

Abstract
Left ventricular (LV) diastolic dysfunction is among others attributed to cardiomyocyte stiffness. Mesenchymal stromal cells (MSC) have cardiac‐protective properties. We explored whether intravenous (i.v.) application of PLacenta‐eXpanded (PLX) MSC‐like cells (PLX) improves LV diastolic relaxation in streptozotocin (STZ)‐induced diabetic mice and investigated underlying mechanisms. Diabetes mellitus was induced by STZ application (50 mg/kg body weight) during five subsequent days. One week after the first STZ injection, PLX or saline were i.v. applied. Two weeks later, mice were hemodynamically characterized and sacrificed. At this early stage of diabetic cardiomyopathy with low‐grade inflammation and no cardiac fibrosis, PLX reduced LV vascular cell adhesion molecule‐1, transforming growth factor‐β1, and interferon‐γ mRNA expression, induced the percentage of circulating regulatory T cells, and decreased the splenic pro‐fibrotic potential in STZ mice. STZ + PLX mice exhibited higher LV vascular endothelial growth factor mRNA expression and arteriole density versus STZ mice. In vitro, hyperglycemic PLX conditioned medium restored the hyperglycemia‐impaired tube formation and adhesion capacity of human umbelical vein endothelial cells (HUVEC) via increasing nitric oxide (NO) bioavailability. PLX further induced the diabetes‐downregulated activity of the NO downstream protein kinase G, as well as of protein kinase A, in STZ mice, which was associated with a raise in phosphorylation of the titin isoforms N2BA and N2B. Concomitantly, the passive force was lower in single isolated cardiomyocytes from STZ + PLX versus from STZ mice, which led to an improvement of LV diastolic relaxation. We conclude that i.v. PLX injection improves diabetes mellitus‐associated diastolic performance via decreasing cardiomyocyte stiffness. Stem Cells Translational Medicine 2017;6:2135–2145
Keywords: Placenta‐expanded cell, Diastole, Diabetes mellitus, Titin, Passive force
Significance Statement.
Impaired left ventricular (LV) diastolic relaxation represents the earliest preclinical manifestation of diabetic cardiomyopathy, a cardiac disorder with changes in cardiac structure and function in the absence of coronary artery disease and hypertension. Mesenchymal stromal cells (MSC) are well known for their cardioprotective features. This study demonstrates that intravenous application of placenta‐derived MSC‐like cells (PLX) improves LV diastolic relaxation and cardiomyocyte stiffness at an early stage of diabetic cardiomyopathy with low‐grade inflammation and no cardiac fibrosis through increased titin phosphorylation. These findings offer new insights into the cardioprotective effects of PLX, advancing their potential use for cardiac therapies.
Introduction
Chronic heart failure in diabetic patients can be caused by a clinical entity referred as “diabetic cardiomyopathy”, which takes place in the absence of hypertension and coronary artery disease, and is among other features characterized by interstitial inflammation 1, interstitial and perivascular fibrosis 2, impaired vascularization 3, endothelial dysfunction 4, and cardiomyocyte stiffness 5. Several lines of evidence demonstrate that impaired left ventricular (LV) diastolic relaxation represents the earliest preclinical manifestation of diabetic cardiomyopathy and indicate that it can progress to symptomatic heart failure 6, 7. Due to the impairment in LV diastolic performance, diabetic patients are more prone to heart failure after myocardial infarction 8 and have a poorer prognosis on heart failure development 9, 10. Impaired LV diastolic performance is attributed to excessive collagen deposition 11, 12 and cardiomyocyte stiffness 13, 14, of which the latter has recently been shown to be sufficient to induce diastolic dysfunction without any involvement of the extracellular matrix 15. Evidence shows that endothelial dysfunction 16 and vascular rarefaction 17 underlie impaired diastolic relaxation and particularly a systemic, low‐grade inflammation contributes to diastolic LV dysfunction 18 through coronary microvascular endothelial activation, which alters paracrine signalling to cardiomyocytes and predisposes them to high diastolic stiffness 19.
Mesenchymal stromal cells (MSC) are an attractive cell type for cell therapy given their immunomodulatory 20, 21, 22, antifibrotic 21, proangiogenic 23, endothelial‐protective features 24 and their ability to be used allogenically 25. With respect to diabetes mellitus, MSC have been shown to exert anti‐diabetic effects and to improve experimental diabetic cardiomyopathy 26, 27. However, the effect of MSC on the onset of diabetic cardiomyopathy and ON its preclinical manifestation—diastolic dysfunction—HAS not been unraveled so far. Placental‐derived MSC‐like cells, termed PLX, are stable adhesive stromal cells isolated from full‐term human placentas, cultured on carriers, and expanded in a bioreactor 28. They can be expanded in vitro without phenotypic or karyotypic changes 28 and have already been successfully used for the treatment of experimental hind limb ischemia 24 and myocardial infarction 23.
The aims of the present study were to investigate whether intravenous (i.v.) application of PLX could improve diabetes mellitus‐associated impaired LV diastolic relaxation at the early onset of diabetic cardiomyopathy and to analyze underlying mechanisms.
Materials and Methods
PLX Cell Preparation
The details of the PLX production process have been described previously 28. In brief, full‐term human donor placentas were processed at Pluristem Ltd., Haifa (Israel). Adherent cells were first selected and grown in two‐dimensional culture flasks for approximately 1 month and later expanded in a dedicated bioreactor system on a fibrous carrier material for 1 week. Cells were shipped frozen, thawed immediately prior to the experiment, and injected in vivo without further cultivation steps as described previously 23.
PLX Characterization
PLX cells were thawed, washed, resuspended in FACS buffer (phosphate buffered saline (PBS); Biochrom, Berlin, Germany) with 1% FBS), and aliquoted for immunostaining (∼200,000 cells per tube). Manufacturer recommended concentrations of fluorescein (FITC)‐ or phycoerythrin (PE)‐conjugated mouse anti‐human monoclonal antibodies (mAbs) (Becton Dickinson, Heidelberg, Germany) of interest (mesenchymal markers endothelial and leukocyte markers or their isotypes) were added, and cells were incubated at room temperature for 15 minutes. Cells were washed twice before they were subjected to flow cytometry in a Beckman Coulter FC‐500 system. Representative flow cytometry charts of PLX are illustrated in Supporting Information Figure 1.
Animals
Diabetes mellitus was induced in 8‐ to 9‐week‐old male C57BL6/J mice from the Forschungseinrichtung für Experimentelle Medizin (FEM; Berlin) by streptozotocin (STZ; Sigma Aldrich, St. Louis, MO) application (i.p.; 50 mg/kg body weight) during 5 subsequent days. 106 PLX or saline were randomly i.v. injected 1 week after the first STZ injection. Nondiabetic controls received PSB instead of STZ. Fourteen days after PLX application or saline injection, mice were hemodynamically characterized and subsequently sacrificed. After euthanization of mice, LV tissues were excised, immediately snap frozen in liquid nitrogen and stored at −80°C for molecular biology and immunohistological analyses. For the evaluation of immunomodulatory mechanisms, blood was collected and the spleen isolated. To evaluate the engraftment of PLX after i.v. injection, the left ventricle, spleen, lung, kidney, liver, and pancreas were collected, snap frozen in liquid nitrogen and stored at −80°C for molecular biology. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published in the European legislation (Directive 2010/63/EU) and was approved by the local ethics committee (Landesamt für Gesundheit und Soziales, Tierschutz, Berlin).
Blood Glucose
Blood glucose levels were measured with a commercial kit (MBL International, Woburn, MA) according to the manufacturer's protocol.
Assessment of PLX Engraftment
The engraftment of PLX in the left ventricle, lung, kidney, liver, spleen, and pancreas after i.v. application was determined according to the method of McBride et al. 29, slightly modified, as described previously 30. In brief, genomic DNA was extracted from frozen tissues as described previously 31. A standard curve was generated using human genomic DNA obtained from HUVEC serially diluted over a 100,000‐fold dilution range, into murine spleen genomic DNA. Real‐time PCR was performed with 800 ng of target DNA, Alu specific primers and a fluorescent probe 29. Values are expressed as percentage of human DNA per 800 ng (phosphate buffered saline (PBS); Biochrom, Berlin, Germany) of murine tissue.
Immunohistology
Immunohistochemistry was carried out on 5 µm‐thick cryosections using following antibodies: anti‐collagen I (Chemicon, Limburg/Lahn, Germany), anti‐collagen III (Merck Millipore, Darmstadt, Germany), anti‐CD68 (Abcam, Cambridge, U.K.), anti‐CD3 (Santa Cruz Biotechnology, Heidelberg, Germany) anti‐CD4 (BD Biosciences, San Jose, CA), and anti‐CD8a (BD Biosciences). Analysis of stained sections was made in a blinded fashion by digital image analysis on a Leica DMRB microscope (Leica Microsystems, Wetzlar, Germay) at ×200 magnification. Artery and arteriole density was measured using immunohistochemistry with an anti‐α‐smooth muscle actin (SMA) antibody (1:200, Abcam). Microscopy was performed on a Leica DM2000 light microscope. Arteries and arterioles were counted in 10 high power fields (hpf) per animal at a magnification of ×100. The density is presented as number of arteries or arterioles per hpf.
Gene Expression Analysis
RNA was isolated using the RNeasy Mini Kit according to the manufacturer's protocol (Qiagen GmbH, Hilden, Germany), followed by cDNA synthesis. To assess the mRNA expression of the target genes vascular cell adhesion molecule (VCAM)‐1, transforming growth factor (TGF)‐β1, interferon (IFN)‐γ, vascular endothelial growth factor (VEGF), and L32, real‐time polymerase chain reaction (PCR) (Eppendorf Mastercycler epgradient realplex, Hamburg, Germany) was performed using gene expression assays for VCAM‐1: Mm01320970_m1, TGF‐ß: Mm00441724_m1, interferon‐γ (IFN‐γ): Mm00801778_m1, and VEGF: Mm01281447_m1 from Applied Biosystems. mRNA expression was normalized to the housekeeping gene L32 and relatively expressed with the control group set as 1.
Cell Culture
PLX were cultured at a density of 6,000 cells per cm2 in DMEM medium containing 10% fetal bovine serum (FBS) (Biochrom, Berlin, Germany). Human cardiac fibroblasts (Cell Applications, Inc. San Diego, CA) were propagated and cultured in Lung/Cardiac Fibroblasts Basal Medium (Cell Applications, Inc. San DiegoCA) plus supplements (Cell Applications). Cardiac fibroblasts at passage 4 or 5 were plated at a cell density of 10,000 cells per 96‐well or 135,000 cells per 6‐well in Lung/Cardiac Fibroblasts Basal Medium (Cell Applications) plus supplements (Cell Applications). Twenty‐four hours later, cells were washed, and cells were cultured in the presence of standard or conditioned PLX medium with or without 10 ng/ml of transforming growth factor‐β1 (TGF‐β1) (R&D Systems) for 24 hours for crystal violet, Sirius Red staining, or α‐SMA flow cytometry.
Human umbilical vein endothelial cells (HUVEC) were propagated and cultured in endothelial basal medium (EBM)‐2 basal medium (Lonza Walkersville, Walkersville, MD) with endothelial cell growth medium (EGM)‐2 supplements (Lonza). HUVEC at passage 4 or 5 were plated at a cell density of 125,000 cells per 6‐well (adhesion assay), 150,000 cells per 6‐well (flow cytometry, Western blot) or 10,000 cells per 96‐well (caspase 3/7 activity assay) in EBM‐2 Medium (Lonza) plus supplements (Lonza). Twenty‐four hours later, cells were washed and cultured in the presence of standard or conditioned normo‐ or hyperglycemic PLX medium for 24 hours for DAF‐FM (NO) or CMH2DCFDA (reactive oxygen species; ROS) flow cytometry, Western blot, or collected for adhesion assay.
Preparation of PLX‐Conditioned Medium
PLX were plated at a density of 150,000 cells per 6‐well in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% FBS. After 24 hours, the medium was removed, cells were washed with PBS, and 1 ml of normo‐or hyperglycemic EBM‐2 medium with supplements (PLX‐conditioned medium for endothelial cells) or 1 ml of standard PLX medium with or without 10 ng/ml of TGF‐β1 (PLX‐conditioned medium for cardiac fibroblasts) was added on the cells. In parallel, normo‐or hyperglycemic EBM‐2 medium or standard PLX medium with or without 10 ng/ml of TGF‐β1 was added to wells without cells. These media served as respective controls. Twenty‐four hours later, the medium was collected, centrifuged to discard cellular debris, and stored at −80°C until use.
Coculture of Fibroblasts with Splenocytes
Murine C4 fibroblasts were plated at a cell density of 10,000 cells per 96‐well in Basal Iscove medium supplemented with 10% FBS and 1% penicillin/streptomycin. Twenty‐four hours after plating, medium was removed and splenocytes isolated from control + PBS, control + PLX, STZ + PBS, and STZ + PLX mice, according to Van Linthout et al. 20, were added to the fibroblasts at a ratio of 10 splenic cells to 1 fibroblast in RPMI medium 10% FBS, 1% penicillin/streptomycin, as described previously 21. After 3 days, splenocytes were removed and Sirius Red staining was performed.
Sirius Red Staining
Sirius Red staining of fibroblasts was performed as described previously 32. In brief, fibroblasts were fixed in methanol overnight at −20°C, washed once with PBS and incubated in 0.1% Direct Red 80 (Sirius Red; Sigma Aldrich) staining solution at room temperature for 1 hour. After a second wash with PBS, the Sirius red staining of the fibroblasts was eluted in 0.1N sodium hydroxide at room temperature for 1 hour on a rocking platform. The optical density representative for the accumulation of collagen I and III was measured at 540 nm on a SpectraMax 340PC microplate reader (Molecular Device GmbH, Biberach an der Riβ, Germany).
Western Blot
Twenty‐four hours after culture in standard or conditioned normo‐ or hyperglycemic PLX medium, HUVEC were lysed in lysis buffer (Thermo Fisher Scientific, Waltham, MA) supplemented with proteinase inhibitors (Roche). An equal amount of protein (40 µg) was loaded into 12% sodium dodecyl sulfate (SDS)‐polyacrylamide gels. Total endothelial nitric oxide synthase ([Enos] BD Biosciences) and β tubulin (Santa Cruz Biotechnology) were detected with each specific antibody, followed by incubation with an IR dye secondary antibody (LI‐COR Biosciences, Lincoln, NE). The blots were visualized with Odyssey (LI‐COR Biosciences). Quantitative analysis of the intensity of the bands was performed with Odyssey V3.0 software, as described previously 33, 34.
Caspase 3/7 Activity Assay
Twenty‐four hours after culture in 100 µl of standard or conditioned normo‐ or hyperglycemic PLX medium, caspase 3/7 activity was determined with a Caspase Glo 3/7 activity kit (Promega, Madison, WI) according to the manufacturer's protocol. Luminescence was measured in a microplate‐reading luminometer (Mithras LB 940, Berthold Technologies GmbH & Co KG, Germany) as described previously 33.
Adhesion Assay
HUVEC, 24 hours cultured in standard or conditioned normo‐ or hyperglycemic PLX medium, were plated at a cell density of 40,000 cells/96‐well in PBS for 2 hours on 96‐well plates pre‐coated with 2.5 µg/ml fibronectin (Sigma Aldrich). Medium and cells were removed and wells were next twice washed with warm PBS, followed by fixation with paraformaldehyde and crystal violet staining. The absorbance was next measured at 495 nm on a SpectraMax 340PC microplate reader (Molecular Device GmbH).
Tube Formation
HUVEC (20,000 cells per well) were resuspended in normo‐ or hyperglycemic EBM‐2 medium, or conditioned full normo‐ or hyperglycemic PLX medium (see preparation of conditioned medium) and plated on top of a matrigel (BD Biosciencs). Twenty‐four hours after seeding, pictures of eight fields per condition were taken at magnification ×5 and send to Wimasis for analysis via WimTube software (Wimasis GmbH, München, Germany).
Flow Cytometry
Flow cytometry analysis of blood mononuclear cells (MNCs) isolated with Histopaque 1083 (Sigma Aldrich) and cardiac fibroblasts was performed using directly conjugated monoclonal antibodies. The α‐SMA antibody was purchased from BD Biosciences (Franklin Lakes, NJ). The mouse Treg detection kit containg CD4, CD25, FoxP3 antibodies, fixation, and wash solutions was purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). Surface staining was performed according to the manufacturer's instructions. MNCs were first stained with anti‐mouse CD4 and CD25 antibodies, and after fixation, intracellular FOXP3 staining was performed. MNC samples were acquired on a MACSQuant Analyzer (Miltenyi Biotec), and cardiac fibroblast samples on fluorescence‐activated cell sorting (FACS) BD FACSCanto II with BD FACSDiva software version 6.1.3. For the analysis of NO and ROS in HUVEC, cells were stained with 5 μM of DAF‐FM Diacetate (4‐amino‐5‐methylamino‐2′,7′‐difluorofluorescein diacetate, Thermo Fisher Scientific) or of CM‐H2DCFDA (5‐(and‐6)‐chloromethyl‐2′,7′‐dichlorodihydrofluorescein diacetate, acety ester) (Thermo Fisher Scientific) in PBS, respectively, and incubated for 30 minutes at 37°C, as described previously 20. Next, cells were washed with 200 µl of PBS and after resuspending in fresh PBS, incubated for an additional 30 minutes at 37°C. Flow cytometry analysis was performed on a MACSQuant Analyzer (Miltenyi Biotec). Analysis of flow cytometry data was performed using FlowJo software version 8.8.6. (Tree Star Inc.).
Titin Isoform Separation
Titin isoforms were separated as described previously 13. Tissue samples were solubilized in 50 mM Tris SDS buffer (pH 6.8) containing 8 μg/ml leupeptin (Peptin Institute, Japan) and phosphatase inhibitor cocktail (PIC [P2880], 10 μl/ml; Sigma Aldrich). Samples were heated for 3 minutes at 96°C and centrifuged. Then, samples (20 μg) were separated on agarose‐strengthened 1.8% sodium dodecyl sulfate‐polyacrylamide gels. The gel was run at 5 mA constant current for 16 hours.
All‐Titin Phosphorylation by Pro‐Q Diamond Stain
The phosphorylation state of cardiac titin isoforms, N2BA and N2B, was determined using Pro‐Q Diamond phosphoprotein stain (Thermo Fisher Scientific). To preserve the endogenous phosphorylation state of the proteins, frozen tissues from LV mouse hearts were solubilized and treated as described above in the Titin Isoform Separation section. The gels were stained for 1 hour with Pro‐Q Diamond phosphoprotein stain. Fixation, washing, and de‐staining were performed according to the manufacturer's guidelines (Thermo Fisher Scientific). To assess total protein content, gels were stained overnight with SYPRO Ruby (Thermo Fisher Scientific). Staining was visualized using the LAS‐4000 Image Reader (460 nm/605 nm Ex/Em; 2 seconds illumination), and signals were analyzed with Multi Gauge V3.2 and AIDA software. Titin‐isoform phosphorylation was expressed in relative numbers, assuming that the combined phosphorylation of N2BA + N2B titin in control samples is 100%.
Myocardial Protein Kinase G Activity
Myocardial protein kinase G (PKG) activity was assessed in homogenized tissue samples. Tissues samples were homogenized in 25 mmol/l Tris (pH 7.4), 1 mmol/l EDTA, 2 mmol/l EGTA, 5 mmol/l dithiothreitol (DTT), 0.05% Triton X‐100, and protease inhibitor cocktail (Sigma Aldrich) and centrifuged for 5 minutes. Supernatants containing equal amounts of total protein were analyzed for PKG activity. The reaction mixture contained (at final concentration): 40 mmol/l Tris‐HCl (pH·7.4), 20 mmol/l magnesium acetate, 0.2 mmol/l [32P] ATP (500–1,000 c.p.m.·pmol−1) (GE Healthcare LifeScience, Little Chalfont, U.K.), 113 mg/ml heptapeptide (RKRSRAE), 3 mmol/l cGMP (Promega, Madison, WI) and a highly specific inhibitor of cAMP‐dependent protein kinase (5–24, Merck Millipore). The reaction mixtures were incubated at 30°C for 10 minutes, followed by termination of the reaction by spotting 70·μl of the reaction mix onto Whatman P‐81 filters, which were then soaked with 75·mmol/l H3PO4 for 5 minutes and washed three times with 75·mmol/l H3PO4 to remove any unbound [32P] ATP. Filters were rinsed with 100% ethanol and air dried before quantification. For quantification of PKG activity, counts were taken in a Wallac 1409 Liquid Scintillation Counter using universal scintillation cocktail. Specific activity of PKG was expressed as pmol of [32P] incorporated into the substrate (pmol/minute/mg protein).
Myocardial Protein Kinase A and C Activity
Protein kinase A (PKA) and protein kinase C (PKC) activity was analyzed using a nonradioactive PKA and PKC kinase activity assay kit (Enzo Life Science). Samples were homogenized in cell lysis buffer (20 mmol/l MOPS, 50 mmol/l β‐glycerolphosphate, 50 mmol/l sodium fluoride, 1 mmol/l sodium vanadate, 5 mmol/l EGTA, 2 mmol/L EDTA, 1% NP40, 1 mmol/l DTT, 1 mmol/l benzamidine, 1 mmol/l phenylmethanesulphonylfluoride, and 10 μg/ml leupeptin and aprotinin, each). Supernatants were collected after centrifugation at 13.000 rpm for 30 minutes. Supernatants containing equal amounts of total protein (30 ng/μl protein aliquots were assayed according to manufacturer's instructions) were added into the appropriate wells of the substrate microliter plate. The kinase reaction was initiated by addition of ATP, and samples were subsequently incubated at 30°C for 90 minutes. Phosphorylated peptide substrates were recognized by phosphospecific substrate antibody. The phosphospecific antibody was subsequently bound by a peroxidase conjugated secondary antibody anti‐rabbit IgG:HRP. The assay was developed with tetramethylbenzidine, and the intensity of the color was measured in a microplate reader at 450 nm. Results of triplicate determinations were averaged, and specific activity of the kinase was expressed as ng/μl.
Ca2+Calmodulin Dependent Protein Kinase II Activity
Ca2+calmodulin dependent protein kinase II (CaMKII) activity was determined using a CycLex CaMKII assay kit (CY‐1173; MBL Corporation, MA) according to the manufacturer's guidelines. Briefly, frozen tissues were homogenized in sample buffer containing 15% glycerol, 62.5 mmol/l Tris; pH 6.8; 1% (wt/vol) SDS, protease inhibitor, and protein phosphatase inhibitor, all prepared in distilled H2O. Homogenates were centrifuged at 10.000g for 15 minutes at 4°C. The supernatant was removed and stored at −80°C. Protein samples were loaded onto microtiter wells (concentration, ∼2.0 μg per well) coated with CaMKII specific substrate, syntide‐2, along with kinase reaction buffer with or without Ca2+/calmodulin. To quantify CaMKII activity, a standard curve correlating the amount of active CaMKII and the level of phosphorylation of syntide‐2 was constructed.
Passive Force
Force measurements were performed on single skinned cardiomyocytes as described 35, 36. Briefly, samples were defrozen in relaxing solution (in mmol/l: free Mg, 1; KCl, 100; EGTA, 2; Mg‐ATP, 4; imidazole, 10; pH 7.0), mechanically disrupted and incubated for 5 minutes in relaxing solution supplemented with 0.5% Triton X‐100. The cell suspension was washed 5 times in relaxing solution. Single cardiomyocytes were selected under an inverted microscope (Zeiss Axiovert 135, ×40 objective, Oberkochen, Germany) and attached with silicone adhesive between a force transducer and a piezoelectric motor as part of a “Permeabilized Myocyte Test System” (1600A; with force transducer 403A; Aurora Scientific, Aurora, Ontario, Canada). Cardiomyocyte Fpassive was measured in relaxing buffer at room temperature within a sarcomere‐length range between 1.9 and 2.4 μm. Force values were normalized to myocyte cross‐sectional area calculated from the diameter of the cells, assuming a circular shape. As a test of cell viability, each cardiomyocyte was also transferred from relaxing to maximally activating solution (pCa4.5), at which isometric force developed. Once a steady state force was reached, the cell was shortened within 1 ms to 80% of its original length to determine baseline force. Only cells developing active forces >20 kN/m2 were included in the analysis. The passive tension values shown were measured at the end of each stretch step and thus consisted of both the viscous and the elastic force component.
Hemodynamic Measurements
Fourteen days after i.v. PLX injection, mice were anesthetized (0.8–1.2 g/kg urethane and 0.05 mg/kg buprenorphine i.p.), intubated, and artificially ventilated with a rodent ventilator type 7025 (Ugo Basile, Comerio VA, Italy). A 1.2F micro‐tip catheter (Scisense Inc., Ontario, Canada) was positioned in the LV through the apex in an open chest model. Heart rate and LV contractility, measured by the peak rate of LV pressure rise dP/dtmax were measured. Diastolic performance was measured by the peak rate of LV pressure decrease dP/dtmin and the time constant of LV relaxation Tau (τ).
Statistical Analysis
Statistical analysis was performed using Prism 7 for Mac OS X (GraphPad Software, Inc., La Jolla, CA). Ordinary one‐way analysis of variance (ANOVA) was used for statistical analysis of the data with correction for multiple comparisons via the Tukey test or as stated elsewhere. Data are presented as mean ± SEM. Differences were considered to be significant when the two‐sided p value was lower than .05.
Results
Engraftment of PLX After Intravenous Injection
We evaluated the presence of PLX in the left ventricle, lung, kidney, liver, spleen, and pancreas at the day of sacrifice, 14 days after i.v PLX application in control and STZ‐induced diabetic mice via the detection of human Alu sequences (Fig. 1). PLX were retrieved in all analyzed tissues. Interestingly, STZ‐induced diabetes mellitus raised the engraftment of PLX to the left ventricle, lung, kidney, pancreas, and tendentiously to the spleen as obviated by 4.5‐fold (p < .005), 19‐fold (p < .005), 4.7‐fold (p < .05), 47‐fold (p < .05), and 2.4‐fold (p = .1694) higher PLX presence in respective tissues of diabetic compared with control mice.
Figure 1.
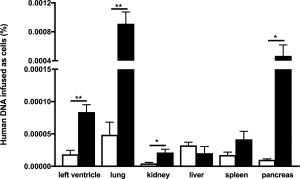
Engraftment of PLacenta‐eXpanded (PLX) 14 days after intravenous injection. Bar graphs represent human DNA infused as cells (%) in the left ventricle, lung, kidney, liver, spleen, and pancreas of control mice (open bars) and streptozotocin (STZ)‐treated mice (closed bars) i.v. injected with PLX. For comparison of PLX presence per respective tissue of control versus STZ mice, an unpaired Student's t test was used with *, p < .05; **, p < .005; n = 4/group.
PLX Exert Anti‐Inflammatory/Immunomodulatory Effects in Streptozotocin‐Induced Diabetic Mice
Given the immunomodulatory properties of MSC 37 and the importance of cardiac inflammation in diabetic cardiomyopathy 38, we next evaluated whether i.v. PLX application decreased cardiac inflammation in STZ‐induced diabetic mice. In this STZ diabetic model of low‐grade inflammation with no increased cardiac presence of CD68 macrophages, CD3+, CD4+, and CD8+ T lymphocytes (Supporting Information Fig. 2), PLX application downregulated the 1.8‐fold (p < .0001) STZ‐induced increase in LV VCAM‐1 expression to levels not significantly different from controls (Fig. 2A) and led to a 1.8‐fold (p < .05) and 3.1‐fold (p < .05) decrease in STZ‐induced transforming growth factor‐β1 (TGF‐β1) and IFN‐γ mRNA expression (Fig. 2B, 2C), respectively. Systemically, PLX induced the percentage of circulating regulatory T cells (CD4 + CD25 + FoxP3+ cells) by 1.6‐fold in STZ mice (Fig. 2D). PLX application did not reduce blood glucose levels (control: 7.5 ± 0.85; STZ: 24 ± 3.0; control PLX: 4.6 ± 0.72; STZ PLX: 24 ± 2.9; p < .0001 STZ vs. control).
Figure 2.
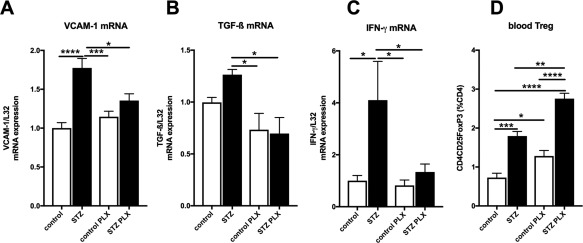
PLX reduce cardiac inflammation and increase blood regulatory T cells in streptozotocin‐induced diabetic mice. Bar graphs represent the mean ± SEM of LV. (A): VCAM‐1, (B): TGF‐β, (C): IFN‐γ, with n = 7–8/PBS groups and n = 5–6/PLX groups, and (D): blood Treg cells, depicted as CD4CD25FoxP3 (% of CD4 cells) with n = 4/group in control (open bars) and STZ (closed bars) mice injected with PBS or PLX, as indicated. For all data: *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001. Abbreviations: IFN‐γ, interferon‐γ; PLX, placenta‐expanded cell; STZ, streptozotocin; TGF‐β1, transforming growth factor‐β1; Treg, regulatory T cells; VCAM, vascular cell adhesion molecule.
PLX Exert Antifibrotic Effects
Given the relevance of cardiac fibrosis in diabetic cardiomyopathy 1 and the antifibrotic potential of MSC 21, we further evaluated whether PLX also exert antifibrotic effects. Therefore, we first examined in vitro whether PLX decrease collagen deposition and the expression of the myofibroblast marker α‐SMA in cardiac fibroblasts in a paracrine manner. Addition of the pro‐fibrotic factor TGF‐β1 to cardiac fibroblasts induced collagen deposition and α‐SMA expression by 1.2‐fold (p < .001) and 1.5‐fold (p < .001), respectively. In contrast, collagen deposition and α‐SMA expression was 1.7‐fold (p < .0001) and 2.3‐fold (p < .0001) lower in cardiac fibroblasts supplemented with PLX + TGF‐β1 conditioned medium compared with TGF‐β1 medium (Fig. 3A, 3B), respectively. Given the link between inflammation and fibrosis 39 and the relevance of the cardiosplenic axis, that is, the homing of immune cells from the spleen toward the heart and their subsequent involvement in cardiac remodeling 40, we next evaluated whether the PLX‐mediated immunomodulatory effect could influence fibroblast collagen production. Therefore, splenocytes of all in vivo experimental groups were cocultured with murine fibroblasts and their impact on collagen deposition of murine fibroblasts was determined (Fig. 3C). Supplementation of splenocytes from control mice did not induce collagen production compared with monocultured fibroblasts. In contrast, addition of STZ splenocytes to fibroblasts raised collagen production by 16% (p < .001) versus monocultured fibroblasts. STZ + PLX splenocytes did not induce collagen production compared with monocultured fibroblasts or fibroblasts cocultured with control splenocytes (Fig. 3D). Finally, we evaluated the impact of i.v. PLX application on cardiac fibrosis in STZ mice via immunohistochemistry. Concomitant with the low cardiac inflammation, cardiac fibrosis was not prominent at this stage of diabetic cardiomyopathy, as indicated by no STZ‐induced accumulation of collagen I and III (Supporting Information Fig. 3).
Figure 3.
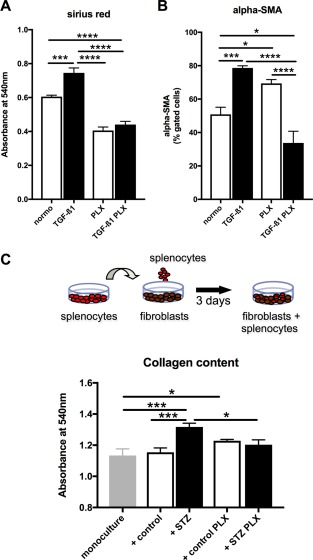
PLX exert antifibrotic effects in vitro. PLX conditioned medium reduces TGF‐β1‐induced collagen deposition and α‐SMA expression in cardiac fibroblasts. Bar graphs represent the mean ± SEM of (A) the absorbance at 540 nm detected after Sirus Red staining, depicting collagen deposition, and (B) α‐SMA expression of cardiac fibroblasts cultured in PLX medium, PLX medium + TGF‐β1, PLX conditioned (cond.) medium, or PLX cond. medium + TGF‐β1 as indicated, with open bars representing without TGF‐β1 and black bars with TGF‐β1 conditions, and n = 6/group. Splenocytes derived from STZ PLX mice induce lower collagen deposition in murine fibroblasts upon coculture compared with splenocytes from STZ mice. (C): Schematic representation of the experimental set‐up. (D): Bar graph represents the mean ± SEM of the absorbance at 540 nm detected after Sirus Red staining of fibroblasts cultured in monoculture or cocultured with splenocytes from control, STZ, control PLX, or STZ PLX mice as indicated, with n = 5–6/group. For all data: *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001. Abbrevaitions: PLX, PLacenta‐eXpanded cell; STZ, streptozotocin; TGF‐ß1, transforming growth factor‐β1.
PLX Improve Vascularization in Streptozotocin‐Induced Diabetic Mice
Endothelial inflammation and impaired vascularization underlie diastolic dysfunction 12, 41. On the other hand, PLX have endothelial‐protective and proangiogenic properties 23, 24. Therefore, we next explored the endothelial‐protective and proangiogenic potential of PLX in STZ‐induced diabetic mice and under hyperglycemic conditions. In STZ mice, LV mRNA expression of VEGF was 2.1‐fold (p < .05) lower versus nondiabetic control mice, whereas STZ + PLX mice exhibited 2.2‐fold (p < .05) higher LV VEGF mRNA expression compared with STZ mice (Fig. 4A). In parallel, LV arteriole density was 1.4‐fold (p < .0001) lower in STZ versus control mice and 1.2‐fold (p < .05) higher in the STZ mice receiving PLX compared with untreated STZ mice (Fig. 4B, 4C). Artery density was comparable in STZ versus control mice (Fig. 4B–4D). In vitro, hyperglycemia resulted in a 1.6‐fold (p < .01) increase in eNOS expression, which was not altered in HUVEC cultured with conditioned hyperglycemic PLX medium (Fig. 4E). The hyperglycemia‐induced eNOS expression did not affect NO production in HUVEC (Fig. 4F), but led to a 1.6‐fold (p < .0001) rise in ROS (Fig. 4G). This increase in oxidative stress was not associated with prominent endothelial apoptosis (Fig. 4H), but reflected in a 1.1‐fold (p < .05) reduced adhesion capacity of HUVEC (Fig. 4I) and impaired tube formation (Fig. 4J–4M) as indicated by 3.0‐fold (p < .0001), 1.7‐fold (p < .0001), and 1.5‐fold (p < .001) decreased total branching points, total tubes, and tube length, respectively. PLX abrogated the hyperglycemia‐triggered ROS in a paracrine manner by 1.2‐fold (p < .0001), which was paralleled by a 1.2‐fold (p = 0.13) lower caspase 3/7 activity (Fig. 4H), a 1.2‐fold (p < .001) higher adhesion capacity (Fig. 4I) and improved tube formation (Fig. 4J–4M) as shown by 1.9‐fold (p < .05), 1.4‐fold (p < .01), and 1.3‐fold (p < .05) higher total branching points, total tubes, and tube length in HUVEC cultured in hyperglycemic PLX condition medium compared with hyperglycemic medium alone, respectively.
Figure 4.
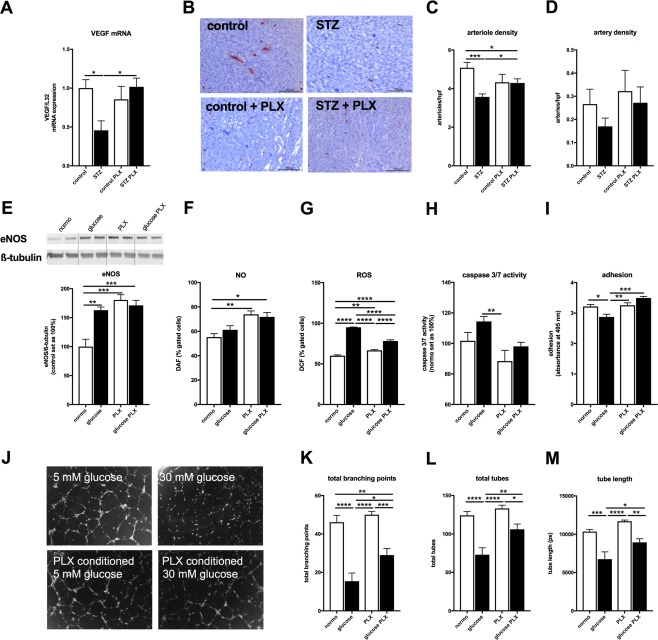
PLX improve hyperglycemia‐impaired vascularization and endothelial function. (A): Bar graphs represent the mean ± SEM of LV VEGF mRNA expression in control (open bars) and STZ (closed bars) mice injected with PBS or PLX, as indicated with n = 7/control and n = 6/STZ and PLX groups. (B): Representative pictures at magnification ×100 of arteries and arterioles in the LV of control, STZ, control + PLX, and STZ + PLX mice as indicated. Bar graphs represent the mean ± SEM of LV (C) arteriole and (D) artery density per high power field (hpf) in control (open bars) and STZ (closed bars) mice injected with PBS or PLX, as indicated with n = 8/control, n = 10/STZ, n = 5/control + PLX, and n = 11/STZ + PLX groups. In vitro, PLX‐conditioned hyperglycemic medium modulate (E) eNOS expression, (F) NO, (G) ROS levels, and (H) caspase 3/7 activity in HUVEC, which is translated in improved (I). adhesion capacity and (J–M) tube formation versus HUVEC cultured under hyperglycemic conditions. Bar graphs represent the mean ± SEM of (E) eNOS expression normalized to β‐tubulin and the control groups set as 100%, (F) % of DAF‐positive, (G) % of DCF‐positive gated cells, with all n = 4/condition, (H) caspase 3/7 activity depicted with the normoglycemia group set as 100% with n = 8–10/condition, and (I) adhered crystal violet‐stained HUVEC depicted as the absorbance at 495 nm with n = 5–6/condition. (J): Representative pictures of HUVEC cultured in normo‐ (5 mM glucose), hyperglycemic (30 mM glucose) medium, or normo‐ (5 mM glucose), hyperglycemic (30 mM glucose) conditioned PLX medium, as indicated. Bar graphs represent the mean ± SEM of (K) total branching points, (L) total tubes, and (M) tube length analyzed from n = 7 to 8 fields/condition. For all data: *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001. Abbrevaitions: eNos, endothelial nitric oxide synthase; NO, nitric oxide; ROS, reactive oxygen species, PLX, placenta‐expanded cell; ROS, reactive oxygen species; STZ, streptozotocin.
PLX Decrease Cardiomyocyte Stiffness and Improve Diastolic Relaxation in Streptozotocin‐Induced Diabetic Mice
Given the importance of cardiac titin dysregulation and cardiomyocyte stiffness in diastolic dysfunction 13, the impact of PLX on cardiac titin phosphorylation and cardiomyocyte passive tension was investigated. PLX elevated the STZ‐impaired phosphorylation state of titin isoforms N2BA and N2B by 3.3‐fold (p < .0005) and 6.1‐fold (p < .0001), respectively, to values not different from controls (Fig. 5A–5C). Concomitantly, PLX restored the diabetes‐downregulated PKA and PKG activity by 1.2‐fold (p < .05) and 1.8‐fold (p < .05) to control values (Fig. 5D, 5E), respectively, and tendencially increased the diabetes‐downregulated CaMKII activity (Fig. 5F). Protein kinase C activity was not different among the groups (Fig. 5G). In parallel, passive force in single isolated cardiomyocytes from STZ + PLX mice was comparable to control mice and was lower than diabetes‐induced passive force of cardiomyocytes from STZ mice (Fig. 5H). In vivo, STZ + PLX mice showed an improved diastolic function compared with STZ diabetic mice as indicated by the heart‐rate independent 1.2‐fold (p < .005) lower time constant of LV relaxation parameter Tau and the 1.2‐fold (p < .05) increase of the relaxation parameter dP/dtmin (Fig. 5I, 5J). As known for the early time point in the STZ animal model 42, LV contractility was slightly reduced in both STZ‐groups versus controls (STZ: 15% and STZ + PLX: 13% versus controls; p = NS), but did not differ significantly between the four groups.
Figure 5.
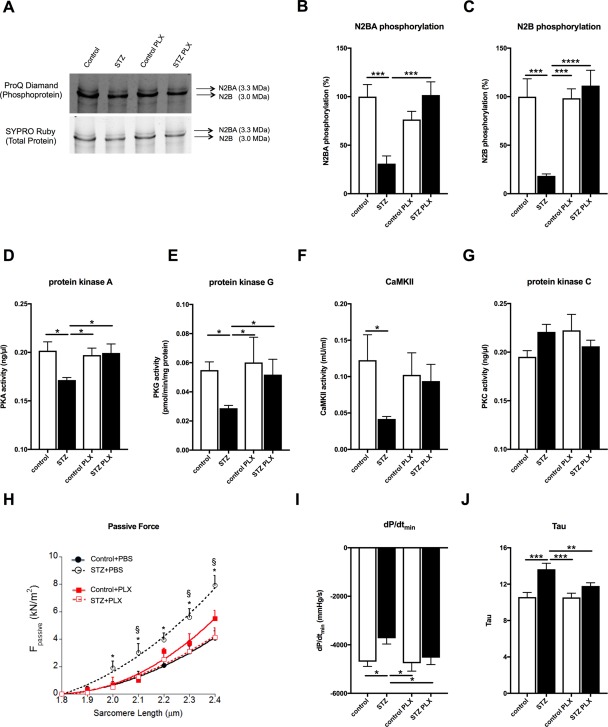
PLX restore cardiomyocyte stiffness and improve diastolic relaxation in streptozotocin‐induced diabetic mice. (A): Representative images of titin N2BA and N2B phosphoproteins and total proteins in control, STZ, control PLX, and STZ PLX mice, as indicated. Bar graphs represent the mean ± SEM of (B) N2BA phosphorylation and (C) N2B phosphorylation depicted as % with the control set as 100% of control (open bars) and STZ (closed bars) mice injected with PBS or PLX, as indicated with n = 7/control, n = 10/STZ, n = 5–7/control + PLX and n = 7–8/STZ + PLX groups and *, p < .05; **, p < .01; ***, p < .0005; ****, p < .0001 of (D) PKA (ng/µl) in control (open bars) and STZ (closed bars) mice injected with PBS or PLX, as indicated with n = 3/group and *, p < .05; (E) PKG (pmol/min/µg protein) in control (open bars) and STZ (closed bars) mice injected with PBS or PLX, as indicated with n = 4/PBS group, n = 3/PLX group and *, p < .05, (F) CaMKII activity (CaMKII; mU/ml) in control (open bars) and STZ (closed bars) mice injected with PBS or PLX, as indicated with n = 5/control group, n = 9/STZ group, and n = 4/PLX groups, and *, p < .05, (G) PKC (ng/µl) in control (open bars) and STZ (closed bars) mice injected with PBS or PLX, as indicated with n = 3/group and *, p < .05, (H) Passive tension Fpassive (kN/m2) versus sarcomere length (µm) in control (‐•‐), STZ (‐○‐), control + PLX (‐▪‐), and STZ + PLX (‐◻‐), as indicated, with n = 15–19 cardiomyocytes/group and *, p < .05 versus control and STZ + PLX and §, p < .05 versus control + PLX. Bar graphs represent the mean ± SEM of (I) dP/dtmin and (J) Tau with n = 8 and n = 11 for the control and STZ groups, respectively, and *, p < .05; **, p < .01; ***, p <.0005 analyzed by one‐way analysis of variance (ANOVA) without correction for multiple comparison. Abbreviations: PKA, protein kinase A; PKC, protein kinase C; PKG, protein kinase G; PLX, placenta‐expanded cell; STZ, streptozotocin.
Discussion
The salient finding of the present study is that i.v. PLX application reduces cardiomyocyte stiffness and improves LV diastolic relaxation in an early phase of STZ‐induced diabetic mice. Endothelial dysfunction 16, impaired vascularization 3, microvascular inflammation 43, cardiac fibrosis 11, 12, and cardiomyocyte stiffness 15 all underlie impaired LV diastolic relaxation, one of the first clinical signs of diabetic cardiomyopathy. In this model of low‐grade STZ‐induced diabetes mellitus, i.v. PLX application improved LV diastolic relaxation and cardiomyocyte stiffness without affecting glucose levels. PLX reduced the diabetes‐associated low‐grade inflammation as indicated by the decrease in diabetes‐induced LV TGF‐β1 and IFN‐γ expression, and the increase of blood regulatory T cells, which have on their own vascular‐protective 44, 45 and antifibrotic features 46. Cardiac fibrosis was not yet pronounced in this STZ‐induced diabetes mellitus model. Nevertheless, splenocytes of STZ mice displayed already a higher pro‐fibrotic potential compared with splenocytes of control mice, which follows from their higher capacity to induce collagen deposition in murine fibroblasts upon coculture relative to control splenocytes, an effect, which could previously also be observed with splenocytes from ob/ob versus control mice 47. The pro‐fibrotic splenocyte activity of STZ mice was abolished in STZ mice receiving PLX, indicating the immunomodulatory and antifibrotic potential of PLX under diabetic conditions. These findings are supported by observations in myocarditis mice, where splenocytes from Coxsackievirus B3‐infected mice injected with MSC induced less collagen production compared with splenocytes from Coxsackievirus B3 mice 21, be it now in a diabetic setting. Given the importance of the cardiosplenic axis 40, these findings foresee that i.v. PLX application may counteract the cardiac fibrogenesis process during the pathogenesis of diabetic cardiomyopathy in this systemic manner, that is, independently from their, as also shown in vitro, direct paracrine antifibrotic potential.
In agreement with previously demonstrated endothelial‐protective and proangiogenic features of PLX in experimental models of hind limb ischemia 24 and myocardial infarction 23, PLX further decreased the LV VCAM‐1 expression and reinstated the diabetes‐downregulated LV VEGF expression and arteriole density in STZ mice. In vitro experiments with hyperglycemic PLX conditioned medium support that PLX restore the hyperglycemia‐impaired vascularization in a paracrine manner as indicated by the increased tube formation (total branching points, tubes and tube length) of HUVEC cultured in hyperglycemic PLX conditioned medium versus hyperglycemic medium. In parallel, hyperglycemic PLX conditioned medium raised the adhesion potential of endothelial cells. These effects were found in the presence of a decrease in hyperglycemia‐induced ROS levels and unaltered NO levels compared with hyperglycemic conditions, indicative for an improved NO bioavailability 33, 48. In agreement with the potential of PLX to increase NO bioavailability, PLX administration in STZ mice raised the low activity of the NO‐downstream target PKG in STZ mice to levels of control mice. Low PKG activity increases resting tension due to hypophosphorylation of titin 12, 13, 49. In addition, low PKA 50 and low CaMKII 36 activities are both associated with hypophosphorylation of titin and increased cardiomyocyte stiffness. In contrast, increased PKC activity and subsequent PKC phosphorylation of titin's PEVK element leads to myocardial stiffness 51. STZ mice displayed reduced cardiac PKA and CaMKII activity compared with control mice, whereas PLX recovered the diabetes‐downregulated PKA activity and tendencially raised CaMKII activity in STZ mice. PKC activity was not different among the groups. In parallel to the restored PKG and PKA activity, the hypophosphorylation of the titin isoforms N2B and N2BA found in STZ mice was repaired in STZ PLX mice and normalized to controls. Consequently, STZ PLX mice exhibited lower cardiomyocyte stiffness compared with STZ mice, which was further translated into ameliorated diastolic relaxation as shown by an improvement in Tau and the relaxation parameter dP/dtmin. Our findings are consistent with the postulated paradigm that comorbidities including diabetes mellitus induce a systemic pro‐inflammatory state, which triggers endothelial inflammation, which in turn reduces NO bioavailability and PKG activity in adjacent cardiomyocytes leading to cardiomyocyte stiffness 41 and/or provoke cardiac fibrosis. In obese, diabetic ZSF1 rats, diastolic dysfunction is present due to cardiomyocyte stiffness in the absence of cardiac fibrosis 13. Similarly, at this early stage of STZ‐induced diabetic cardiomyopathy without signs of cardiac fibrosis, cardiomyocyte stiffness underlied the diastolic stiffness, which could be improved via i.v. PLX application. Conform to the capacity of MSC to home to damaged tissue upon i.v. application 21, cardiac engraftment of PLX was higher in STZ compared with control mice. In contrast to previous reports with MSC 52, no anti‐diabetic effect of PLX could be observed in the STZ mice. We suggest that the cardioprotective outcome of PLX in STZ mice are a consequence of their paracrine endothelial‐protective and proangiogenic effects upon cardiac engraftment, and their systemic extracardiac immunomodulatory effects 53, which may be of extra relevance in this model of systemic low‐grade inflammation.
Conclusion
In conclusion, i.v. PLX application improves LV diastolic relaxation and cardiomyocyte stiffness at an early stage of diabetic cardiomyopathy through increased titin phosphorylation. These findings offer new insights into the cardioprotective effects of PLX, advancing their potential use for cardiac therapies.
Author Contributions
S.V.L.: conception and design, administrative support, collection and/or assembly of data, data analysis and interpretation, manuscript writing; N.H.: conception and design, collection and/or assembly of data; data analysis and interpretation; K.M., A.K., I.M., and L.P.: collection and/or assembly of data, data analysis and interpretation; Z.A.: financial support, final approval of manuscript; K.P.: collection and/or assembly of data; W.A.L.: data analysis and interpretation, final approval of manuscript; C.T.: conception and design, data analysis and interpretation, financial support, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
L.P. is an employee of Pluristem Therapeutics Inc. and holds stock and stock options. Z.A. is an employee and shareholder of Pluristem Therapeutics Inc. and has intellectual property rights. The other authors indicated no potential conflicts of interest.
Supporting information
Supporting Information
Acknowledgments
We thank Alejandro Cabezas‐Cruz, Josefine Dunst, Ulrike Fritz, Kerstin Puhl, and Norbert G. Zingler for excellent technical assistance. We acknowledge the assistance of the BCRT Flow Cytometry Lab. This study was supported by the Deutsche Forschungsgemeinschaft (DFG) funding through the Berlin‐Brandenburg School for Regenerative Therapies, and by the Berlin‐Brandenburg Center for Regenerative Therapies – BCRT (Bundesministerium für Bildung und Forschung – 0313911) and MEDIA (The MEtabolic Road to DIAstolic Heart Failure) consortium from the Framework Programme 7 of the European Commission to C.T. and Z.A. N.H. and W.A.L. were supported by the German Research Foundation grant HA 7512/2‐1. S.V.L. was supported by the Dr. Marija Orlovic Stiftung.
References
- 1. Van Linthout S, Spillmann F, Riad A et al. Human apolipoprotein A‐I gene transfer reduces the development of experimental diabetic cardiomyopathy. Circulation 2008;117:1563–1573. [DOI] [PubMed] [Google Scholar]
- 2. Tschope C, Walther T, Königer J et al. Prevention of cardiac fibrosis and left ventricular dysfunction in diabetic cardiomyopathy in rats by transgenic expression of the human tissue kallikrein gene. FASEB J 2004;18:828–835. [DOI] [PubMed] [Google Scholar]
- 3. Chen JX, Zeng H, Reese J et al. Overexpression of angiopoietin‐2 impairs myocardial angiogenesis and exacerbates cardiac fibrosis in the diabetic db/db mouse model. Am J Physiol Heart Circ Physiol 2012;302:H1003–H1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Farhangkhoee H, Khan ZA, Kaur H et al. Vascular endothelial dysfunction in diabetic cardiomyopathy: Pathogenesis and potential treatment targets. Pharmacol Ther 2006;111:384–399. [DOI] [PubMed] [Google Scholar]
- 5. Kruger M, Babicz K, von Frieling‐Salewsky M et al. Insulin signaling regulates cardiac titin properties in heart development and diabetic cardiomyopathy. J Mol Cell Cardiol 2010;48:910–916. [DOI] [PubMed] [Google Scholar]
- 6. Raev DC. Which left ventricular function is impaired earlier in the evolution of diabetic cardiomyopathy? An echocardiographic study of young type I diabetic patients. Diabetes Care 1994;17:633–639. [DOI] [PubMed] [Google Scholar]
- 7. Cosson S, Kevorkian JP. Left ventricular diastolic dysfunction: An early sign of diabetic cardiomyopathy?. Diabetes Metab 2003;29:455–466. [DOI] [PubMed] [Google Scholar]
- 8. Stone PH, Muller JE, Hartwell T et al. The effect of diabetes mellitus on prognosis and serial left ventricular function after acute myocardial infarction: Contribution of both coronary disease and diastolic left ventricular dysfunction to the adverse prognosis. The MILIS Study Group. J Am Coll Cardiol 1989;14:49–57. [DOI] [PubMed] [Google Scholar]
- 9. MacDonald MR, Petrie MC, Varyani F et al. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: An analysis of the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) programme. Eur Heart J 2008;29:1377–1385. [DOI] [PubMed] [Google Scholar]
- 10. Falcao‐Pires I, Hamdani N, Borbély A et al. Diabetes mellitus worsens diastolic left ventricular dysfunction in aortic stenosis through altered myocardial structure and cardiomyocyte stiffness. Circulation 2011;124:1151–1159. [DOI] [PubMed] [Google Scholar]
- 11. Westermann D, Lindner D, Kasner M et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 2011;4:44–52. [DOI] [PubMed] [Google Scholar]
- 12. Tschope C, Van Linthout S. New insights in (inter)cellular mechanisms by heart failure with preserved ejection fraction. Curr Heart Fail Rep 2014;11:436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hamdani N, Franssen C, Lourenço A et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail 2013;6:1239–1249. [DOI] [PubMed] [Google Scholar]
- 14. Hamdani N, Paulus WJ. Myocardial titin and collagen in cardiac diastolic dysfunction: Partners in crime. Circulation 2013;128:5–8. [DOI] [PubMed] [Google Scholar]
- 15. Chung CS, Hutchinson KR, Methawasin M et al. Shortening of the elastic tandem immunoglobulin segment of titin leads to diastolic dysfunction. Circulation 2013;128:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tschope C, Bock CT, Kasner M et al. High prevalence of cardiac parvovirus B19 infection in patients with isolated left ventricular diastolic dysfunction. Circulation 2005;111:879–886. [DOI] [PubMed] [Google Scholar]
- 17. Mohammed SF, Hussain S, Mirzoyev SA et al. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015;131:550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Linthout S, Tschope C. Inflammation—Cause or consequence of heart failure or both? Curr Heart Fail Rep 2017;14:251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Franssen C, Chen S, Unger A et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail 2016;4:312–324. [DOI] [PubMed] [Google Scholar]
- 20. Van Linthout S, Savvatis K, Miteva K et al. Mesenchymal stem cells improve murine acute coxsackievirus B3‐induced myocarditis. Eur Heart J 2011;32:2168–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Savvatis K, van Linthout S, Miteva K et al. Mesenchymal stromal cells but not cardiac fibroblasts exert beneficial systemic immunomodulatory effects in experimental myocarditis. PLoS One 2012;7:e41047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miteva K, Pappritz K, El‐Shafeey M et al. Mesenchymal stromal cells modulate monocytes trafficking in Coxsackievirus B3‐induced myocarditis. Stem Cells Translational Medicine 2017;6:1249–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roy R, Brodarac A, Kukucka M et al. Cardioprotection by placenta‐derived stromal cells in a murine myocardial infarction model. J Surg Res 2013;185:70–83. [DOI] [PubMed] [Google Scholar]
- 24. Prather WR, Toren A, Meiron M et al. The role of placental‐derived adherent stromal cell (PLX‐PAD) in the treatment of critical limb ischemia. Cytotherapy 2009;11:427–434. [DOI] [PubMed] [Google Scholar]
- 25. Hare JM, Traverse JH, Henry TD et al. A randomized, double‐blind, placebo‐controlled, dose‐escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol 2009;54:2277–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang N, Li J, Luo R et al. Bone marrow mesenchymal stem cells induce angiogenesis and attenuate the remodeling of diabetic cardiomyopathy. Exp Clin Endocrinol Diabetes 2008;116:104–111. [DOI] [PubMed] [Google Scholar]
- 27. Li Q, Turdi S, Thomas DP et al. Intra‐myocardial delivery of mesenchymal stem cells ameliorates left ventricular and cardiomyocyte contractile dysfunction following myocardial infarction. Toxicol Lett 2008;195:119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramot Y, Meiron M, Toren A et al. Safety and biodistribution profile of placental‐derived mesenchymal stromal cells (PLX‐PAD) following intramuscular delivery. Toxicol Pathol 2009;37:606–616. [DOI] [PubMed] [Google Scholar]
- 29. McBride C, Gaupp D, Phinney DG. Quantifying levels of transpolanted murine and human mesenchymal stem cells in vivo via real‐time PCR. Cytotherapy 2003;5:7–18. [DOI] [PubMed] [Google Scholar]
- 30. Miteva K, Haag M, Peng J et al. Human cardiac‐derived adherent proliferating cells reduce murine acute Coxsackievirus B3‐induced myocarditis. PLoS One 2011;6:e28513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Linthout S, Lusky M, Collen D et al. Persistent hepatic expression of human apo A‐I after transfer with a helper‐virus independent adenoviral vector. Gene Ther 2002;9:1520–1528. [DOI] [PubMed] [Google Scholar]
- 32. Spillmann F, Miteva K, Pieske B et al. High‐density lipoproteins reduce endothelial‐to‐mesenchymal transition. Arterioscler Thromb Vasc Biol 2015;35:1774–1777. [DOI] [PubMed] [Google Scholar]
- 33. Spillmann F, Van Linthout S, Miteva K et al. LXR agonism improves TNF‐alpha‐induced endothelial dysfunction in the absence of its cholesterol‐modulating effects. Atherosclerosis 2014;232:1–9. [DOI] [PubMed] [Google Scholar]
- 34. Miteva K, Van Linthout S, Pappritz K et al. Human endomyocardial biopsy specimen‐derived stromal cells modulate angiotensin II‐induced cardiac remodeling. Stem Cells Translational Medicine 2016;5:1707–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hamdani N, Bishu KG, von Frieling‐Salewsky M et al. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res 2013;97:464–471. [DOI] [PubMed] [Google Scholar]
- 36. Hamdani N, Krysiak J, Kreusser MM et al. Crucial role for Ca2(+)/calmodulin‐dependent protein kinase‐II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res 2013;112:664–674. [DOI] [PubMed] [Google Scholar]
- 37. Ren G, Zhang L, Zhao X et al. Mesenchymal stem cell‐mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008;2:141–150. [DOI] [PubMed] [Google Scholar]
- 38. Tschope C, Walther T, Escher F et al. Transgenic activation of the kallikrein‐kinin system inhibits intramyocardial inflammation, endothelial dysfunction and oxidative stress in experimental diabetic cardiomyopathy. FASEB J 2005;19:2057–2059. [DOI] [PubMed] [Google Scholar]
- 39. Van Linthout S, Miteva K, Tschope C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res 2014;102:258–269. [DOI] [PubMed] [Google Scholar]
- 40. Ismahil MA, Hamid T, Bansal SS et al. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: Critical importance of the cardiosplenic axis. Circ Res 2014;114:266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- 42. Riad A et al. Reduced cardiac performance after differential pharmacological stress in streptozotocin‐induced diabetic rats. J Clinic Experiment Cardiol 2010;1:108. [Google Scholar]
- 43. van Dijk CG, Oosterhuis NR, Xu YJ et al. Distinct endothelial cell responses in the heart and kidney microvasculature characterize the progression of heart failure with preserved ejection fraction in the obese ZSF1 rat with cardiorenal metabolic syndrome. Circ Heart Fail 2016;9:e002760. [DOI] [PubMed] [Google Scholar]
- 44. Kasal DA, Barhoumi T, Li MW et al. T regulatory lymphocytes prevent aldosterone‐induced vascular injury. Hypertension 2012;59:324–330. [DOI] [PubMed] [Google Scholar]
- 45. Mian MO, Barhoumi T, Briet M et al. Deficiency of T‐regulatory cells exaggerates angiotensin II‐induced microvascular injury by enhancing immune responses. J Hypertens 2016;34:97–108. [DOI] [PubMed] [Google Scholar]
- 46. Cao Y, Xu W, Xiong S. Adoptive transfer of regulatory T cells protects against Coxsackievirus B3‐induced cardiac fibrosis. PLoS One 2013;8:e74955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Spillmann F, De Geest B, Muthuramu I et al. Apolipoprotein A‐I gene transfer exerts immunomodulatory effects and reduces vascular inflammation and fibrosis in ob/ob mice. J Inflamm (Lond) 2016;13:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van Linthout S, Spillmann F, Lorenz M et al. Vascular‐protective effects of high‐density lipoprotein include the downregulation of the angiotensin II type 1 receptor. Hypertension 2009;53:682–687. [DOI] [PubMed] [Google Scholar]
- 49. van Heerebeek L, Hamdani N, Falcão‐Pires I et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012;126:830–839. [DOI] [PubMed] [Google Scholar]
- 50. Yamasaki R, Wu Y, McNabb M et al. Protein kinase A phosphorylates titin's cardiac‐specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res 2002;90:1181–1188. [DOI] [PubMed] [Google Scholar]
- 51. Hidalgo C, Hudson B, Bogomolovas J et al. PKC phosphorylation of titin's PEVK element: A novel and conserved pathway for modulating myocardial stiffness. Circ Res 2009;105:631–638, 617 p following 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Linthout SV, Spillmann F, Schultheiss HP et al. Effects of mesenchymal stromal cells on diabetic cardiomyopathy. Curr Pharm Des 2011;17:3341–3347. [DOI] [PubMed] [Google Scholar]
- 53. Lee RH, Pulin AA, Seo MJ et al. Intravenous hmscs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti‐inflammatory protein tsg‐6. Cell Stem Cell 2009;5:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information