

Abstract
Prediction of benefit from combined chemotherapy and the antiepidermal growth factor receptor cetuximab is a not yet solved question in head and neck squamous cell carcinoma (HNSCC). In a selected series of 14 long progression-free survival (PFS) and 26 short PFS patients by whole gene and microRNA expression analysis, we developed a model potentially predictive of cetuximab sensitivity. To better decipher the “omics” profile of our patients, we detected transcript fusions by RNA-seq through a Pan-Cancer panel targeting 1385 cancer genes. Twenty-seven different fusion transcripts, involving mRNA and long noncoding RNA (lncRNA), were identified. The majority of fusions (81%) were intrachromosomal, and 24 patients (60%) harbor at least one of them. The presence/absence of fusions and the presence of more than one fusion were not related to outcome, while the lncRNA-containing fusions resulted enriched in long PFS patients (P = 0.0027). The CD274-PDCD1LG2 fusion was present in 7/14 short PFS patients harboring fusions and was absent in long PFS patients (P = 0.0188). Among the short PFS patients, those harboring this fusion had the worst outcome (P = 0.0172) and increased K-RAS activation (P = 0.00147). The associations between HNSCC patient's outcome following cetuximab treatment and lncRNA-containing fusions or the CD274-PDCD1LG2 fusion deserve validation in prospective clinical trials.
1. Introduction
The therapeutic opportunities for recurrent and/or metastatic (RM) head and neck squamous cell carcinoma (HNSCC) may be divided into 3 modalities: (i) potentially salvageable treatments, like (re)irradiation or salvage surgery; (ii) palliative systemic therapies, such as chemotherapy and/or targeted agents; and (iii) the best supportive care. Salvage therapies are the first options, but their feasibility is limited by the patient's performance status or by other prognostic factors as disease-free interval as well as by technical aspects such as the site and extension of disease or to the previous administered treatments [1–3].
First line palliative systemic therapy is represented by the combination of platinum-based chemotherapy and cetuximab, an antiepidermal growth factor receptor (EGFR) agent. This combination, as shown in the pivotal EXTREME trial, is able to achieve a clinical response in more than one-third of the patient, and it is able to statistically improve overall survival (OS) and progression-free survival (PFS) and it improves the patient's quality of life, when compared to chemotherapy alone [4]. However, the median OS is of 10.1 months with more than 80% of the patients experiencing one grade 3 or 4 adverse event. Thus, the presence of multiple mechanisms of intrinsic resistance to this therapeutic combination exposes some patients to the double-negative effect of drug toxicity and disease unresponsiveness.
Therefore, the issue of predicting which patient will benefit from this approach is an outstanding question in head and neck oncology. In fact, all the efforts in identifying specific alteration in EGFR status (studied by immunohistochemistry, amplification, or mutation) did not reach their purpose [5–7].
A solution might, however, lie with a molecular approach [8, 9], judging also from the encouraging results of our predictive models, developed to test cetuximab sensitivity in RM-HNSCC patients [10, 11].
Specifically, we implemented a model of cetuximab and chemotherapy (CT) sensitivity analyzing the 2 extremities of responsiveness to the drugs, represented by patients achieving a long PFS, defined to be more than 1 year, and patients showing a short PFS, defined to be less than 5.6 months, that is, the median PFS of the EXTREME trial. Using these selected patient cohorts and applying, as first, gene expression analysis [10] and in a second study an integrative analysis of miRNA and mRNA expression [11], we identified specific profiles corresponding to the long and short PFS [10] and a height miRNA gene-integrated signature with an excellent accuracy in predicting treatment response [11]. Trying to better decipher the different biological molecular characteristics of the extremities of the response curve to cetuximab CT, we decided to explore the new area of fusion transcripts in the search of other complementing genomics.
Gene fusions could occur by structural rearrangements or by transcription read-through of neighboring genes, being the second mechanism responsible for a large proportion of gene fusions (see [12] for a recent computationally oriented literature review); their clinical utility in cancer as biomarkers for prognosis or diagnosis is proven, and some fusion proteins are promising therapeutic targets (see [13] for the landscape of cancer-associated transcript fusions). At present, 300 samples of the TCGA-HNSCC dataset were characterized for the presence of transcript fusions [12], and among the identified fusion events, FGFR3-TACC3 fusion was detected in two HPV-positive tumors. Subsequently, in a HNSCC cell model system [14], the signaling by FGFR3-TACC3 fusion protein was further characterized as a novel mechanism of resistance to EGFR/ERBB3 inhibition. A limited number of other reports focused on gene fusions in HNSCC samples [15] or cell lines [16–17].
No data are presently available in a clinical setting about a possible association between gene fusion presence and response to a targeted therapy, such as the one with EGFR inhibitors. Taking again the advantage of the RM-HNSCC clinical material already genomically characterized by us [10, 11], we looked for the expression of fusion transcripts derived from 1385 different genes, selected on the basis of their putative role in cancer, as potential markers of intrinsic sensitivity/resistance to cetuximab CT.
2. Materials and Methods
2.1. Patients and Study Design
Forty formalin-fixed paraffin-embedded (FFPE) tumor specimens from RM-HNSCC patients treated between 2008 and 2012 with first-line platinum and cetuximab-based combination were collected and divided according to PFS following cetuximab CT treatment in long (14 patients) and short PFS (26 patients) as detailed in [10]. Briefly, the two groups were balanced for known prognostic factors [18] (primary tumor site, performance status, weight loss, prior radiotherapy, tumor grade, residual disease at primary tumor site, age, and gender). Long PFS had a median PFS of 19 months (range 12–36) while short PFS had a median PFS of 3 months (range 1–5.5).
2.2. Transcript Fusion Detection
To detect transcript fusions in RM-HNSCC, the TruSight RNA Pan-Cancer panel (Illumina) targeting 1385 cancer genes, including 507 known genes involved in fusions and 878 genes either mutated or deregulated in cancers, was used according to the provider's protocol. The panel design covers all exons and 160 bp at the 5′ and 3′ UTR of every gene. Briefly, cDNA is generated from 50 ng of total RNA from the FFPE specimens using random priming. After second strand synthesis, sequencing adapters are ligated to the double-stranded cDNA fragments. The coding regions of expressed cancer-associated genes were captured from 200 ng of this library using sequence-specific probes to create the final sequencing library. Quality check was performed using 4200 TapeStation and D1000 ScreenTape Assays (Agilent) yielding libraries with a band peak at ~250–300 bp. Samples were equimolarly pooled and sequenced on a NextSeq500 sequencer using the NextSeq500 High Output Kit v2 (150 cycles) chemistry (Illumina) to obtain 40 M/sample paired end reads of length 2 × 75 bps.
The data processing was performed on BaseSpace Sequence Hub, a dedicated genomics computing environment for data management and analysis applying TopHat Alignment v1.0. TopHat Alignment workflow allows the following functions: (i) read mapping on homo sapiens UCSC hg19 through the TopHat 2 aligner and (ii) fusion calling with TopHat-Fusion [19]. After the alignment of sequencing reads within the exon regions, the reads not entirely aligned were divided into multiple segments of 25 bp. It is expected that the initially unmapped reads contain sequencing portions residing on different chromosomes or on the same chromosome but, after rearrangement, representing potential fusion candidates. The first and last 25 bp portions were aligned on the genome through Bowtie. When an alignment pattern is detected, the entire read sequence is used to identify the fusion point by stitching segments to obtain the full read alignment. The oligo capture approach of the TruSight RNA Pan-Cancer panel allows pulling down one target gene among the 1385 genes in the panel and the partner fusion not necessarily included in the panel. Since the TopHat-fusion algorithm works independently of the information about known genes, it can also lead to the identification of novel fusion products. To avoid false positive calls, candidate gene fusions were filtered out imposing the following parameters: (i) intrachromosome fusions have to be separated by 100.000 bp distance; (ii) spanning reads on both sides should have at least 13 bp; and (iii) reads map to multiple locations (>2). The annotated gene fusions were then displayed using the OmicCircos software package [20] with respect to genomic position using the hg19 reference.
2.3. Characteristics of Genes Present in Transcript Fusion
The information on genes/lncRNA was retrieved from https://www.ncbi.nlm.nih.gov/gene and https://lncipedia.org/ [21] (version 4.1, May 4, 2017, containing 146,742 human-annotated lncRNAs) and http://cancer.sanger.ac.uk/cosmic and https://cancergenome.nih.gov/.
The presence of fusion transcripts in cancers was searched in the following websites: Pubmed (https://www.ncbi.nlm.nih.gov/pubmed) and TCGA fusion gene data portal (http://54.84.12.177/PanCanFusV2/).
2.4. Functional Analysis
To disclose the molecular pathways associated with CD274/PDCD1LG2 fusion, we retrieved gene expression data from Bossi et al. [10] deposited on GEO repository (GSE65021). Gene set enrichment was investigated by gene set enrichment analysis (GSEA) [22] analyzing seven oncogenic signatures found in our previous studies [10, 11] and including β-catenin, E2F3, EGFR, KRAS, MYC, NOTCH, and p53. To graphically represent the significant gene sets and to display their enrichment significance, we used Enriched Map implemented as a Java plugin for the freely available Cytoscape network visualization and analysis software [23].
2.5. Statistical Analysis
The presence of fusions in long and short PFS as well as of specific fusions was evaluated using the Fisher exact test through GraphPad Prism software package. A P value equal or <0.05 was considered to indicate statistical significance. Differences in PFS between patients harboring or not CD274/PDCD1LG2 fusion among the 26 short PFS patients were assessed using log-rank test and R package survival [24].
3. Results and Discussion
With the aim to disclose the biological features associated with cetuximab sensitivity in RM-HNSCC, we applied an RNA-seq approach through a Pan-Cancer panel to a selected cohort [40 patients treated with platinum- and cetuximab-based combination and having long PFS (n = 14) and short PFS (n = 26)].
Based on the applied workflow of analysis, 27 different fusion transcripts were identified; Figure 1 shows the genomic landscape of the identified transcript fusions that is further detailed in Supplementary Table 1 available online at https://doi.org/10.1155/2017/6870614. Twenty-two out of twenty-seven (81%) fusion transcripts were intrachromosomal and located in neighboring genes while 5 resulted from structural rearrangements and translocations to a different chromosome. The identified transcript fusions involved rearrangements in all, but Chr7, Chr10, Chr18, Chr20, and ChrY, chromosomes; Chr3, Chr11, and Chr22 harbor three different fusions and high level of gains (ratio > 1.5) at 11q13, and their association with poor survival has been described in HNSCC (see [25]); Chr1, Chr2, Chr8, Chr9, Chr14, and Chr19 harbor two fusions. The total number and chromosomal distribution of the identified fusion transcripts are essentially in agreement with data already reported in HNSCC samples [13–15] even if the comparison is difficult due to the different approach adopted (targeted versus whole genome) (see below). In all the HNSCC studies on clinical samples, including the present one, fusion transcripts resulting from translocations are relatively rare while the majority is generated by nonstructural rearrangement mechanisms, such as transcription read-through of neighboring genes or splicing of mRNA molecules. This type of gene fusions is reported to be preferentially derived by genomic instability (see [12] for review of the mechanisms).
Figure 1.

Circos plot of the genomic landscape of gene fusions identified by RNA-seq in our 40 RM-HNSCC samples. The outer ring displays the chromosome ideograms. The fusion transcripts are shown as line arcs linking the two genomic loci.
Twenty-four patients (60%) harbor at least one of the 27 identified transcript fusions; the presence and main characteristics of gene fusions detected in each RM-HNSCC patient of our selected case material are reported in Table 1 (see [10] for clinical pathologic characteristics of the patients). We investigated whether the presence of transcript fusions is associated with long or short PFS under cetuximab treatment: 10/14 (71%) long PFS cases and 14/26 (54%) short PFS harbor at least one fusion; however, the presence or absence of fusions is not significantly related with outcome (Table 2). The chromosomal rearrangements in cancer cells could also lead to multiple fusion events. Fifteen and 9 cases harbor only one transcript fusion and more than one, respectively (Table 2); in detail, 3 patients harbor 2 fusions, 4 patients 3 fusions, 1 patient 4 fusions, and 1 patient 6 fusions; nine fusions are present in two or more patients (Table 1). Since the coexistence of multiple fusions might mirror the extent of aberrations present in the tumor, we investigated whether the presence of more than one fusion is associated with outcome under cetuximab. Five out of the 10 long PFS and 4/14 short PFS cases presented more than one fusion, but this difference did not reach a significant level (Table 2). The accumulation of transcript fusions may be associated with tumor progression; since in our case material the RNA was obtained in 9 cases from samples taken at recurrence/metastasis, we analyzed whether in this subgroup of patients (9/40) the presence/number of transcript fusions was higher. Eight out of 9 recurrent/metastatic cases (89%) harbor at least one fusion compared to 16/31 (52%) cases from primary lesions; although the difference did not reach a significant level (P = 0.06), a trend was clearly appreciable.
Table 1.
Presence and main characteristics of gene fusions detected in each RM-HNSCC patient of our selected case material; see [10] for clinical pathologic characteristics of the patients.
| Sample ID | Gene fusion | “Left” partner | “Right” partner | ||
|---|---|---|---|---|---|
| Gene | Chromosome | Gene | Chromosome | ||
| Short PFS under chemotherapy-cetuximab treatment | |||||
| GU05 | No | ||||
| GU09 | No | ||||
| GU10 | Yes | DLG2 | Chr11 | PICALM | Chr11 |
| NUMA1 | Chr11 | GRIA3 | ChrX | ||
| ZMYM2 | Chr13 | TRIM28 | Chr19 | ||
| GU11 | No | ||||
| GU13 | Yes | CLTC | Chr17 | RPS6KB1 | Chr17 |
| GU14 | Yes | CD274 | Chr9 | PDCD1LG2 | Chr9 |
| GU15 | No | ||||
| GU17 | No | ||||
| GU18 | No | ||||
| GU20 | Yes | CD274 | Chr9 | PDCD1LG2 | Chr9 |
| GU21 | No | ||||
| GU22 | No | ||||
| GU23 | No | ||||
| GU24 | Yes | BMS1P20 | Chr22 | IGLL5 | Chr22 |
| GU25 | Yes | CD274 | Chr9 | PDCD1LG2 | Chr9 |
| GU26 | Yes | FGF12 | Chr3 | MB21D2 | Chr3 |
| GU27 | No | ||||
| GU28 | Yes | METTL13 | Chr1 | DNM3 | Chr1 |
| CTNNA2 | Chr2 | HES1 | Chr3 | ||
| RPS6KA2 | Chr6 | RNASET2 | Chr6 | ||
| MUSK | Chr9 | LPAR1 | Chr9 | ||
| CD274 | Chr9 | PDCD1LG2 | Chr9 | ||
| TRAF3 | Chr14 | ENSG00000259717 | Chr14 | ||
| GU29 | Yes | CD274 | Chr9 | PDCD1LG2 | Chr9 |
| GU30 | Yes | RCSD1 | Chr1 | MPZL1 | Chr1 |
| GU31 | Yes | CD274 | Chr9 | PDCD1LG2 | Chr9 |
| PPP6R3 | Chr11 | MLL | Chr11 | ||
| GU34 | Yes | NUMA1 | Chr11 | GRIA3 | ChrX |
| ZMYM2 | Chr13 | TRIM28 | Chr19 | ||
| GU38 | No | ||||
| GU40 | No | ||||
| GU41 | Yes | PVT1 | Chr8 | ENSG00000253288 | Chr8 |
| GU43 | Yes | CD274 | Chr9 | PDCD1LG2 | Chr9 |
| Long PFS under chemotherapy cetuximab treatment | |||||
| GU04 | Yes | FLNB | Chr3 | ENSG00000245384 | Chr4 |
| GU06 | No | ||||
| GU07 | Yes | METTL13 | Chr1 | DNM3 | Chr1 |
| MUSK | Chr9 | LPAR1 | Chr9 | ||
| ENSG00000231669 | ChrX | MSN | ChrX | ||
| GU08 | Yes | ENSG00000231669 | ChrX | MSN | ChrX |
| GU12 | Yes | C9 | Chr5 | RCOR1 | Chr14 |
| ENSG00000259446 | Chr15 | RYR3 | Chr15 | ||
| IGLV1-40 | Chr22 | IGLL5 | Chr22 | ||
| GU16 | Yes | WDR90 | Chr16 | RHOT2 | Chr16 |
| GU19 | Yes | ANK1 | Chr8 | KAT6A | Chr8 |
| ZBTB7A | Chr19 | MAP2K2 | Chr19 | ||
| GU32 | Yes | ZBTB7A | Chr19 | MAP2K2 | Chr19 |
| TPTE | Chr21 | BAGE2 | Chr21 | ||
| ENSG00000231669 | ChrX | MSN | ChrX | ||
| GU33 | Yes | ENSG00000231121 | Chr12 | NAV3 | Chr12 |
| GU35 | No | ||||
| GU36 | Yes | ZC3H15 | Chr2 | ITGAV | Chr2 |
| PPP6R3 | Chr11 | MLL | Chr11 | ||
| ENSG00000231121 | Chr12 | NAV3 | Chr12 | ||
| PI4KA | Chr22 | CRKL | Chr22 | ||
| GU37 | Yes | ENSG00000231121 | Chr12 | NAV3 | Chr12 |
| GU39 | No | ||||
| GU42 | No | ||||
Table 2.
Summary of the gene fusions detected in patients treated with cetuximab and chemotherapy and selected for the extremities of response (see [10]).
| Patients harboring gene fusions | N (%) | P value§ | |
|---|---|---|---|
| Long PFS (14) | Short PFS (26) | ||
| Absence | 4/14 | 12/26 | 0.3295§ |
| Presence | 10/14 | 14/26 | |
| 1 for each patient | 5/10 | 10/14 | 0.4028§ |
| >1 for each patient | 5/10 | 4/14 | |
| Only mRNA in the fusion | 6/10 | 13/14 | 0.1222§ |
| LncRNA in the fusion | 8/10 | 2/14 | 0.0027§ |
| CD274/PDCD1LG2 fusion | 0/10 | 7/14 | 0.0188§ |
§The P values are reported as the two-sided Fisher exact test.
The 27 transcript fusions involve both mRNA and lncRNA, being 21 mRNA-mRNA, 3 lncRNA-mRNA, 2 mRNA-lncRNA, and 1 lncRNA-lncRNA. The lncRNA-containing fusions are enriched in long PFS patients with 8/10 and 2/14 in long and short PFS cases, respectively, harboring a lncRNA fusion (P = 0.0027). Two fusions involving lncRNA, ENSG00000231669-MSN, and ENSG00000231121-NAV3, were each detected in three long PFS patients. At present, while lncRNAs are relatively well-characterized, being involved in the regulation of numerous cellular processes and being associated with cancer development and progression [26], little is known about their role in lncRNA-containing fusions. The current fusion-detection algorithms and bioinformatics pipelines are focused on recognizing fusion candidates mapping to protein-coding mRNA systematically omitting lncRNA [12]. As a result, only a handful of gene fusions containing lncRNAs has been reported [27]. Some studies highlighted a biological, functional, and even clinical relevance of specific mRNA/lncRNA fusions proving that these lncRNAs might contribute to the aberrant regulation of their partner [12]. The identification of lncRNA-containing fusions was achieved in our case material due to the adopted targeted approach. We selected this approach, instead of RNA-seq used with the 300 HNSCC [13] and the 47 oral squamous cell carcinoma (OSCC) [15], due to the availability of only archival FFPE samples whose RNA-seq analysis may result limited, as recently highlighted in another cancer type by direct comparison of paired frozen and FFPE samples [28].
Several studies of gene fusion networks have found that the majority of fusion genes partner with a single other gene, but it is known that some genes might recombine with multiple partners being the MLL the extreme example, described to fuse with over 60 different partner genes [29]. In our study, MLL was present in a single fusion while IGLL5 recombined with BMS1P20 and IGL1-40 (see Supplementary Table 1).
The distribution and the gene partners of our 27 fusion transcripts were compared with the 382 and the 282 fusions detected by a whole genome approach in HNSCC [13] and OSCC [15], respectively; we recorded a 33% of fusions shared among more patients and no overlap with fusions previously identified in HNSCC/OSCC. The different results could be mainly attributed to the use of a panel that enabled a higher sequencing depth but that was biased toward cancer genes. Despite these differences, one or both gene partners of our 27 fusions was/were present in association with other genes in the 7887 high confidence fusion transcripts identified in 4366 primary tumor samples from 13 tumor types including HNSCC (Supplementary Table 1).
We analyzed the characteristics of the gene partners (see details in Supplementary Table 1) to potentially define the molecular functions of the identified fusions, and we observed that chromatin modifiers (KMTA2-MLL, RCOR1, and KAT6A), kinases (RPS6KA, MUSK, TRIM28, MPZL1, and MAP2K2), and phosphates (LPAR1, PICALM, RPS6KB1, DLG2, PPP6RB, TPTA, and PI4KA) were frequently present.
Worth mentioning, the fusion CD274-PDCD1LG2 was present as single fusion (5/14) or associated with other fusions (2/14) in short PFS patients while it was absent in all long PFS patients (P = 0.0188). CD274-PDCD1LG2 fusion defined a subgroup into short PFS; in fact, by Kaplan-Meier analysis, the median estimates of PFS in patients harboring or not the fusion were 2.2 and 3.4 months, respectively (P = 0.0172 by the log-rank test) (Figure 2). Thus, we investigated the biology behind the CD274-PDCD1LG2 fusion in the short PFS cases analyzing molecular pathways through GSEA. The oncogenic signatures reported in our previous studies [10, 11] were tested for their enrichment in cases harboring CD274-PDCD1LG2 fusion, and as reported in the enrichment map (Figure 3), KRAS (P = 0.00147; NES = 1.62) is enriched in cases with the fusion, while EGFR (P = 10E‐04; NES = −2.04), p53 (P = 10E‐04; NES = −1.83), NOTCH (P = 10E‐04; NES = −1.82), and β-catenin (P = 10E‐04; NES = −2.04) onco-signatures are enriched in cases without the fusion. E2F3 and MYC are not significantly different between cases with the presence or absence of fusion.
Figure 2.
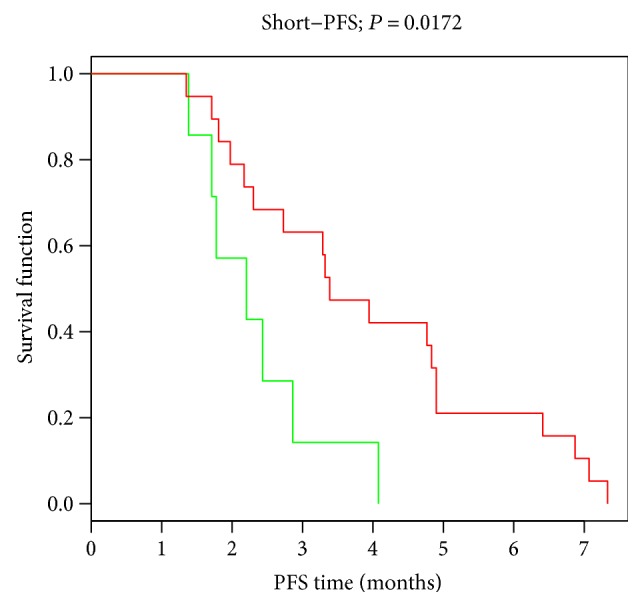
Kaplan-Meier curves showing PFS among patients with the presence or absence of the CD274-PDCD1LG2 fusion transcript. Median PFS: 2.2 months in the group with fusion (n = 7) and 3.4 months in the group not harboring the fusion (n = 19) (P = 0.0172).
Figure 3.
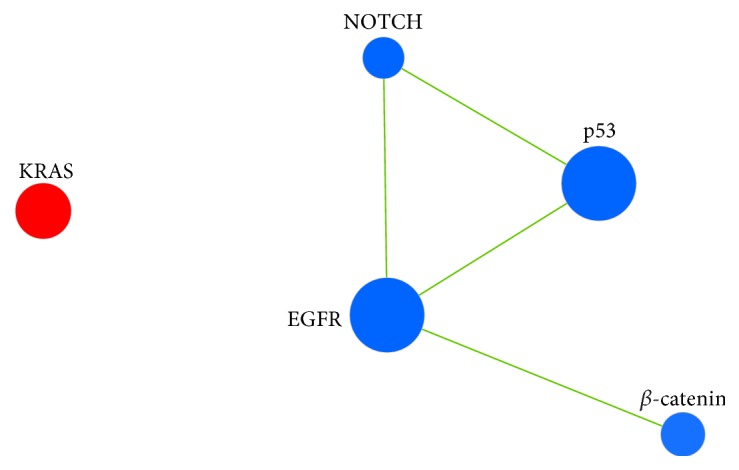
Enrichment map visualizing results of GSEA analysis for cases with the presence/absence of the CD274-PDCD1LG2 fusion. Seven oncogenic signatures inferred from our previous studies [10, 11] were tested, and five resulted a significant difference (oncogenic signature = node). Node size: number of genes in the gene set. Node color: red = enriched in cases harboring the CD274-PDCD1LG2 fusion. Blue = enriched in cases not harboring the fusion. Edges: connect significantly overlapping gene sets (width reflects the degree of the overlap).
Both partner genes are in the 9p24.1 locus, a region of recurrent structural and copy number alterations in hematologic tumors, and this fusion was present in 20% of primary mediastinal large B-cell lymphoma and in lower percent in other lymphomas [30]. Furthermore, in lymphomas, the rearrangement was significantly correlated with overexpression of PDL transcripts [30]. The products of the gene partners of our most frequent fusion transcript, CD274 and PDCD1LG2, also known as programmed death ligand-1 and 2 (PD-L1 and PD-L2), respectively, have been implicated in promoting tumor cell immune evasion acting as negative regulators of antitumor immunity by binding their cognate receptor, PD-1, on cytotoxic T-cells. No data are presently available about this fusion in other types of solid tumors, and our observation that RM-HNSCC cases harboring CD274-PDCD1LG2 fusion have poor prognosis and resistance to cetuximab deserve further analysis and validation in wider series of patients entered/entering in anti-EGFR-targeted trials.
Recently, high PD-L1 expression was identified as a strong prognostic factor of HNSCC patient's worse outcome [31]. Within clinical trials with immune checkpoint inhibitors (CPIs) in RM-HNSCC, higher response rates were noted in patients with higher PD-L1 expression [32, 33]. However, other PD-1 ligands could be crucial in determining the efficacy of CPIs. As it has been recently showed, the coexpression of both PD-L1 and PD-L2 in tumoral specimens of patients treated with pembrolizumab correlated with higher responsiveness to this drug [34]. Further investigation is required into the role of the CD274-PDCD1LG2 fusion as pharmacogenomics biomarker not only as a prognosticator in RM-HNSCC patients but also as a possible predictive biomarker of immunotherapy response to better select patients for a tailored treatment approach.
4. Conclusions
Transcript fusions resulting from chromosomal rearrangements are genetic alterations well-known from decades and they can in oncology (i) serve as diagnostic markers, (ii) provide insight into tumor biology, and (iii) serve as specific therapeutic targets. By an RNA-seq approach through a dedicated Pan-Cancer panel, we investigated the presence and role of transcript fusions as potential pharmacogenomic markers of RM-HNSCC patients' response to cetuximab and platinum-based chemotherapy. We identified 27 different fusion transcripts and observed significant associations between lncRNA-containing fusions and patient's better outcome and the presence of the CD274-PDCD1LG2 fusion and worst outcome. These observations deserve the testing in clinical trials but if confirmed, as seen with other gene fusions in other tumor entities, they could open the way to a more tailored therapeutic approach. Further on, combination treatments with only immune therapeutic approaches or with targeted agents or classic chemotherapy could be of major importance to increase efficacy and outcome in RM-HNSCC patients. In this regard, our findings are important to be acknowledged and could lead to further researches and new trial designs.
Supplementary Material
Supplementary Table 1. Details of the 27 different fusion transcripts that identified based on the applied workflow of analysis.
Acknowledgments
This work was partially supported by Associazione Italiana Ricerca sul Cancro (AIRC IG 14750 to Silvana Canevari and AIRC IG 18519 to Loris De Cecco). This project was supported by European Union's Horizon 2020 Research and Innovation Programme under Grant Agreement no. 689715. The authors thank Illumina's bioinformatics and technical personnel for the support in the analysis on BaseSpace.
Contributor Information
Silvana Canevari, Email: silvana.canevari@istitutotumori.mi.it.
Loris De Cecco, Email: loris.dececco@istitutotumori.mi.it.
Conflicts of Interest
The authors certify that there is no conflict of interests with any financial organizations regarding the material discussed in this study.
Authors' Contributions
Paolo Bossi and Marco Siano equally contributed as first authors. Lisa Licitra and Loris De Cecco equally contributed as last authors.
References
- 1.Cacicedo J., Navarro A., Alongi F., et al. The role of re-irradiation of secondary and recurrent head and neck carcinomas. Is it a potentially curative treatment? A practical approach. Cancer Treatment Reviews. 2014;40(1):178–189. doi: 10.1016/j.ctrv.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Liao C. T., Chang J. T., Wang H. M., et al. Salvage therapy in relapsed squamous cell carcinoma of the oral cavity: how and when? Cancer. 2008;112(1):94–103. doi: 10.1002/cncr.23142. [DOI] [PubMed] [Google Scholar]
- 3.Zafereo M. Surgical salvage of recurrent cancer of the head and neck. Current Oncology Reports. 2014;16(5):p. 386. doi: 10.1007/s11912-014-0386-0. [DOI] [PubMed] [Google Scholar]
- 4.Vermorken J. B., Mesia R., Rivera F., et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. The New England Journal of Medicine. 2008;359(11):1116–1127. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 5.Bossi P., Resteghini C., Paielli N., Licitra L., Pilotti S., Perrone F. Prognostic and predictive value of EGFR in head and neck squamous cell carcinoma. Oncotarget. 2016;7(45):74362–74379. doi: 10.18632/oncotarget.11413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Licitra L., Mesia R., Rivera F., et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Annals of Oncology. 2011;22(5):1078–1087. doi: 10.1093/annonc/mdq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Licitra L., Störkel S., Kerr K. M., et al. Predictive value of epidermal growth factor receptor expression for first-line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: analysis of data from the EXTREME and CRYSTAL studies. European Journal of Cancer. 2013;49(6):1161–1168. doi: 10.1016/j.ejca.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 8.Kang H., Kiess A., Chung C. H. Emerging biomarkers in head and neck cancer in the era of genomics. Nature Reviews Clinical Oncology. 2015;12(1):11–26. doi: 10.1038/nrclinonc.2014.192. [DOI] [PubMed] [Google Scholar]
- 9.Tonella L., Giannoccaro M., Alfieri S., Canevari S., De Cecco L. Gene expression signatures for head and neck cancer patient stratification: are results ready for clinical application? Current Treatment Options in Oncology. 2017;18(5):p. 32. doi: 10.1007/s11864-017-0472-2. [DOI] [PubMed] [Google Scholar]
- 10.Bossi P., Bergamini C., Siano M., et al. Functional genomics uncover the biology behind the responsiveness of head and neck squamous cell cancer patients to cetuximab. Clinical Cancer Research. 2016;22(15):3961–3970. doi: 10.1158/1078-0432.CCR-15-2547. [DOI] [PubMed] [Google Scholar]
- 11.De Cecco L., Giannoccaro M., Marchesi E., et al. Integrative miRNA-gene expression analysis enables refinement of associated biology and prediction of response to cetuximab in head and neck squamous cell cancer. Genes. 2017;8(1):p. 35. doi: 10.3390/genes8010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Latysheva N. S., Babu M. M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Research. 2016;44(10):4487–4503. doi: 10.1093/nar/gkw282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshihara K., Wang Q., Torres-Garcia W., et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2015;34(37):4845–4854. doi: 10.1038/onc.2014.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daly C., Castanaro C., Zhang W., et al. FGFR3-TACC3 fusion proteins act as naturally occurring drivers of tumor resistance by functionally substituting for EGFR/ERK signaling. Oncogene. 2017;36(4):471–481. doi: 10.1038/onc.2016.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo T., Gaykalova D. A., Considine M., et al. Characterization of functionally active gene fusions in human papillomavirus related oropharyngeal squamous cell carcinoma. International Journal of Cancer. 2016;139(2):373–382. doi: 10.1002/ijc.30081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng Y., Wang Y., Li J., Chang I., Wang C. Y. A novel read-through transcript JMJD7-PLA2G4B regulates head and neck squamous cell carcinoma cell proliferation and survival. Oncotarget. 2017;8(2):1972–1982. doi: 10.18632/oncotarget.14081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Persson M., Andrén Y., Mark J., Horlings H. M., Persson F., Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(44):18740–18744. doi: 10.1073/pnas.0909114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Argiris A., Li Y., Forastiere A. Prognostic factors and long-term survivorship in patients with recurrent or metastatic carcinoma of the head and neck. Cancer. 2004;101(10):2222–2229. doi: 10.1002/cncr.20640. [DOI] [PubMed] [Google Scholar]
- 19.Kim D., Salzberg S. L. TopHat-fusion: an algorithm for discovery of novel fusion transcripts. Genome Biology. 2011;12(8, article R72) doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y., Yan C., Hsu C. H., et al. OmicCircos: a simple-to-use R package for the circular visualization of multidimensional omics data. Cancer Informatics. 2014;13:13–20. doi: 10.4137/CIN.S13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Volders P. J., Helsens K., Wang X., et al. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Research. 2013;41(D1) Database issue:D246–D251. doi: 10.1093/nar/gks915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian A., Tamayo P., Mootha V. K., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merico D., Isserlin R., Stueker O., Emili A., Bader G. D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5(11, article e13984) doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Therneau T. M., Grambsch P. M. Modeling Survival Data: Extending the Cox Model. New York, NY, USA: Springer; 2000. [Google Scholar]
- 25.Chen Y., Chen C. DNA copy number variation and loss of heterozygosity in relation to recurrence of and survival from head and neck squamous cell carcinoma: a review. Head & Neck. 2008;30(10):1361–1383. doi: 10.1002/hed.20861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolha L., Ravnik-Glavač M., Glavač D. Long noncoding RNAs as biomarkers in cancer. Disease Markers. 2017;2017:14. doi: 10.1155/2017/7243968.7243968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu Y., Ren S., Jing T., et al. Clinical utility of a novel urine-based gene fusion TTTY15-USP9Y in predicting prostate biopsy outcome. Urologic Oncology: Seminars and Original Investigations. 2015;33:384.e9–384.e20. doi: 10.1016/j.urolonc.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 28.Esteve-Codina A., Arpi O., Martinez-García M., et al. A comparison of RNA-seq results from paired formalin-fixed paraffin-embedded and fresh-frozen glioblastoma tissue samples. PLoS One. 2017;12(1, article e0170632) doi: 10.1371/journal.pone.0170632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer C., Hofmann J., Burmeister T., et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013;27(11):2165–2176. doi: 10.1038/leu.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Twa D. D., Chan F. C., Ben-Neriah S., et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood. 2014;123(13):2062–2065. doi: 10.1182/blood-2013-10-535443. [DOI] [PubMed] [Google Scholar]
- 31.Müller T., Braun M., Dietrich D., et al. PD-L1: a novel prognostic biomarker in head and neck squamous cell carcinoma. Oncotarget. 2017;8(32):52889–52900. doi: 10.18632/oncotarget.17547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bauml J., Seiwert T. Y., Pfister D. G., et al. Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: results from a single-arm, phase II study. Journal of Clinical Oncology. 2017;35(14):1542–1549. doi: 10.1200/JCO.2016.70.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferris R. L., Blumenschein G., Jr., Fayette J., et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. The New England Journal of Medicine. 2016;375(19):1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yearley J. H., Gibson C., Yu N., et al. PD-L2 expression in human tumors: relevance to anti-PD-1 therapy in cancer. Clinical Cancer Research. 2017;23(12):3158–3167. doi: 10.1158/1078-0432.CCR-16-1761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Details of the 27 different fusion transcripts that identified based on the applied workflow of analysis.