

Abstract
Treatment of preterm human infants with high oxygen can result in disrupted lung alveolar and vascular development. Local or systemic administration of endothelial progenitor cells (EPCs) is reported to remedy such disruption in animal models. In this study, the effects of both fresh (enriched for KDR) and cultured bone marrow (BM)‐derived cell populations with EPC characteristics were examined following hyperoxia in neonatal mouse lungs. Intraperitoneal injection of fresh EPCs into five‐day‐old mice treated with 90% oxygen resulted in full recovery of hyperoxia‐induced alveolar disruption by 56 days of age. Partial recovery in septal number following hyperoxia was observed following injection of short‐term cultured EPCs, yet aberrant tissue growths appeared following injection of long‐term cultured cells. Fresh and long‐term cultured cells had no impact on blood vessel development. Short‐term cultured cells increased blood vessel number in normoxic and hyperoxic mice by 28 days but had no impact on day 56. Injection of fresh EPCs into normoxic mice significantly reduced alveolarization compared with phosphate buffered saline‐injected normoxic controls. These results indicate that fresh BM EPCs have a higher and safer corrective profile in a hyperoxia‐induced lung injury model compared with cultured BM EPCs but may be detrimental to the normoxic lung. The appearance of aberrant tissue growths and other side effects following injection of cultured EPCs warrants further investigation. Stem Cells Translational Medicine 2017;6:2094–2105
Keywords: Hyperoxia, Cell therapy, Endothelial progenitor cells, Bone marrow, Lung injury, Alveolarization, Fresh cells, Cultured cells, Side effects
Significance Statement.
This study describes cell‐based therapies for the potential treatment of very preterm babies following lung injury from high‐oxygen treatments. Results showed that fresh, enriched bone marrow (BM) cell fractions effectively differentiate into endothelial cells in vitro and promote lung recovery following high oxygen‐induced lung injury. It was also discovered that prolonged cell culture caused a gradual decrease in therapeutic outcome and occasionally promoted unwanted growths. It is suggested that long‐term cell culture of BM cells should be avoided and that fresh enriched progenitor cells may provide a preferential source of cells for treatment of the postnatal deficits of high‐oxygen‐induced lung injury in preterm infants.
Introduction
Human premature birth, defined as delivery at less than 37 weeks gestation, has been estimated to occur in 11.1% of all births worldwide and leads to immaturity of the lung causing inefficient oxygen delivery to the circulatory system 1. Treatments include exogenous surfactant, glucocorticoids, ventilation, and/or oxygen therapy to accelerate lung maturation and assist normal lung function. In the case of very premature birth (<32 weeks of gestation), a greater level of intervention unavoidably injures the lung, resulting in chronic lung disease characterized by bronchopulmonary dysplasia (BPD) 2. In preterm infants who require oxygen therapy, the severity of BPD often correlates with the level of oxygen administered. Evidence from rodent studies suggests that relative to lower levels, higher percentage oxygen treatment (>90% O2) results in detrimental effects to essential developmental processes of late‐stage lung maturation including alveolarization and angiogenesis 3, 4, 5. The shorter and early timing of treatment in this study was adapted from previous studies aimed to mimic oxygen exposure in premature infants, also limiting this to the saccular stage of lung development to avoid an increase of animal morbidity, which occurs after 6 days of high oxygen treatment 6, 7.
Previous studies in mouse models of preterm birth have demonstrated that hyperoxia‐mediated changes to vascularization can be temporary, whereas alterations to alveolarization are more persistent 5. Interventions that potentially improve alveolarization defects following hyperoxia include cell, targeted chemokine, and/or conditioned media therapies 8, 9, 10, 11, 12, 13, 14. However, such interventions still require significant optimization and experimental evaluation before possible clinical use.
Several populations of endogenous stem cells that may have clinical utility for lung repair following injury have been described 4, 15, 16. Bronchoalveolar stem cells are reported to possess regenerative potential but are not readily accessible from donors 17, 18. On the other hand, exogenous bone marrow (BM)‐derived stem cells, a more readily accessible stem cell population, have been reported to possess reparative properties relevant to various lung disease models 10, 19, 20, 21, 22, 23. BM stem cell populations comprise hematopoietic, endothelial, and mesenchymal cell stem/progenitor populations, each of which is reported as supportive of lung regeneration following injury 17, 24.
Reduced lung endothelial progenitor cell (EPC) numbers and an associated deficit in neo‐vascularization are observed in BPD following neonatal respiratory hyperoxia 4, 25. Furthermore, transplantation of EPCs to various injury models, including hind limb or myocardial ischemia, as well as hyperoxic lung injury, is reported to result in their engraftment into blood vessels supporting neoangiogenesis 26, 27, 28, 29. It is, therefore, hypothesized that application of exogenous EPCs before, during, or following hyperoxia may improve associated alveolar lung injury.
In this study, the regenerative capacity of marker‐specific BM‐derived EPC subpopulations was investigated in a model of hyperoxia‐mediated lung injury in neonatal mice. Freshly isolated, as well as short‐term and long‐term cultured EPCs were tested for their potential to: (a) stimulate blood vessel growth in vitro and (b) repair injured postnatal lung architecture, following 90% hyperoxia treatment. Our results suggest that fresh, rather than cultured EPCs are preferred for mitigating aberrations to lung blood vessel and alveolar architecture following hyperoxia‐mediated lung injury.
Materials and Methods
Animal Procedures
All animal experimentation was approved by the School of Biomedical Sciences Animal Ethics Committee at Monash University, Melbourne, Australia. Adult C57BL/6J male mice were used for BM cell isolation, either wildtype mice (for EPC analysis only), or transgenic mice expressing enhanced green fluorescent protein via the chicken beta‐actin promoter (Jackson Labs, CA, https://www.jax.org/). Pregnant C57Bl/6J females were treated with 90% oxygen from E17.5 as previously described 5. Pups born remained in high oxygen for four consecutive days, until postnatal day four (D4). On D5, samples from seven normoxic and seven hyperoxic pups were collected for analysis as uninjected controls. A pilot injection of long‐term cultured cells was performed on normoxic and hyperoxic groups using cultured BM cells from 56 days in culture (C56) to be analyzed on D28 (morphometric test only). Cells were diluted at 5 × 102 cells per µl of phosphate buffered saline (PBS; without calcium and magnesium). Pups were weighed before injection. A low number of cells (1–1.5 × 104 for pups weighing 2–3 g) was tested in the pilot study due to generally low number of KDR+ cells present in fresh BM. After this number was demonstrated to be efficient, it was further used in the main study. Ten groups (five normoxic for testing cell side effects on normal lung, and five hyperoxic, up to 24 pups per group) were injected intraperitoneally with a 10 µl/g body weight of cell suspension of either freshly‐isolated KDR‐enriched EPCs (Fr) or EPCs expanded in culture from male green fluorescent protein positive (GFP+) mice. Precultured cells were obtained from either early passages (7 days) or late passages, that is, 77 (and frozen and thawed) or 91–98 days, referred to as C7, C77, and C91‐98. The fifth group was injected with PBS (vehicle) alone. Injected mice were sacrificed on D28 and D56 for analysis of the lung as previously described 5. On D14 and D21, injected mice were sacrificed for analysis of GFP expression in blood, liver, and lung by fluorescent‐activated cell sorting (FACS).
Cell Culture Media
Endothelial differentiation culture medium (EDCM) was optimized using unsorted BM. EDCM contained Iscove's Modified Dulbecco's Medium (Gibco, Carlsbad, CA, https://www.thermofisher.com/us/en/home/brands/gibco.html), and supplementary factors: 20% fetal calf serum (FCS) (Thermo Scientific, Waltham, MA, https://www.thermofisher.com/se/en/home.html), 2 mM l‐glutamine, 1% insulin‐transferrin‐selenite, 1% nonessential aminoacids, 0.5% penicillin‐streptomycin, 0.1% β‐mercaptoethanol (Gibco), 15 IU/ml heparin (Fisons Pharmaceuticals, Ipswich, U.K.), 50 ng/ml vascular endothelial growth factor (Sigma Aldrich, MO, http://www.sigmaaldrich.com), 75 µM ascorbic acid (Sigma Aldrich), 1 µM hydrocortisone (Sigma Aldrich), and 5 ng/ml basic fibroblast growth factor (Sigma Aldrich). For plate‐adherent cell isolation, dulbecco modified eagle medium (DMEM) with supplementary factors: 20% FCS, 2 mM l‐glutamine, 1% insulin‐transferrin‐selenite, 1% non‐essential amino acids and 0.5% penicillin‐streptomycin was used (all reagents from Gibco). Long‐term culture growth medium contained DMEM, 10% FCS, 1,000 U/ml leukemia inhibitory factor, 10 ng/ml epidermal growth factor, and 10 ng/ml platelet‐derived growth factor (Sigma Aldrich).
EPC Isolation, Differentiation, and Branching Analysis
BM was flushed from the femur with sterile PBS containing 10% fetal calf serum (PBS FCS). Red blood cell lysis buffer (Sigma Aldrich) was mixed for 1 minute, and cells washed with PBS FCS. Remaining cells were tested for the following EPC markers: prominin (PROM1), melanoma cell adhesion molecule (MCAM), tyrosine kinase receptor (KDR), and a receptor tyrosine kinase (TEK) 30, 31, 32, 33 by FACS. Filtered cell preparations were blocked for 30 minutes at 4°C in 10% FCS. Fluorophore‐conjugated antibodies (fluorescein isothiocyanate, FITC; or phycoerythrin, PE) PE‐KDR, FITC‐PROM1, FITC‐PECAM1, and PE‐TEK (eBioscience, San Diego, CA, https://www.thermofisher.com/se/en/home/life-science/antibodies/ebioscience.html), and FITC‐MCAM (Miltenyi Biotec, Bergisch Gladbach, Germany, http://www.miltenyibiotec.com) were applied according to the manufacturers’ recommendation. Cells were washed, sorted with a FACSCalibur, and analyzed using CellQuest software (BD, San Jose, CA, https://www.bdbiosciences.com). Magnetic‐activated cell sorting (MACS) was performed on pre‐stained cells. Anti‐FITC and anti‐PE Microbeads (Miltenyi Biotec) were applied for 10 minutes at 4°C and washed in PBS. Cells were magnetically sorted using an AutoMACS Separator (Miltenyi Biotec) in positive selection sensitive mode. Both positive and negative fractions were counted and plated in 0.1% Attachment Factor‐ (Life Technologies, Carlsbad, CA, https://www.thermofisher.com/us/en/home/brands/life-technologies.html) coated plastic 48‐well plates at 2.0–3.0 × 105 cells per well containing EDCM. FCS was reduced to 2% on day six. After 2 weeks of culture, cells were fixed for 10 minutes at room temperature in situ with 1% paraformaldehyde (PFA). For extended culture, cells were maintained in supplemented DMEM media prior to culture in EDCM 34. Fixed differentiated EPCs were analyzed for endothelial markers PECAM1 and von Willebrand factor (VWF). The number of PECAM1‐immunoreactive branches was counted regardless of their size or thickness (Fig. 1A) 35.
Figure 1.
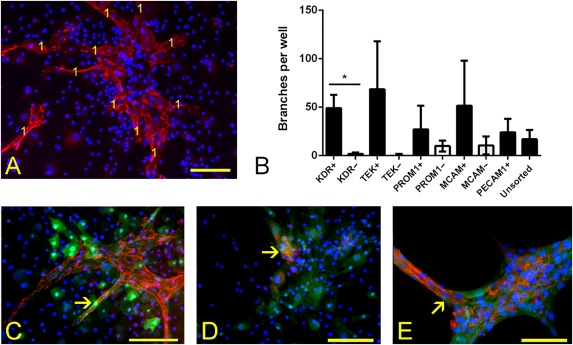
Analysis of endothelial differentiation in vitro. (A): An example of vessel‐like structure quantification, being based on the number of branches displaying immunoreactivity to a PECAM1 antibody (red), nuclei—blue. Each branch scored is marked with the digit 1. This example is taken from a KDR‐enriched culture following 7 days in endothelial differentiation culture medium. (B): A histogram depicting vessel‐like branch number per well in selected fractions of EPCs (data were normalized according to the number of plated cells). A statistical difference in branch numbers formed between specific enriched (+) and depleted (–) fractions is observed only between the KDR‐enriched and KDR‐depleted fractions (asterisk; p < .05; error bars represent SEM; n = 3). (C–E): Presence of green fluorescent protein (GFP) in PECAM1‐immunoreactive structures is marked with arrows; immunoreactivity to the PECAM1 antibody (red), nuclear reactivity to Hoechst (blue), and GFP fluorescence at 488 nm (green) ×20 magnification. (C): Fresh KDR‐enriched cells at passage 0 (P0), (D) Unsorted cells at P0 (short‐term culture, C7), (E) Unsorted cells at P12 after 77 days of culture (long‐term culture, C77). Scale bar = 100 µm. Abbreviations: KDR, tyrosine kinase receptor; MCAM, melanoma cell adhesion molecule; PROM1, prominin; TEK, receptor tyrosine kinase; PECAM1, platelet and endothelial cell adhesion molecule 1.
FACS Analysis
The following antibodies were applied on cell suspensions as described above: PE‐KDR (eBiosciences, San Diego, CA), allophycocyanin‐KDR (APC‐KDR, BD Pharmingen, San Jose, CA), FITC‐PROM1 (eBioscience), PE‐PROM1 (eBioscience), PE‐TEK (eBioscience), FITC‐MCAM (Miltenyi Biotec, Bergisch Gladbach, Germany), APC‐PROM1 (eBioscience), FITC‐Pecam1 (eBioscience), and AlexaFluor647‐ChIIIA4 (AF647‐EPHA3) antibody (Monash University, VIC, Australia). EPHA3 antibody was used as an additional marker of cells that can promote vascularization, also reported to be a tumor endothelial‐associated marker playing an active role in vascular growth 36. Two unconjugated rabbit polyclonal antibodies were also used: anti‐PECAM1 (Abcam, Cambridge, U.K., http://www.abcam.com/) and anti‐PROM1 (Abcam). Cell suspensions were washed three times in PBS. For the unconjugated antibodies, AF568‐conjugated goat anti‐rabbit IgG (Invitrogen/Life technologies, Carlsbad, CA) was applied for 30 minutes and washed three times in PBS. For this study, LSRII flow cytometer (BD) and FlowJo software were used.
Immunohistochemistry and Fluorescent In Situ Hybridization
Fixed in vitro BM cells were washed with PBS, incubated in 0.01% Triton (for PECAM1 antibody only) for 5 minutes. Cells were blocked with 5% goat serum for 1 hour at room temperature (RT). All antibodies were diluted in 5% goat serum and applied for 1 hour at RT: PECAM1 (Abcam; 1:50), VWF (Santa Cruz Biotechnology, Dallas, Texas, https://www.scbt.com/; 1:50), green fluorescent protein, or GFP (Abcam; 1:1,000) and labeled with AF568‐conjugated goat anti‐rabbit IgG (Invitrogen; 1:1,000).
In order to detect injected cells, lung sections were stained for GFP immunofluorescence using the tyramide signal‐enhancement method. For GFP signal enhancement, biotinylated secondary antibody (goat anti‐rabbit IgG, Invitrogen, at 1:1,000) was applied for 1 hour. Streptavidin‐HRP (Perkin Elmer, Waltham, MA, http://www.perkinelmer.com/) was applied at 1:1,000 for 30 minutes, followed by Biotin‐Tyramide (Perkin Elmer) at 1:50 for 7 minutes, and fluorophore‐conjugated Streptavidin (Invitrogen) at 1 µg/ml for 30 minutes (washes applied in between). Hoechst‐33342 was applied for 2 minutes and mounted using fluorescent mounting medium (DAKO/Agilent Technologies, Santa Clara, CA, http://www.agilent.com/). Immunoreactivity was analyzed using fluorescent microscopy (Olympus, Tokyo, Japan, https://www.olympus-lifescience.com/).
For Y chromosome detection, lungs from female mice injected with male EPCs were processed as described 37 and detected with the mouse IDetect Y chromosome paint probe (IDLabs, ON, Canada, https://www.empiregenomics.com/shop/idlabs.html), counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) and visualized using an Olympus confocal microscope (FluoView FV500, Tokyo, Japan).
Preparation of Cells for IP Injection
BM was isolated as described above from GFP‐positive male mice. Cells were enriched for EPCs by MACS using the KDR antibody and injected fresh into hyperoxic or normoxic mice. Other BM cells were plated according to a standard marrow stromal cell (MSC) isolation procedure 34, 38, then trypsinized and injected after 7–10 days in cell culture. For long‐term culture, cells were plated on plastic dishes in growth medium. Cells were cultured in 10% CO2 and repassaged once a week. Cells were expanded and frozen for further analysis. All injected cell fractions were analyzed for GFP and KDR and for vessel‐like structure forming capacity.
Morphometric Analysis of the Lung
Lungs were pressure‐fixed in situ as described earlier 5. Lung was then dissected, weighed, and measured by water displacement, then postfixed for 24 hours with 4% PFA at 4°C, embedded in paraffin and cut 5. Sections were randomly selected for analysis and stained using hematoxylin and eosin and Weigert's elastin stain 5. Five photographs were taken per section at ×20 magnification (area of each image being 0.564 mm2) to assess lung morphology. Mean linear intercept (MLI), tissue area, the number of alveoli, the number of blood vessels surrounded by elastin per tissue area, and the number of secondary septa per tissue area were analyzed on each lung image 5.
Collection of Tissues for GFP FACS Analysis
Lung and liver tissues were placed into a prewarmed (37°C) collagenase solution (4 mg of collagenase I, 9 mg of collagenase IV in 45 ml of PBS without calcium and magnesium; Gibco), chopped and incubated on a shaker at 37°C for 20 minutes, filtered and washed with PBS‐EDTA. Blood samples were collected from the heart into KCl hypotonic solution using a 19G needle inserted in the heart post mortem, incubated at 37°C for 20 minutes, and then washed with PBS‐EDTA.
Western Blot Analysis
Proteins were extracted from snap‐frozen left lobes of the lung and analyzed according to previously described Western blot procedures 5. The antibodies were used at the following concentrations for 1 hour at RT: vascular endothelial growth factor A (VEGFA, 1:200, Santa Cruz), β‐tubulin (1:1,000, TUBB3, Thermo Scientific), and Pro‐surfactant protein C (SFTPC, 1:2,000, Millipore, Billerica, MA). Secondary HRP‐conjugated goat anti‐rabbit IgG antibody (1:15,000, GE Healthcare, Buckinghamshire, U.K., http://www.gehealthcare.com/) was used for 1 hour at RT.
Statistical Analyses
ANOVA was used to determine any significant differences between measurements, p < .05, α = 0.05. A univariate general linear model was applied for morphometric analysis, where “group” (for example, normoxia‐PBS) was the main factor, and “litter” (values from one litter) and “animal” (values from a single mouse) allocated to be random factors (800 measurements per mouse for MLI, five measurements per mouse for other morphometric tests, with two to four mice per litter analyzed at each time point). ANOVA was followed by a Dunnett's post hoc test against hyperoxia‐ and normoxia‐PBS controls. Error bars were standard error of the mean (SEM). For Western blot analysis samples were compared within one gel, control samples present in each gel. Three to four animals were analyzed per group. Data analysis was performed using PASW statistical software (IBM, Armonk, NY, http://www.ibm.com).
Results
Characterization of EPC Fractions by FACS Analysis
BM cells positive for endothelial progenitor markers (KDR+, PROM1+, TEK+, and MCAM+) accounted on average for 2.4%, 10.4% 3.0%, and 34.9%, respectively, of all freshly isolated mononuclear cells analyzed by FACS (data not shown). PROM1+, TEK+, MCAM+, and KDR+ fractions were gated and then these populations were individually characterized for their relative immunoreactivity to KDR, TEK, PROM1, MCAM, PECAM1, and EPHA3 antibodies before cell culture. Tested EPC marker incidences overlapped to various degrees (4.1%–100.0%) depending on the sample in freshly‐isolated cells; only EPHA3 being less than 1.0% (data not shown). After 7 days in culture, unsorted plate‐adherent cells (C7) still displayed immunoreactivity to all of the above markers (data not shown), consistent with the phenotype of the freshly isolated EPC subgroups. Long‐term cultured cells at C77 displayed immunoreactivity to cell markers consistent with freshly‐isolated uncultured EPC subgroups, as well as low PECAM1 (1.9%) and high EPHA3 (58.9%) profiles. The incidence of EPHA3 immunoreactivity in the C56 and in C91‐98 cell populations was 86.3% and 30.0%, respectively, (data not shown).
Identification of a Fresh BM Subpopulation with Vessel‐Like Branch Forming Capability
Prior to injection into hyperoxia‐treated mice, the capacity of isolated EPCs to form blood vessel‐like branches in vitro was verified. To select a marker that could prospectively identify an EPC subpopulation with branch forming capacity, whole BM was first magnetically sorted on the basis of KDR, MCAM, PROM1, or TEK immunoreactivity. Marker‐enriched and ‐depleted cell fractions were then cultured separately, and PECAM1‐positive vessel‐like bifurcations were counted to determine branch‐forming capacity (Fig. 1A). Unlike other marker‐enriched or unsorted fractions, only vessels generated from the KDR‐enriched fraction produced a significantly increased number of branches compared with the fraction depleted for the same marker (49.1 ± 13.7 vs. 1.8 ± 1.4 branches; n = 3; p < .05; Fig. 1B). Therefore, freshly isolated KDR‐enriched EPCs were used for subsequent analyses and referred to as fresh cultured EPCs or Fr cells.
Analysis of Cells Prior to Injection
In order to compare fresh EPCs to those expanded in culture, plate‐adherent cell fractions, that is, whole unsorted short‐term (C7) and long‐term (C77, C91, C98) cultured GFP BM cells, were used in this study and tested in a similar fashion. According to FACS analysis, the total number of GFP+ cells in Fr cells enriched magnetically for KDR was on average 29.9%, in cultured C7 cells it was 85.9%, in C77 it was 82.3%, and in C91–98 it was <50.0%. All cell fractions were analyzed for the presence of KDR by FACS before differentiation in order to determine its percentage contribution. KDR+ cells were represented in a range from 15.1% to 31.4% of all cells in respective fractions (data not shown).
All tested cell fractions organized to form branches when transferred from growth medium to EDCM. The time to vessel‐like branch appearance in EDCM decreased as the culture time in DMEM growth medium increased: 6 to 7 days for Fr cells and 7‐day precultured cells (C7); compared with 2 to 3 days for 77‐day precultured cells (C77). Distinct Pecam1‐immunoreactive cells were present in these three fractions (Fig. 1C–1E). Cells in branches displayed various intensity of GFP fluorescence in association with immunoreactivity to PECAM1 (arrows, Fig. 1C, 1E). Limited GFP was observed in PECAM1‐immunoreactive branches formed from Fr cells (Fig. 1C) and C7 cells (Fig. 1D), whereas in C77 branches GFP was clearly present (Fig. 1E). Relative to Fr and C77 cells, C7 cell branches appeared less structured (Fig. 1D). Relative to Fr, C7, and C77 cells, C91‐98 cells (passage 13–15) showed an increasing tendency to detach, float and form aggregates (data not shown).
Pilot Injection of Cultured Cells After Neonatal Hyperoxia
Cultured cells (C56) were intraperitoneally injected into normoxic and hyperoxic mice in a pilot study to observe whether morphometric parameters of the lung would be altered by the cells in this time period and whether such experimentation could be acceptably tolerated by the subject animals. During the 4 weeks of treatment, injected mice did not display any signs of discomfort and no significant differences in weight or growth compared with PBS‐injected controls. Morphometric analysis of the lungs on D28 demonstrated that MLI was increased in both injected groups, compared with normoxia‐PBS group, but decreased, compared with hyperoxia‐PBS group. All other parameters, such as lung tissue area, number of alveoli per area, number of secondary septa per tissue area, and number of blood vessels, were not significantly different from normoxia‐PBS group, whereas hyperoxia‐PBS group displayed less alveoli and more secondary septa. Considering these results, cultured BM cells were determined to be suitable for further analysis and, therefore, were expanded and frozen for further injections. These cells were also kept in culture without freezing at various stages until passage 15 for subsequent intraperitoneal injection.
Injection and Tracking of Cultured EPCs Following Neonatal Hyperoxia
Fresh or cultured EPCs were intraperitoneally injected into normoxic or hyperoxic neonatal mice at postnatal day 5 (D5). Injected groups included (i) normoxia‐PBS (control), (ii) hyperoxia‐PBS (hyperoxia only); and (iii) hyperoxia or normoxia: (a) Fr KDR‐enriched (uncultured), (b) C7 (one passage), (c) C77 (11 passages and freezing) (d) C91 (13 passages) or (e) C98 cells (15 passages). All mice injected with C91 and C98 cells displayed apparent discomfort while moving, and developed small red spots at the site of injection that developed into small lumps by D21–28 (∼15 mm in diameter) containing GFP+ cells (Fig. 2A and data not shown). Histological analysis of lungs from mice injected with C91‐C98 cells demonstrated alterations in lung architecture, characterized by condensed cellular structures (Fig. 2B). Donor GFP signals were observed throughout lung sections in condensed and alveolar areas (Fig. 2C). A high number of nucleated cells was observed in the pulmonary blood vessel lumen following C91–98 cell injection (Fig. 2D). Aberrant growth formation was only observed on one occasion in a single cell batch with limited cell numbers, and therefore could not properly be studied further.
Figure 2.
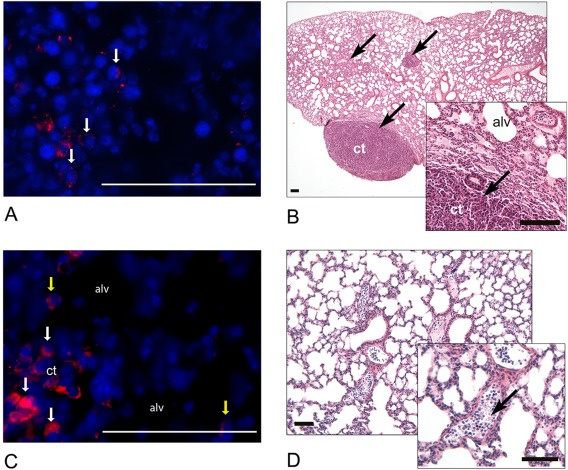
Histological analysis of tissues following C91–98 cell injections. (A): Presence of GFP‐immunoreactivity (red, arrows) in the lumps at the site of injection, nuclei—blue. (B): A representative lung section demonstrating areas of ct on D28 (Mayer's hematoxylin and eosin). Ct areas are indicated with arrows. (C): A representative lung section demonstrating the presence of GFP immunoreactivity within the ct (white arrows) and in the alveolar region (alv, yellow arrows) on D28: GFP (red), Hoechst (blue). (D): Nucleated cells in lung blood vessel lumena (Mayer's hematoxylin and eosin). The presence of nucleated cells is marked with an arrow within the enlarged inset. Scale = 100 µm. Abbreviations: alv, alveolus; ct, condensed tissue; GFP, green fluorescent protein.
PBS, Fr, or C7/77 injected mice displayed no signs of discomfort, and the injection site did not show any small red spots or lumps (data not shown). No aberrant structures or high numbers of nucleated cells in blood vessels were observed in the lung (data not shown; lung architecture is described in detail below in “Morphometric analyses”). The number of GFP+ cells detected in the lung and blood of Fr, C7, or C77 injected mice compared with non‐GFP+ PBS‐injected normoxia controls on D14 and D21 (D9 and D16 after injection) was below 0.5%, and in the liver below 1.7% (Fig. 3A). Plated aliquots of representative cell populations for injection retained GFP fluorescence during 14 days of culture (Fig. 3B) and beyond (data not shown), but on D28 no GFP cell fluorescence was detected in lung sections from EPC‐injected mice (data not shown). This was verified by fluorescent in situ hybridization (FISH) where no Y‐chromosome signals were observed in sections from D28 female lung that had been injected with male GFP+ EPCs (Fig. 3C, 3D).
Figure 3.
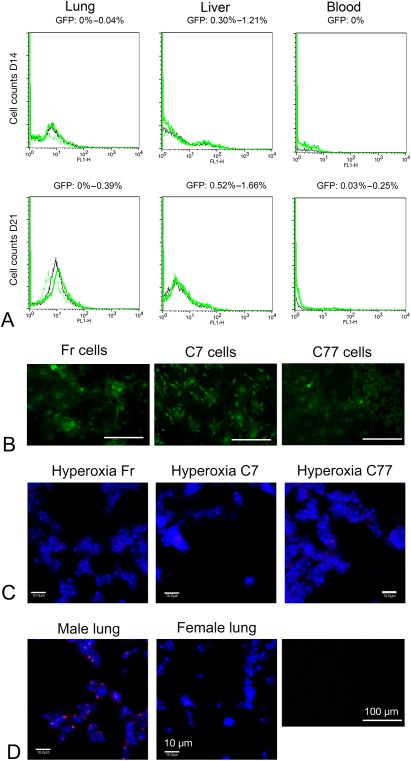
Tracking of donor cells following injection. (A): Fluorescent‐activated cell sorting analysis for the presence of GFP+ cells in the lung, liver, and blood, nine (D14) and 16 (D21) days after cell injection on D5. The negative control (PBS‐injected normoxic mouse lung) is indicated with a black line. Representative samples injected with GFP+ cells are indicated with green lines. The percentage of GFP+ cells is labeled above the charts: 0%–0.04% on D14 and 0%–0.39% on D21 in the lung, 0.30%–1.21% on D14 and 0.52%–1.66% on D21 in the liver, and 0% on D14 and 0.03%–0.25% on D21 in blood. (B): Injected Fr, C7, and C77 cell populations retain GFP signal (green) in culture for 14 days. Scale bar—100 µm. (C): Fluorescent in situ hybridization (FISH) for Y chromosome signals (red), nuclei are counterstained blue with DAPI. No FISH positive Y signal was observed in female lungs injected with Fr, C7 or C77 male donor cells on D28 (n = 1–2; one random section per animal analyzed). Scale bar—10 µm. (D): Control samples for FISH and GFP. Y chromosome red signals were observed in positive control lung sections from a male mouse. No FISH positive Y chromosome signal was observed in negative control section from a female mouse. No GFP signal was observable in GFP‐ control cells isolated from wild type mouse. Abbreviation: GFP, green fluorescent protein; DAPI, 4′,6‐diamidino‐2‐phenylindole.
Animal Weight and Length
Female mice from hyperoxic groups injected with PBS had significantly lower weight and length than normoxic groups at various time points; however, male mice maintained healthy weight and length (Fig 4). Compared with PBS injected normoxia controls, C91–98 hyperoxia male and female mice displayed significant reductions in weight and length at various stages up to their day of sacrifice (varying from 3–5 weeks of age; Fig 4). Death was determined according to a humane endpoint. Compared with PBS injected normoxia controls, C77 injected male and female hyperoxia groups displayed a significant reduction in weight and length at most time‐points examined (Fig. 4A–4D). Out of all hyperoxic groups, hyperoxia‐Fr group had significantly higher weight and length than hyperoxia‐PBS group at a number of time points, except for male length at weeks seven and eight (Fig. 4B). All other groups, except for the normoxia‐Fr group, displayed sporadic reductions compared with normoxia‐PBS in either weight or length, but these reductions were not generally maintained, apart from the reductions in weight and length of female hyperoxia‐C7 and hyperoxia‐PBS treated groups (Fig. 4D).
Figure 4.
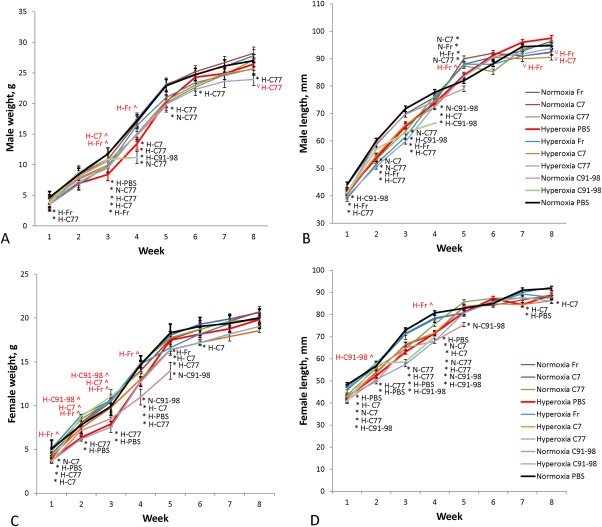
Mouse weight and length indices. (A): Average male animal weight per group. (B): Average male animal length per group. (C): Average female animal weight per group. (D): Average female animal length per group. Hyperoxia‐C91–98 and normoxia‐C91–98 groups were only observed for the first 4 to 5 weeks, and then sacrificed due to animal distress. Error bars indicate SEM, n = 3–7. Significant differences to control (normoxia‐PBS) at each time point are marked with an asterisk (*) and abbreviated group name in black (e.g., at 3 weeks male animal weight in the hyperoxia‐PBS group was significantly lower than in the normoxia‐PBS control group; p < .05). Significant differences to hyperoxia‐PBS (in hyperoxic groups only) are marked with a circumflex accent (^) in case of increase or letter V (V) in case of decrease and abbreviated group name in red.
Morphometric Analyses of Hyperoxic Lungs Following Cell Injection
Lungs from hyperoxic and normoxic mice before (D5) and following injection with PBS, Fr, C7, and C77 cell fractions were analyzed for alveolar size, secondary septal crest number, tissue area, and blood vessel quantity on D28 and D56 (Table 1). All treatment groups showed comparable lung weight and volume at all examined time points, relative to PBS‐normoxia controls (no significant difference).
Table 1.
Morphometric parameters of lungs. Means with standard errors are presented for normoxic (NormO2) and hyperoxic (HypO2) lungs before (D5) and after (D28, D56) the injection of (PBS) or cells (fresh or cultured for 7 or 77 days)
| Day of test | Cells/PBS | MLI | Alveoli | Tissue Area (*102) | Sec Septa per tissue (×10−1) | Blood vessels | Blood Vessels per tissue | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NormO2 | HypO2 | NormO2 | HypO2 | NormO2 | HypO2 | NormO2 | HypO2 | NormO2 | HypO2 | NormO2 | HypO2 | ||
| D5 | None | 45.0^ ± 2.7 | 57.1** ±2.2 | 16.2^ ± 0.2 | 13.7 ± 0.3 | 16.4^ ±1.2 | 14.5* ±0.8 | 37.7^ ± 2.8 | 26.0** ± 1.7 | 14.6^ ± 0.6 | 12.4* ± 0.5 | 88.6 ± 5.4 | 85.8 ± 6.2 |
| D28 | PBS | 26.7^ ± 0.9 | 32.0** ± 0.9 | 21.8^ ± 0.8 | 19.9** ± 0.4 | 19.5 ± 0.9 | 19.2 ± 0.8 | 25.6^ ± 4.3 | 36.5** ± 2.9 | 19.3 ± 1.1 | 21.5 ± 1.1 | 114.3 ± 3.5 | 116.5 ±0.7 |
| Fresh | 28.4^ ± 0.5 | 29.8^** ± 1.0 | 21.5^ ± 0.6 | 19.9** ± 0.5 | 20.3 ± 0.6 | 18.7 ± 0.5 | 27.8^ ± 4.3 | 30.2^ ± 2.6 | 20.5 ± 0.9 | 20.4 ± 1.1 | 111.2 ± 4.9 | 119.6 ±3.0 | |
| C7 | 27.0^ ± 0.6 | 31.1** ± 0.6 | 21.2 ± 03 | 20.0** ± 0.4 | 20.1 ± 1.2 | 20.9^ ± 0.6 | 23.1^ ± 2.7 | 26.5^ ± 2.1 | 24.6** ± 0.5 | 25.2**^ ± 1.0 | 131.9* ± 3.7 | 108.0 ± 3.3 | |
| C77 | 29.5^** ± 1.0 | 32.3** ± 1.1 | 20.4 ± 0.5 | 20.0* ± 0.6 | 18.3 ± 0.7 | 18.1 ± 0.9 | 34.5** ± 5.6 | 44.4**^ ± 11.7 | 16.4^ ± 2.1 | 19.2 ± .2.3 | 108.0^ ± 22.2 | 118.3 ± 15.4 | |
| D56 | PBS | 30.4^ ± 0.6 | 32.8** ± 1.3 | 20.4 ± 0.2 | 19.6 ± 0.6 | 18.4 ± 0.7 | 17.5 ± 0.6 | 28.3 ± 2.9 | 31.4 ± 1.3 | 22.2^ ± 0.5 | 17.8** ± 1.1 | 108.4 ± 2.8 | 113.1 ± 4.9 |
| Fresh | 32.6** ± 1.9 | 30.1^ ± 1.1 | 19.7 ± 0.9 | 20.3 ± 0.8 | 17.4 ± 1.5 | 18.0 ± 0.8 | 36.3 ± 2.2 | 31.1 ± 3.0 | 20.4 ± 1.9 | 20.0 ± 1.0 | 99.5 ±4.5 | 121.2 ± 4.4 | |
| C7 | 30.9^ ± 1.5 | 32.8* ± 2.6 | 20.0 ± 0.5 | 18.7 ± 0.9 | 17.8 ± 1.3 | 17.4 ± 1.4 | 30.0 ± 3.6 | 34.0 ± 3.7 | 19.1 ± 1.0 | 19.1 ± 1.0 | 112.8 ± 7.0 | 99.7 ± 3.3 | |
| C77 | 31.3 ± 0.8 | 33.0** ± 0.8 | 20.5 ± 1.1 | 18.9 ± 1.0 | 17.7 ± 0.6 | 17.5 ± 0.7 | 35.1 ± 0.8 | 43.1**^ ± 4.9 | 19.4 ± 0.9 | 19.8 ± 1.3 | 110.2 ± 6.1 | 114.5 ± 10.1 | |
Significant differences to respective normoxic controls (dark gray cells) are marked with two asterisks (**, p < .01, n = 4–5) or one asterisk (*, p < .05, n = 4–5). Significant differences to respective hyperoxic controls (light gray cells) are marked with circumflex accent (^, p < .05, n = 4–5).
Abbreviations: alveoli, number of alveoli was counted per 3.63 mm line (vertical and horizontal) drawn across an image of each lung sections, and is presented in airspaces per 1 mm; blood vessels, number of elastin‐positive blood vessels per 0.564 mm2 of lung image; blood vessels per tissue, number of these vessels by 1 mm2 of tissue area; MLI, mean linear intercept (in µm); sec septa, number of secondary septa per 1 mm2 of lung tissue area; tissue area, lung tissue area in mm2 per 0.564 mm2 of lung image.
First, a ratio of lung tissue to airspace showed reduced tissue content in hyperoxic groups on D5 (p < .05) but not on D28 or D56 (Table 1, Fig. 5A–5E). Second, alveolar enlargement was evident in all hyperoxic groups compared with normoxic‐PBS group on D28; however, hyperoxic mice injected with Fr cells displayed significantly lower alveolar size compared with hyperoxic PBS‐injected mice (Table 1, MLI). On D56, all hyperoxic groups retained a relatively increased alveolar size, except for hyperoxic mice injected with Fr cells, which displayed sizes comparable to normoxic controls and significantly differed from hyperoxic controls (Table 1; Fig. 5F MLI). The number of alveoli was significantly decreased in hyperoxic groups relative to normoxia‐PBS group on D5 and D28 (Table 1, Alveoli count, Fig. 5G). All hyperoxic groups returned to control levels on D56, including hyperoxia PBS‐injected group. Alveoli were also significantly enlarged in the normoxia‐C77 group on D28 and in the normoxia‐Fr group on D56, suggesting a deleterious effect from injected cells in the absence of hyperoxic injury. The number of secondary septa was significantly reduced on D5 (Table 1, Sec Septa per Tissue, Fig. 5H). By D28, the number of crests was reduced in normoxia‐PBS controls, but remained elevated in hyperoxia‐PBS and ‐C77 groups, as well as the normoxia‐C77 group. Hyperoxic groups injected with Fr and C7 cells had significantly lower septal number compared with hyperoxic PBS‐injected group and did not differ from normoxic control (Table 1, Sec Septa per Tissue, Fig. 5H). On D56, with the exception of the hyperoxia‐C77 group which exhibited a marked increase in crest number, no group showed any statistically significant difference to controls (Table 1, Fig. 5A–5E, 5H).
Figure 5.
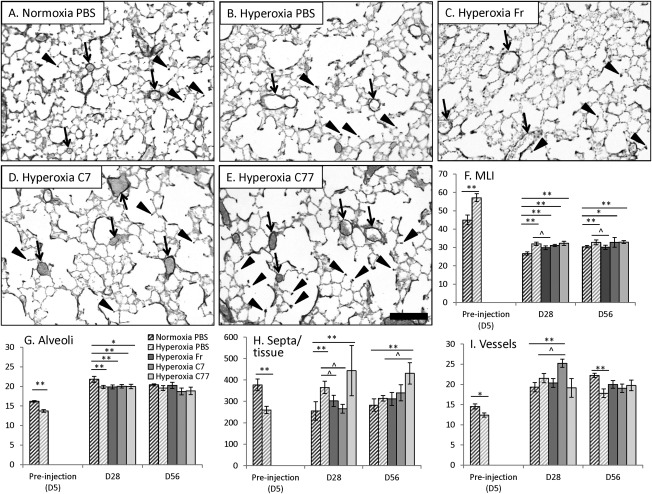
Morphometric parameters of lung sections. (A–E): Histology of representative mouse lungs from control (A, normoxia‐PBS) and hyperoxic groups (B–E) on D56 (Weigert's elastin staining, elastin—black). Example blood vessels are labeled with arrows, example secondary septa are labeled with arrowheads. Scale bar = 100 μm. (F–I): Statistical analysis of morphometric parameters of the lung in hyperoxic groups injected with cells (fresh or cultured for 7 or 77 days) in comparison to normoxia‐PBS and hyperoxia‐PBS. (F): Alveolar size based on MLI (in µm), (G) Number of alveoli was counted per 3.63 mm line, in airspaces per 1 mm, (H) Number of secondary septa per 1 mm2 of lung tissue area, (I) Number of elastin‐positive blood vessels per 0.564 mm2 of lung image. Significant differences to respective normoxic controls (dark upward diagonal lines) are marked with two asterisks (**, p < .01, n = 4–5) or one asterisk (*, p < .05, n = 4–5). Significant differences to respective hyperoxic controls (light upward diagonal lines) are marked with circumflex accent (^, p < .05, n = 4–5). Abbreviations: MLI, mean linear intercept; PBS, phosphate buffered saline.
Finally, pulmonary vascularization was significantly reduced on D5, yet in PBS, fresh‐ and C77‐injected groups it returned to control levels by D28 (Table 1, Fig. 5I). There was no significant difference in vessels/tissue ratio on D5 or D56 between all groups and control. On D28 and compared with controls, however, both normoxic and hyperoxic C7‐injected groups displayed a significantly higher vessel number, and normoxia‐C7 group displayed a significantly higher vessels/tissue ratio (Table 1). By D56 these values had returned to control levels. The hyperoxia‐PBS group, however, maintained a significantly lower vessel number compared with normoxia controls (Table 1, Fig. 5A–5E, 5I).
Analysis of Protein Levels by Western Blot
Western blot analysis was performed to determine whether VEGFA levels are affected in the lung following injections of EPCs positive for KDR (VEGFA receptor); and to determine whether cell injection has adverse effect on surfactant production (SFTPC) essential for normal lung function. When dimer and monomer forms of VEGFA were compared separately, dimer (42 kDa) levels in normoxia‐C77 were reduced; however, in hyperoxia‐Fr and hyperoxia‐C7, they were increased, and monomer (21 kDa) levels in normoxia‐Fr and normoxia‐C7 groups were significantly reduced on D28 compared with control levels (p < .05; Fig. 6A, 6B). When the sums of monomer and dimer bands were compared, there was only a reduction in normoxia‐C77 group, but all other groups retained control levels. No repetitive differences in VEGFA monomer or dimer levels between groups on D5 and D56 were detected (data not shown). There was no significant difference at any time point in the level of SFTPC precursor or cleaved forms (21 and 25 kDa) compared with controls (see Fig. 6A, 6C for D28; data not shown for D5 and D56). No significant differences to hyperoxia‐PBS group were detected at any time point in either VEGFA or SFTPC levels.
Figure 6.
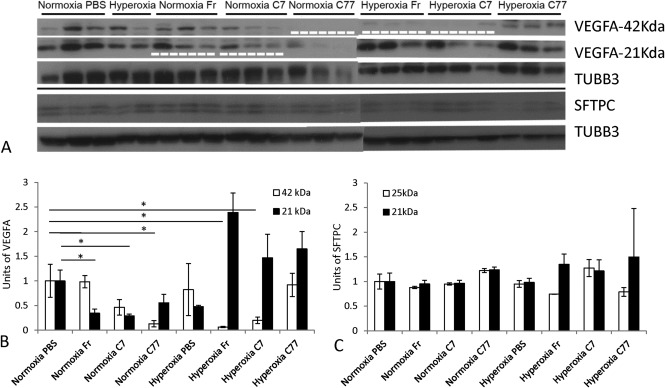
Western blot protein analysis of lungs processed on D28. (A): A representative protein Western blot in different groups. TUBB3 was used as loading control. Loading amount: 8–10 µg of protein. Film exposure time: 3–15 seconds. Significant reductions in protein levels compared with normoxia‐PBS controls are indicated by a white dashed line (p < .05; n = 3). (B): Chart representing VEGFA levels. VEGFA levels were reduced in Fr‐ and C7‐injected groups: 21 kDa VEGFA in normoxic and 42 kDa VEGFA in hyperoxic mice, as well as in normoxia‐C77 group (42 kDa). Significant differences to normoxic controls are marked with asterisks (p < .05; n = 3). No significant differences to hyperoxic controls were observed. (C): Chart representing SFTPC levels, no significant differences to normoxic and hyperoxic controls were observed. Abbreviations: PBS, phosphate buffered saline; SFTPC, surfactant protein C; TUBB3, Tubulin beta 3; VEGFA, vascular endothelial growth factor A.
Discussion
In this study, the regenerative potential of BM‐isolated marker‐specific EPCs for repairing aspects of neonatal mouse lung hyperoxia injury were analyzed. First, these data indicate that in this system KDR‐enriched cells represent preferred EPC subpopulation for subsequent in vivo evaluation of repair studies given their advantageous ability to form vessel‐like structures in vitro. Only the fresh KDR‐enriched cell‐injected group (hyperoxia‐Fr) demonstrated a restored alveolarization. Other parameters were also restored to normoxic control levels in this treatment group. However, increased alveolar size in normoxic group on Day 56 indicates that the injection of fresh BM derivatives into the uninjured lung can affect respiratory morphogenesis. The physiological significance of this effect remains unclear, but does suggest caution in the use of BM‐derived cells in clinical studies, and demonstrates the importance of determining the best time and dose of oxygen exposure for which such treatments would be effective. Determination of the best treatment regime requires further investigation.
Previously, the injection of early passage plate‐adherent cells into hyperoxia newborn mice (intravenously) and rats (intratracheally) was reported to confer a therapeutic benefit 13, 14. Further, cultured EPCs (not immunoreactive to KDR, but expressing Tie2) and injected into the right ventricle of the heart were demonstrated to improve lung architecture after neonatal hyperoxia 10. Although administration of cultured cells to hyperoxia‐injured lung here effectively restored blood vessel number to control levels (or above these levels), no benefit on alveolar size or number was observed. The combined results of these studies may highlight the potential importance of administration route and population purity with respect to alveolar therapeutic outcome when using cultured BM cells. Further, the length of time in culture and/or cell freezing appeared to correlate with the outcome, with longer term cultured cells (C77, frozen) inducing significant increases in secondary septal formation on D28 and D56, an effect not observed with cells cultured for a shorter time (C7, C56). The transient significant increase in blood vessel number following injection of early culture cells (C7) could be compared with the improvement of vasculature observed recently by another group after injection of passage four BM‐derived MSCs into hyperoxic animals 39. However, in contrast to current study, the similar effect was not observed in normoxic animals. Whether overfilling the marrow MSC/EPC pool or influence of injected cell signaling has affected this, remains to be explored.
In previous studies, it was demonstrated that hyperoxia increases pulmonary vessel/tissue ratio on D28 but has no effect of blood vessel number on D56 [5]. Yet, after PBS injection here, these parameters in normoxic lung were different, that is, high blood vessel values and low tissue area values (compared with the animals that were not injected), masking the previously observed D28 difference and creating a significant difference by D56. Other previous studies observed normal vasculature at 8 weeks of age after 4 days of neonatal oxygen treatment 6. The effects of intraperitoneal injection of fluid (such as PBS) on lung vascularization remain under question, but can be addressed in future studies, while the number of animals per group could be increased and the number of experimental groups decreased.
Reduction in VEGFA levels observed in this study appeared sporadically after cell injections, occasionally coinciding with MLI increases, when the functional dimer (42 kDa) was affected (normoxic‐C77 and hyperoxic‐Fr and ‐C7 groups). Similar to previous studies, it was restored by day 56 4. VEGFA is produced by a fraction of alveolar epithelial type II cells (AECII) and was reported to be linked to septa formation 40. Since there was no systematic reduction of this protein in any injected group, the changes could indicate various stages of protein assembly and dimerization in the injected groups at 28 days. This combined with the demonstration that levels of SFTPC (produced by AECII) remained unchanged in all groups, suggests that no significant decrease in general AECII function was exerted by four days of hyperoxia or from cell injection.
Injecting longer term cultured cells (C91‐C98) led to aberrant tissue formations. Donor cell‐derived GFP signals were detected in condensed tissue areas in the lungs indicating that intraperitoneally injected cells had reached the lung and participated in forming local structures. The observed high amount of nucleated cells in the blood vessels could indicate inflammation. Previous studies demonstrated, that the longer cells are expanded in culture, the higher is their chance to potentially undergo chromosomal mutations 41. It has been demonstrated that after similar culture conditions a significant number of human MSCs undergo malignant transformations spontaneously, which could explain the effect observed here 42. The use of long‐term cultured cells for therapeutic purposes should therefore be avoided. EPHA3+ cells were observed at very low levels in freshly‐isolated BM cells (<1%). The injection of EPHA3+ cells (C56, C77) into mice did not consistently reduce hyperoxic damage to the lung, and therefore, the role of such cells in ameliorating deficits to alveolarization and vascularization during this type of injury does not appear significant. A possible connection between the presence of high EPHA3 immunoreactivity in the C91–98 cell populations and aberrant growth and condensed tissue in the lung also remains to be determined.
In this study, the effects of cultured and fresh noncultured cells on lung parameters varied significantly and indicate that different cell populations of the same origin can affect the development of alveolarization differently. The therapeutic efficacy of fresh KDR‐enriched cells could be originating from a fraction that was further lost in culture, for example nonadherent cells. However, this hypothesis remains to be investigated. The conclusion about avoiding long‐term culture of BM cells due to loss of their potential has previously been observed 43. Usually, cell culture is a necessary step required for the expansion of cell numbers for further transplantation. However, in this study we demonstrate that small cell numbers can be sufficient for the regenerative processes after hyperoxia. The therapeutic effects of human cells in animal hyperoxia models, including large animal models, are now widely studied. Further steps toward improving or replacing cell treatment to reduce potential side effects warrant further experiments.
Conclusion
Fresh KDR‐enriched BM cell fractions effectively differentiated into endothelial cells in vitro, and promoted alveolarization recovery following hyperoxia‐induced lung injury. Prolonged cell culture however caused a gradual decrease in therapeutic outcome, and occasionally promoted the growth of aberrant cell masses. It is concluded therefore that long‐term cell culture of enriched EPCs is of less potential therapeutic value and is perhaps clinically dangerous, and that freshly enriched EPCs may provide a preferred source of cells for the treatment of the postnatal deficits of hyperoxia‐induced lung injury.
Author Contributions
A.F.: design, provision of study material, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; D.B.: data analysis and interpretation, final approval of manuscript; D.A. and J.N.: provision of study materials, collection and assembly of data, data analysis and interpretation, final approval of manuscript; R.M.: conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript; T.C.: financial support, administrative support, data interpretation, final approval of the manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Acknowledgments
We would like to thank Prof. Richard Harding and his team for support with methodology of neonatal hyperoxia, as well as all necessary equipment for oxygen treatment; Dr. Sharon Flecknoe, Ian Boundy, Stefania Tombs, Prof. Jeffrey Kerr, and Dr. Jonathan Bensley for consulting in histological protocols and morphometric lung analysis; Prof. Aidan Sudbury and Dr. Robert De Matteo for consulting in statistical analysis; Prof. Martin Lackmann and Dr. Mary Vail for collaborating on long‐term cultured cells, providing access to EPHA3+ cells, EPHA3 antibody, and necessary equipment; Irene Hatzinisiriou for technical assistance on microscopy and FACS; Monash University Animal Facilities staff members for supervising animal welfare; Paula Stoddart (Miltenyi) for providing antibody samples for MACS; Dr. Nathanael Raschzok for the in situ hybridization protocol; all groups in the Department of Anatomy and Developmental Biology (Monash University) for sharing reagents, equipment, and methodologies; and Dr. Mei Ling Lim for manuscript revision. This project was supported by the NHMRC Program Grant 384100.
Contributor Information
Alexandra B. Firsova, Email: alexandra.firsova@gmail.com.
Richard Mollard, Email: rmollard@unimelb.edu.au.
References
- 1. Blencowe H, Cousens S, Oestergaard MZ et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: A systematic analysis and implications. Lancet 2012;379:2162–2172. [DOI] [PubMed] [Google Scholar]
- 2. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med 2001;163:1723–1729. [DOI] [PubMed] [Google Scholar]
- 3. Thebaud B, Abman SH. Bronchopulmonary dysplasia: Where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med 2007;175:978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Balasubramaniam V, Mervis CF, Maxey AM et al. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2007;292:L1073–L1084. [DOI] [PubMed] [Google Scholar]
- 5. Firsova AB, Cole TJ, Mollard R. Transient vascular and long‐term alveolar deficits following a hyperoxic injury to neonatal mouse lung. BMC Pulm Med 2014;14:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yee M, White RJ, Awad HA et al. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am J Pathol 2011;178:2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McGrath‐Morrow SA, Stahl J. Apoptosis in neonatal murine lung exposed to hyperoxia. Am J Respir Cell Mol Biol 2001;25:150–155. [DOI] [PubMed] [Google Scholar]
- 8. Panos RJ, Bak PM, Simonet WS et al. Intratracheal instillation of keratinocyte growth factor decreases hyperoxia‐induced mortality in rats. J Clin Invest 1995;96:2026–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kunig AM, Balasubramaniam V, Markham NE et al. Recombinant human VEGF treatment enhances alveolarization after hyperoxic lung injury in neonatal rats. Am J Physiol Lung Cell Mol Physiol 2005;289:L529–L535. [DOI] [PubMed] [Google Scholar]
- 10. Balasubramaniam V, Ryan SL, Seedorf GJ et al. Bone marrow‐derived angiogenic cells restore lung alveolar and vascular structure after neonatal hyperoxia in infant mice. Am J Physiol Lung Cell Mol Physiol 2010;298:L315–L323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim T‐H, Chow Y‐H, Gill SE et al. Effect of IGF blockade on hyperoxia‐induced lung injury. Am J Respir Cell Mol Biol 2012;47:372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deng H, NicholasMason S, Auten RL. Lung inflammation in hyperoxia can be prevented by antichemokine treatment in newborn rats. Am J Respir Crit Care Med 2000;162:2316–2323. [DOI] [PubMed] [Google Scholar]
- 13. Aslam M, Baveja R, Liang OD et al. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med 2009;180:1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Haaften T, Byrne R, Bonnet S et al. Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am J Respir Crit Care Med 2009;180:1131–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dauger S, Ferkdadji L, Saumon G et al. Neonatal exposure to 65% oxygen durably impairs lung architecture and breathing pattern in adult mice. Chest 2003;123:530–538. [DOI] [PubMed] [Google Scholar]
- 16. Zimova‐Herknerova M, Myslivecek J, Potmesil P. Retinoic acid attenuates the mild hyperoxic lung injury in newborn mice. Physiol Res 2008;57:33–40. [DOI] [PubMed] [Google Scholar]
- 17. Warburton D, Perin L, Defilippo R et al. Stem/progenitor cells in lung development, injury repair, and regeneration. Proc Am Thorac Soc 2008;5:703–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tropea KA, Leder E, Aslam M et al. Bronchioalveolar stem cells increase after mesenchymal stromal cell treatment in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell and Mol Physiol 2012;302:L829–L837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abe S, Boyer C, Liu X et al. Cells derived from the circulation contribute to the repair of lung injury. Am J Respir Crit Care Med 2004;170:1158–1163. [DOI] [PubMed] [Google Scholar]
- 20. Anjos‐Afonso F, Siapati EK, Bonnet D. In vivo contribution of murine mesenchymal stem cells into multiple cell‐types under minimal damage conditions. J Cell Sci 2004;117:5655–5664. [DOI] [PubMed] [Google Scholar]
- 21. Ishizawa K, Kubo H, Yamada M et al. Bone marrow‐derived cells contribute to lung regeneration after elastase‐induced pulmonary emphysema. FEBS Lett 2004;556:249–252. [DOI] [PubMed] [Google Scholar]
- 22. Spees JL, Whitney MJ, Sullivan DE et al. Bone marrow progenitor cells contribute to repair and remodeling of the lung and heart in a rat model of progressive pulmonary hypertension. FASEB J 2008;22:1226–1236. [DOI] [PubMed] [Google Scholar]
- 23. Yamada M, Kubo H, Kobayashi S et al. Bone marrow‐derived progenitor cells are important for lung repair after lipopolysaccharide‐induced lung injury. J Immunol 2004;172:1266–1272. [DOI] [PubMed] [Google Scholar]
- 24. Albera C, Polak JM, Janes S et al. Repopulation of human pulmonary epithelium by bone marrow cells: a potential means to promote repair. Tissue Eng 2005;11:1115–1121. [DOI] [PubMed] [Google Scholar]
- 25. Qi Y, Jiang Q, Chen C et al. Circulating endothelial progenitor cells decrease in infants with bronchopulmonary dysplasia and increase after inhaled nitric oxide. PLoS One 2013;8:e79060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoder MC, Mead LE, Prater D et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 2007;109:1801–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Asahara T, Masuda H, Takahashi T et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res 1999;85:221–228. [DOI] [PubMed] [Google Scholar]
- 28. Kalka C, Masuda H, Takahashi T et al. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci USA 2000;97:3422–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Young PP, Hofling AA, Sands MS. VEGF increases engraftment of bone marrow‐derived endothelial progenitor cells (EPCs) into vasculature of newborn murine recipients. Proc Natl Acad Sci USA 2002;99:11951–11956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dumont DJ, Gradwohl G, Fong GH et al. Dominant‐negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev 1994;8:1897–1909. [DOI] [PubMed] [Google Scholar]
- 31. Shibuya M. Differential roles of vascular endothelial growth factor receptor‐1 and receptor‐2 in angiogenesis. J Biochem Mol Biol 2006;39:469–478. [DOI] [PubMed] [Google Scholar]
- 32. Kang Y, Wang F, Feng J et al. Knockdown of CD146 reduces the migration and proliferation of human endothelial cells. Cell Res 2006;16:313–318. [DOI] [PubMed] [Google Scholar]
- 33. Yin AH, Miraglia S, Zanjani ED et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997;90:5002–5012. [PubMed] [Google Scholar]
- 34. Rochefort GY, Vaudin P, Bonnet N et al. Influence of hypoxia on the domiciliation of Mesenchymal Stem Cells after infusion into rats: possibilities of targeting pulmonary artery remodeling via cells therapies? Respir Res 2005;6:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tata DA, Anderson BJ. A new method for the investigation of capillary structure. J Neurosci Methods 2002;113:199–206. [DOI] [PubMed] [Google Scholar]
- 36. Vail ME, Murone C, Tan A et al. Targeting EphA3 inhibits cancer growth by disrupting the tumor stromal microenvironment. Cancer Res 2014;74:4470–4481. [DOI] [PubMed] [Google Scholar]
- 37. Raschzok N, Teichgräber U, Billecke N et al. Monitoring of liver cell transplantation in a preclinical swine model using magnetic resonance imaging. Cell Med 2010;1:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rojas M, Xu J, Woods CR et al. Bone marrow‐derived mesenchymal stem cells in repair of the injured lung. Am J Respir Cell Mol Biol 2005;33:145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sammour I, Somashekar S, Huang J et al. The effect of gender on mesenchymal stem cell (MSC) efficacy in neonatal hyperoxia‐induced lung injury. PLoS One 2016;11:e0164269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yamamoto H, Yun EJ, Gerber HP et al. Epithelial‐vascular cross talk mediated by VEGF‐A and HGF signaling directs primary septae formation during distal lung morphogenesis. Dev Biol 2007;308:44–53. [DOI] [PubMed] [Google Scholar]
- 41. Ahrlund‐Richter L, De Luca M, Marshak DR et al. Isolation and production of cells suitable for human therapy: Challenges ahead. Cell Stem Cell 2009;4:20–26. [DOI] [PubMed] [Google Scholar]
- 42. Rosland GV, Svendsen A, Torsvik A et al. Long‐term cultures of bone marrow‐derived human mesenchymal stem cells frequently undergo spontaneous malignant transformation. Cancer Res 2009;69:5331–5339. [DOI] [PubMed] [Google Scholar]
- 43. Bonab MM, Alimoghaddam K, Talebian F et al. Aging of mesenchymal stem cell in vitro. BMC Cell Biol 2006;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]