibly paradoxical responses between PTHLH deletion and overexpression in cultured ICC cells appear to facilitate a compromise between maximal mitogenic stimulation and the avoidance of antiproliferative defenses.
Fig. 6.
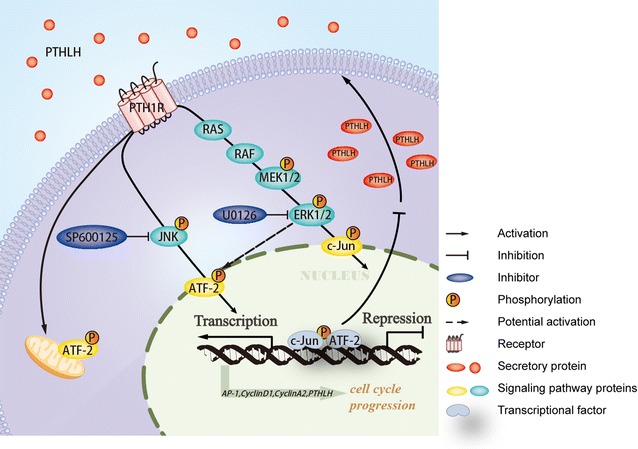
A model of the PTHLH/PTH1R-ATF2 negative-feedback loop. PTHLH/PTH1R signaling activated the expression and transcriptional activity of ATF2 via the ERK1/2 and JNK signaling pathways to promoted ICC cell growth, and ATF2 positively regulated CyclinD1 expression and negatively regulated PTHLH expression. Treatment with ERK1/2 and JNK signaling pathway inhibitors abolished ATF2 transcriptional activity
Another interesting finding of this study is the negative regulated role of ATF2 in RBE cells (Fig. 6). ATF2 is an important transcription factor that can facilitate malignant proliferation. In our previous study, we found that ATF2 promoted growth of ICC cells and was correlated with a poor prognosis for ICC patients. In vitro evidence indicated that the upregulation of ATF2 phosphorylation and activity promotes cancer progression via facilitating cell proliferation-related gene expression. In our study, we demonstrated that PTHLH can promote oncogenic functions of ATF2 by activating ERK/JNK pathways. And a previous report revealed PTHLH can activate PKC pathway [35]. Eric Lau [36] previously reported that PKCε promotes oncogenic functions of ATF2 in the nucleus while blocking its apoptotic function at mitochondria. Thus, we believe that PTHLH promotes nuclear translocation and transcriptional function in the oncogenic functions of ATF2. Interestingly, another novel finding of our study is the negative role of ATF2 in PTHLH production in REB cells. And we also found autocrine activation of the PTHLH promoter by c-Jun (data not show). Collectively, these findings indicated that ATF2 limits PTHLH transcriptional output to maintain specific concentration by forming a homodimer or a heterodimer with JUN. It is possible that cancer cells may have negative feedback loops that are essential for survival.
Conclusions
In summary, we report that ICC-secreted PTHLH acts in an autocrine manner in intrahepatic cholangiocarcinoma progression by activating the canonical ERK/JNK signaling pathway. However, our findings focus on only PTHLH-mediated ICC cell proliferation, not provide new insight into the ICC metastasis. Despite the importance of PTHLH tumorigenic role, our knowledge of the PTH1R that mediate changes in the tumor progression and interaction with PTHLH in ICC is still limited. Based on our findings, further investigation for interfering with PTH1R, which mediate signaling in cancer cells, may serve as effective treatment approaches to ICC patients. And we will improve the mechanisms of PTHLH/PTH1R-mediated ICC progression and involve the interaction of the transcription factors CREB and AP-1 (c-JUN, c-FOS and ATF2) in ICC development.
Authors’ contributions
The first five authors contribute equally to this paper. JT assisted in the design of study, performed experiments, analyzed/interpreted data, and drafted the manuscript; YL, SH and LP contributed to study design, interpreted data, and helped with manuscript revision; JS helped draft the manuscript; XX, FX, JH, XL and JX provided technical support and helped to revise the manuscript; ZL, CL, ND, KL, JM, GW, JL, DZ, CZ and QX helped performed some of the experiments and supported materials; LB contributed to study design, revision of the manuscript and provided funding. All authors read and approved the final manuscript.
Acknowledgements
We thank AJE team for providing language help and writing assistance. This work was supported by the National Natural Science Foundation of China (Grants 81170354, 81470790, and 81500398), Natural Science Foundation of Guangdong Province (2015A030313295 and 2015A030310480), Guangzhou Pilot Project of Clinical and Translational Research Center (early gastrointestinal cancers, No. 7415696196402), and Guangdong Provincial Bioengineering Research Center for Gastroenterology Diseases.
Competing interests
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study. Please contact author for data requests.
Consent for publication
The participants gave informed consent before taking part in this study. All samples were de-identified.
Ethics approval and consent to participate
NanFang Hospital Institutional Review Board.
Institutional Animal Care Use Committee.
Funding
This work was supported by the National Natural Science Foundation of China (Grants 81170354, 81470790, and 81500398), Natural Science Foundation of Guangdong Province (2015A030313295 and 2015A030310480), Guangzhou Pilot Project of Clinical and Translational Research Center (early gastrointestinal cancers, No. 7415696196402), and Guangdong Provincial Bioengineering Research Center for Gastroenterology Diseases.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ICC
intrahepatic cholangiocarcinoma
- PTHLH
parathyroid hormone-like hormone
- PTHrP
parathyroid hormone-related protein
- IHC
immunohistochemistry
- ChIP
chromatin immunoprecipitation
- ATF2
activating transcription factor-2
- PTH1R
PTH type 1 receptor
- ECC
extrahepatic cholangiocarcinoma
- IF
immunofluorescence
- PI
propidium iodide
- CK19
cytokeratin19
- CHO
cholangiocarcinoma
- KD
knockdown
Additional file
Footnotes
Jing Tang, Yan Liao, Shuying He, Jie Shi and Liang Peng contributed equally to this work
Electronic supplementary material
The online version of this article (10.1186/s12967-017-1342-1) contains supplementary material, which is available to authorized users.
Contributor Information
Jing Tang, Email: 465805529@qq.com.
Yan Liao, Email: 378169367@qq.com.
Shuying He, Email: 869180341@qq.com.
Jie Shi, Email: 416190784@qq.com.
Liang Peng, Email: 199255047@qq.com.
Xiaoping Xu, Email: 673749662@qq.com.
Fang Xie, Email: 272978167@qq.com.
Na Diao, Email: 962218580@qq.com.
Jinlan Huang, Email: 504575084@qq.com.
Qian Xie, Email: 1053307805@qq.com.
Chuang Lin, Email: 605905270@qq.com.
Xiaoying Luo, Email: 287063849@qq.com.
Kaili Liao, Email: 1175865120@qq.com.
Juanjuan Ma, Email: 418367868@qq.com.
Jingyi Li, Email: 307484423@qq.com.
Daichao Zhou, Email: 331071165@qq.com.
Zhijun Li, Email: 853232238@qq.com.
Jun Xu, Email: 775650429@qq.com.
Chao Zhong, Email: 745327707@qq.com.
Guozhen Wang, Email: 2320052799@qq.com.
Lan Bai, Phone: +862061641539, Email: bailan_99@yeah.net, Email: bailan99@hotmail.com.
References
- 1.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383(9935):2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.John MT. Parathyroid hormone-related protein, its regulation of cartilage and bone development, and role in treating bone diseases. Physiol Rev. 2016;96:831–871. doi: 10.1152/physrev.00031.2015. [DOI] [PubMed] [Google Scholar]
- 4.Kim W, Takyar FM, Swan K, Jeong J, VanHouten J, Sullivan C, et al. Calcium-sensing receptor promotes breast cancer by stimulating intracrine actions of parathyroid hormone-related protein. Cancer Res. 2016;76(18):5348–5360. doi: 10.1158/0008-5472.CAN-15-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urosevic J, Garcia-Albéniz X, Planet E, Real S, Céspedes MV, Guiu M, et al. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signaling and PTHLH. Nat Cell Biol. 2014;16(7):685–694. doi: 10.1038/ncb2977. [DOI] [PubMed] [Google Scholar]
- 6.Alvaro D, Gigliozzi A, Attili AF. Regulation and deregulation of cholangiocyte proliferation. J Hepatol. 2000;33(2):333–340. doi: 10.1016/S0168-8278(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 7.Lee JK, Chuang MJ, Lu CC, Hao LJ, Yang CY, Han TM, et al. Parathyroid hormone and parathyroid hormone related protein assays in the investigation of hypercalcemic patients in hospital in a Chinese population. J Endocrinol Invest. 1997;20(7):404–409. doi: 10.1007/BF03347992. [DOI] [PubMed] [Google Scholar]
- 8.Aizawa M, Koshiyama H, Inoue D, Fukunaga Y, Katakami H, Miki M, et al. Postoperative aggravation of hypercalcemia–leukocytosis syndrome in a case of squamous cell type cholangiocarcinoma. Intern Med. 1997;36(3):232. doi: 10.2169/internalmedicine.36.232. [DOI] [PubMed] [Google Scholar]
- 9.Davis JM, Sadasivan R, Dwyer T, Van Veldhuizen P. Case report: cholangiocarcinoma and hypercalcemia. Am J Med Sci. 1994;307(5):350–352. doi: 10.1097/00000441-199405000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, et al. Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281:10856–10864. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- 11.Gardella TJ, Luck MD, Jensen GS, Schipani E, Potts JT, Jr, Jüppner H. Inverse agonism of amino-terminally truncated parathyroid hormone (PTH) and PTH-related peptide (PTHrP) analogs revealed with constitutively active mutant PTH/PTHrP receptors. Endocrinology. 1996;137(9):3936–3941. doi: 10.1210/endo.137.9.8756569. [DOI] [PubMed] [Google Scholar]
- 12.Gesty-Palmer D, Flannery P, Yuan L, Corsino L, Spurney R, Lefkowitz RJ, et al. A β-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med. 2009;1:1–9. doi: 10.1126/scitranslmed.3000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouwens DM, de Ruiter ND, van der Zon GC, Carter AP, Schouten J, van der Burgt C, et al. Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras-MEK-ERK pathway and of Thr69 through RalGDS-Src-p38. EMBO J. 2002;21(14):3782–3793. doi: 10.1093/emboj/cdf361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta S, Campbell D, Dérijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267(5196):389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura T, Okuyama S, Okamoto S, Nakajima T, Sekiya S, Oda K. Down-regulation of the cyclin A promoter in differentiating human embryonal carcinoma cells is mediated by depletion of ATF-1 and ATF-2 in the complex at the ATF/CRE site. Exp Cell Res. 1995;216:422–430. doi: 10.1006/excr.1995.1053. [DOI] [PubMed] [Google Scholar]
- 16.Vale-Cruz DS, Ma Q, Syme J, LuValle PA. Activating transcription factor-2 affects skeletal growth by modulating pRb gene expression. Mech Dev. 2008;125:843–856. doi: 10.1016/j.mod.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 17.Beier F, Taylor AC, LuValle P. Activating transcription factor 2 is necessary for maximal activity and serum induction of the cyclin A promoter in chondrocytes. J Biol Chem. 2000;275:12948–12953. doi: 10.1074/jbc.275.17.12948. [DOI] [PubMed] [Google Scholar]
- 18.Lewis JS, Vijayanathan V, Thomas TJ, Pestell RG, Albanese C, Gallo MA, et al. Activation of cyclin D1 by estradiol and spermine in MCF-7 breast cancer cells: a mechanism involving the p38 MAP kinase and phosphorylation of ATF-2. Oncol Res. 2005;15(3):113–128. doi: 10.3727/096504005776367924. [DOI] [PubMed] [Google Scholar]
- 19.Lee CH, Chang CJ, Lin YJ, Yeh CN, Chen MF, Hsieh SY. Viral hepatitis-associated intrahepatic cholangiocarcinoma shares common disease processes with hepatocellular carcinoma. Br J Cancer. 2009;100(11):1765–1770. doi: 10.1038/sj.bjc.6605063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaib YH, El-Serag HB, Davila JA, Morgan R, McGlynn KA. Risk factors of intrahepatic cholangiocarcinoma in the United States: a case–control study. Gastroenterology. 2005;128:620–626. doi: 10.1053/j.gastro.2004.12.048. [DOI] [PubMed] [Google Scholar]
- 21.Thinkhamrop K, Khuntikeo N, Phonjitt P, Chamadol N, Thinkhamrop B, Moore MA, et al. Association between diabetes mellitus and fatty liver based on ultrasonography screening in the world’s highest cholangiocarcinoma incidence region, Northeast Thailand. Asian Pac J Cancer Prev. 2015;16(9):3931–3936. doi: 10.7314/APJCP.2015.16.9.3931. [DOI] [PubMed] [Google Scholar]
- 22.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10(11):656–665. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 23.Ho PW, Goradia A, Russell MR, Chalk AM, Milley KM, Baker EK, et al. Knockdown of PTHR1 in osteosarcoma cells decreases invasion and growth and increases tumor differentiation in vivo. Oncogene. 2015;34(22):2922–2933. doi: 10.1038/onc.2014.217. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Yu C, Dai J, Keller JM, Hua A, Sottnik JL, et al. Parathyroid hormone-related protein inhibits DKK1 expression through c-Jun-mediated inhibition of β-catenin activation of the DKK1 promoter in prostate cancer. Oncogene. 2013;33(19):2464–2477. doi: 10.1038/onc.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walia MK, Ho PM, Taylor S, Ng AJ, Gupte A, Chalk AM, et al. Activation of PTHrP-cAMP-CREB1 signaling following p53 loss is essential for osteosarcoma initiation and maintenance. elife. 2016;5:e13446. doi: 10.7554/eLife.13446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhatia V, Saini MK, Falzon M. Nuclear PTHrP targeting regulates PTHrP secretion and enhances LoVo cell growth and survival. Regul Pept. 2009;27:149–155. doi: 10.1016/j.regpep.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lv Z, Wu X, Cao W, Shen Z, Wang L, Xie F, et al. Parathyroid hormone related protein serves as a prognostic indicator in oral squamous cell carcinoma. J Exp Clin Cancer Res. 2014;18(33):100. doi: 10.1186/s13046-014-0100-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 29.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11(8):558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 30.Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22(4):438–451. doi: 10.1016/j.ccr.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwong LN, Costello JC, Liu HY, Jiang S, Helms TL, Langsdorf AE, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18(10):1503–1510. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cole AM, Myant K, Reed KR, Ridgway RA, Athineos D, Van den Brink GR, et al. Cyclin D2-cyclin-dependent kinase 4/6 is required for efficient proliferation and tumorigenesis following Apc loss. Cancer Res. 2010;70(20):8149–8158. doi: 10.1158/0008-5472.CAN-10-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seong YS, Min C, Li L, Yang JY, Kim SY, Cao X, et al. Characterization of a novel cyclin-dependent kinase 1 inhibitor, BMI-1026. Cancer Res. 2003;63(21):7384–7391. [PubMed] [Google Scholar]
- 34.Satinover DL, Stukenberg PT, Gorbsky GJ. The reversibility of mitotic exit in vertebrate cells. Nature. 2006;440:954–958. doi: 10.1038/nature04652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fortino V, Torricelli C, Gardi C, Valacchi G, Rossi Paccani S, Maioli E. ERKs are the point of divergence of PKA and PKC activation by PTHrP in human skin fibroblasts. Cell Mol Life Sci. 2002;59(12):2165–2171. doi: 10.1007/s000180200015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lau E, Kluger H, Varsano T, Lee K, Scheffler I, Rimm DL, et al. PKCε promotes oncogenic functions of ATF2 in the nucleus while blocking its apoptotic function at mitochondria. Cell. 2012;148(3):543–555. doi: 10.1016/j.cell.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study. Please contact author for data requests.