

Abstract
Increasing evidence is available showing the importance of the FAK (focal adhesion kinase) protein level in the migration and homeostasis of intestinal cells. TGFβ (transforming growth factor beta) modulates FAK protein expression in a complex fashion not only by inducing the activation of p38 and Smad signaling resulting in increased fak promoter activity and increased FAK protein levels, but also by activating ERK (extracellular signal regulated kinases), p38, and the Smad pathway. We show that the blockade of ERK signaling by a specific MEK (MAPK kinase) inhibitor attenuates TGFβ–induced FAK mRNA stability and reduces FAK protein levels in rat IEC-6 intestinal epithelial cells. The mTOR (mammalian target of rapamycin)-specific inhibitor rapamycin and small interfering RNAs for mTOR and p70S6 kinase also block TGFβ–induced FAK protein synthesis. Furthermore, we have found that a TGFβ–induced increase in wound closures in monolayers of these cells is abolished in the presence ERK or mTOR inhibition. Thus, TGFβ also modulates FAK protein levels in cultured rat IEC-6 intestinal epithelial cells via ERK activation, acting at the transcriptional level to complement Smad signaling and at on the translational level via the mTOR pathway downstream of ERK, which in turn promotes intestinal epithelial cell migration.
Keywords: Transforming growth factor β (TGFβ), Focal adhesion kinase(FAK), Extracellular signal regulated kinases (ERK), Mammalian target of rapamycin (mTOR), mRNA, Rat IEC-6 intestinal epithelial cells
Introduction
The mammalian intestinal epithelium constantly regulates the balance of its cell production and cell loss, with complete mucosal renewal in less than a week (Hall et al. 1994). Such mechanisms are integral not only to normal mucosal biology, but also to pathological processes such as ulcer healing and neoplastic transformation in the gastrointestinal mucosa. The transforming growth factor β (TGFβ) pathway plays a major role in the maintenance of epithelial homeostasis (Tanigawa et al. 2005; Sturm and Dignass 2008).
TGFβ has been reported to promote the migration and arrest of the growth of proliferating intestinal cells and to induce their terminally differentiated state, acting here as a tumor suppressor (Barnard et al. 1989, 1991; Sancho et al. 2004). This factor may also promote epithelial cell monolayer restitution after injury by stimulating migration (Tanigawa et al. 2005). TGFβ classically signals through the canonical Smad signal pathway to elicit transcriptional responses. However, increasing evidence demonstrates the importance of non-Smad TGFβ-induced signaling mechanisms that complement the canonical Smad pathway (Moustakas and Heldin 2005; Lamouille and Derynck 2007). Such non-Smad signals include the extracellular signal regulated kinases (ERK) and p38 mitogen-activated protein kinases (MAPK) in intestinal cells (Hartsough and Mulder 1995) with accumulating evidence for cross-talk between them and with the Smad-mediated pathway (Yue et al. 1999; Walsh et al. 2003).
The intracellular tyrosine kinase FAK (focal adhesion kinase) is found prominently within focal adhesions and serves as a convergent integration point in adhesion and cell migration between integrin signaling from the extracellular matrix (ECM) and receptor-mediated signaling activated by growth factors such as TGFβ (Parsons et al. 2000; Mitra et al. 2005). Tyrosine phosphorylation of FAK enables FAK binding to other signaling molecules via SH2 and SH3 (Src homology 2 and 3) domains and has been linked to the regulation of diverse cellular events including cell survival, motility, and cell cycle progression (Hanks et al. 2003; Parsons 2003). Integrin-mediated FAK phosphorylation may be necessary for the facilitation of human differentiated intestinal epithelial cell survival by ERK signaling (Bouchard et al. 2007). FAK phosphorylation is also able to suppress apoptosis induced by TNF-α (tumor necrosis factor alpha) and cycloheximide in rat intestinal IEC-6 cells (Zhang et al. 2006). Thus, most studies have focused on understanding the activation of FAK at the posttranslational level. However, increasing evidence demonstrates the importance of small changes in FAK mRNA and protein levels in, for example, migrating intestinal epithelial cells (Yu et al. 2000; Basson et al. 2006), human gastric and colonic ulcers (Walsh et al. 2008b), and human colon cancer cells (Nakagawa et al. 1998; Agochiya et al. 1999).
TGFβ increases FAK at the protein level via independent Smad and p38 MAPK signals (Walsh et al. 2008b). Indeed, TGFβ seems to induce fak transcription effect by inducing fak promoter activity through several Smad-binding elements in the fak promoter sequence in a Smad-dependent fashion. However, whether ERK signaling by TGFβ also influences FAK levels is not known.
The ERK pathway is invoked in various cell types by diverse extracellular stimuli. Stimulation of this pathway, downstream of Ras, activates a series of protein kinases and finally activates ERK1/2, which in turn phosphorylates downstream targets involved in cellular events such as cell proliferation, differentiation, survival, and motility (Kohno and Pouyssegur 2006). TGFβ rapidly induces ERK1 activity in intestinal epithelial cells, and this inhibits growth (Hartsough and Mulder 1995). However, the final growth outcome of epithelial cells may be modulated by synergistic pathways (Hartsough and Mulder 1995). Recent studies suggest that ERK modulates translation, in addition to its more classical influence on transcription through the mammalian target of rapamycin (mTOR) pathway (Ma et al. 2005). Targets for mTOR are proteins involved in controlling mRNA translation, including the ribosomal protein S6 kinases (p70S6K) and the initiation factor 4E-binding proteins (4E-BPs; Averous and Proud 2006). mTOR signaling is inhibited by the tumor suppressors TSC1/2, and ERK-dependent phosphorylation of TSC1/2 leads to its dissociation from mTOR enabling translation in cooperation with the PI3K (phosphatidylinositol 3-kinase)/Akt pathway (Ma et al. 2005). Furthermore, recent studies link mTOR signaling with intestinal cell migration by amino acid stimulation in a PI3K-dependent fashion (Rhoads et al. 2006). Interestingly, another recent study has connected the overexpression of the adaptor protein Eps8 to the upregulation of FAK expression via mTOR activation in human colon cancer cell lines (Maa et al. 2007).
In light of previous data showing that TGFβ stimulates fak promoter activity and FAK protein expression, and against the background of current studies demonstrating mechanistic links between TGFβ, ERK, and mTOR in the translational control of the regulation of gene expression, which in turn is crucial for cell growth, survival, and homeostasis (Mamane et al. 2006), we have sought to determine whether ERK and mTOR are involved in the TGFβ regulation of FAK expression in cultured rat IEC-6 intestinal epithelial cells. We have observed that TGFβ treatment results in the rapid phosphorylation of mTOR and its effectors p70S6K and 4E-BP1 (initiation factor 4E-binding proteins), whereas inhibition of mTOR activation results in a reduced FAK protein level and wound closures. Consistent with this observation, ERK inhibition blocks mTOR activation, which in turn abolishes the stimulation of FAK protein levels and wound closures by TGFβ. Therefore, the TGFβ-induced FAK protein level modulation via ERK/mTOR promotes rat intestinal epithelial IEC-6 cell migration. Furthermore, we have found that FAK mRNA stability increases with TGFβ treatment in an ERK-dependent manner. Taken together with our previous observations (a, Walsh et al. 2008b), these results trace divergent and reconvergent TGFβ pathways inducing transcriptional regulation through ERK complementing Smad signaling and translational regulation through ERK/mTOR signaling for the modulation of FAK expression implicated as being important for epithelial intestinal cell homeostasis and migration.
Materials and methods
Cell culture
IEC-6 cells, originally derived from rat jejunum (Quaroni et al. 1979), were purchased from the American Tissue Culture Collection (Rockville, Md.) and maintained by following ATCC recommendations. Cells were cultured in tissue culture flasks and were studied in dishes pre-coated with type I Collagen (Sigma, St. Louis, Mo.) as previously described (Basson et al. 1992). Subconfluent cells were serum-deprived for 6 h before treatment with TGFβ1 (1 ng/ml: R&D Systems, Minneapolis, Minn.; Sigma) in medium containing 0.5% FBS (fetal bovine serum) for a further 24 h before lysis. For pharmacological signal inhibition, cells were pre-treated with PD98059 (10 μM), actinomycin D (10 μg/ml), cycloheximide (5 μg/ml), rapamycin (5–100 ng/ml), or appropriate vehicle controls for 1 h before the addition of TGFβ. In all cases, appropriate vehicle controls were used where appropriate. TGFβ was dissolved in a stock solution of 4 mM HCl, 0.2% BSA (bovine serum albmunin) solution following manufacturer’s suggestion and then used at a 1:20,000 dilution in cell culture medium for actual experimentation. This resulted in changing the composition of the cell culture medium by effectively adding 0.08 μM HCl and 0.00004% BSA. The pH of the HEPES-buffered cell culture medium did not change discernibly after the addition of this vehicle in preliminary studies, and the addition of 0.00004% BSA to the culture medium, which contained 0.5% FBS, was felt not to be biologically significant. Accordingly, a separate vehicle control was not used for the addition of TGFβ.
Migration assay
A circular wound model was used for the quantification of epithelial sheet migration. We used a pipette tip to create small circular wounds in confluent IEC-C cell monolayers on collagen I substrates and measured wound closure by quantification of photomicrographs of the original wound and of the same wound after 24 h. Images obtained were analyzed with the Kodak 1D Scientific Imaging computer program.
Western blot analysis
Cells were rinsed twice with ice-cold phosphate-buffered saline and then lysed in buffer containing 10 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg/ml aprotinin, and 1 mM sodium ortho vanadate (all from Sigma). Equal amounts of protein (10–20 μg; BCA, Pierce, Rockford, Ill.) were resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. After being blocked, membranes were incubated with the following antibodies overnight: total FAK (clone 4.47, Upstate Biotechnolgy, Mass.), total p70S6K (Upstate Biotechnolgy), total mTOR, total p44/42 MAP kinase, phospho-mTOR, phospho- p70S6K, and phospho-p44/42 MAPK (all Cell Signaling Technology, Mass.), with α-tubulin (EMD Chemicals, Gibbstown, N.J.), or D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Biodesign, Saco, Me.) as loading controls. After incubation with the appropriate secondary antibody, protein bands were visualized by enhanced chemiluminescence (ECL, GE Health Care, Little Chalfont, Buckinghamshire, England). Imaging and analysis of band density were performed on a Kodak 440CF Image Station. All exposures used for densiometric analysis were within the linear range of the system.
Transfection of small interfering RNA
To inhibit mTOR and p70S6K expression, IEC-6 cells were transfected with small interfering RNA (siRNA). The siRNA duplexes targeted to rat mTOR (simTOR: 5′CCC AGC UUU GUC AUG CCU dT dT 3′) and to p70S6K (GGA GAA CUA TTT AUG CAG UUA dT dT 3′) and their complementary RNA strands harboring a dT dT tail at the 3′end were synthesized by Dharmacon (Lafayette, Colo.). Non-targeted siRNA duplex (NT1) used as a control was purchased from Dharmacon. One day prior transfection, cells were seeded in type-I-collagen-coated six-well plates (2×105 cells/well). Transfection of siRNAs was performed by using Oligofectamine and Plus Reagent (Invitrogen) according to the manufacturer’s protocol. At 24 h after transfection, cells were subjected to Western blot analysis.
Dual luciferase reporter assay for FAK promoter activity
To assemble the FAK promoter-luciferase construct, we first obtained a human genomic DNA-BAC clone from BACPAC Resources (Children’s Hospital of the Oakland Research Institute, Oakland, Calif.) with an insert containing the human FAK promoter, exon 1, and intron 1 sequences as indicated in the GenBank data base. The BAC plasmid was cut with SacI and StuI (New England Biolabs, Ipswich, Mass.) releasing a fragment of 1.65 kb that was subcloned into the pBluescript II SK (+) vector (Stratagene, La Jolla, Calif.) for amplification. The pBSK-FAK-promoter clone was subsequently cut with NarI, end-filled, and then cut with KpnI (New England Biolabs) to produce a 1.3-kb fragment that was subcloned into a pGL2 Basic vector (Promega, Madison, Wis.) to generate the FAK-promoter-luciferase construct used in the Dual Luciferase Reporter Assay (Promega). The 1.3-kb fragment was sequenced by using T3 and T7 primers (New England Biolabs) to verify that it matched the FAK genomic sequence in GenBank (not shown). For transfections, cells were seeded in type-I-collagen-coated six-well plates (2×105 cells/well) one day before transfection with the pGL2-fused FAK promoter construct by using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. For normalization of luciferase activity, a pRL-TK contol vector containing the Herpes simplex virus thymidine kinase promoter encoding Renilla luciferase was used together with the pGL2 basic and recombinant construct. After 24 h, cells were analyzed by using the dual luciferase reporter assay system kit according to the manufacturer’s instructions (Promega). Luminescence was measured with an AutoLumat Plus LB 953 Luminometer (Berthold Technologies, Germany).
Real-time reverse transcription with polymerase chain reaction for FAK mRNA expression
Reverse transcription with polymerase chain reaction (RT-PCR) for FAK was performed as described previously (Basson et al. 2006). Briefly, RNA was isolated from IEC-6 cells, after treatment with PD98059 (10 μM), actinomycin D (10 μg/ml), and respective control agent for 1 h before the addition of TGFβ, by using Trizol reagent (Invitrogen). Total RNA was used for the synthesis of first-strand cDNA by using the Super Script III First Strand Synthesis Super Mix for qRT-PCR (Invitrogen). RT-PCR was performed with rat FAK primer pairs giving a 167-bp product and for ribosomal gene (18S) designed with the help of MIT Prime3 online primer designer protocol. The primers for FAK, viz., sense: 5′ ATT GCT GCC TCG GAA TGT TCT 3′ and antisense: 5′ GCT GAG GTA AAA CGT CGA AAA 3′, and the primers for 18S, viz., sense: 5′ CGG CTA CCA CAT CCA AGG AA 3′and antisense: 5′ GCT CGA ATT ACC GCG GCT 3′ (Integrated DNA Technologies, Coralville, Iowa), were used for the PCR amplification with SYPR Green PCR Master Mix (Applied Biosystems) by using the ABI 7700 Sequence Detection System. Relative FAK mRNA levels were determined by the comparative CT Method (Applied Biosystems User Manual) after ascertaining that the efficiencies of the control 18S and FAK primers were similar. The resulting number of mRNA molecules was calculated by negative log transformation of the differences between CT for 18S and FAK.
Statistical analysis
Results are expressed as mean±SEM. The statistical significance of differences between means of samples in each experiment was assessed by a Student’s t-test. P values less than 0.05 were considered significant.
Results
TGFβ-mediated increase of FAK protein level is abolished by the MEK inhibitor PD98059, but not fak promoter activity in IEC-6 cells
Although TGFβ alters FAK levels in IEC-6 cells via a Smad-dependent and p38-dependent induction of fak promoter activity, TGFβ can also signal via ERK (Blanchette et al. 2001; Sanchez-Capelo 2005). We therefore examined the effect of ERK blockade on TGFβ induction of FAK, pretreating subconfluent cells for 1 h with the ERK kinase (MEK) inhibitor PD98059 at various concentrations. As shown in Fig. 1a, FAK protein levels increased similarly after a 24-h treatment with 0.1, 1, or 10 ng/ml TGFβ compared with dimethylsulfoxide (DMSO)-treated controls (143–162±17–23%, *P=0.03, n=3). TGFβ stimulated ERK phosphorylation similarly over the range of 0.1, 1, and 10 ng/ml (Fig. 1b, bars 1–4, *P≤0.01, n=3). ERK blockade by the MEK inhibitor PD98059 over the range of 1–20 μM prevented TGFβ-stimulated ERK phosphorylation (Fig. 1b, bars 5–10, n=3), suggesting that ERK contributed to the effects of TGFβ on FAK protein levels.
Fig. 1.
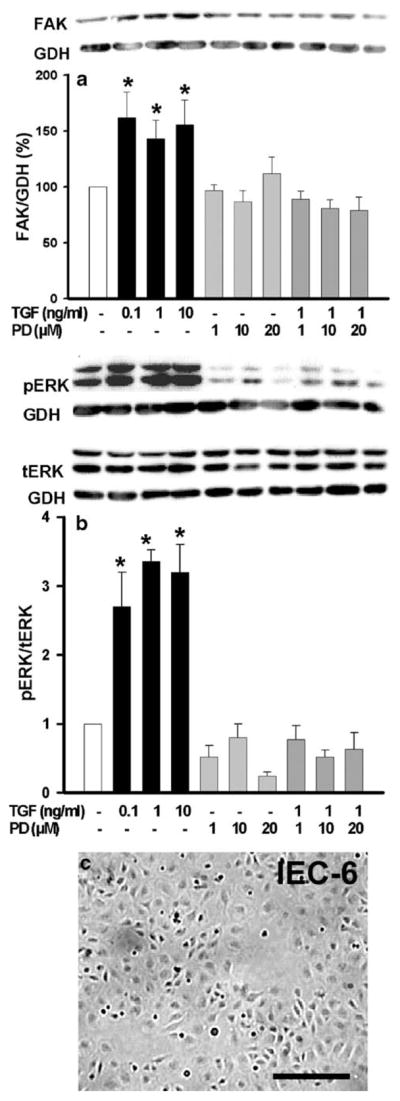
MEK signal inhibition abolishes TGFβ–mediated increase of ▶ immunoreactive FAK protein levels. a After a 24-h exposure to TGFβ at 0.1 ng/ml, 1 ng/ml, or 10 ng/ml and a 1-h pretreatment with the MEK inhibitor PD98059 at 1 μM, 10 μM, or 20 μM, immunoreactive FAK protein was analyzed in IEC-6 cells (*P≤0.03, n=3). A typical Western blot for total FAK and GAPDH (GDH) is shown above. b TGFβ-stimulated ERK phosphorylation (*P≤0.03, n=3) is abolished by pretreatment with the MEK inhibitor PD98059 at 1 μM, 10 μM, or 20 μM. A typical Western blot probed for phosphorylated ERK, total ERK, and GAPDH (GDH) is shown above. c Typical microscopic image of a subconfluent rat intestinal epithelial IEC-6 monolayer used for TGFβ treatments. Bar 150 μm
Since the blocking of Smad or p38 signaling prevented the induction of fak promoter activity by TGFβ, we further investigated whether ERK signaling was also necessary for FAK induction at the transcriptional level. IEC-6 cells transfected with a pGL2-FAK promoter luciferase construct exhibited an ~1.5-fold increase in luciferase activity after TGFβ treatment, which was in the range (less than twofold) of another published observation of TNF-alpha-induced FAK promoter activity (Golubovskaya et al. 2004). However, transfected cells pretreated with the MEK inhibitor exhibited a similar ~1.6-fold increase in luciferase activity in response to TGFβ (Fig. 2, P=0.007 vs. non-treated respective control, n=4 for each). Therefore, the induction of fak promoter activity under TGFβ control seemed to be ERK-independent, whereas FAK protein expression required ERK activity.
Fig. 2.
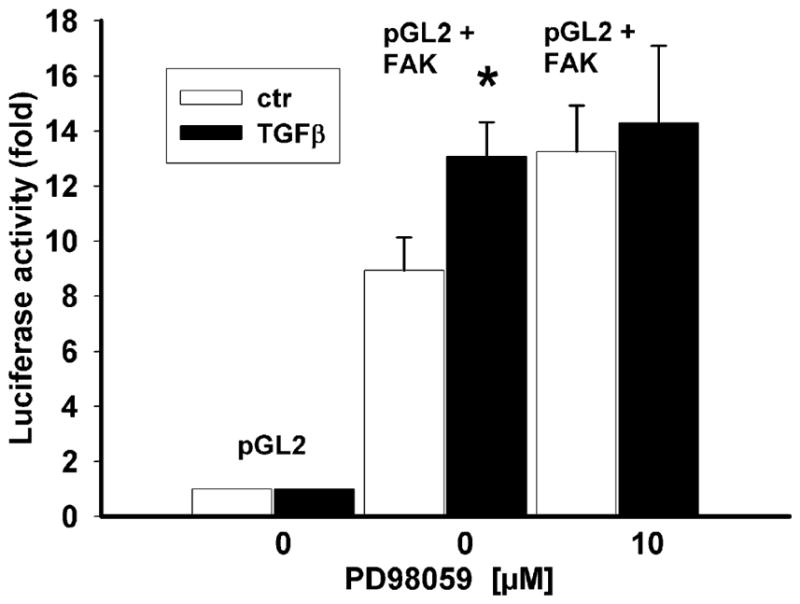
TGFβ-induced fak gene transcription and role of ERK signaling. Activity of the fak promoter-luciferase construct (pGL2+FAK), normalized to rinella and expressed as percent of the respective non-treated control, is significantly higher in TGFβ–treated cells (P=0.007 vs. non-treated respective controls, n=4 for each) and in cells pretreated with the MEK-specific inhibitor PD98059 (ctr control)
FAK mRNA stability, but not protein stability, was rescued by TGFβ treatment dependent on ERK signaling in IEC-6 cells
TGFβ regulates the mRNA stability of genes such as Furin (Blanchette et al. 2001), PTHrP (Sellers et al. 2002), and PTEN (Chow et al. 2007). We therefore investigated whether TGFβ affected FAK mRNA stability. FAK mRNA stability in IEC-6 cells was measured after treatment with TGFβ, the MEK inhibitor, and actinomycin D, a transcriptional inhibitor. RT-PCR analysis of the samples (Fig. 3a) demonstrated that the addition of TGFβ resulted in a twofold (2.3-fold) increase in FAK mRNA stability, increasing the half-life from 22 min to 50 min. The amount of FAK mRNA at 30 min after transcriptional blockade with actinomycin D was significantly higher in DMSO-vehicle-treated cells exposed to TGFβ compared with the amount of residual FAK mRNA in control cells treated with the vehicle alone (n=4, *P=0.003). In contrast, the amount of residual FAK mRNA in cells treated with PD98059 did not differ from vehicle control values regardless of TGFβ treatment. Thus, inhibition of MEK signaling negated this TGFβ effect on FAK mRNA stability. We next examined whether TGFβ also affected FAK protein stability. As shown in Fig. 3b,, FAK protein level was highly reduced after a 12-h cycloheximide treatment. Neither TGFβ nor PD98059 treatment altered the rate of protein decay after cycloheximide blockade of new protein synthesis. These results suggested that TGFβ-mediated ERK signaling contributed to an increase in FAK mRNA stability, but did not prevent FAK protein degradation.
Fig. 3.

Regulation of FAK mRNA stability and translation by TGFβ and the MEK-specific inhibitor PD98059 (PD). a FAK mRNA stability was increased by TGFβ. After treatment with the transcription inhibitor actinomycin D, TGFβ was added to cell cultures, and RNA was extracted at the indicated time-points. The amount of transcript was analyzed by RT-PCR in four separate experiments (ctr control). The amount of transcript after TGFβ–treatment was significantly different from controls at the 30-min time point (*P=0.003). Ribosomal gene (18S) transcript was utilized as an internal control for amplification efficiency. b Cells were treated with TGFβ and PD98059 as indicated in the presence of cycloheximide for 12 h. FAK and α–tubulin were detected by Western blotting. FAK expression was significantly lowered (n=4, P<0.00005) at 12 h after cycloheximide treatment, but no differences were observed in FAK decay among the cells treated with TGFβ and PD98059
TGFβ-mediated increase in FAK protein level is attenuated by inhibition of the mTOR signal pathway in IEC-6 cells
ERK signaling activates the mTOR pathway in some cell systems (Ma et al. 2005; Bermudez et al. 2008). Furthermore, evidence has been provided for a potential translational regulatory effect of mTOR on FAK protein synthesis in the absence of TGFβ (Maa et al. 2007). We therefore investigated the effect of mTOR blockade on the TGFβ induction of FAK. Although TGFβ-treated control cells in this experiment exhibited a 33±2% increase in FAK protein (n=6, P=0.05), the mTOR-specific blocker rapamycin (5–100 nM) prevented the TGFβ stimulation of FAK protein expression (Fig. 4a). We next utilized specific siRNA to reduce mTOR or its common downstream effector p70S6K (Fig. 4b–c). Transfection of IEC-6 cells with 100 nM siRNA for mTOR decreased mTOR protein to 47±4% of that in the non-targeting siRNA (NT1)-treated control cells (P=0.0004), whereas transfection with 100 nM siRNA for p70S6K decreased p70S6K to 46±9% of control levels (P=0.007). As shown in Fig. 4d–e, TGFβ treatment in control transfections (NT1 siRNA) increased FAK protein levels by 60±4% (n=5, *P=0.008) over the basal level. Reduction of mTOR or its effector p70S6K with 100 nM of specific siRNA completely prevented the increase in FAK protein expression caused by TGFβ.
Fig. 4.

Inhibition of the mTOR signaling pathway abolishes TGFβ mediated FAK protein expression. a Treatment of IEC-6 cells with the mTOR-specific inhibitor rapamycin completely inhibits the TGFβ-induced increase in FAK expression at the indicated concentrations. Bottom Densiometric analysis of FAK, expressed as a ratio to α-tubulin and then as a percent of the DMSO control (n=6, *P<0.05). Top Typical Western blots probed for FAK and α–tubulin. b, c Typical Western blots probed for FAK, mTOR and p70S6K. Reducing mTOR and p70S6K protein by specific siRNAs (100 nM) attenuates TGFβ–mediated FAK protein expression. Treatment with TGFβ in the control transfection increased immunoreactive FAK protein in IEC-6 cells. mTOR and p70S6K reduction completely inhibited the TGFβ–induced increase in FAK expression (NT non-targeting). d, e Densitometric analysis of FAK, expressed as a ratio of α-tubulin and as a percentage of the NT1 control
TGFβ-stimulated phosphorylation of mTOR and its targets p70S6 kinase and 4E-BP1 kinase
Increasing evidence demonstrates that TGFβ can increase protein synthesis though the mTOR pathway (Le Pabic et al. 2005; Lamouille and Derynck 2007), and indeed, mTOR signaling in response to other stimuli has been implicated in the control of protein synthesis through the phosphorylation of p70S6 kinase and 4E-BP1 kinase (Averous and Proud 2006; Mamane et al. 2006). Because TGFβ increases FAK protein synthesis, we explored the connection of TGFβ with mTOR and its effectors p70S6 kinase and 4E-BP1 kinase. TGFβ induced activation of mTOR, p70S6 kinase, and 4E-BP1 (Fig. 5) within 5 min, with maximum phosphorylation (1.19-fold increase) of mTOR (Fig. 5a) within 5 min (n=14, *P<0.05, #P=0.001). TGFβ induced the phosphorylation of 4E-BP1 (Fig. 5b) with a peak (1.19-fold increase) at 15 min (n=13, *P<0.04), and the phosphorylation of p70S6 kinase (Fig. 5c) with a peak (1.18-fold increase) at 30 min (n=14, *P<0.02), corroborating the downstream signaling effect on translation for mTOR controlled by TGFβ.
Fig. 5.
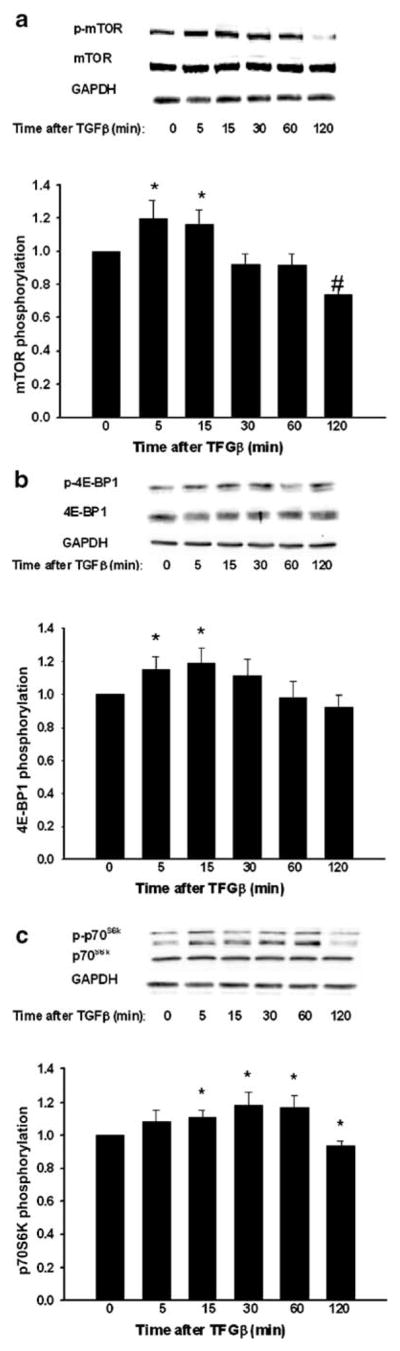
TGFβ stimulates the phosphorylation of mTOR, p70S6K, and 4E-BP1 in cells treated for variable lengths of time before lysis. a Top Typical Western blot probed for phosphorylated mTOR, stripped, and reprobed for total mTOR and GAPDH. Bottom Densitometric results as the ratio of phosporylated to total mTOR normalized to GAPDH (n=10, *P<0.05, #P<0.001). Control phosphorylation was defined at 1.0. b Top Typical Western blot probed for phosphorylated 4E-BP1, stripped, and reprobed for total 4E-BP1 and GAPDH. Bottom Densitometric results as the ratio of phosphorylated to total 4E-BP1 normalized to GAPDH (n=10, *P<0.04). Control phosphorylation was defined at 1.0. c Top Typical Western blot probed for phosphorylated p70S6K, stripped, and reprobed for total p70S6K and GAPDH. Bottom Densitometric results as the ratio of phosporylated to total p70S6K normalized to GAPDH (n=10, *P<0.02). Control phosphorylation was defined at 1.0
TGFβ-stimulated activation of mTOR signaling is mediated by ERK signaling in IEC-6 cells
To investigate further the involvement of the ERK pathway in TGFβ-stimulated mTOR signaling, we added the MEK inhibitor PD98059 to cultured IEC-6 cells treated with TGFβ for 5 min. As shown in Fig. 6, the MEK inhibitor significantly blocked the increase of mTOR, p70S6 kinase, and 4E-BP1 kinase phosphorylation induced by TGFβ treatment. These results suggested that TGFβ-mediated ERK-signaling contributed to an increased FAK protein level via de novo protein synthesis by mTOR signaling.
Fig. 6.

TGFβ stimulated mTOR, p70S6K, and 4E-BP1 phosphorylation in cells treated for 5 min is abolished by pre-treatment with the MEK inhibitor PD98059 (10 μM) for 1 h. a Typical Western blot probed for phosphorylated mTOR, phosphorylated p70S6K, phosphorylated 4E-BP1, and GAPDH for the indicated times. b–d Densitometric results as the ratio of phosporylated protein normalized to GAPDH (n≥4, *P<0.05). Control phosphorylation (Ctr) at time point 0 was defined at 1.0
TGFβ-stimulated wound closure requires ERK and mTOR signaling in IEC-6 cells
TGFβ has been reported to promote epithelial cell monolayer migration (Tanigawa et al. 2005), and mTOR has been suggested as a downstream effector of this effect (Lamouille and Derynck 2007). Therefore, to investigate further the functional impact of the TGFβ-stimulated increase of FAK protein levels mediated by ERK/mTOR signaling, we studied the migration of cultured rat IEC-6 cells on collagen I. As shown in Fig. 7, TGFβ treatment (1 ng/ml) significantly stimulated wound closure after 24 h by 40±7.6% compared with similarly wounded monolayers not treated with TGFβ (n=24 pooled from three separate experiments with similar results, *P=0.05; Fig. 7b, c, f, g). ERK blockade with PD98059 or blocking the mTOR pathway with rapamycin abolished this stimulation of wound closure by TGFβ (Fig. 7d, e, h, i) This suggested that TGFβ promoted the migration of cells in monolayers of rat intestinal epithelial cell line IEC-6 via ERK and mTOR signaling.
Fig. 7.

TGFβ (1 ng/ml)-stimulated migration of cultured rat intestinal epithelial IEC-6 cells on collagen I is abolished by 1-h pretreatment with 10 μM PD98059 or 100 ng/ml rapamycin. a TGFβ was added before wounding, and migration was quantified after 24 h (n=3 for each circular wound, *P=0.05). b–i Typical microscopic images of cell migration with the original wound (b–e) delineated by a black dashed line and the final wound at 24 h (f–i) by a white line. Bars 150 μm
Discussion
Although TGFβ is known to activate pre-existing FAK, our present results suggest that TGFβ induces fak transcription in a manner not apparently dependent on ERK. In contrast, TGFβ seems likely to act via ERK on FAK mRNA stability and FAK translation in rat IEC-6 intestinal epithelial cells, since the blocking of the ERK pathway completely abolishes the ability of TGFβ to increase FAK mRNA stability, protein expression, and migration. Furthermore, the rapid activation of the mTOR translational machinery by TGFβ–stimulated ERK activation leads to an increase of de novo FAK protein synthesis in cultured rat intestinal cells.
TGFβ increases the expression of numerous proteins involved in growth inhibition, transcription, ECM homeostasis, and motility consistent with the distinct biological responses of TGFβ signaling in epithelial cells (Hartsough and Mulder 1995). For instance, TGFβ increases protein levels of type-1 inhibitor of plasminogen activator, collagen I, TGFβ, and the TGFβ–converting enzyme furin in various epithelial cell lines via ERK (Hayashida et al. 1999; Yue et al. 1999; Blanchette et al. 2001; Kutz et al. 2001). FAK is also a critical kinase involved in cell-matrix interactions during adhesion and motility, and a convergent point for trans-activation by many growth factors that regulate proliferation and transcription. Thus, unsurprisingly, TGFβ upregulates FAK protein. Even small changes in FAK protein levels may be critical for the amount of FAK available to be activated by other regulatory stimuli such as cytokines or growth factors. For instance, EGF-stimulated cell motility correlates with 45% increased FAK tyrosine phosphorylation in human gastric epithelial monolayers (Szabo et al. 2002).
Although non-transformed rat IEC-6 intestinal epithelial cells are a common model for the study of intestinal epithelial biology (Rhoads et al. 2006; Nicola et al. 2008), manifest differences exist between the behavior of an established cell line in culture and the responses of cells in vivo subjected to a complex array of neurohumoral, matrix-driven, and other stimuli (Basson et al. 1992). However, in vivo observations suggest that efficient ulcer healing requires the interplay of diverse growth factors including TGFβ (Tarnawski et al. 2001). Interrupted TGFβ–mediated signaling in Smad3 null mice delays ulcer healing (Owen et al. 2008), whereas human ulcers are associated with distinct patterns of TGFβ immunoreactivity that colocalize with similar patterns of immunoreactivity for FAK and phosphorylated FAK (Walsh et al. 2008b). Similar changes in TGFβ, FAK, and activated FAK immunoreactivity are also found in migrating intestinal epithelial IEC-6 cell monolayers (Walsh et al. 2008b). Although in vitro observations should only be extrapolated to in vivo biology with caution, such findings, together with our present in vitro results, support a role for TGFβ in FAK protein induction in vivo and in vitro and the potential significance of such mechanisms in ulcer healing.
TGFβ may influence FAK at several levels. For example, TGFβ stimulates FAK397 phosphorylation in hepatocarcinoma cells (Cai et al. 2000), keratinocytes (Jeong and Kim 2004), and intestinal epithelial cells (Walsh et al. 2008a, 2008b). However, TGFβ also influences FAK expression in intestinal epithelial cells by stimulating fak promoter activity via Smad and p38 MAPK (Walsh et al. 2008a, 2008b). In the present study, we have confirmed that TGFβ stimulates the fak promoter and demonstrate this stimulation to be independent of ERK.
ERK is rapidly activated by TGFβ in intestinal epithelial cells (Hartsough and Mulder 1995). However, the TGFβ induction of fak transcription appears to be independent of ERK, since the MEK inhibitor does not prevent TGFβ stimulation of fak promoter activity. On the contrary, inhibition of the ERK pathway alone stimulates fak promoter activity equivalent to that of TGFβ alone. Although these results make it difficult to rule out a small effect of TGFβ-mediated ERK activation on FAK promoter activity, we have been unable to demonstrate such an effect, whereas our results clearly demonstrate an effect of TGFβ-mediated ERK activation at other regulatory levels. The increase in basal FAK promoter activity observed here with ERK inhibition is consistent with another recent study from our laboratory examining the complex regulation of FAK promoter activity (Walsh et al. 2008a). One explanation may be that ERK activation increases Smad phoshorylation at sites that prevent the nuclear localization of Smad2/3. Inhibition of this cytosolic restraint by ERK blockade might explain the increase of basal FAK promoter activity independent of TGFβ treatment (Kretzschmar et al. 1999; Cushing et al. 2008).
The regulation of mRNA turnover is a crucial mechanism of the post-transcriptional control of gene expression (Mitchell and Tollervey 2000; Newbury 2006). TGFβ can modulate mRNA stability in various ways that may involve cis-acting or trans-acting elements with a range of transcription factors, depending upon the mRNA in question (Beelman and Parker 1995). For instance, TGFβ may enhance parathyroid-hormone-related protein mRNA stability in squamous carcinoma cells via the binding of numerous unidentified mRNA-binding proteins to the terminal coding region (Sellers et al. 2002). Our results suggest that TGFβ delays FAK mRNA degradation in an ERK-dependent manner in IEC-6 cells. The FAK mRNA half-life is short, suggesting that the strict control of fak gene expression is important biologically. TGFβ also influences the mRNA stability of urokinase-type plasminogen activator mRNA stability in transformed epidermal keratinocytes by an ERK-dependent mechanism (Santibanez et al. 2000).
Whereas TGFβ influences FAK mRNA stability, our study suggests that TGFβ also increases FAK protein levels by stimulating de novo protein synthesis. The mTOR pathway controls de novo protein synthesis in response to signals from mitogenic factors, nutrients, cellular energy levels, and stress. TGFβ treatment does not significantly inhibit the protein degradation observed in the presence of cycloheximide. Furthermore, the downstream targets of mTOR signaling, viz., p70S6 kinase and 4E-BP1 kinase, are phosphorylated in response to TGFβ in an ERK-dependent manner. Treating our cells with the mTOR inhibitor rapamycin and specific siRNA for mTOR and p70S6K completely blocks the TGFβ stimulation of FAK protein expression. Furthermore, the mTOR inhibitor rapamycin blocks migration. Despite the TGFβ modulation of protein synthesis via mTOR not having previously been reported, this model is consistent with a previous report that ERK signaling is implicated in translational control upstream of mTOR via phosphorylation of TSC2 enabling mTOR signaling (Ma et al. 2005), and with the observation that another cytokine, Cyr61, upregulates hypoxia-inducing factor-1α protein in gastric cancer cells via mTOR-dependent stimulation of de novo protein synthesis (Lin et al. 2008). Although we have not specifically studied the adaptor protein Eps8 here, Eps8 has also been linked to the upregulation of FAK expression via mTOR activation in human colon cancer cell lines (Maa et al. 2007).
Despite other transcription factors possibly playing a role in the TGFβ-independent regulation of FAK, we have observed the rapid activation of mTOR, p70S6, and 4E-BP1 kinase in 5–15 min after TGFβ treatment. Such rapid activation is not without precedent. TGFβ activates ERK itself in 5 min in epithelial cells (Hartsough and Mulder 1995), activates p70S6 kinase within 15 min in cultured human hepatic stellate cells (Le Pabic et al. 2005), and activates mTOR, p70S6, and 4E-BP1 kinase within 1 h in murine mammary epithelial cells (Lamouille and Derynck 2007).
Indeed, this rapid activation of mTOR and its downstream effectors may contribute to the TGFβ stabilization of the otherwise highly labile FAK mRNA. Such a model would be consistent with the previous demonstration that IGF-1 stimulation increases survivin protein expression in prostate cancer cells by stabilization and translation of a pool of survivin mRNA though the mTOR pathway (Vaira et al. 2007). mRNA stability is determined by the balance between mRNA translation in polysomes and mRNA degradation in P-bodies. p53 has been associated with mTOR signaling in a human embryonic stem cell line in which rapid inhibition of protein synthesis caused by activated tumor suppressor p53 can be counteracted by mTOR-induced translation of anti-apoptotic proteins (Constantinou and Clemens 2007). However, the nature of a possible interplay between mTOR and p53 signaling in regulating FAK protein levels remains unclear.
Together with our previous observations, our present data delineate distinct and complementary pathways by which TGFβ modulates FAK protein. Although TGFβ-induced activation of p38 and Smad signaling is required to induce fak transcription (a, Walsh et al. 2008b), TGFβ activation of ERK might increase the availability of the resulting FAK mRNA, whereas mTOR signaling further potentiates the TGFβ response by enhancing FAK protein synthesis. Acute exposure to TGFβ also rapidly phosphorylates and activates FAK protein (a, Walsh et al. 2008b), potentially further synergistically activating FAK pathway signaling in response to chronic exposure to TGFβ. The complex modulation of FAK transcription, mRNA stability, protein synthesis, and activation might represent an important convergent point for integrin-driven signals and those of growth factors such as TGFβ, which might be crucial in promoting motility and maintaining homeostasis in intestinal epithelial cells.
Acknowledgments
This work was supported in part by NIH RO1 DK067257 and a VA Merit Research Award (both to M.D.B.).
We thank Matt Sanders for helpful comments on the manuscript.
Contributor Information
Silke Suer, Department of Surgery, Michigan State University, 1200 East Michigan Avenue, Suite #655, Lansing MI 48912, USA. Department of Surgery, John D. Dingell VAMC, Detroit, Mich., USA.
Dinakar Ampasala, Department of Surgery, Michigan State University, 1200 East Michigan Avenue, Suite #655, Lansing MI 48912, USA. Department of Surgery, John D. Dingell VAMC, Detroit, Mich., USA.
Mary F. Walsh, Department of Surgery, Michigan State University, 1200 East Michigan Avenue, Suite #655, Lansing MI 48912, USA. Department of Surgery, John D. Dingell VAMC, Detroit, Mich., USA. Department of Surgery, John D. Dingell VAMC, Detroit, Mich., USA
Marc D. Basson, Department of Surgery, Michigan State University, 1200 East Michigan Avenue, Suite #655, Lansing MI 48912, USA. Department of Surgery, John D. Dingell VAMC, Detroit, Mich., USA
References
- Agochiya M, Brunton VG, Owens DW, Parkinson EK, Paraskeva C, Keith WN, Frame MC. Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene. 1999;18:5646–5653. doi: 10.1038/sj.onc.1202957. [DOI] [PubMed] [Google Scholar]
- Averous J, Proud CG. When translation meets transformation: the mTOR story. Oncogene. 2006;25:6423–6435. doi: 10.1038/sj.onc.1209887. [DOI] [PubMed] [Google Scholar]
- Barnard JA, Beauchamp RD, Coffey RJ, Moses HL. Regulation of intestinal epithelial cell growth by transforming growth factor type beta. Proc Natl Acad Sci USA. 1989;86:1578–1582. doi: 10.1073/pnas.86.5.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard JA, Polk WH, Moses HL, Coffey RJ. Production of transforming growth factor-alpha by normal rat small intestine. Am J Physiol. 1991;261:C994–C1000. doi: 10.1152/ajpcell.1991.261.6.C994. [DOI] [PubMed] [Google Scholar]
- Basson CT, Kocher O, Basson MD, Asis A, Madri JA. Differential modulation of vascular cell integrin and extracellular matrix expression in vitro by TGF-beta 1 correlates with reciprocal effects on cell migration. J Cell Physiol. 1992;153:118–128. doi: 10.1002/jcp.1041530116. [DOI] [PubMed] [Google Scholar]
- Basson MD, Sanders MA, Gomez R, Hatfield J, Vanderheide R, Thamilselvan V, Zhang J, Walsh MF. Focal adhesion kinase protein levels in gut epithelial motility. Am J Physiol Gastrointest Liver Physiol. 2006;291:G491–G499. doi: 10.1152/ajpgi.00292.2005. [DOI] [PubMed] [Google Scholar]
- Beelman CA, Parker R. Degradation of mRNA in eukaryotes. Cell. 1995;81:179–183. doi: 10.1016/0092-8674(95)90326-7. [DOI] [PubMed] [Google Scholar]
- Bermudez O, Marchetti S, Pages G, Gimond C. Post-translational regulation of the ERK phosphatase DUSP6/MKP3 by the mTOR pathway. Oncogene. 2008 doi: 10.1038/sj.onc.1211040. (in press) [DOI] [PubMed] [Google Scholar]
- Blanchette F, Rivard N, Rudd P, Grondin F, Attisano L, Dubois CM. Cross-talk between the p42/p44 MAP kinase and Smad pathways in transforming growth factor beta 1-induced furin gene transactivation. J Biol Chem. 2001;276:33986–33994. doi: 10.1074/jbc.M100093200. [DOI] [PubMed] [Google Scholar]
- Bouchard V, Demers MJ, Thibodeau S, Laquerre V, Fujita N, Tsuruo T, Beaulieu JF, Gauthier R, Vezina A, Villeneuve L, Vachon PH. Fak/Src signaling in human intestinal epithelial cell survival and anoikis: differentiation state-specific uncoupling with the PI3-K/Akt-1 and MEK/Erk pathways. J Cell Physiol. 2007;212:717–728. doi: 10.1002/jcp.21096. [DOI] [PubMed] [Google Scholar]
- Cai T, Lei QY, Wang LY, Zha XL. TGF-beta 1 modulated the expression of alpha 5 beta 1 integrin and integrin-mediated signaling in human hepatocarcinoma cells. Biochem Biophys Res Commun. 2000;274:519–525. doi: 10.1006/bbrc.2000.3177. [DOI] [PubMed] [Google Scholar]
- Chow JY, Quach KT, Cabrera BL, Cabral JA, Beck SE, Carethers JM. RAS/ERK modulates TGFbeta-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis. 2007;28:2321–2327. doi: 10.1093/carcin/bgm159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinou C, Clemens MJ. Regulation of translation factors eIF4GI and 4E-BP1 during recovery of protein synthesis from inhibition by p53. Cell Death Differ. 2007;14:576–585. doi: 10.1038/sj.cdd.4402045. [DOI] [PubMed] [Google Scholar]
- Cushing MC, Mariner PD, Liao JT, Sims EA, Anseth KS. Fibro-blast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells. FASEB J. 2008;22:1769–1777. doi: 10.1096/fj.07-087627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678:111–125. doi: 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Hall PA, Coates PJ, Ansari B, Hopwood D. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J Cell Sci. 1994;107:3569–3577. doi: 10.1242/jcs.107.12.3569. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Ryzhova L, Shin NY, Brabek J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front Biosci. 2003;8:d982–d996. doi: 10.2741/1114. [DOI] [PubMed] [Google Scholar]
- Hartsough MT, Mulder KM. Transforming growth factor beta activation of p44MAPK in proliferating cultures of epithelial cells. J Biol Chem. 1995;270:7117–7124. doi: 10.1074/jbc.270.13.7117. [DOI] [PubMed] [Google Scholar]
- Hayashida T, Poncelet AC, Hubchak SC, Schnaper HW. TGF-beta1 activates MAP kinase in human mesangial cells: a possible role in collagen expression. Kidney Int. 1999;56:1710–1720. doi: 10.1046/j.1523-1755.1999.00733.x. [DOI] [PubMed] [Google Scholar]
- Jeong HW, Kim IS. TGF-beta1 enhances betaig-h3-mediated keratinocyte cell migration through the alpha3beta1 integrin and PI3K. J Cell Biochem. 2004;92:770–780. doi: 10.1002/jcb.20110. [DOI] [PubMed] [Google Scholar]
- Kohno M, Pouyssegur J. Targeting the ERK signaling pathway in cancer therapy. Ann Med. 2006;38:200–211. doi: 10.1080/07853890600551037. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutz SM, Hordines J, McKeown-Longo PJ, Higgins PJ. TGF-beta1-induced PAI-1 gene expression requires MEK activity and cell-to-substrate adhesion. J Cell Sci. 2001;114:3905–3914. doi: 10.1242/jcs.114.21.3905. [DOI] [PubMed] [Google Scholar]
- Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. 2007;178:437–451. doi: 10.1083/jcb.200611146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pabic H, L’Helgoualc’h A, Coutant A, Wewer UM, Baffet G, Clement B, Theret N. Involvement of the serine/threonine p70S6 kinase in TGF-beta1-induced ADAM12 expression in cultured human hepatic stellate cells. J Hepatol. 2005;43:1038–1044. doi: 10.1016/j.jhep.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Lin MT, Kuo IH, Chen CC, Chu CY, Chen HY, Lin BR, Sureshbabu M, Shih HJ, Kuo ML. Involvement of HIF-1alpha-dependent PAI-1 up-regulation in Cyr61/CCN1 induced gastric cancer cell invasion. J Biol Chem. 2008 doi: 10.1074/jbc.M708933200. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Maa MC, Lee JC, Chen YJ, Chen YJ, Lee YC, Wang ST, Huang CC, Chow NH, Leu TH. Eps8 facilitates cellular growth and motility of colon cancer cells by increasing the expression and activity of focal adhesion kinase. J Biol Chem. 2007;282:19399–19409. doi: 10.1074/jbc.M610280200. [DOI] [PubMed] [Google Scholar]
- Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416–6422. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- Mitchell P, Tollervey D. mRNA stability in eukaryotes. Curr Opin Genet Dev. 2000;10:193–198. doi: 10.1016/s0959-437x(00)00063-0. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- Nakagawa K, Sogo S, Hioki K, Tokunaga R, Taketani S. Acquisition of cell adhesion and induction of focal adhesion kinase of human colon cancer Colo 201 cells by retinoic acid-induced differentiation. Differentiation. 1998;62:249–257. doi: 10.1046/j.1432-0436.1998.6250249.x. [DOI] [PubMed] [Google Scholar]
- Newbury SF. Control of mRNA stability in eukaryotes. Biochem Soc Trans. 2006;34:30–34. doi: 10.1042/BST20060030. [DOI] [PubMed] [Google Scholar]
- Nicola JP, Basquin C, Portulano C, Reyna-Neyra A, Paroder M, Carrasco N. The Na+/I− symporter (NIS) mediates active iodide uptake in the intestine. Am J Physiol Cell Physiol. 2008 doi: 10.1152/ajpcell.00509.2008. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen CR, Yuan L, Basson MD. Smad3 knockout mice exhibit impaired intestinal mucosal healing. Lab Invest. 2008 doi: 10.1038/labinvest.2008.77. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19:5606–5613. doi: 10.1038/sj.onc.1203877. [DOI] [PubMed] [Google Scholar]
- Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80:248–265. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads JM, Niu X, Odle J, Graves LM. Role of mTOR signaling in intestinal cell migration. Am J Physiol Gastrointest Liver Physiol. 2006;291:G510–G517. doi: 10.1152/ajpgi.00189.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Capelo A. Dual role for TGF-beta1 in apoptosis. Cytokine Growth Factor Rev. 2005;16:15–34. doi: 10.1016/j.cytogfr.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Sancho E, Batlle E, Clevers H. Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Biol. 2004;20:695–723. doi: 10.1146/annurev.cellbio.20.010403.092805. [DOI] [PubMed] [Google Scholar]
- Santibanez JF, Iglesias M, Frontelo P, Martinez J, Quintanilla M. Involvement of the Ras/MAPK signaling pathway in the modulation of urokinase production and cellular invasiveness by transforming growth factor-beta(1) in transformed keratinocytes. Biochem Biophys Res Commun. 2000;273:521–527. doi: 10.1006/bbrc.2000.2946. [DOI] [PubMed] [Google Scholar]
- Sellers RS, Capen CC, Rosol TJ. Messenger RNA stability of parathyroid hormone-related protein regulated by transforming growth factor-beta1. Mol Cell Endocrinol. 2002;188:37–46. doi: 10.1016/s0303-7207(01)00752-3. [DOI] [PubMed] [Google Scholar]
- Sturm A, Dignass AU. Epithelial restitution and wound healing in inflammatory bowel disease. World J Gastroenterol. 2008;14:348–353. doi: 10.3748/wjg.14.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo IL, Pai R, Jones MK, Ehring GR, Kawanaka H, Tarnawski AS. Indomethacin delays gastric restitution: association with the inhibition of focal adhesion kinase and tensin phosphorylation and reduced actin stress fibers. Exp Biol Med (Maywood) 2002;227:412–424. doi: 10.1177/153537020222700607. [DOI] [PubMed] [Google Scholar]
- Tanigawa T, Pai R, Arakawa T, Higuchi K, Tarnawski AS. TGF-beta signaling pathway: its role in gastrointestinal pathophysiology and modulation of ulcer healing. J Physiol Pharmacol. 2005;56:3–13. [PubMed] [Google Scholar]
- Tarnawski A, Szabo IL, Husain SS, Soreghan B. Regeneration of gastric mucosa during ulcer healing is triggered by growth factors and signal transduction pathways. J Physiol (Paris) 2001;95:337–344. doi: 10.1016/s0928-4257(01)00046-8. [DOI] [PubMed] [Google Scholar]
- Vaira V, Lee CW, Goel HL, Bosari S, Languino LR, Altieri DC. Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene. 2007;26:2678–2684. doi: 10.1038/sj.onc.1210094. [DOI] [PubMed] [Google Scholar]
- Walsh MF, Thamilselvan V, Grotelueschen R, Farhana L, Basson M. Absence of adhesion triggers differential FAK and SAPKp38 signals in SW620 human colon cancer cells that may inhibit adhesiveness and lead to cell death. Cell Physiol Biochem. 2003;13:135–146. doi: 10.1159/000071864. [DOI] [PubMed] [Google Scholar]
- Walsh MF, Ampasala DR, Rishi AK, Basson MD. TGF-beta1 modulates focal adhesion kinase expression in rat intestinal epithelial IEC-6 cells via stimulatory and inhibitory Smad binding elements. Biochim Biophys Acta. 2008a doi: 10.1016/j.bbagrm.2008.11.002. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh MF, Ampasala DR, Hatfield J, Vander Heide R, Suer S, Rishi AK, Basson MD. Transforming growth factor-beta stimulates intestinal epithelial focal adhesion kinase synthesis via smad- and p38-dependent mechanisms. Am J Pathol. 2008b;173:385–399. doi: 10.2353/ajpath.2008.070729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CF, Sanders MA, Basson MD. Human caco-2 motility redistributes FAK and paxillin and activates p38 MAPK in a matrix-dependent manner. Am J Physiol Gastrointest Liver Physiol. 2000;278:G952–G966. doi: 10.1152/ajpgi.2000.278.6.G952. [DOI] [PubMed] [Google Scholar]
- Yue J, Frey RS, Mulder KM. Cross-talk between the Smad1 and Ras/MEK signaling pathways for TGFbeta. Oncogene. 1999;18:2033–2037. doi: 10.1038/sj.onc.1202521. [DOI] [PubMed] [Google Scholar]
- Zhang HM, Keledjian KM, Rao JN, Zou T, Liu L, Marasa BS, Wang SR, Ru L, Strauch ED, Wang JY. Induced focal adhesion kinase expression suppresses apoptosis by activating NF-kappaB signaling in intestinal epithelial cells. Am J Physiol Cell Physiol. 2006;290:C1310–C1320. doi: 10.1152/ajpcell.00450.2005. [DOI] [PubMed] [Google Scholar]