

Abstract
Hematopoiesis is the process of blood cell formation starting from hematopoietic stem/progenitor cells (HSPCs). The understanding of regulatory networks involved in hematopoiesis and their impact on gene expression is crucial to decipher the molecular mechanisms that control hematopoietic development in physiological and pathological conditions, and to develop novel therapeutic strategies. An increasing number of epigenetic studies aim at defining, on a genome‐wide scale, the cis‐regulatory sequences (e.g., promoters and enhancers) used by human HSPCs and their lineage‐restricted progeny at different stages of development. In parallel, human genetic studies allowed the discovery of genetic variants mapping to cis‐regulatory elements and associated with hematological phenotypes and diseases. Here, we summarize recent epigenetic and genetic studies in hematopoietic cells that give insights into human hematopoiesis and provide a knowledge basis for the development of novel therapeutic approaches. As an example, we discuss the therapeutic approaches targeting cis‐regulatory regions to reactivate fetal hemoglobin for the treatment of β‐hemoglobinopathies. Epigenetic studies allowed the definition of cis‐regulatory sequences used by human hematopoietic cells. Promoters and enhancers are targeted by transcription factors and are characterized by specific histone modifications. Genetic variants mapping to cis‐regulatory elements are often associated with hematological phenotypes and diseases. In some cases, these variants can alter the binding of transcription factors, thus changing the expression of the target genes. Targeting cis‐regulatory sequences represents a promising therapeutic approach for many hematological diseases. Stem Cells Translational Medicine 2017;6:2106–2114
Keywords: Epigenetics, Hematopoiesis, Transcriptional regulation, Anemia
Significance Statement.
This review summarizes the epigenetic and genetic studies identifying genes and cis‐regulatory regions involved in normal and pathological hematopoiesis. Novel potential therapeutic approaches targeting cis‐regulatory sequences, which hold great promise for the treatment of many hematological diseases, are discussed. Readers will gain an overview of the epigenetic mechanisms regulating hematopoiesis and acquire knowledge about genome editing‐based approaches for the treatment of β‐hemoglobinopathies.
Introduction
Different cell types from the same organism or tissue are genetically identical but functionally heterogeneous because of the differential expression of genes. Epigenetic modifications are responsible for changes in gene activity that are not strictly dependent on the DNA sequence. Epigenetic markers can be used to identify cis‐regulatory DNA elements (e.g., promoters and enhancers) that mediate developmental stage‐ and tissue‐specific gene expression. High‐throughput sequencing technologies have been extensively used to study the epigenetic regulation of gene expression programs in a wide range of hematopoietic cell types. The definition of regulatory regions controlling cell‐specific gene expression programs is fundamental to understand the molecular mechanisms underlying hematopoiesis in health and disease.
Human Hematopoiesis
Human blood contains several different cell types with specific functions. Erythrocytes, also known as red blood cells (RBCs), transport oxygen from the lungs to the tissues and remove carbon dioxide. Leukocytes, also known as white blood cells (WBCs), are involved in inflammatory reaction and immune response. WBCs comprise granulocytes (neutrophils, basophils, eosinophils, and mast cells), lymphocytes (T cells, B cells, and natural killer [NK] cells), monocytes/macrophages, and dendritic cells. Platelets are cell fragments derived from megakaryocytes and play an essential role in the maintenance of hemostasis (Fig. 1).
Figure 1.
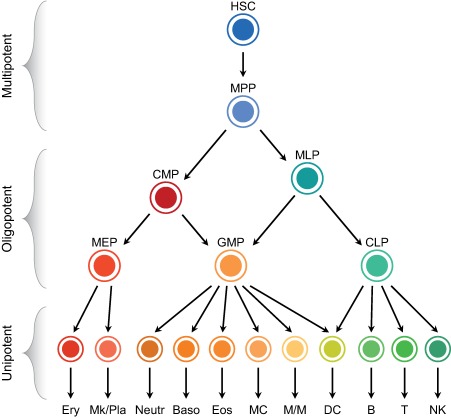
The current model of human hematopoiesis. Schematic representation of the hematopoietic hierarchical tree, composed of multipotent, oligopotent, and unipotent cell types. Abbreviations: B, B cells; Baso, basophils; CLP, common lymphoid progenitor; CMP, common myeloid progenitor; DC, dendritic cells; Eos, eosinophils; Ery, erythrocytes; GMP, granulocyte/macrophage progenitor; HSC, hematopoietic stem cell; MC, mast cells; MEP, megakaryocyte/erythroid progenitor; Mk/Pla, megakaryocytes/platelets; MLP, multilymphoid progenitor; M/M, monocytes/macrophages; MPP, multipotent progenitor cell; Neutr, neutrophils; NK, natural killer cells; T, T cells.
Hematopoietic stem cells (HSCs) sustain the life‐long production of all blood cells thanks to their ability to self‐renew and differentiate toward multiple lineages. The hierarchical model of hematopoiesis places HSCs at the root of the tree composed of a series of multipotent, oligopotent, and unipotent progenitors that finally differentiate toward mature hematopoietic cells. Multipotent progenitor cells (MPPs), the immediate progeny of HSCs, have a full lineage potential but a limited self‐renewal ability. MPPs give rise to oligopotent progenitors: the common myeloid progenitors (CMPs) and the multilymphoid progenitors (MLPs) 1. CMPs differentiate into the granulocyte/macrophage progenitors (GMPs) and the megakaryocyte/erythroid progenitors (MEPs), which then give rise to unilineage progenitors and mature precursors. MLPs are able to generate both GMPs and the common lymphoid progenitors, which differentiate into B, T, and NK cell precursors (Fig. 1).
This current model of hematopoiesis (reviewed in 2) is based on prospective isolation of hematopoietic cells carrying specific surface markers and evaluation of the lineage output using in vitro or in vivo assays. However, several studies at single‐cell level challenged the classical hierarchy of human hematopoiesis. Combining new sorting strategies, optimized single‐cell functional assays and single‐cell gene expression profiling, Notta et al. showed that, in adult bone marrow, classically defined CMPs and MEPs are heterogeneous populations, composed mostly of unilineage committed cells 3. Therefore, multipotent cells, such as HSCs and MPPs, give rise to committed unipotent progenitors, without an intermediate oligopotent CMP and MEP stage. Furthermore, the study of Velten et al. showed that HSCs and their immediate progeny, such as MPPs and MLPs, do not represent discrete cell types, but are included in a continuum of low‐primed undifferentiated (CLOUD)‐hematopoietic stem/progenitor cells (HSPCs) 4. In the CLOUD, HSCs gradually acquire continuous transcriptomic lineage priming into lympho/myeloid or megakaryocytic/erythroid major branches.
Despite these recent single‐cell studies suggesting a revision of the hierarchical organization of human hematopoiesis, the current model is suitable and widely used to investigate the molecular mechanisms that drive HSPCs commitment and differentiation.
Epigenetic Control of Gene Expression
Gene expression is regulated by transcription factors and cofactors binding cis‐regulatory DNA sequences, such as promoters and enhancers. Promoters are located at the 5′ end of the genes and consist of multiple DNA motifs for transcription factors that recruit the transcriptional machinery and define the transcription start site (TSS) 5. Enhancers are clusters of transcription factors binding sites and can increase the transcription of the target promoters 5, 6, 7. Enhancers function at various distances from their target genes and can be located upstream or downstream of genes or within introns. Chromatin looping is thought to bring promoters and enhancers in close proximity to activate gene transcription. Enhancers are fundamental for the spatial‐ and temporal‐specific expression of genes. These regulatory elements can be cell type‐specific: distinct enhancer elements can act on the same gene at different cellular stages or in different tissues, as well as in response to different stimuli. More recently, super‐enhancers have been defined as clusters of cell‐specific enhancers densely occupied by transcription factors and cofactors, and involved in the regulation of genes specifying cell identity 8.
Cis‐regulatory elements present characteristic epigenetic features. They are typically devoid of nucleosomes to allow the binding of transcription factors to their DNA motifs 9, 10. Nucleosomes surrounding these highly accessible DNA sequences are characterized by specific histone modifications. Histone H3 lysine 4 trimethylation (H3K4me3) and monomethylation (H3K4me1) are preferentially associated with promoters and enhancers, respectively 9, 11. Lysine 27 acetylation of histone H3 (H3K27ac) marks highly active promoters and enhancers, and super‐enhancers 8, 11, 12 (Fig. 2). Histone modifying enzymes that act as transcriptional coactivators, such as the histone acetyltransferase p300 and CREB binding protein (CBP), are localized in active regulatory elements, in particular enhancers 9, 13.
Figure 2.
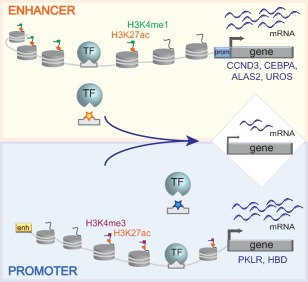
Genetic variants in cis‐regulatory elements affect target gene expression. Genetic variants mapping to enhancers or promoters (indicated by yellow and blue stars, respectively) can prevent the binding of transcription factors and result in decreased expression of the target genes (e.g., CCND3, CEBPA, ALAS2, UROS, PKLR, and HBD). Active enhancers are characterized by H3K4me1 and H3K27ac histone marks, whereas H3K27ac and H3K4me3 mark active promoters. Abbreviations: prom, promoter; enh, enhancer; TF, transcription factor.
In the last 10 years, epigenetic features characteristic of transcriptional regulatory regions (DNA accessibility, histone marks, transcription factors, and cofactors binding) have allowed the mapping of cis‐regulatory elements genome‐wide in a multitude of human cell lines, primary cells, and tissues, thanks to different high‐throughput technologies based on next‐generation sequencing (NGS) 7, 9. Chromatin accessibility can be directly assessed with different approaches: DNase‐seq, formaldehyde‐assisted isolation of regulatory elements (FAIRE)‐seq, and assay for transposase‐accessible chromatin using sequencing (ATAC‐seq) 9. DNase‐seq detects accessible DNA by DNaseI enzymatic digestion of nucleosome‐depleted sites, also known as DNaseI hypersensitive (HS) sites, which represent cis‐regulatory elements 14. FAIRE‐seq (formaldehyde assisted isolation of regulatory elements coupled with NGS) determines open chromatin regions by sequencing protein‐free DNA, after removal of formaldehyde crosslinked protein‐DNA complexes 15. ATAC‐seq is based on the ability of the hyperactive Tn5 transposase to fragment DNA and integrate sequencing adapters into open chromatin regions 16. Chromatin immunoprecipitation (ChIP) combined with NGS (ChIP‐seq) 17, 18 is also widely used to study chromatin modifications typical of cis‐regulatory elements. ChIP‐seq profile of H3K4me3 allows the identification of promoter regions 9, 17, 18, while H3K4me1 analysis defines enhancer regions 9. H3K27ac can be used alone or integrated with H3K4 methylation profiles to identify highly active promoters and enhancers, and super‐enhancers 8, 19, 20. ChIP‐seq analysis of binding sites of multiple transcription factors and/or coactivator proteins, such as p300 and CBP acetyltransferase, can also be used to identify enhancer regions 9, 21, 22.
Human hematopoiesis is one of the most established cell differentiation systems and is amenable to the study of gene transcription and chromatin structure. The transition through the hematopoietic hierarchy is regulated at transcriptional level. Master transcription factors control the activation of lineage‐specific transcriptional programs through the binding of cis‐regulatory elements. The identification of enhancers is a key step to understand how gene expression is finely regulated during hematopoiesis and how it is altered in pathological conditions 23. Enhancers establish not only the transcription level, but also when and where a gene is expressed, thus determining cell identity. Most of the epigenetic studies characterizing the regulatory landscape of human hematopoietic cells rely on the prospective isolation of the different cell types defined using specific panels of surface markers (Fig. 1). In the last years, large international consortia, such as ENCODE (http://www.encodeproject.org), Roadmap Epigenomics (http://www.roadmapepigenomics.org), and Blueprint Epigenome (http://www.blueprint-epigenome.eu) collected epigenetic data of several human hematopoietic cell types, including rare blood cells 24, 25, 26. These data represent an important public resource for basic biology and disease‐oriented research. Several studies investigated the regulatory landscape of HSPCs in comparison with lineage‐restricted progenitors or mature cells and described changes in enhancer dynamics during erythroid 27, 28, 29, 30, 31, myeloid 30, 31, 32, and lymphoid 28, 31, 32 commitment and differentiation. These studies showed that each individual cell type, within the hematopoietic hierarchy, displayed a set of cell‐specific cis‐regulatory regions associated to genes involved in cellular functions related to the given cell type. Interestingly, enhancer and super‐enhancer landscape better define cell identity, compared with transcriptomic profile and promoter usage 12, 30, 31.
Insights into Human Hematopoiesis from Genetic and Epigenetic Studies
Human genetic studies allow the identification of DNA variants mapping to epigenetically defined regulatory regions and influencing gene expression. Genome‐wide association studies (GWASs) allow the identification of sequence variations (single nucleotide polymorphisms [SNPs]) associated with human phenotypes and diseases, which are collected in large databases, such as the GWAS Catalog 33 and GWASdb 34. An increasing number of GWASs identified genetic variants linked to a variety of traits, ranging from height and hair color to metabolic and hematological phenotypes. SNPs can be also associated to disease susceptibility and represent genetic modifiers of diseases. Interestingly, numerous trait‐ and disease‐associated sequence variations occur in cis‐regulatory elements, such as enhancers 24, 25, 26, 35, 36. Genes potentially targeted by regulatory elements associated with these SNPs are then identified as: (a) the nearest genes, (b) genes whose expression is correlated with the specific sequence variation (expression quantitative trait loci SNPs), (c) genes potentially implicated in the phenotype.
Several GWASs identified SNPs associated with hematological phenotypes 37, 38, 39, 40. Large studies described 68 genetic loci linked with platelet number and size 38, and 10 SNPs associated with WBCs number and subtypes 40. Seventy‐five genetic loci have been associated with RBC traits, such as RBC number and size (mean corpuscular volume [MCV]), hematocrit, and hemoglobin concentration 37. In this study, a large fraction of SNPs co‐mapped with epigenetically defined erythroid enhancers 37. SNPs associated with cis‐regulatory elements have in general small effects on gene transcription and disease phenotypes, explaining less than 10% of phenotypic variance, but can give us insights into human hematopoiesis in vivo. Indeed, they allow the discovery of new candidate genes involved in hematopoiesis 37, 38, 40, 41 and hematopoietic disorders 42, 43. Interestingly, mutations in regulatory elements have been shown to be responsible for several genetic and nongenetic hematological diseases 44, 45, 46, 47, 48, 49, 50, 51.
Genetic variants mapping to cis‐regulatory elements can be experimentally validated to unravel the molecular mechanisms underlying the observed phenotypes. They can affect enhancer and promoter activity, for example, by either disrupting or creating de novo binding sites for transcription factors, thus deregulating the expression of the target genes 46, 52, 53, 54 (Figs. 2, 3). Functional and mechanistic studies can be focused on the genes associated with the cis‐regulatory regions and potentially involved in hematopoiesis. As an example, GWASs identified a SNP associated with low RBC number and high MCV in an enhancer region located 15 kb upstream of the cyclin D3 gene (CCND3) (Fig. 2) 39, 55. Starting from this observation, Sankaran et al. unraveled the role of cyclin D3 in the regulation of erythrocyte number and size through gene disruption studies 41. Other studies dissected the regulatory elements harboring genetic variants associated with particular phenotypes 46, 56. A recent GWAS identified a SNP, associated with low basophil counts, 39 kb downstream of the gene encoding CCAAT/enhancer binding protein alpha (CEBPA), a master regulator implicated in basophil specification 56, 57. This SNP overlaps with an enhancer targeted by the myeloid master regulators GATA2 and RUNX1 and identified specifically in CMPs, which give rise to several lineages including basophils. CRISPR‐Cas9‐mediated mutagenesis of this region in HSPCs led to CEBPA downregulation and impairment of basophil production and maturation (Fig. 2) 56. Interestingly, disease‐causing variants affecting the binding sites of the erythroid master regulator GATA1 were identified in regulatory elements of the ALAS2 44, 45, UROS 47, PKLR 48, and HBD 49 genes. Loss of GATA1 binding at promoter or enhancer regions was associated with decreased expression of the target gene (Fig. 2), which can lead to severe clinical phenotypes (e.g., X‐linked sideroblastic anemia for ALAS2, congenital erythropoietic porphyria for UROS and pyruvate kinase deficiency for PKLR). Initial validation studies based on electrophoretic mobility shift and luciferase assays showed that these mutations impair GATA1 binding and the enhancer or promoter activity 45, 47, 49. More recently, CRISPR‐Cas9‐mediated in situ disruption of the GATA1 motifs present in the ALAS2, UROS, and PKLR regulatory regions reproduced the gene downregulation observed in patients harboring mutation of these binding sites 46. Interestingly, the disruption of the GATA1 motifs seems to affect the binding of the GATA1‐dependent activation complex 46.
Figure 3.
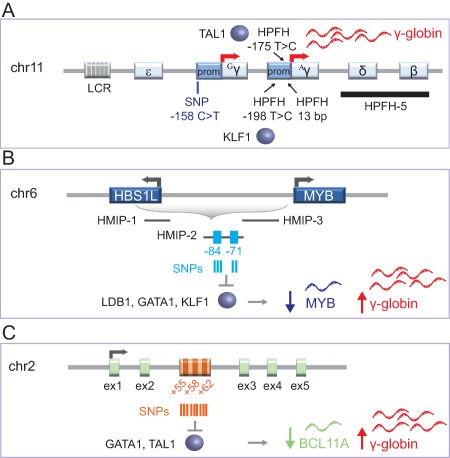
Genetic variants in the β‐globin locus, HBS1L‐MYB, and BCL11A loci influence fetal hemoglobin (HbF) levels. (A): Schematic representation of the β‐globin locus on chromosome 11. Point mutations (HPFH −175 T > C and HPFH −198 T > C upstream of the γ‐globin transcription start sites [TSSs]) and deletions (HPFH 13‐bp in Aγ‐globin promoter and HPFH‐5) are associated with HPFH. The −175 T > C and −198 T > C mutations create binding sites for TAL1 and KLF1 transcriptional activators. A common SNP at position −158 bp of the Gγ‐globin promoter is associated with moderately high levels of HbF. LCR and β‐like globin genes (embryonic ɛ, fetal Aγ and Gγ, and adult δ and β) are indicated. (B): The region between HBS1L and MYB genes on chromosome 6 contains three HMIP blocks 1, 2, and 3. Five SNPs associated with higher HbF levels map to the −84 and −71 kb MYB enhancers in HMIP‐2. These variants reduce LDB1, GATA1, and KLF1 occupancy, thus decreasing MYB expression and, as a consequence, increasing HbF expression. (C): Representation of the BCL11A gene on chromosome 2. Several SNPs, associated with high HbF levels map to three erythroid‐specific intronic enhancers located 55, 58, and 62 kb downstream of the BCL11A TSS. A SNP within the +62 kb enhancer impairs GATA1 and TAL1 binding, thus leading to a reduction of BCL11A expression and increase of HbF levels. Targeted disruption of a GATA1 binding site in the +58 kb enhancer is also associated with decreased BCL11A levels and high HbF expression. Abbreviations: HMIP, HBS1L‐MYB intergenic polymorphism; HPFH, hereditary persistence of fetal hemoglobin; LCR, locus control region; SNP, single nucleotide polymorphism.
cis‐Regulatory Regions as Genetic Modifiers of Fetal Hemoglobin
Hemoglobin (Hb) is a tetrameric protein containing two α‐like and two β‐like globins. The β‐like globin genes (embryonic ɛ, fetal Aγ and Gγ, adult δ and β) are located on chromosome 11 within the β‐globin gene cluster. Here, these genes are under the control of the β‐globin locus control region (LCR), composed of DNase I HSs. The LCR is able to recruit transcription factors and interact with the different β‐like globin gene promoters to regulate their expression during development. A γ‐to‐β globin switching occurs shortly after birth. In adulthood, the expression of fetal hemoglobin (HbF, α2γ2) is reduced to <1% of the total Hb output and the major Hb type is HbA (α2β2). β‐hemoglobinopathies are the most common genetic disorders worldwide and are characterized by reduced or abnormal production of adult β‐chains. In β‐thalassemia, mutations affecting β‐globin chain production cause the precipitation of unpaired α‐globin chains within erythroid precursors, leading to their death and thereby causing ineffective production of RBCs. In sickle cell disease (SCD), a single amino acid substitution (E6V) in the β‐globin chain causes the production of sickle hemoglobin (HbS). HbS has the propensity to polymerize and precipitate under deoxygenated conditions, resulting in RBC deformation and vaso‐occlusions.
β‐thalassemia and SCD display a remarkable variability in the clinical severity. However, reasons explaining this heterogeneity are not fully understood. Different studies have shown that inter‐individual variation in HbF expression may influence the clinical outcome of these pathologies, with high HbF levels correlated with less severe complications and longer life expectancy 58, 59, 60. In β‐thalassemia, γ‐globin compensates the β‐globin deficiency, whereas in SCD it exerts a potent anti‐sickling effect thanks to a critical amino acid blocking the lateral contacts between β‐like globin chains required for the formation of HbS polymers.
A benign syndrome, referred to as “hereditary persistence of fetal hemoglobin” (HPFH), is characterized by increased HbF levels (up to 90% of the total Hb) in the adult life without major impairment of RBC indices 61. Molecular studies have identified two different types of HPFH, either caused by large deletions encompassing the β‐ and δ‐genes (13–106 kb; e.g., HPFH5) or due to point mutations in the γ‐globin promoters (Fig. 3A). Deletional HPFH are thought to either juxtapose the γ‐globin promoters to enhancers normally located far away from the γ‐globin genes or remove γ‐globin inhibitory sequences 61, 62. Non‐deletional HPFH may alter the binding of transcription factors to critical regions of the γ‐globin promoters. These mutations occur in three distinct regions of the highly similar γ‐globin promoters: (a) approximately 200 bp upstream of the TSS of the γ‐globin genes. The −198 bp T > C mutation in the Aγ‐globin promoter has been recently shown to generate a de novo binding site for the erythroid transcriptional activator KLF1 63, (b) at position −175 bp where the mutation T > C in both the γ‐promoters creates a binding site for TAL1, a transcription factor activating the expression of many erythroid‐specific genes 52, (c) between −117 and −102 bp within the CCAAT box and direct repeat (DR) elements targeted by potential HbF repressors, such as NR2F2 (COUP‐TFII), NR2C1 (TR2), and NR2C2 (TR4) 64. A 13‐bp HPFH deletion (−114 to −102 bp) disrupting the CCAAT box and the DR elements of the Aγ‐globin gene has also been described 61, 65 (Fig. 3A).
HPFH large deletions and point mutations are frequently associated with a pancellular HbF distribution among the erythrocytes. Interestingly, pancellular HbF expression in compound heterozygous with SCD and HPFH traits results in absent or milder SCD symptoms 61. As an example, SCD‐HPFH individuals have no features of SCD, including vaso‐occlusive events and hemolytic anemia 66. Beside HPFH mutations, other genetic factors can moderately raise HbF levels above 1% of the total Hb and are associated with heterocellular HbF distribution. Patients showing heterocellular HbF expression still display anemia and vaso‐occlusive complications, albeit less severe than in SCD 67. Notably, this condition was associated with reduced pain crisis rate in SCD patients 68. Genetic studies identified sequence variations at three genomic loci (β‐globin locus, HBS1L‐MYB, and BCL11A) that account for >30% of the variance in HbF levels 69, 70, 71.
In the β‐globin locus, a common SNP (C > T; commonly referred to as the “XmnI site”) at position −158 bp of the Gγ‐globin promoter (chromosome 11p15) was linked with moderately high HbF levels 71, 72, 73 (Fig. 3A). Other SNPs associated with HbF levels were mapped in the β‐globin locus 74, 75. However, the functional cis‐regulatory elements targeted by these SNPs have not been identified yet. Polymorphisms of the β‐globin locus alone cannot explain the considerable variance in HbF levels, as demonstrated by studies showing that high HbF determinant segregates independently of the β‐globin gene cluster 68, 76, 77, thus suggesting the involvement of other QTLs in the modulation of γ‐globin expression.
Craig and colleagues demonstrated in a large kindred that another major determinant of HbF production is located on chromosome 6q23‐q24 78. Further studies identified multiple genetic variants on chromosome 6q23 that are strongly associated with HbF levels 70. These SNPs are distributed in three genomic regions (referred to as HBS1L‐MYB intergenic polymorphism [HMIP] blocks 1, 2, and 3) that span a 79‐kb segment from the guanosine triphosphate (GTP)‐binding elongation factor HBS1L gene to 45 kb upstream of the myeloblastosis oncogene MYB gene (Fig. 3B). HMIP block 2 contains SNPs that are strongly associated with HbF and erythroid traits. Among these variants, five SNPs were mapped in two epigenetically defined erythroid‐specific enhancers, located 84 and 71 kb upstream of the MYB TSS 79. These variants impaired the binding of the looping factor LDB1 and GATA1, TAL1 and KLF1 erythroid master regulators (Fig. 3B). As a consequence, the long‐range interaction between the erythroid‐specific enhancers and MYB promoter were decreased, as well as MYB expression 79. CRISPR‐Cas9 mutagenesis confirmed the genetic association of the −84 and −71 kb enhancer regions with MYB expression levels 80. MYB downregulation was associated with moderately increased HbF levels 79. Lower MYB levels lead to a slow cell‐cycle progression, which is associated with increased F‐cell production. Alternatively, increased HbF levels can be ascribed to the failed activation of genes encoding HbF repressors (e.g., BCL11A) by MYB 79 (Fig. 3B).
A GWAS performed in individuals with contrasting extreme F‐cell distribution (below the 5th or above the 95th percentile) mapped an additional QTL in the gene encoding the zinc‐finger transcription factor BCL11A on chromosome 2p15 69. Other GWASs validated the association between several SNPs in the BCL11A locus and higher HbF levels 68, 81, 82, 83. These SNPs lie in an erythroid‐specific intronic super‐enhancer composed of three constituent enhancers located 55, 58, and 62 kb downstream of the BCL11A TSS (+55, +58, and +62 DNase HS) 84, 85 (Fig. 3C). A sequence variant in the +62 DNase HS affects the binding of GATA1 and TAL1 transcription factors, leading to a modest reduction of BCL11A expression and increased HbF levels in a SCD cohort 84 (Fig. 3C). These observations suggested a role of BCL11A in the repression of HbF expression in adult life. Further studies showed that shRNA‐knockdown of BCL11A in primary erythroid cells increased γ‐globin expression 86. Notably, inactivation of BCL11A led to the correction of the murine SCD phenotype by inducing pancellular HbF expression 87.
Therapeutic Strategies for β‐Hemoglobinopathies Targeting cis‐Regulatory Regions
Current treatments for SCD and β‐thalassemia involve symptomatic care and RBC transfusions, which, however, can lead to iron overload and organ damage. The only definitive cure for β‐hemoglobinopathies is the allogeneic HSC transplantation, which is available to a small proportion (30%) of the patients with an human leukocyte antigen (HLA)‐compatible donor 88. Transplantation of autologous genetically corrected HSCs is considered an attractive therapeutic alternative for patients lacking a suitable donor. Gene therapy trials based on the use of lentiviral vectors expressing a β‐globin transgene are currently ongoing 89, 90, 91, 92, 93. Alternative promising approaches aim at reactivating therapeutic HbF expression 54. Many of these strategies are based on targeted genome editing of cis‐regulatory elements 85, 94, 95, 96.
Several groups attempted to mimic the beneficial HPFH mutations in the Aγ‐ and/or Gγ‐globin gene promoters. HPFH mutations in the γ‐globin promoters may disrupt binding sites for γ‐globin silencers 94 or generate new binding sites for γ‐globin activators 52, 63 (Fig. 3A). Traxler et al. reproduced the 13‐bp HPFH deletion in the γ‐globin promoters using the CRISPR/Cas9 system, thereby inducing HbF levels sufficient to inhibit HbS polymerization in SCD HSPC‐derived erythrocytes. This deletion is thought to reactivate fetal genes by removing the CCAAT box and the DR element, thus disrupting the binding sites for γ‐globin transcriptional repressors 97. The introduction of HPFH point mutations represents a potential strategy to reactivate HbF expression. In erythroid cell lines, the insertion of −175T > C or the −198T > C substitutions in the γ‐globin promoters created de novo binding sites for transcriptional activators, and induced loop formation between the LCR and γ‐globin promoters, thus increasing HbF expression (Fig. 3A) 52, 63. This potential therapeutic approach has not yet been explored in human HSPCs. Another strategy aims at recreating deletional HPFH, removing the β‐ and δ‐globin genes and the putative γ‐globin inhibitory sequences. Ye et al. explored a CRISPR/Cas9 strategy in normal donor‐derived HSPCs to excise a 12.9‐kb region deleted in HPFH individuals (HPFH‐5; Fig. 3A) 95, 98. This approach resulted in a significant γ‐globin induction and downregulation of the β‐globin expression in HSPC‐derived erythrocytes.
Downregulation of factors responsible for fetal γ‐globin repression can be explored to achieve therapeutic HbF expression. Over the past decades, molecular studies unraveled the role of several nuclear factors in the regulation of the fetal‐to‐adult hemoglobin switching 99, 100. However, interfering with the expression of HbF repressors, such as MYB, leukemia/lymphoma‐related factor (LRF), KLF1, and BCL11A, can impair terminal erythropoiesis and the development of multiple hematopoietic lineages. MYB is essential for the hematopoietic system development 101 and its downregulation in human HSPCs determines a cell‐cycle arrest and impairs erythroid differentiation 102. The transcription factor LRF, encoded by the ZBTB7A gene, is necessary for HSC maintenance and B cell commitment 103. In mice, loss of LRF leads to lethal anemia, due to increased apoptosis of erythroid precursors 104, and LRF knockdown increases HbF expression but delays human erythroid differentiation 105, 106. KLF1 is a transcriptional regulator promoting erythroid lineage development at the expense of the megakaryocytic compartment 107, 108 and KLF1 knockout mice die in utero from severe anemia 109. Although KLF1 haploinsufficiency can induce HbF expression 110, KLF1 is not an ideal target to develop a safe therapy for β‐hemoglobinopathies, given its essential role in erythropoiesis. BCL11A is essential for proper development of B cells and BCL11A‐deficient HSCs showed cell cycle and multi‐lineage differentiation defects 111. Therefore, erythroid‐restricted BCL11A knockdown is mandatory. Interestingly, erythroid‐specific expression of a shRNA targeting BCL11A induced HbF reactivation in normal donor and SCD erythrocytes circumventing HSC toxicity 112. Recently, Chang et al. showed that complete BCL11A knockdown impair RBC enucleation 113. However, in humans, BCL11A haploinsufficiency is associated with HbF persistence with normal hematological functions 114, suggesting that a fine modulation of BCL11A expression is required to develop a safe therapeutic strategy for β‐hemoglobinopathies. An alternative strategy is based on the disruption of erythroid‐specific BCL11A enhancers, allowing a fine tuning of BCL11A expression. CRISPR/Cas9 dissection of BCL11A intronic enhancers identified a potential GATA1 binding site in the +58 DNase HS critical for erythroid expression of BCL11A (Fig. 3C) 85. CRISPR/Cas9 and zinc finger nuclease‐mediated disruption of this GATA1 motif led to reasonably decreased BCL11A expression and increased HbF levels in human erythrocytes 85, 96. Importantly, targeting of the BCL11A erythroid‐specific enhancer did not affect RBC enucleation 113.
For a potential clinical application of these novel approaches aimed at reactivating HbF expression, further studies are required to: (a) assess the frequency of genome edited bona fide human HSCs. To this aim, the delivery and the activity of the genome editing tools have to be optimized; (b) evaluate the potential toxicity due to the delivery and the on‐ and off‐target activity of the genome editing tools in HSCs and their progeny; (c) perform a proper comparison of the different therapeutic strategies in terms of Hb content and functional correction of the patient phenotype.
Conclusion
In summary, the combination of genetic and epigenetic studies enabled the identification of genes and cis‐regulatory regions involved in normal and pathological hematopoiesis. Cis‐regulatory elements represent in some cases potential therapeutic targets. The recent advent of genome editing technologies offers great promise for the development of targeted therapeutic approaches. Future studies will address the efficiency, efficacy, and safety of novel therapeutic strategies aimed at modulating gene expression by targeting cis‐regulatory regions.
Author Contributions
C.A. and O.R.: conception and design, manuscript writing; A.M.: conception and design, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Acknowledgments
This work was supported by grants from AFM‐Telethon (17224), Agence Nationale de la Recherché (ANR‐16‐CE18‐0004 and ANR‐10‐IAHU‐01 “Investissements d'avenir” program). We thank Maxime de Kouchkovsky for the critical review of the manuscript.
References
- 1. Doulatov S, Notta F, Eppert K et al. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol 2010;11:585–593. [DOI] [PubMed] [Google Scholar]
- 2. Doulatov S, Notta F, Laurenti E et al. Hematopoiesis: A human perspective. Cell Stem Cell 2012;10:120–136. [DOI] [PubMed] [Google Scholar]
- 3. Notta F, Zandi S, Takayama N et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016;351:aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Velten L, Haas SF, Raffel S et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol 2017;19:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet 2006;7:29–59. [DOI] [PubMed] [Google Scholar]
- 6. Pennacchio LA, Bickmore W, Dean A et al. Enhancers: Five essential questions. Nat Rev Genet 2013;14:288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: From properties to genome‐wide predictions. Nat Rev Genet 2014;15:272–286. [DOI] [PubMed] [Google Scholar]
- 8. Whyte WA, Orlando DA, Hnisk D et al. Master transcription factors and mediator establish super‐enhancers at key cell identity genes. Cell 2013;153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heintzman ND, Stuart RK, Hon G et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007;39:311–318. [DOI] [PubMed] [Google Scholar]
- 10. Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem 1988;57:159–197. [DOI] [PubMed] [Google Scholar]
- 11. Ernst J, Kheradpour P, Mikkelsen TS et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011;473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hnisz D, Abraham BJ, Lee TI et al. Super‐enhancers in the control of cell identity and disease. Cell 2013;155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heintzman ND, Hon CG, Hawkins RD et al. Histone modifications at human enhancers reflect global cell‐type‐specific gene expression. Nature 2009;459:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crawford GE, Holt IE, Whittle J et al. Genome‐wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res 2006;16:123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giresi PG, Kim J, McDaniell RM et al. FAIRE (Formaldehyde‐Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 2007;17:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buenrostro JD, Giresi PG, Zaba LC et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nat Methods 2013;10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barski A, Cuddapah S, Cui K et al. High‐resolution profiling of histone methylations in the human genome. Cell 2007;129:823–837. [DOI] [PubMed] [Google Scholar]
- 18. Mikkelsen TS, Ku M, Jaffe DB et al. Genome‐wide maps of chromatin state in pluripotent and lineage‐committed cells. Nature 2007;448:553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Z, Zang C, Rosenfeld JA et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nature Genet 2008;40:897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Creyghton MP, Cheng AW, Welstead CG et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 2010;107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Su MY, Steiner LA, Bogardus H et al. Identification of biologically relevant enhancers in human erythroid cells. J Biol Chem 2013;288:8433–8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dogan N, Wu W, Morrissey CS et al. Occupancy by key transcription factors is a more accurate predictor of enhancer activity than histone modifications or chromatin accessibility. Epigenetics Chromatin 2015;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cico A, Andrieu‐Soler C, Soler E. Enhancers and their dynamics during hematopoietic differentiation and emerging strategies for therapeutic action. FEBS Lett 2016;590:4084–4104. [DOI] [PubMed] [Google Scholar]
- 24. ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roadmap Epigenomics Consortium , Kundaje A, Meuleman W et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Astle WJ, Elding H, Jiang T et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 2016;167:1415–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu J, Shoa Z, Glass K et al. Combinatorial assembly of developmental stage‐specific enhancers controls gene expression programs during human erythropoiesis. Dev Cell 2012;23:796–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abraham BJ, Cui K, Tang Q et al. Dynamic regulation of epigenomic landscapes during hematopoiesis. BMC Genomics 2013;14:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang J, Liu X, Li D et al. Dynamic control of enhancer repertoires drives lineage and stage‐specific transcription during hematopoiesis. Dev Cell 2016;36:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Romano O, Peano C, Tagliazucchi GM et al. Transcriptional, epigenetic and retroviral signatures identify regulatory regions involved in hematopoietic lineage commitment. Sci Rep 2016;6:24724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Corces MR, Buenrostro JD, Wu B et al. Lineage‐specific and single‐cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 2016;48:1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gonzalez AJ, Setty M, Leslie CS. Early enhancer establishment and regulatory locus complexity shape transcriptional programs in hematopoietic differentiation. Nat Genet 2015;47:1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. MacArthur J, Bowler E, Cerezo M et al. The new NHGRI‐EBI Catalog of published genome‐wide association studies (GWAS Catalog). Nucleic Acids Res 2017;45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li MJ, Li Z, Wang P et al. GWASdb v2: An update database for human genetic variants identified by genome‐wide association studies. Nucleic Acids Res 2016;44:D869–D876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maurano MT, Humbert R, Rynes E et al. Systematic localization of common disease‐associated variation in regulatory DNA. Science 2012;337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paul DS, Albers CA, Rendon A et al. Maps of open chromatin highlight cell type‐restricted patterns of regulatory sequence variation at hematological trait loci. Genome Res 2013;23:1130–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van der Harst P, Zhang W, Mateo Leach I et al. Seventy‐five genetic loci influencing the human red blood cell. Nature 2012;492:369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gieger C, Radhakrishnan A, Cvejic A et al. New gene functions in megakaryopoiesis and platelet formation. Nature 2011;480:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kamatani Y, Matsuda K, Okada Y et al. Genome‐wide association study of hematological and biochemical traits in a Japanese population. Nat Genet 2010;42:210–215. [DOI] [PubMed] [Google Scholar]
- 40. Nalls MA, Couper DJ, Tanaka T et al. Multiple loci are associated with white blood cell phenotypes. PLoS Genet 2011;7:e1002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sankaran VG, Ludwig LS, Sicinska E et al. Cyclin D3 coordinates the cell cycle during differentiation to regulate erythrocyte size and number. Genes Dev 2012;26:2075–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benyamin B, Ferreira MA, Willemsen G et al. Common variants in TMPRSS6 are associated with iron status and erythrocyte volume. Nat Genet 2009;41:1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Finberg KE, Heeney MM, Campagna DR et al. Mutations in TMPRSS6 cause iron‐refractory iron deficiency anemia (IRIDA). Nat Genet 2008;40:569–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Campagna DR, de Bie CI, Schmitz‐Abe K et al. X‐linked sideroblastic anemia due to ALAS2 intron 1 enhancer element GATA‐binding site mutations. Am J Hematol 2014;89:315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kaneko K, Furuyama K, Fujiwara T et al. Identification of a novel erythroid‐specific enhancer for the ALAS2 gene and its loss‐of‐function mutation which is associated with congenital sideroblastic anemia. Haematologica 2014;99:252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wakabayashi A, Ulirsch JC, Ludwig LS et al. Insight into GATA1 transcriptional activity through interrogation of cis elements disrupted in human erythroid disorders. Proc Natl Acad Sci USA 2016;113:4434–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Solis C, Aizencang GI, Astrin KH et al. Uroporphyrinogen III synthase erythroid promoter mutations in adjacent GATA1 and CP2 elements cause congenital erythropoietic porphyria. J Clin Invest 2001;107:753–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Manco L, Riberio ML, Maximo V et al. A new PKLR gene mutation in the R‐type promoter region affects the gene transcription causing pyruvate kinase deficiency. Br J Haematol 2000;110:993–997. [DOI] [PubMed] [Google Scholar]
- 49. Matsuda M, Sakamoto N, Fukumaki Y. Delta‐thalassemia caused by disruption of the site for an erythroid‐ specific transcription factor, GATA‐1, in the delta‐globin gene promoter. Blood 1992;80:1347–1351. [PubMed] [Google Scholar]
- 50. Mansour MR, Abraham BJ, Anders L et al. Oncogene regulation. An oncogenic super‐enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014;346:1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Navarro JM, Touzart A, Pradel LC et al. Site‐ and allele‐specific polycomb dysregulation in T‐cell leukaemia. Nat Commun 2015;6:6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wienert B, Funnell AP, Norton LJ et al. Editing the genome to introduce a beneficial naturally occurring mutation associated with increased fetal globin. Nat Commun 2015;6:7085. [DOI] [PubMed] [Google Scholar]
- 53. Weiss MJ. Gene editing—Not just CRISPR. Paper presented at: ASH 58th Annual Meeting; December 3–6, 2016; San Diego, CA. [Google Scholar]
- 54. Cavazzana M, Antoniani C, Miccio A. Gene therapy for beta‐hemoglobinopathies. Mol Ther 2017;25:1142–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ganesh SK, Zakai NA, van Rooij FJ et al. Multiple loci influence erythrocyte phenotypes in the CHARGE consortium. Nat Genet 2009;41:1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guo MH, Nandakumar SK, Ulirsh JC et al. Comprehensive population‐based genome sequencing provides insight into hematopoietic regulatory mechanisms. Proc Natl Acad Sci USA 2017;114:E327–E336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang H, Li Y. Mechanisms controlling mast cell and basophil lineage decisions. Curr Allergy Asthma Rep 2014;14:457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Platt OS, Brambilla DJ, Rosse WF et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–1644. [DOI] [PubMed] [Google Scholar]
- 59. Platt OS, Thorington BD, Brambilla DJ et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11–16. [DOI] [PubMed] [Google Scholar]
- 60. Weatherall DJ. Phenotype‐genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat Rev Genet 2001;2:245–255. [DOI] [PubMed] [Google Scholar]
- 61. Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci 1998;850:38–44. [DOI] [PubMed] [Google Scholar]
- 62. Sankaran VG, Xu J, Byron R et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med 2011;365:807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wienert B, Martyn GE, Kurita R et al. KLF1 drives the expression of fetal hemoglobin in British HPFH. Blood 2017;130:803–807. [DOI] [PubMed] [Google Scholar]
- 64. Tanabe O, McPhee D, Kobayasia S et al. Embryonic and fetal beta‐globin gene repression by the orphan nuclear receptors, TR2 and TR4. EMBO J 2007;26:2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Steinberg MH, Forget BG, Higgs DR et al. Disorders of Hemoglobin Genetics, Pathophysiology, and Clinical Management. Cambridge, U.K: Cambridge University Press, 2009. [Google Scholar]
- 66. Akinsheye I, Solovieff N, Ngo D et al. Fetal hemoglobin in sickle cell anemia: Molecular characterization of the unusually high fetal hemoglobin phenotype in African Americans. Am J Hematol 2012;87:217–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Steinberg MH, Chui DH, Dover GJ et al. Fetal hemoglobin in sickle cell anemia: A glass half full? Blood 2014;123:481–485. [DOI] [PubMed] [Google Scholar]
- 68. Lettre G, Sankaran VG, Bezerra MA et al. DNA polymorphisms at the BCL11A, HBS1L‐MYB, and beta‐globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA 2008;105:11869–11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Menzel S, Garner C, Gut I et al. A QTL influencing F cell production maps to a gene encoding a zinc‐finger protein on chromosome 2p15. Nat Genet 2007;39:1197–1199. [DOI] [PubMed] [Google Scholar]
- 70. Thein SL, Menzel S, Peng X et al. Intergenic variants of HBS1L‐MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci USA 2007;104:11346–11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Thein SL, Wainscoat JS, Sampietro M et al. Association of thalassaemia intermedia with a beta‐globin gene haplotype. Br J Haematol 1987;65:367–373. [DOI] [PubMed] [Google Scholar]
- 72. Labie D, Dunda‐Belkhodja O, Rouabhi F et al. The −158 site 5' to the G gamma gene and G gamma expression. Blood 1985;66:1463–1465. [PubMed] [Google Scholar]
- 73. Labie D, Pagnier J, Lapoumeroulie C et al. Common haplotype dependency of high G gamma‐globin gene expression and high Hb F levels in beta‐thalassemia and sickle cell anemia patients. Proc Natl Acad Sci USA 1985;82:2111–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Galarneau G, Palmer CD, Sankaran VG et al. Fine‐mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 2010;42:1049–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Akinsheye I, Alsultan A, Solovieff N et al. Fetal hemoglobin in sickle cell anemia. Blood 2011;118:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Giampaolo A, Mavilio F, Sposi NM et al. Heterocellular hereditary persistence of fetal hemoglobin (HPFH). Molecular mechanisms of abnormal gamma‐gene expression in association with beta thalassemia and linkage relationship with the beta‐globin gene cluster. Hum Genet 1984;66:151–156. [DOI] [PubMed] [Google Scholar]
- 77. Martinez G, Novelletto A, Di Rienzo A et al. A case of hereditary persistence of fetal hemoglobin caused by a gene not linked to the beta‐globin cluster. Hum Genet 1989;82:335–337. [DOI] [PubMed] [Google Scholar]
- 78. Craig JE, Rochette J, Fischer CA et al. Dissecting the loci controlling fetal haemoglobin production on chromosomes 11p and 6q by the regressive approach. Nat Genet 1996;12:58–64. [DOI] [PubMed] [Google Scholar]
- 79. Stadhouders R, Aktuna S, Thongjuea S et al. HBS1L‐MYB intergenic variants modulate fetal hemoglobin via long‐range MYB enhancers. J Clin Invest 2014;124:1699–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Canver MC, Lessard S, Pinello L et al. Variant‐aware saturating mutagenesis using multiple Cas9 nucleases identifies regulatory elements at trait‐associated loci. Nat Genet 2017;49:625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Uda M, Galanello R, Sanna S et al. Genome‐wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta‐thalassemia. Proc Natl Acad Sci USA 2008;105:1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sedgewick AE, Timofeev N, Sebastiani P et al. BCL11A is a major HbF quantitative trait locus in three different populations with beta‐hemoglobinopathies. Blood Cells Mol Dis 2008;41:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sebastiani P, Farrell JJ, Alsultan A et al. BCL11A enhancer haplotypes and fetal hemoglobin in sickle cell anemia. Blood Cells Mol Dis 2015;54:224–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bauer DE, Kamran SC, Lessard S et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013;342:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Canver MC, Smith EC, Sher F et al. BCL11A enhancer dissection by Cas9‐mediated in situ saturating mutagenesis. Nature 2015;527:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sankaran VG, Menne TF, Xu J et al. Human fetal hemoglobin expression is regulated by the developmental stage‐specific repressor BCL11A. Science 2008;322:1839–1842. [DOI] [PubMed] [Google Scholar]
- 87. Xu J, Bauer DE, Kerenyi MA et al. Corepressor‐dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci USA 2013;110:6518–6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lucarelli G, Andreani M, Angelucci E. The cure of thalassemia by bone marrow transplantation. Blood Rev 2002;16:81–85. [DOI] [PubMed] [Google Scholar]
- 89. Cavazzana‐Calvo M, Payen E, Negre O et al. Transfusion independence and HMGA2 activation after gene therapy of human beta‐thalassaemia. Nature 2010;467:318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cavazzana M. Gene therapy studies in hemoglobinopathies: Successes and challenges. Paper presented at: ASH 58th Annual Meeting; December 3–6, 2016; San Diego, CA. [Google Scholar]
- 91. Kanter J, Walters MC, Hsieh MM et al. Interim results from a phase 1/2 clinical study of lentiglobin gene therapy for severe sickle cell disease. Blood 2016;128:1176. [Google Scholar]
- 92. Thompson AA, Kwiatkowski J, Rasko J et al. Lentiglobin gene therapy for transfusion‐dependent β‐Thalassemia: Update from the northstar HGB‐204 phase 1/2 clinical study. Paper presented at: ASH 58th Annual Meeting; December 3–6, 2016; San Diego, CA. [Google Scholar]
- 93. Ferrari G. Gene therapy for hemoglobinopathies. Paper presented at: ASGCT Annual Meeting; May 4–7, 2016; Washington, DC. [Google Scholar]
- 94. Traxler E, Yao Y, Li C et al. Genome editing recreates hereditary persistence of fetal hemoglobin in primary human erythroblasts. Paper presented at: ASH 57th Annual Meeting; December 5–8, 2015; Orlando, FL. [Google Scholar]
- 95. Ye L, Wang J, Tan Y et al. Genome editing using CRISPR‐Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and beta‐thalassemia. Proc Natl Acad Sci USA 2016;113:10661–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Vierstra J, Reik A, Chang KH et al. Functional footprinting of regulatory DNA. Nat Methods 2015;12:927–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Traxler EA, Yao Y, Wang YD et al. A genome‐editing strategy to treat beta‐hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 2016;22:987–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Camaschella C, Serra A, Gottardi E et al. A new hereditary persistence of fetal hemoglobin deletion has the breakpoint within the 3' beta‐globin gene enhancer. Blood 1990;75:1000–1005. [PubMed] [Google Scholar]
- 99. Wilber A, Nienhuis AW, Persons DA. Transcriptional regulation of fetal to adult hemoglobin switching: New therapeutic opportunities. Blood 2011;117:3945–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sankaran VG. Targeted therapeutic strategies for fetal hemoglobin induction. Hematol Am Soc Hematol Educ Program 2011;2011:459–465. [DOI] [PubMed] [Google Scholar]
- 101. Mucenski ML, McLain K, Kier AB et al. A functional c‐myb gene is required for normal murine fetal hepatic hematopoiesis. Cell 1991;65:677–689. [DOI] [PubMed] [Google Scholar]
- 102. Bianchi E, Zini R, Salati S et al. c‐myb supports erythropoiesis through the transactivation of KLF1 and LMO2 expression. Blood 2010;116:e99–e110. [DOI] [PubMed] [Google Scholar]
- 103. Lee SU, Maeda M, Ishikawa Y et al. LRF‐mediated Dll4 repression in erythroblasts is necessary for hematopoietic stem cell maintenance. Blood 2013;121:918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Maeda T, Ito K, Merghoub T et al. LRF is an essential downstream target of GATA1 in erythroid development and regulates BIM‐dependent apoptosis. Dev Cell 2009;17:527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Masuda T, Wang X, Maeda M et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 2016;351:285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sankaran VG, Menne TF, Scepanovic D et al. MicroRNA‐15a and −16‐1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Natl Acad Sci USA 2011;108:1519–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Frontelo P, Manwani D, Galdass M et al. Novel role for EKLF in megakaryocyte lineage commitment. Blood 2007;110:3871–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Siatecka M, Xue L, Bieker JJ. Sumoylation of EKLF promotes transcriptional repression and is involved in inhibition of megakaryopoiesis. Mol Cell Biol 2007;27:8547–8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nuez B, Michalovich D, Bygrave A et al. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature 1995;375:316–318. [DOI] [PubMed] [Google Scholar]
- 110. Borg J, Papadopoulos P, Georgitsi M et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet 2010;42:801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tsang JC, Yu Y, Burke S et al. Single‐cell transcriptomic reconstruction reveals cell cycle and multi‐lineage differentiation defects in Bcl11a‐deficient hematopoietic stem cells. Genome Biol 2015;16:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Brendel C, Guda S, Renella R et al. Lineage‐specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J Clin Invest 2016;126:3868–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chang KH, Smith SE, Sullivan T et al. Long‐term engraftment and fetal globin reactivation upon genome editing of BCL11A in bone marrow‐derived CD34+ hematopoietic stem and progenitor cells. Mol Ther Methods Clin Dev 2017;4:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Basak A, Hancarova M, Ulirsch JC et al. BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J Clin Invest 2015;125:2363–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]