

Abstract
The major neuropsychiatric conditions of schizophrenia, affective disorders, and infantile autism are characterized by chronic symptoms of episodic, stable, or progressive nature that result in significant morbidity. Symptomatic treatments are the mainstay but do not resolve the underlying disease processes, which are themselves poorly understood. The prototype psychotropic drugs are of variable efficacy, with therapeutic mechanisms of action that are still uncertain. Thus, neuropsychiatric disorders are ripe for new technologies and approaches with the potential to revolutionize mechanistic understanding and drive the development of novel targeted treatments. The advent of methods to produce patient‐derived stem cell models and three‐dimensional organoids with the capacity to differentiate into neurons and the various neuronal cellular lineages mark such an advance. We discuss numerous techniques involved, their applications, and areas that require further optimization. Stem Cells Translational Medicine 2017;6:2062–2070
Keywords: Induced pluripotent stem cells, Nervous system, Cell biology, Reprogramming
Significance Statement.
Rapid progress in stem cell biology has the potential to revolutionize various areas of medicine. The goal of this review is to highlight the potential use of induced pluripotent stem cells to investigate and treat neuropsychiatric conditions and discuss pitfalls that need to be resolved along the way.
Introduction
Neuropsychiatric conditions present a substantial challenge to study for a variety of reasons. Psychiatric phenotypes may be sporadic or episodic, with normal periods of baseline functioning in between episodes (in cyclothymic, bipolar, and major depressive disorders), which can present an obstacle to accurate characterization 1. Neuropsychiatric manifestations often involve higher cognitive processes that are difficult to study in nonhuman models. In tandem with the brain's complexity, the pathology of the major psychiatric disorders has been subtle and elusive. The vast genetic, pathologic, and neuroimaging literature have yielded several conflicting and nonspecific findings. In contrast to monogenic disorders in which a single gene with high penetrance is disease‐causing, the majority of neuropsychiatric disorders are polygenic, involving multiple alleles with relatively small individual effect sizes, and multifactorial, involving the effects of multiple genes in combination with environmental factors, which further complicates their elucidation 2. Given this range of challenges, it is perhaps unsurprising that relatively slow progress has been made in our understanding of the biologic basis of psychiatric conditions relative to other diagnostic conditions.
Recent progress has come from large‐scale genome‐wide association and whole genome sequencing studies which have begun to reveal the genetic substrate in the major psychiatric conditions. However, pathophysiological insight beyond the identification of risk genes requires elucidation of functional networks and pathways to unravel disease mechanisms. Until recently, animal models and post‐mortem human brain provided the primary models for comprehensive study of neuropsychiatric disorders, despite their overt limitations. However, over the last decade, human induced pluripotent stem cell (hiPSC) technology has emerged to advance the study of neuropsychiatric disease and hold significant potential to define pathophysiological mechanisms and identify potential therapeutic targets.
Stem cells possess both self‐renewal and pluripotent capabilities. After the identification of embryonic stem cells (ESCs) and embryonic germ cells derived from the inner cell mass of blastocysts and post implantation embryos, respectively, the next advance came with the generation of induced pluripotent stem cells (Fig. 1) from adult somatic cells from individuals with particular diseases by combinatorial expression of reprogramming factors (3–5). The ability of these cells to produce previously inaccessible tissue such as neurons lies at the heart of their remarkable benefit to the neurobiology of psychiatric disorders. Crucially, patient‐derived hiPSCs provide an alternative to primary human brain tissue which can be difficult to obtain and which may lack cell types that can be critical for disease pathology, solving a previously intractable constraint in the study of neuropsychiatric disorders 3. Furthermore, while the exact etiological mechanisms of clinically and genetically heterogeneous neuropsychiatric conditions might be unknown or difficult to model, patient‐derived hiPSCs inherently recapitulate the genetic heterogeneity typically found in human patients, and developmental phenotypes in these hard‐to‐model cases can be determined in hiPSC‐derived neural progenitor cells (NPCs) and neurons. Similarly, rare Mendelian disorders caused by single‐gene mutations can be modeled using patient‐derived hiPSCs. Since hiPSCs are amenable to CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 gene editing, isogenic cell lines that share a genetic background while differing in the presence or absence of disease‐specific mutations can also be developed to model these single‐gene disorders 4. Controlling for the genetic background in this way represents a powerful tool to elucidate gene function in a human model system (Fig. 1). Altogether, these attributes translate into significant potential to revolutionize the direction of psychiatric research and treatment.
Figure 1.
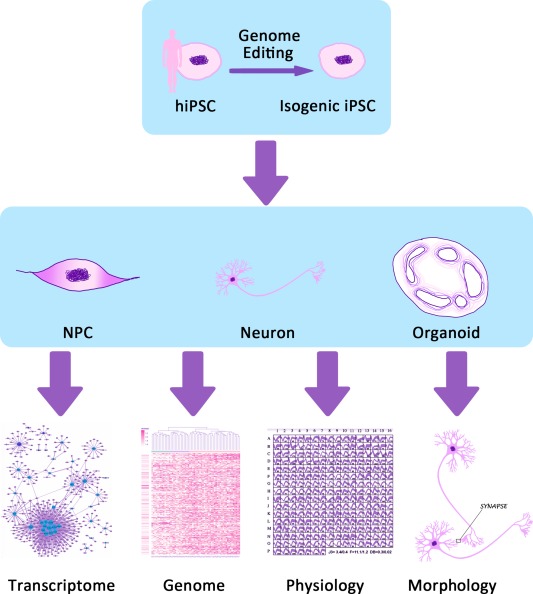
Patient‐derived somatic cells from a patient with neuropsychiatric disease are reprogrammed into hiPSCs via Yamanaka factors. Patient‐specific hiPSCs are then induced to differentiate into NPCs, neurons, and organoids. These disease‐relevant cells provide a platform for research approaches such as transcriptomic, genomic, and biochemical analyses, as well as analyses of neuronal circuit integration, neuronal ion channel properties, neuronal morphology, and other neuronal phenotypes. Abbreviations: hiPSCs, human induced pluripotent stem cells; NPCs, neural progenitor cells.
Human Induced Pluripotent Stem Cells to Produce Progenitors, Neurons, and Organoids
Somatic cells can be converted to a pluripotent state via co‐expression of a variety of reprogramming factors 5, 6. Initial somatic cell populations available to generate hiPSCs include fibroblasts, peripheral blood cells, and dental pulp cells 6, 7, 8. While fibroblasts are efficiently reprogrammed, the alternative cells types require less invasive procedures to acquire, offering a practical advantage when obtaining cells from young patients.
To generate neurons, hiPSCs are typically differentiated into NPCs, with subsequent differentiation of these cells into neurons (although direct conversion of hiPSCs to neurons is possible) 9. The generation of a heterogeneous population of NPCs and neurons may be desirable to assess mutant phenotypes in a variety of cell types. Recently, techniques to generate neurons with specific terminal identities have been described via treatment of hiPSCs with recombinant proteins and/or small molecules that mimic in vivo patterning in the developing nervous system. Various cell types that have been generated through this process include excitatory and inhibitory cortical neurons 10, 11, dopaminergic neurons 12, serotonergic neurons 13, Purkinje cells 14, peripheral motor neurons 15, and hippocampal granule neurons 16.
Recently, a number of methods for generating three‐dimensional (3D) culture systems that mimic early aspects of brain development have also been established 17, 18, 19. These are variously known as cerebral organoids, cortical spheroids, or forebrain organoids (hereafter referred to as “organoids”). The different names have arisen due to slight variations between different organoid‐generating protocols. These differences mainly lie in the initial patterning step (or lack thereof) that induces hiPSCs to become brain cells. This is typically accomplished by pharmacologically inhibiting or enhancing various molecular signaling pathways that have been linked with brain development. The addition of these patterning factors has been used to model specific brain regions including the cerebellum, hippocampus, and both the dorsal and ventral cerebral cortex 17, 20, 21, 22. Organoids generated without these factors are typically more heterogeneous and can contain a variety of different brain regions within the same organoid, as shown by immunohistochemistry and single‐cell RNA‐sequencing 18, 23. However, more homogeneous organoids have recently been produced without extensive drug addition by using bioengineering techniques 18, 24. Heterogeneous organoids might also be desirable in their own right to analyze the interactions between cells from a variety of brain regions.
Both patterned and nonpatterned organoids recapitulate many aspects of embryonic brain development, such as the formation of stereotypical stem cell niches of the developing brain with corresponding production of various NPC subtypes and the migration of neurons away from these proliferative niches (Fig. 2) 26. Spatial relationships between neurons and progenitor cells are maintained, allowing for analysis of critical aspects of embryonic brain development, such as proliferative zone formation, neuronal migration, and the proliferative capacity of multiple different NPC subtypes. Importantly, organoids from hiPSCs are capable of producing outer subventricular zone radial glia (oRG), a recently identified class of progenitors that is greatly expanded in humans but rare in rodents 27.
Figure 2.
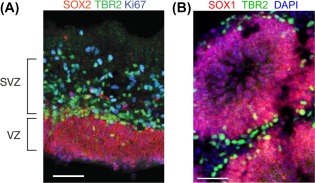
Organoids can recapitulate normal and abnormal brain development. (A): A human cortical brain section from gestational week 11.5 displays a SOX2+ VZ and TBR2+ SVZ with Ki67+ proliferative cells present in both areas 26. (B): An organoid section labeled with SOX1 and TBR2 antibodies indicates the presence of progenitor regions with the stereotypical structure found in the embryonic brain. Note that organoids do not perfectly replicate the structure of the developing brain, as indicated by the presence of multiple, smaller proliferative zones in the organoid compared with one large proliferative region in the developing cortex. (A, B) Scale bars = 50 μm. Abbreviations: SVZ, subventricular zone; VZ, ventricular zone.
Molecular and Cellular Approaches to Determine Pathophysiological Mechanisms
The full complement of molecular and cellular approaches/techniques can be applied to hiPSC‐derived NPCs and neurons in order to identify mechanisms underlying neuropsychiatric disease (Table 1). Morphological, histological, physiological and transcriptional characteristics of hiPSC‐derived cells can be elucidated by gene expression profiling (RNA‐Seq), chromatin studies (Chip‐Seq), and proteomic analyses, as well as specific analyses of neuronal phenotypes such as neuronal identity, morphology, and electrophysiology.
Table 1.
Approaches used in the investigation of human induced pluripotent stem cells (hiPSCs) and hiPSC‐derived tissue relating to neuropsychiatric disorders
| Assay | hiPSC | NPC | Neuron | Organoid |
|---|---|---|---|---|
| Immunohistochemistry / protein assay | [28], [29], [30], [31], [32], [33], [34],[35] | [36], [29], [30], [31], [32], [35] | [36], [29], [30], [31], [32], [33], [35] | [34] |
| Transcriptomic and epigenomic profiling | [28], [30], [31] | [36], [31], [25], [34] | [36], [28], [29], [30], [31], [32] | [34] |
| Growth / proliferation | [34], [35] | [29], [31], [35] | ||
| Neurite / synapse morphology | [36], [31], [32], [33], [35] | [34] | ||
| Electrophysiology | [36], [31], [32], [33], [34], [35] | [34] | ||
| Biochemical assay / response | [31], [33] | [36], [32], [37], [35] |
Numbers in table refer to references.
Color Legend: Green: Bipolar Disorder.
Blue: Schizophrenia.
Red: Autism.
Orange: Rett syndrome (monogenic, syndromic autism).
Light blue: Fragile X syndrome (monogenic, syndromic ASD).
Purple: Timothy syndrome (monogenic, syndromic ASD).
Abbreviation: NPCs, neural progenitor cells.
It is difficult to accurately measure transcriptional alterations in primary postmortem tissue due the paucity of available brain tissue and postmortem RNA degradation. hiPSC models, on the other hand, can be generated from a large number of patients and the cells of interest can immediately be processed for sequencing experiments, ameliorating concerns associated with post‐mortem tissue. Transcriptional alterations and correlated disease pathology can also be assessed in disease‐relevant cell types such as inhibitory and excitatory neurons, oligodendrocytes, and so forth. 31, 37, 38. In addition, culturing and analyzing homogeneous cell populations can reduce some of the heterogeneity typically found in the brain, which can complicate analysis of transcriptomic data from whole brain tissue.
Another challenge is that brain tissue is not available during critical times of pathophysiology of neuropsychiatric disorders, or the timing is unknown. For example, it is likely that many of the pathophysiological changes in autism occur prenatally 25. hiPSCs can be used to generate cell types that are present at high numbers during embryonic development, such as NPCs, whose transcriptomic analysis is of great interest for neurodevelopmental disorders of prenatal development. hiPSC models represent an efficient and effective way to detect transcriptomic and proteomic changes in these relevant but difficult‐to‐obtain cell types.
ChIP‐seq analysis can validate transcriptomic profiling by revealing chromatin signatures and epigenetic changes in pathways consistent with altered signaling. Epigenomic profiling may also be used to investigate downstream targets of mutated genes 39. Similarly, proteomic profiling has been undertaken using hiPSC‐derived cells 40. The small volume and high homogeneity of cells relative to tissue from animal models constitute an advantage to using hiPSCs.
Importantly, morphological and physiological analysis of relevant cell types can be correlated with any transcriptomic/proteomic/epigenomic alterations that might be identified. For instance, whole cell patch clamp recordings can be used to characterize neuronal circuit formation and electrical properties of hiPSC‐derived neurons 33. The patch‐seq approach combines whole‐cell patch‐clamp recording of individual neurons with single‐cell RNA sequencing to correlate electrical activity encoding unique patterns of neuron activation and connectivity with individual gene expression signatures 37. This can distinguish neuron classes and assess differential cell‐specific expression patterns for risk genes, which can be correlated with individual neuronal phenotypes. Morphological characteristics of patient‐derived hiPSCs can be determined using various assays such as neurite outgrowth assays and immunolabeling 31, 38. Developmental phenotypes of hiPSC‐derived neurons and NPCs, such as proliferation and differentiation, can also be characterized by various in vitro assays 31. Analysis of these phenotypes can then be combined with other molecular techniques described above to identify molecular pathways that might be responsible for altered disease physiology.
Relevant differentiated cell types can also be used in various screens to probe for chemical modifiers of a specific signaling pathway or cellular phenotype (see below). These screens can be valuable for finding modulators of particular salience to neuronal behavior 41. Microfluidic approaches, which enable precise control of the cellular microenvironment and allow the systematic presentation of cues, have various applications conducive to hiPSC study, such as assays to interrogate the effect of delivered morphogen gradients on developmental trajectories, and assays to screen for chemotaxic and haptotaxic cues. These microfluidic chemotaxis assays can reciprocally be used to assess migratory properties of cells under specific conditions 42, and can also help distinguish between cell‐autonomous and non‐cell autonomous phenotypic effects. In addition, hiPSCs are amenable to gene editing techniques, enabling the deletion or modification of candidate target genes and subsequent analysis of their effects on neurological phenotypes (Table 1). The concise nature of this review does not allow detailed exposition of newer techniques such as fluorescent in situ sequencing 43, which have potential applications in answering additional questions in neuron biology.
In general, approaches to organoids mirror those to hiPSCs, but the 3D nature of organoids and their developmental trajectory may suggest other investigations. For example, some organoids may generate functional photosensitive cells that can respond to light‐based stimulation 44, which suggests a possible mechanism to query neuronal responses to exogenous sensory stimuli, in concert with engineered optogenetic systems.
Stem Cell Models of Neuropsychiatric Disorders
Schizophrenia
Schizophrenia (SCZD), a non‐Mendelian disorder with complex genetics, is a major psychiatric condition of young adulthood with a 1% prevalence. The pathogenesis remains elusive and involves a wide range of neuroanatomic findings affecting the phenotypes of various neuronal and glial populations, none of which have yet proven to be pathognomonic. Attention has particularly focused on alterations in cellular morphology in the hippocampus and dorsolateral prefrontal cortex including smaller pyramidal neuron cell bodies, decreased presynaptic protein markers, diminished dendritic spine density, and decreased markers of parvalbumin‐positive GABAergic neurons 45, 46.
SCZD is an ideal disorder to model through stem cell approaches due to its heterogeneous, complex genetics that are hard to recapitulate in animal models. In one of the first illustrations of the hiPSC approach in neuropsychiatry, Brennand et al. 38 directly reprogrammed fibroblasts from SCZD patients (one childhood onset disorder; two sibling pairs each with a schizophrenic father) into hiPSCs and differentiated these into neurons. Neuronal connectivity, neurite outgrowth and differential gene expression were assayed in disease‐specific hiPSC neurons. A recombinant labeled form of the neurotropic rabies virus was used as a transsynaptic tracer to show decreased neuronal connectivity in SCZD hiPSC neurons, in addition to decreased neurite number and decreased levels of the excitatory postsynaptic protein PSD95. In addition, gene expression profiles of SCZD hiPSC neurons identified altered expression of many components of the cAMP and WNT signaling pathways. However, levels of other synaptic proteins were unaffected, and electrophysiological activity in SCZD hiPSC neurons was normal.
When compared with previously described SCZD pathologies, hiPSC SCZD neurons correctly modeled some of these traits (reduced neural outgrowth and connectivity) but not others (no observed decrease in synaptic density or function). It remains to be seen whether these differential findings were patient‐specific to the individuals in the study, or generalizable.
Note that the five patients in this study were comprised of an individual with childhood SCZD, which is an atypical presentation of the condition, and two sibling pairs selected from pedigrees where three of four members were affected with a SCZD spectrum disorder (father plus two siblings). Thus, rare Mendelian forms of SCZD cannot be ruled out. Another study 29 used hiPSCs derived from carriers of a genetic lesion (translocation or microdeletion) in DISC1, a locus that segregates in an autosomal dominant fashion with a broad diagnosis of neuropsychiatric disease, including SCZD, bipolar disorder (BPD) and major depression, as well as a narrow diagnosis of SCZD. hiPSC‐derived forebrain glutamatergic neurons revealed decreased density of SV2+ synaptic boutons, reduction in the presynaptic marker synapsin 1 and the postsynaptic marker PSD95, and widespread transcriptional dysregulation affecting synaptic, nervous system, and dendritic transcripts, as well as DISC1‐interacting factors, among others. These findings were correlated with functional synaptic transmission deficits in DISC1 mutant neurons, including decreased frequency of excitatory spontaneous synaptic currents and DISC1‐dependent presynaptic release defects. DISC1 knock‐in of a control line recapitulated the synaptic and electrophysiological deficits, while isogenic correction of the DISC1 lesion in patient‐derived neurons reversed the deficits. While the generalizability of the findings needs to be further established, the authors posit that a common molecular mechanism underlies a wide spectrum of psychiatric illness. This will need to be reconciled with the very different phenotypic presentations, course, and longitudinal outcomes of these various neuropsychiatric conditions.
Bipolar Disorder
BPD presents with cyclical episodes of mania and depression, with intervening periods of return to baseline stability 1. The cause of BPD is not well understood but is likely to involve neurotransmitter dysfunction and defects in critical signal transduction pathways. There is substantial variability in disease course and treatment response of BPD. For example, lithium chloride (LiCl) has been shown to be effective for some BPD patients, but not so for others 47.
Recently, hiPSCs derived from individuals with BPD were differentiated into mostly glutamatergic dentate gyrus neurons and investigated via patch‐clamp recording. This revealed a number of abnormalities in action potential (AP) firing consistent with hyper‐excitability, such as decreased threshold for APs, and increased AP number and maximal amplitude 33. Gene expression profiling of these neurons showed upregulated mitochondrial gene expression compared with control neurons. BPD neurons also revealed enhanced mitochondrial function and smaller mitochondria. LiCl partly rescued mitochondrial dysfunction by increasing the mitochondria size in lithium‐responsive neurons. RNA‐Seq was performed to detect genes important for the differences in drug response. This study provides an example of how multilevel (physiological, cellular, transcriptomic, pharmacologic) approaches may converge on the hiPSC model to provide an integrated understanding of disease.
Autism
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by persistent deficits in social communication across multiple contexts and restricted, repetitive patterns of behavior, with or without intellectual disability or language impairment 1. Similar to SCZD, ASD is characterized by both phenotypic and genetic heterogeneity. Identifiable mutations in a single gene or set of genes account for a minority of ASD cases (classified as “syndromic” forms of ASD) 48, 49. Most instances of ASD are thought to be caused by a combination of interacting genetic and environmental factors (referred to as “non‐syndromic” or “idiopathic” ASD); genetic factors include de novo mutations in risk genes, copy number variations, and deleterious combinations of common genetic variants 48, 50, 51, 52. More than 100 de novo risk genes have been identified, that only account for a fraction of causality 53, and up to half of autism is caused by interaction of small effect variants in several genes 54.
hiPSC models can effectively recapitulate the heterogeneous genetic background typical of nonsyndromic ASD that would be difficult or impossible to generate/study in traditional animal models. In a recent study from our group, NPCs and neurons were generated from hiPSC lines derived from nonsyndromic ASD patients with comorbid macrocephaly 31. NPCs differentiated from ASD cell lines displayed increased proliferation associated with dysregulation of a novel transcriptional cascade, indicating a potential mechanism for the brain overgrowth observed in the patients from which these cells were derived. In addition, neurons derived from ASD cell lines displayed aberrant synaptogenesis and network synchrony, which resemble physiological alterations/aberrations typically observed in ASD 34. This study illustrates how a clinically relevant phenotype can be used to elucidate unifying pathology at the cellular level that is mechanistically relevant to disease.
Syndromic forms of ASD, such as Rett syndrome (RTT), Fragile X syndrome, and Timothy Syndrome have also been effectively modeled with hiPSCs 55. For instance, hiPSCs generated from RTT patients were able to recapitulate a variety of neurological phenotypes previously identified in this disease, such as smaller soma size, fewer dendritic spines, lower spontaneous Ca2+ transient frequency, abnormal excitatory synaptic transmission, and fewer excitatory synapses 35. Because these clinically relevant phenotypes were observed, the authors were then able to test potential treatments on these cells to determine whether any phenotypes could be reversed. In this case, candidate drugs, such as IGF‐1, improved the neuronal growth and synaptogenesis in these derived cells 35.
Initial studies using hiPSCs to model neurological disorders relied on monolayer culture systems to assess molecular and cellular phenotypes. These two‐dimensional (2D) systems lack the in vivo spatial relationships between proliferating NPCs and post‐mitotic neurons, and typically do not produce the variety of NPCs found in the developing brain. As noted above, 3D organoid culture systems offer a unique opportunity to study embryonic ASD pathophysiology in human development. In addition to recapitulating many of the cellular aspects of early brain development, gene expression in organoids is analogous to gene expression in the cortex during mid‐gestation in humans 19, 23, 34, 56, precisely the time period of enhanced ASD‐risk gene expression 57, 58.
Recently, various groups have used organoids to model cortical development in syndromic ASD due to PTEN, MECP2, and CACNA1C mutations 20, 59, 60. In these studies, hiPSC or human embryonic stem cell (hESC) lines from either affected or nonaffected individuals were generated and differentiated into organoids, along with isogenic hiPSC/hESC lines that either corrected or introduced deleterious mutations into the original lines, respectively. In one study, PTEN deletion resulted in extensive increases in progenitor cell proliferation and subsequent increases in size and surface folding, mimicking the brain enlargement that is often observed in PTEN‐associated ASD 60, 61. This size increase was likely caused by enhanced proliferation of NPCs within these organoids, including an aberrantly expanded population of outer radial glial (oRG) cells, a cell population thought to be critical for expansion of the cerebral cortex in higher‐order mammals 60, 62. Note, however, that excessive cortical folding is not found in human patients with PTEN mutations. Importantly, organoids derived from mouse Pten knockout ES cells failed to produce folding, did not generate oRG cells, and displayed only minor and transient increases in progenitor cell proliferation compared with control organoids 61.
Nonsyndromic ASD has also been modeled in organoids 34. In this study, hiPSCs generated from four patients with nonsyndromic ASD and comorbid macrocephaly along with first‐degree family members were differentiated into organoids. While the ASD organoids did not recapitulate the macrocephalic phenotypes found in ASD patients, the transcription factor FOXG1 was overexpressed, which was linked to overproduction of inhibitory neurons and synapses, illustrating the excitatory/inhibitory imbalance often associated with ASD 34. The lack of mutations in individual causative genes prevented the development of isogenic cell lines in this study. However, transcriptomic analysis of these organoids identified novel molecular pathways and individual genes that might play a role development of nonsyndromic ASD.
The ability to detect early developmental phenotypes that cannot be observed in animal models or monolayer human cultures (such as aberrant proliferation of oRG progenitor cells), along with the ability to generate relevant nonsyndromic ASD phenotypes highlight the potential that organoids have to model human ASD.
Prospects for hiPSC Treatment of Neuropsychiatric Disorders
It is likely that hiPSCs will be useful in the development of future therapies for neuropsychiatric conditions by providing a platform in which to both derive and test novel hypotheses. In the therapeutic development process, hiPSCs derived from patients with well‐characterized genetic, phenotypic, and drug response histories are differentiated into relevant cell populations such as NPCs or neurons and subsequently subjected to various chemical treatments. Assaying the reaction of these cells to treatment can then facilitate the determination of drug responses in an individual patient and/or enables the screening of novel compounds for efficacy, rendering unprecedented power to drug discovery. Initial studies have highlighted the potential of this process, although clearly much additional work is needed to efficiently yield novel therapeutics.
In one initial effort 38, neurons derived from SCZD hiPSC lines were treated with various antipsychotic medications and assayed for improvement in neuronal connectivity after 3 weeks of drug administration. Loxapine, a first‐generation antipsychotic, was the only compound to significantly increase neuronal connectivity in hiPSC‐derived neurons from all five study patients. Two newer generation antipsychotics (Olanzapine and Risperidone) and an antipsychotic considered the therapeutic gold standard, Clozapine, did not show this response. This illustrates a current challenge: choosing the correct phenotype and cell type to measure drug response. Loxapine is thought to exert its main action by blocking dopamine 2 receptors to reduce positive symptoms of psychosis, and is also a potent serotonin 2A antagonist. However, the SCZD‐derived neurons included <10% dopaminergic neurons. Although neuronal connectivity was decreased in SCZD‐derived neurons, the overall assay may not have completely captured the appropriate cell type and/or phenotype to accurately determine drug activity. Note also that schizoid personality disorder, the specific phenotype in one patient, is not clinically treated with antipsychotic therapy. Optimization of concentration and timing of drug administration, in addition to measuring phenotypes in relevant cell populations, will expand the power of similar studies.
In another study, hiPSC lines were generated from individuals with BPD that either did or did not respond to LiCl. Neurons derived from both of these groups displayed a hyperexcitability phenotype. This was only ameliorated via 1‐week chronic application of LiCl in cells derived from patients that responded clinically to LiCl treatment, not those that were unresponsive 33. Specifically, in “responder” neurons only, LiCl significantly reduced Na+/K+ currents, the total number of evoked APs and the frequency of spontaneous APs. Although neuronal hyperexcitability has not been causally linked with BPD pathology, these results suggest that this phenotype in hiPSC‐derived neurons may be predictive of treatment response. In this case, selection of a phenotype that is predictive of clinical response to medication was critical, and could be used in the future to test novel therapeutic treatments of BPD.
This approach has been extended to other neuropsychiatric disorders, as well. For example, studies have reported that the candidate molecule IGF‐1 can rescue aberrant neuronal phenotypes in a number of syndromic forms of ASD, such as Phelan‐McDermid syndrome 63 and RTT 35 as well as in nonsyndromic ASD 31.
High throughput screening can also be used to predict outcomes of treatment, as well as to identify novel therapies. For example, a screen using hiPSC‐derived dopaminergic neurons evaluated compounds previously documented to be neuroprotective to 1‐methyl‐4‐phenylpyridinium or rotenone in nonprimary cells of human origin or in vivo rodent models 64. Findings in this assay broadly correlated with outcomes in human Parkinson's disease clinical trials, suggesting the efficacy of small molecule screens in hiPSC‐derived cells to identify novel therapeutic compounds. Other screens have assessed inhibitors of amyloid‐β toxicity in forebrain GABAergic and glutamatergic neurons 65 and changes in transcript levels of the target gene (IKBKAP) in familial dysautonomia 66. In addition, neurons derived from mouse stem cells have been used to identify positive allosteric modulators of the AMPA receptor 67 and neuroprotective compounds for amyotrophic lateral sclerosis 68.
While displaying a great deal of promise, human hiPSC‐derived platforms used for drug screening require further improvement. Cells that require time‐consuming culturing and differentiation procedures may present practical limitations for high‐throughput drug screening. In addition, high‐throughput screening may require homogeneity of cell types, as heterogeneity would cause a decline in consistency and meaningfulness of a positive hit. The hiPSC differentiation process may result in significant downstream variation in the types of cells that are generated, depending on the protocol that is used. Also, as described above, selection of a relevant assay/phenotype is critical in generating meaningful results. Furthermore, the in vivo development of neurological disorders is almost certainly driven by a confluence of genetic, environmental, and stochastic processes which subtly alter the establishment of brain circuits and connectivity. While these exact conditions are difficult to model precisely in an in vitro system, microfluidic devices will likely be useful in future models that attempt to more comprehensively capture the global suite of in vivo interactions encompassing autocrine, paracrine, and physical communications between cells. Altogether, the benefits of using stem cells to model human neurological disorders offers new and exciting opportunities to refine exiting therapies in addition to generating new ones.
Current Challenges
A number of additional salient challenges remain in using hiPSCs to model neuropsychiatric disease. One of the primary difficulties arises from the natural genetic heterogeneity observed in cell lines derived from unique individuals. This diversity/heterogeneity can result in difficulties reprogramming somatic cells from individual patients to hiPSCs and might also affect the ability of established hiPSC lines to differentiate into desired cell populations 69. Numerous protocols must often be attempted in order to successfully generate desired cell populations from individual hiPSC lines. 3D cultures can be even more heterogeneous, with high variability in the structure and composition of individual organoids between cell lines, between individual experiments using the same cell lines, and even between individual organoids of the same cell line in the same experiment 70. Perhaps as a result, organoids have been shown to recapitulate some phenotypes of neurological disease but not all of them (e.g., see Autism section above). More defined differentiation procedures for both monolayer and 3D culture can alleviate some of these problems. In addition, the use of genomic editing techniques to generate isogenic cell lines with the addition or subtraction of causative mutations in identical genetic backgrounds can also reduce heterogeneity in hiPSC systems 71.
The ability of hiPSC derived cells/tissue/cultures to accurately model relevant neurological phenotypes represents another challenge in using this technology. For example, the interaction of endocrine and other bodily systems in modulating various phenotypes cannot be replicated in vitro. Diseases that are caused by epigenetic mechanisms may not be modeled accurately since the epigenetic status of cells changes during the reprogramming process 72. In addition, DNA methylation patterns of hiPSCs can also resemble their source cells, thus pushing hiPSC differentiation toward the cells of origin 73. Finally, the 2D culturing environment is limited in its ability to precisely model conditions that require cell–cell interactions and the extracellular milieu. Newly developed 3D‐culturing procedures can recapitulate at least some of these interactions. However, because organoids lack the precise organization of the animal brain, and do not possess key features of the brain such as the neurovasculature, current advances in human hiPSC‐based technologies cannot completely replace animal models for disease modeling.
Neurological disorders of adult onset/aging are particularly difficult to model with hiPSCs; hiPSC‐derived neurons have transcriptional profiles that are reflective of embryonic‐stage neurons 74, 75, which may make them more suitable for prenatal onset or neurodevelopmental conditions rather than late onset disease, although efforts to induce aging‐related hiPSC phenotypes have begun (for example, with progerin) 76. hiPSC‐derived tissue may not adequately model the adult epigenetic state, as the epigenetic landscape of hiPSC lines is converted to an early developmental stage during the reprogramming process 6. In addition, the manifestation of adult onset neurological disorders might require environmental insults accumulated over many years to initiate the pathological process. Direct differentiation of somatic cells to neurons has been shown to maintain the epigenetic landscape of older cells, which may help alleviate at least some of the problems associated with hiPSC reprogramming 77. However, neuronal cultures directly differentiated from somatic cells cannot be expanded, due to their post‐mitotic nature. Expansion of somatic cell cultures (e.g., primary fibroblast culture) is possible but is more limited than hiPSC culture, restricting the number of experiments that can be performed from one individual's cells. In addition, some neurodevelopmental processes that do not typically occur in the mature brain, such as neurite outgrowth and synapse formation, must be undertaken after neuronal differentiation occurs. This might lead to aberrant phenotypes associated with these abnormal events while having little to do with relevant disease phenotypes. Furthermore, it can be very challenging or even impossible to model long‐term environmental insults in culture. New techniques are needed to model these environmental effects in vitro, which often play a major role in neurological pathology.
Conclusion
Work with hiPSCs promises to generate novel insights into pathophysiology that will generate mechanistic insights into therapeutic strategies; concurrently, hiPSCs will provide a platform for analyses of potential treatments, via testing compounds with known pharmacologic profiles on human disease‐relevant cells and analyzing the range of consequent biological responses. Further applications for hiPSCs can be expected in phenotypic drug screens, as an analysis platform for high throughput screening; and in personalized in vitro therapeutic trials. We are only just beginning to scratch the surface of these various possibilities. Additional development of more refined hiPSC models of neurological disorders should tap the potential of this technology.
Author Contributions
A.A., L.B., C.F., M.Z., and A.W.‐B.: manuscript writing, final approval of the manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Acknowledgments
A.W.‐B. is supported by a grant from the Simons Foundation. L.B. is supported by a postdoctoral fellowship from The Hartwell Foundation.
References
- 1. American Psychiatric Association . DSM‐5 Task Force: Diagnostic and Statistical Manual of Mental Disorders: DSM‐5, Fifth Edition. Arlington, VA: American Psychiatric Association, 2013. [Google Scholar]
- 2. O'Donovan MC, Owen MJ. The implications of the shared genetics of psychiatric disorders. Nat Med 2016;22:1214–1219. [DOI] [PubMed] [Google Scholar]
- 3. Lee SH, Lumelsky N, Studer L et al. Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol 2000;18:675–679. [DOI] [PubMed] [Google Scholar]
- 4. Smith C, Gore A, Yan W et al. Whole‐genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN‐based genome editing in human iPSCs. Cell Stem Cell 2014;15:12–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663–676. [DOI] [PubMed] [Google Scholar]
- 6. Buganim Y, Faddah DA, Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nat Rev Genet 2013;14:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeRosa BA, Van Baaren JM, Dubey GK et al. Derivation of autism spectrum disorder‐specific induced pluripotent stem cells from peripheral blood mononuclear cells. Neurosci Lett 2012;516:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Griesi‐Oliveira K, Acab A, Gupta AR et al. Modeling non‐syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry 2015;20:1350–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vierbuchen T, Ostermeier A, Pang ZP et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010;463:1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shi Y, Kirwan P, Smith J et al. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci 2012;15:477–486. S471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maroof AM, Keros S, Tyson JA et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 2013;12:559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kriks S, Shim JW, Piao J et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 2011;480:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu J, Zhong X, Liu H et al. Generation of serotonin neurons from human pluripotent stem cells. Nat Biotechnol 2016;34:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang S, Wang B, Pan N et al. Differentiation of human induced pluripotent stem cells to mature functional Purkinje neurons. Sci Rep 2015;5:9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sances S, Bruijn LI, Chandran S et al. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat Neurosci 2016;19:542–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu DX, Di Giorgio FP, Yao J et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Reports 2014;2:295–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kadoshima T, Sakaguchi H, Nakano T et al. Self‐organization of axial polarity, inside‐out layer pattern, and species‐specific progenitor dynamics in human ES cell‐derived neocortex. Proc Natl Acad Sci USA 2013;110:20284–20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lancaster MA, Renner M, Martin CA et al. Cerebral organoids model human brain development and microcephaly. Nature 2013;501:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pasca AM, Sloan SA, Clarke LE et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods 2015;12:671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Birey F, Andersen J, Makinson CD et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017;545:54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Muguruma K, Nishiyama A, Kawakami H et al. Self‐organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep 2015;10:537–550. [DOI] [PubMed] [Google Scholar]
- 22. Sakaguchi H, Kadoshima T, Soen M et al. Generation of functional hippocampal neurons from self‐organizing human embryonic stem cell‐derived dorsomedial telencephalic tissue. Nat Commun 2015;6:8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Camp JG, Badsha F, Florio M et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci USA 2015;112:15672–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lancaster MA, Corsini NS, Wolfinger S et al. Guided self‐organization and cortical plate formation in human brain organoids. Nat Biotechnol 2017;35:659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stoner R, Chow ML, Boyle MP et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med 2014;370:1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hansen DV, Lui JH, Parker PR et al. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 2010;464:554–561. [DOI] [PubMed] [Google Scholar]
- 27. Bershteyn M, Nowakowski TJ, Pollen AA et al. Human iPSC‐derived cerebral organoids model cellular features of Lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell 2017;20:435–449. e434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Urbach A, Bar‐Nur O, Daley GQ et al. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010;6:407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wen Z, Nguyen HN, Guo Z et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014;515:414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen HM, DeLong CJ, Bame M et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl Psychiatry 2014;4:e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marchetto MC, Belinson H, Tian Y et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry 2017;22:820–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang P, Mokhtari R, Pedrosa E et al. CRISPR/Cas9‐mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol Autism 2017;8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mertens J, Wang QW, Kim Y et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015;527:95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mariani J, Coppola G, Zhang P et al. FOXG1‐dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 2015;162:375–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marchetto MC, Carromeu C, Acab A et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010;143:527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pasca SP, Portmann T, Voineagu I et al. Using iPSC‐derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med 2011;17:1657–1662. [Database [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cadwell CR, Palasantza A, Jiang X et al. Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch‐seq. Nat Biotechnol 2016;34:199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brennand KJ, Simone A, Jou J et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011;473:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sugathan A, Biagioli M, Golzio C et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci USA 2014;111:E4468–E4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tobe BTD, Crain AM, Winquist AM et al. Probing the lithium‐response pathway in hiPSCs implicates the phosphoregulatory set‐point for a cytoskeletal modulator in bipolar pathogenesis. Proc Natl Acad Sci USA 2017;114:E4462–E4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao WN, Cheng C, Theriault KM et al. high‐throughput screen for Wnt/beta‐catenin signaling pathway modulators in human iPSC‐derived neural progenitors. J Biomol Screen 2012;17:1252–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brennand K, Savas JN, Kim Y et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry 2015;20:361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee JH, Daugharthy ER, Scheiman J et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat Protoc 2015;10:442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Quadrato G, Nguyen T, Macosko EZ et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017;545:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Glantz LA, Lewis DA Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 2000;57:65–73. [DOI] [PubMed] [Google Scholar]
- 46. Lewis AD, Curley AA., Glausier J et al. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 2012;35:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miklowitz DJ, Johnson SL. The psychopathology and treatment of bipolar disorder. Annu Rev Clin Psychol 2006;2:199–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abrahams BS, Geschwind DH. Advances in autism genetics: On the threshold of a new neurobiology. Nat Rev Genet 2008;9:341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Geschwind DH. Genetics of autism spectrum disorders. Trends Cogn Sci 2011;15:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Berg JM, Geschwind DH. Autism genetics: Searching for specificity and convergence. Genome Biol 2012;13:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klei L, Sanders SJ, Murtha MT et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism 2012;3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev 2012;22:229–237. [DOI] [PubMed] [Google Scholar]
- 53. Yuen RK, Merico D, Cao H et al. Genome‐wide characteristics of de novo mutations in autism. NPJ Genom Med 2016;1:160271–1602710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gaugler T, Klei L, Sanders SJ et al. Most genetic risk for autism resides with common variation. Nat Genet 2014;46:881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ardhanareeswaran K, Mariani J, Coppola G et al. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat Rev Neurol 2017;13:265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luo C, Lancaster MA, Castanon R et al. Cerebral organoids recapitulate epigenomic signatures of the human fetal brain. Cell Rep 2016;17:3369–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Parikshak NN, Luo R, Zhang A et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013;155:1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Willsey AJ, Sanders SJ, Li M et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013;155:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mellios N, Feldman DA, Sheridan SD et al. MeCP2‐regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry 2017. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li Y, Muffat J, Omer A et al. Induction of expansion and folding in human cerebral organoids. Cell Stem Cell 2017;20:385–396. e383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Butler MG, Dasouki MJ, Zhou XP et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 2005;42:318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nowakowski TJ, Pollen AA, Sandoval‐Espinosa C et al. Transformation of the radial glia scaffold demarcates two stages of human cerebral cortex development. Neuron 2016;91:1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shcheglovitov A, Shcheglovitova O, Yazawa M et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013;503:267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Peng J, Liu Q, Rao MS et al. Using human pluripotent stem cell‐derived dopaminergic neurons to evaluate candidate Parkinson's disease therapeutic agents in MPP+ and rotenone models. J Biomol Screen 2013;18:522–533. [DOI] [PubMed] [Google Scholar]
- 65. Xu X, Lei Y, Luo J et al. Prevention of beta‐amyloid induced toxicity in human iPS cell‐derived neurons by inhibition of Cyclin‐dependent kinases and associated cell cycle events. Stem Cell Res 2013;10:213–227. [DOI] [PubMed] [Google Scholar]
- 66. Lee G, Ramirez CN, Kim H et al. Large‐scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol 2012;30:1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McNeish J, Roach M, Hambor J et al. High‐throughput screening in embryonic stem cell‐derived neurons identifies potentiators of alpha‐amino‐3‐hydroxyl‐5‐methyl‐4‐isoxazolepropionate‐type glutamate receptors. J Biol Chem 2010;285:17209–17217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yang YM, Gupta SK, Kim KJ et al. A small molecule screen in stem‐cell‐derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 2013;12:713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hu BY, Weick JP, Yu J et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci USA 2010;107:4335–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Renner M, Lancaster MA, Bian S et al. Self‐organized developmental patterning and differentiation in cerebral organoids. EMBO J 2017;36:1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Koo T, Lee J, Kim JS. Measuring and Reducing Off‐Target Activities of Programmable Nucleases Including CRISPR‐Cas9. Mol Cells 2015;38:475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol 2010;28:1079–1088. [DOI] [PubMed] [Google Scholar]
- 73. Kim K, Doi A, Wen B et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010;467:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mariani J, Simonini MV, Palejev D et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci USA 2012;109:12770–12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nicholas CR, Chen J, Tang Y et al. Functional maturation of hPSC‐derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 2013;12:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Miller JD, Ganat YM, Kishinevsky S et al. Human iPSC‐based modeling of late‐onset disease via progerin‐induced aging. Cell Stem Cell 2013;13:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mertens J, Paquola AC, Ku M et al. Directly reprogrammed human neurons retain aging‐associated transcriptomic signatures and reveal age‐related nucleocytoplasmic defects. Cell Stem Cell 2015;17:705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]