

Abstract
Cytotoxic chemotherapies are used to treat breast cancer, but are limited by systemic toxicity. The key to addressing this important issue is the development of a nontoxic, tissue selective, and molecular specific delivery system. In order to potentially increase the therapeutic index of clinical reagents, we designed an Aminopeptidase P (APaseP) targeting tissue-specific construct conjugated to a homing peptide for selective binding to human breast-derived cancer cells. Homing peptides are short amino acid sequences derived from phage display libraries that have the unique property of localizing to specific organs. Our molecular construct allows for tissue-specific drug delivery, by binding to APaseP in the vascular endothelium. The breast homing peptide evaluated in our studies is a cyclic nine-amino-acid peptide with the sequence CPGPEGAGC, referred to as PEGA. We show by confocal microscopy that the PEGA peptide and similar peptide conjugates distribute to human breast tissue xenograft specifically and evaluate the interaction with the membrane-bound proline-specific APaseP (KD = 723 ± 3 nM) by binding studies. To achieve intracellular breast cancer cell delivery, the incorporation of the Tat sequence, a cell-penetrating motif derived from HIV, was conjugated with the fluorescently labeled PEGA peptide sequence. Ultimately, tissue specific peptides and their conjugates can enhance drug delivery and treatment by their ability to discriminate between tissue types. Tissue specific conjugates as we have designed may be valuable tools for drug delivery and visualization, including the potential to treat breast cancer, while simultaneously minimizing systemic toxicity.
Graphical abstract
INTRODUCTION
Protein molecular targets are the predominant focus of drug discovery and delivery, as they offer a selective approach to cancer therapy. Targeted molecular therapy, such as transtuzumab (Herceptin), has made major advances for breast cancer; however, its efficacy has been limited by resistance, and inefficient cellular penetration at the tumor site.1,2 These limitations have prompted scientists to find better ways to deliver selective treatments that would not succumb to resistance and enhance the therapeutic index of current and future small molecule therapies for breast cancer. One way to address this concern is to design a drug delivery conjugate containing a chemical entity that will display site specificity and selectivity built into its molecular structure, such as to target diseased tissue and diminish toxicity to surrounding, nonpathological tissues.3 To deliver therapy to a diseased tissue site, a drug delivery conjugate was engineered with tissue specificity, selectivity, and retention. We have utilized this strategy to develop a selective drug delivery conjugate for breast cancer by using a breast tissue homing peptide derived from in vivo phage display.
In our approach, we sought to develop tissue specificity, cell penetration, and tumor delivery in our peptide-based design. To address tissue specificity we employed homing peptides, short amino acid sequences derived from phage display libraries that have the unique property of homing to specific organs vascular endothelium.4,5 Specifically, the breast homing peptide used in our study was a cyclic nine-amino-acid peptide with the sequence CPGPEGAGC, referred to as PEGA.4 Shown to selectively localize to breast tissue, the PEGA sequence is a critical component to the development of a breast tissue specific drug delivery conjugate. The PEGA sequence has been suggested to interact with the membrane-bound proline-specific aminopeptidase P (APaseP).4
APaseP is 96.3 kDa expressed in two subtypes, a membrane-bound form encoded by a gene on the human X chromosome and the cytosolic protein encoded by a gene on human chromosome 10.7 The membrane-bound form can be found on the plasma membrane of endothelial and epithelial cells.7 APaseP catalyzes the removal of any unsubstituted N-terminal amino acid adjacent to the penultimate proline residue.6 The nine-amino-acid bradykinin is a known ligand for APaseP.7 The PEGA peptide which is similar to bradykinin has been reported to bind to APaseP by column affinity chromatography.4
Aminopeptidases are ubiquitously expressed in a variety of human tissues, and are named by the amino acid they preferentially cleave, and at least 11 aminopeptidases (subtypes N, M, A, and L) besides APaseP have been reported.8 Aminopeptidase N (APaseN) is a ubiquitous (100 kDa) peptidase found mainly on the epithelial cells of the small intestine and kidneys, monocytes, and granulocytes, and in endothelial cells in a variety of tissues.9,10 APaseN, also known as CD13, plays an important role in angiogenesis and peptide motifs of APaseN conjugated to fluorescent photoprobes, cytotoxic agents, and Tc99-radiolabels have been reported.9–11 Aminopeptidase M (APaseM), a 280 kDa peptidase widely distributed in various human tissues, such as the kidneys, intestines, and liver,12 has been pharmacologically linked to cell growth, angiogenesis, and tumor progression, and has been used to determine pharmacological markers of cancer.13 Alanine aminopeptidase (236 kDa) found in several tissues is used as a marker of kidney function, and can help detect kidney disorders following urinalysis.14 Fluorescent probes for 326 kDa leucine aminopeptidase have been synthesized for a variety of biological and pathological studies, in addition to fullerene C60-peptide probes for immunomodulatory activity.15,16
The expression level of APaseP is 100-fold increased in breast tissue, and PEGA has been shown to bind the vasculature of hyperplastic and malignant lesions in transgenic breast cancer mice via phage display.4 Therefore, to directly target breast tissue we focused our conjugate design efforts on APaseP, along with the addition of a transcription-activating factor (Tat) derived from HIV-1 for cell penetration of conjugate. Since the interaction between PEGA and APaseP may be limited to the cell’s surface, an improved drug delivery conjugate may be realized with cellular internalization.4
Tat is a transcription-activating factor derived from HIV-1, and is essential for viral gene expression.17 We reasoned that conjugation of the cell-penetrating sequence Tat (amino acids 49–57) to the PEGA peptide would increase cell penetration of the conjugate into breast cells.18–22 Tat, unlike other cell penetrating peptides (CPP), does not induce any cellular leakage at the cell membrane and cellular contents at concentrations up to 20 uM.18 Cellular leakage would compromise retention at the tissue site, a critical component of the success of a drug delivery conjugate. Additionally, cell penetrating peptide toxicity is another biological obstacle considering that any leakage from the tissue site may allow for deleterious cellular effects. In vitro studies have shown that CPPs can exert toxic effects on membranes of cells or organelles by disrupting their membrane, or by interaction with cellular components; for example, CPPs binding to polyanions like nucleic acids or heparin proteoglycan.18,19 Therefore, using more efficient cell penetrating peptides like model amphipathic peptide (MAP) and transportan that cause cellular leakage was not ideal.18 MAP and transportan have been shown to disrupt the cellular membrane, organelles, and interact with cellular components causing toxicity, while Tat has been shown to be non-cytotoxic.18 Another cell penetrating peptide considered was pVEC, an amphipathic 18-amino-acid peptide derived from murine vascular endothelial-cadherin protein.23 Although pVEC had shown efficacy in the delivery of nitrogen mustard, chlorambucil, to breast-derived cells when coupled to PEGA, pVEC itself has been shown to exhibit hemolytic properties.5,24 In addition, Tat has higher protein transduction efficiency and fluorescent readout when compared to pVEC.24,25 Tat has also been successfully coupled to nanoparticles to enhance their cellular penetration; and doxorubicin, an anti-neoplastic drug, via an N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer with a fluorescein label and effectively allowed for cell entry.26 Hence, we hypothesized that Tat would be an effective CPP for the breast tissue specific drug delivery conjugate. Ultimately, specificity and penetration at the tissue site were both addressed in the design of the drug delivery conjugate, by PEGA and Tat, respectively.
Another important component in determining the delivery of the conjugate in vitro and in vivo systems is the visualization of the conjugate in a biological system. To address tumor delivery, an imaging agent needs to provide a clear and distinguishable signal in a biological environment. We chose tetramethylrhodamine, TAMRA, because it is excited in the visible range at 550 nm, allowing for in vivo visualization of tissue penetration in addition to visualization in cells.27 Other fluorophores investigated were traditional fluorescein FITC, and dansyl fluorophores. Both stable in solid phase, the FITC and dansyl fluorophore were not ideal for in vivo imaging due to their lack of depth penetration. In addition, their fluorescence signal is within the range of autofluorescence from 300 to 500 nm rendering signal interpretation problematic.27 For in vitro studies TAMRA is potentially ideal for live cell and in vivo imaging because of its stability, and its detection would not be affected by autofluorescence.
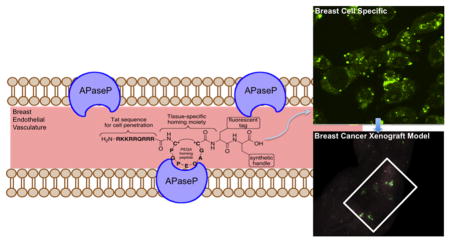
In order to test this hypothesis, we designed a breast tissue specific drug delivery conjugate with the PEGA, TAT, and TAMRA moieties, including a modified C-terminal synthetic handle with an amino acid suitable for bioconjugation (Figure 1A). As a proof of principle, each component of the conjugate was synthesized in order to test the specificity, selectivity, and retention of the conjugate in human carcinoma cell lines and tissue.
Figure 1.

Design and structures of breast tissue specific conjugates. (A) Composed of four components as follows: (1) TAT, cell penetrating sequence; (2) tissue specific homing moiety; (3) fluorescent tag; and (4) synthetic handle. (B) Solid phase peptide synthesis was used to synthesize the drug delivery conjugates with a tetramethylrhodamine fluorophore functionalized lysine. C* refers to cysteine amino acid.
RESULTS
Synthesis of the Breast Tissue Specific Conjugate and Fluorescently Labeled Amino Acids
We utilized solid phase peptide synthesis (SPPS) with Fmoc-protected amino acids (Figure 1B). These amino acids were preferred as they undergo coupling and deprotection under milder reaction conditions needed for facile synthesis. A Wang resin was used for its low loading capability when the sequences were synthesized individually.28 The addition of the fluorophore-functionalized lysine was easily introduced into the peptide synthesis. Upon completion of the desired peptide sequence a cleavage cocktail of TFA/TIS/H2O was added to the final peptide on Wang resin. Cyclization of the S–S bond is accomplished by exposure to air during the acid induced cleavage of the peptide.28 After 3 h the beads were filtered and the solution dried and collected. Following a cold ether/water extraction, the aqueous phase was collected, stored at −20 °C, and then lyophilized. HPLC-MS analysis confirmed synthesis of the final product in high yield (88–91%). Synthetic handles used in our studies were a C-terminal carboxylic acid or proparglycine. Both groups allow for varying synthetic methods for conjugation of a therapeutic; particularly, 1B, 2B, and 3B can be used as a substrate for [3+ 2] cycloaddition, also known as click chemistry.29,30
Tissue Specific Expression of APaseP in Breast Cancer Derived Cell Lines
To achieve an effective and specific drug delivery conjugate utilizing the PEGA binding partner, APaseP, differential expression of APaseP in particular tissues is essential. For an effective drug delivery system, differential expression of APaseP would be critical. In order to probe this protein’s expression in various tissue-specific cancer-derived cell lines, immunocytochemical staining of APaseP was performed on fixed breast cancer cells (MCF-7 and MDA-MB-231), prostate cancer cells (PC-3), and lung cancer cells (A-549). Confocal images of treated anti-APaseP mAb demonstrated the differential expression of APaseP in MCF-7 and MDA-MB-231 cells versus PC-3 cells and A-549 cells (Figure 2). The breast cancer cell lines showed robust staining for APaseP, indicating the tissue-specific binding properties of PEGA to breast-derived tissues.
Figure 2.

Immunocytostaining of the PEGA binding partner, Aminopeptidase P, in breast, lung, and prostate cancer cell lines. Fluorescent microscopy image of MCF-7, MDA-MB-231, PC-3, and A-549 cells upon incubation with an anti-APaseP mouse mAb followed by Alexafluor 488 labeled anti-IgG secondary antibody: (A) fluorescent image of cells; (B) bright field image; and (C) DAPI stain. The green color in panel A suggests the presence of Aminopeptidase P in breast-derived cancer cell lines.
Binding Affinity of Breast Tissue Drug Delivery Conjugate for Aminopeptidase P (APaseP)
A Biacore assay was used to determine the binding affinity of APaseP for the breast tissue specific drug delivery conjugate. During Biacore analysis, APaseP was immobilized directly onto the flow cell (FC).27,31,32 The breast tissue specific drug delivery conjugate was run in the mobile phase in HBS-P (10 mM Hepes/150 mM NaCl/0.05% P-20). These analytes were injected over the FC at varying concentrations. Assuming a (1:1) analyte binding model, the breast tissue specific drug delivery conjugate showed binding affinity to APaseP, KD = 723 ± 3 nM. Bradykinin, a physiological ligand to APaseP, bound APaseP at KD = 15.3 ± 8 μM. The observed KD demonstrated nanomolar binding between construct 3B and APaseP (Figure 1B and Table 1).
Table 1.
Binding Affinity of the Breast Tissue-Specific Conjugate (3B) for APasePa
| Peptide Conjugate | KD (M) |
|---|---|
| (3B) | 723 ± 3 nM |
| Bradykinin | 15.3 ± 8 μM |
| (1B) | 430 ± 5 nM |
| Brain Homing Peptide | NB |
Linked reactions were performed with the breast tissue specific conjugate injected over a C1-sensor chip with immobilized XPNEP1, APaseP human recombinant protein. A 0–20 μM titration of the breast tissue specific conjugate (3B) in solution was injected over the prepared biosensor surface at a flow rate of 100 μL/min. The KD results are presented as mean ± SEM. NB = no binding
Cytotoxicity of Breast Tissue-Specific Conjugate in the MCF-7 Breast Cancer Derived Cell Line
To determine the toxicity of the conjugate, the PEGA peptide and Tat peptide were treated with serial dilutions of each for 96 h, and cell viability was measured by MTT assay. Due to the known toxicity of other cell penetrating peptides, and their effective ability to disrupt cellular membranes, it was critical to determine the toxicity of the breast tissue specific conjugate for potential therapeutic applications. From the cellular viability data collected neither the PEGA nor Tat TAMRA peptide was toxic to cells. The conjugate itself did not decrease cell viability when compared to control up to concentrations of 25 μM (Figure 3). It had been previously shown that the Tat peptide does not induce membrane leakage up to a concentrations of 20 uM, suggesting that the peptide building blocks and conjugate showed no toxicity to cells.18,33
Figure 3.

Cell viability of breast tissue-specific conjugate (3B), PEGA TAMRA, and TAT TAMRA conjugate in MCF-7 cells. Cells were exposed to different concentrations of each peptide conjugate or media alone for 96 h. Survival percentages in the treated cells were normalized to values from untreated cells. The results are presented as mean ± SEM. Data are analyzed using Excel and GraphPad Prism software.
Fluorescence Detection and Quantification of the Breast Tissue Specific Drug Conjugate
In order to determine the selectivity of the breast tissue specific drug conjugate for breast-derived cells, fluorescence detection of the conjugate was tested using confocal microscopy. The conjugate and its constructs were treated at 10 μM in media for 1 h in carcinoma cell lines. The cells were washed and imaged live under confocal microscopy. Treatment of breast, lung, and prostate cancer cell lines with the conjugate demonstrated selectivity of breast tissue over lung and prostate as shown in Figure 4. The Tat-TAMRA labeled peptide entered all cell types, whereas the conjugate only entered breast-derived cells.
Figure 4.

Fluorescence detection and quantification of the breast tissue specific drug conjugate. Fluorescence detection of breast tissue specific conjugate and peptide conjugates in MCF-7 (A), MDA-MB-231 (B), A-549 (C), and PC-3 (D) cells by confocal laser scanning microscopy. Cells were incubated with 10 μM breast tissue specific conjugate for 60 min followed by imaging in fluorescence mode (green; left column) and bright field (right column). (E) Corrected fluorescence intensity of breast tissue specific conjugate and peptide conjugates in MDA-MB-231, A-549, and PC-3 cells by confocal laser scanning microscopy. Corrected fluorescence intensity is the subtraction of background signal from fluorescence intensity. The results are presented as mean ± SEM. Data are analyzed using Excel and ImageJ software. *P ≤ 0.05, ** P ≤ 0.01. (F) Flow cytometric histogram of the TAMRA labeled breast tissue specific conjugate does response in MCF-7 cells. Cells were incubated 0, 0.1 (yellow), 1 (green), and 10 (purple) μM of the conjugate for 60 min and analyzed by flow cytometry.
As shown in Figure 4, the breast tissue specific conjugate conferred tissue selectivity that overcame Tat’s nonspecific cell penetration. Quantification of the fluorescent intensity revealed similar results that the breast tissue specific conjugate was more selective for breast cells than lung or prostate (Figure 4D). To explore the specificity of the PEGA sequence for breast tissue, two PEGA modified sequences were designed. PEGA modified 1 replaced flexible alanine and glycine, with proline, and glutamic acid, respectively (Figure 1B). The PEGA modified 1 peptide sequence was highly selective for the breast-derived cell type, significantly more selective for breast over prostate and lung. The PEGA modified 1 modification potentially made the breast homing sequence more rigid due to the increased torsional strain of proline, which in turn could have induced a conserved 3D structure. Furthermore, this could result in the PEGA modified 1 sequence interacting with the APaseP protein more effectively than flexible PEGA. The PEGA modified 2 sequence introduced a proline and glutamine amino acid in place of alanine and glycine, in addition to another glutamine amino acid instead of glutamic acid. The PEGA modified 2 sequence resulted in the loss of breast tissue selectivity. The distribution of the PEGA modified 2 sequence fluorescence, and fluorescence intensity was similar to that of the Tat-TAMRA peptide. This would be expected if the PEGA moiety no longer showed tissue specific properties, as seen in the PEGA modified 2 sequence.
In other confocal studies the breast tissue specific drug delivery conjugate was stable for over 96 h post treatment in cells. Cells showed no signs of stress after treatment. In addition, the PEGA peptide alone did not enter breast-derived cells (Figure S1). This result suggests that the PEGA-APaseP interaction may be limited to the cell surface and PEGA alone does not allow for cellular entry. Z-stacking data was also collected in breast and lung cancer cell lines post treatment of the breast tissue specific conjugate. In breast-derived cell lines, fluorescence was detected at each micron section of cells, as opposed to lung cells where no fluorescence was detected in Z-stack studies. This further validated the cell specific nature of the conjugate. It also revealed that the distribution of the conjugate was not limited to the nucleus or cytoplasm, but was found in both compartments. Flow cytometry studies also showed a dose dependent increase in fluorescence in breast-derived cells with the increasing concentrations of the 3B conjugate. In an effort to determine the specificity and selectivity of 3B, the experiments were performed on live cells. Careful consideration was taken when performing these studies, because fixation has been known to distort localization studies and give false positive CPP localization in the cytosol and nucleus.34 Determining the correct localization and distribution of the conjugate is critical in determining its efficacy as a vehicle for tissue specific drug delivery. Overall, the breast tissue specific conjugate distributed in a tissue specific, cell selective, concentration dependent manner in breast-derived adenocarcinoma cell lines.
In Vivo Localization of the Breast Tissue Specific in a Breast Cancer Xenograft Model
An in vivo model was established by implanting MCF-7 expressing human breast adenocarcinoma cell line in nude mice with estrogen supplementation.35 In an effort to form subcutaneous tumors, a MCF-7 cell cancer xenograft was implanted on the left side of the animal. This particular cell line was chosen because previous in vitro studies showed significant localization of the conjugate in MCF-7 breast-derived cell line. To determine the distribution of the conjugate in vivo, fluorescent whole animal images were collected upon intravenous administration of the conjugate in phosphate buffered saline (PBS). Fluorescence signal was detected in the tumor margins, less than 5 min after administration (data not shown). After 10 min there was noticeable distribution in the tumor and mammary tissue (Figure 5B). In addition, fluorescence was detected in the bladder of the animal (Figure 5C), suggesting that urinary excretion may be the main route of elimination of the conjugate.
Figure 5.

In vivo fluorescence localization of the breast tissue specific conjugate in MCF-7 tumor xenograft model. (A) Nude mice were injected intravenously with the breast tissue specific conjugate 3B (10 mg/kg), and were examined using a MAESTRO in vivo fluorescence imaging system. (B) Fluorescence was detected in the MCF-7 tumor xenograft immediately after the administration of 3B. (C) Main route of elimination of the nontoxic drug delivery conjugate may be through urinary excretion.
DISCUSSION
A breast tissue specific conjugate was designed to target mammary tissue. A conjugate of this nature may be useful for the tissue specific delivery of therapeutic agents. The designed conjugate and its modified versions were evaluated as possible Aminopeptidase P targeting, fluorescently labeled drug delivery agents for breast tissue. From our in vitro studies and in vivo MCF-7 breast adenocarcimona cell model, the TAMRA labeled conjugate allowed for the best visualization of cellular entry into breast-derived cells, and detection of the tumor xenograft and mammary tissue in mouse models. By using a TAMRA fluorophore, fluorescence detection and in vivo depth penetration of the conjugate was demonstrated. In an effort to home the conjugate to breast issue, the tumor homing peptide PEGA was shown to distribute to breast-derived cell lines and tissues with detectable expression of its binding partner, Aminopeptidase P, in breast-derived tissues. The designed drug delivery conjugate was shown to have a binding affinity greater than its physiological ligand bradykinin. Further elucidation of its drug delivery potential, by in vitro and in vivo fluorescence studies and quantification further demonstrated its feasibility as a drug delivery vehicle. As a proof of concept, each component of the conjugate was synthesized in order to test the specificity, selectivity, and retention of the conjugate in breast-derived cell lines. The coupling of a cell penetrating peptide, Tat, to the PEGA sequence, did not compromise specificity or selectivity for the breast. In addition, toxicity did not result from the cell penetrating sequence, and no indicators of cellular leakage were determined from our studies.6 From our work we found the construct to be stable up to 96 h, and nontoxic for up to 96 h. The nontoxic nature of the conjugate may allow the mechanism of cytotoxicity to arise from the therapeutic agent alone. The successful design of the conjugate also allows for a variety of synthetic handles to be coupled to the C-terminus of the amino acid, allowing for a variety of therapeutic agents to be conjugated involving different chemical couplings.13,14 Solid phase peptide synthesis also proved to be very effective means of synthesis, and addition of any modified amino acids could be easily introduced into the synthetic method in high yields. All the peptides and conjugates synthesized were water-soluble and highly stable in physiological saline. Possible limitations from our studies reside in the breast cancer xenograft model. It is possible that the breast adenocarcinoma model was not adequate to visualize the full potential of the tissue specific delivery. Lack of significant mammary pad tissue in nude mice may have limited the efficiency of distribution of the breast tissue specific drug delivery conjugate in vivo. Ultimately, further studies are necessary to determine the efficacy of a therapeutic conjugate 3B using the design proposed. Nonetheless, the purpose of this study was to evaluate the use of an APaseP targeting moiety as a homing agent to breast-derived tissues and further distribute in breast-derived cells, elucidating its binding kinetics and tumor visualization properties, which were accomplished within the scope of this paper. This proof-of-concept study showed that Aminopeptidase P is a promising target for breast tissue specific drug delivery.
MATERIALS AND METHODS
All studies were performed at Georgetown University Center for Drug Discovery (Washington, DC). All chemical reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. Peptides 1–5 were also purchased from Biomatik (Wilmington, DE).
Solid Phase Peptide Synthesis
The dry Wang resin (0.35 mmol/g) was placed in an 50 mL reaction vessel. The reaction vessel was then filled with 30 mL of DCM until all the resin beads were immersed. After 30 min the DCM was removed by filtration under vacuum. The reaction vessel was then filled with DMF (25 mL). The beads were allowed to soak for 1 min, and then manually shaken for 30 s. The solvent was then removed by filtration. The screw cap and edges of the reaction vessel were also rinsed with DMF (25 mL). The DMF washing and rinsing steps were repeated three times. After properly conditioning and swelling the resin, the resin was treated to a 20% piperidine solution in DMF (30 mL) and shaken for 5 min on a shaker table. After 5 min the solution was eluted and the beads were washed with 15 mL of DMF (10×). A ninhydrin test was performed on a small aliquot of beads (~5 beads). After successfully completing Fmoc deprotection, the addition of each N-α Fmoc protected amino acid was prepared as follows: (1) Reaction mixture of 3.5 equiv of amino acid as a powder, 3.4 equiv of PyBOP, and 3.4 equiv of HOBt were added to the reaction vessel; (2) the reaction vessel was filled with DMF (at least 2/3 of volume); (3) 7.0 equiv of DIEA was then added to the vessel; (4) the reaction vessel was then capped and placed on a shaker table and allowed to react for 75 min; (5) upon completion, the solvent was eluted from the vessel; (6) the beads were then rinsed with 20 mL of DMF (5×), 20 mL of DCM (3×), 20 mL of DMF (4×); (7) a small aliquot of beads was used to perform a Kaiser test (5–10 beads), and upon successful amino acid coupling a 20% piperidine in DMF solution (30 mL) was added to the beads for 15 min; (8) the beads were then rinsed with 20 mL of DMF (10×), and a ninhydrin test was performed again on a small aliquot of beads (5–10 beads). After successful deprotection of the Fmoc group, steps 1–8 were repeated for each addition of amino acid in the desired sequence.28 The beads were taken up in 30 mL of methanol and the peptide was cyclized by air oxidation in solution. The beads were then dried over vacuum overnight at room temperature. A cleavage cocktail was prepared (TFA 9.5 mL/TIS 0.25 mL/H2O 0.25 mL) and added to the final peptide sequence on resin in the reaction vessel. The vessel was swirled every 30 min for 5 min intervals, and the cocktail was allowed to react with the beads for 3 h. The beads were then filtered off, and the reaction solution was collected in a round-bottom flask. The solution was then evaporated to one-fourth of its original volume and allowed to stir overnight to promote cyclization of the peptide by air oxidation. Cold ether (25 mL) was then added to the solution and an aqueous extraction (30 mL) was performed. The aqueous phase was then collected and frozen in −20 °C. The peptide was then lyophilized, and product was collected. To ensure cyclization of the peptide, some batches were taken up in 0.1 M solution of NH4HCO3 in water (5–10 mL) to allow for disulfide formation between cysteine residues. After stirring overnight, the reaction mixture was concentrated and cyclic product was isolated via HPLC. Purity was confirmed by HPLC (VYDAC protein and peptide 5 μm C18 100 × 2.1 mm column; detection 220 nm; LC-MS positive mode). Solvents 0.1% TFA in 100% acetonitrile (A) and 0.1% TFA in 100% water (B) were used. The flow rate was 1.0 mL/min, 5–10 μL injection, and 0–100% gradient over 25 min, and then 100% for 5 min was used. The product was analyzed with LC-MS. The expected MH+/ found m/z were as follows: (1A) 2093.41/[M+2H]2+ = 1047.29; 91% yield; (2A) 1541.69/ [M + H]2+ = 783.54; 89% yield; (3A) 2863.27/ [M+3H]3+ = 954.84; 88% yield.
Synthesis of TAMRA Labeled Lysine
A solution of TAMRA succinimide ester (0.08 mmol; 0.050 g) in 30 mL of DMF was added to Fmoc-lysine (1 equiv) in a round-bottom flask and placed in an ice bath. DIEA (2 equiv) was immediately added to the reaction. The mixture was allowed to react for 30 min, and then removed from the ice bath. The solution was then allowed to stir for 2 h and warmed to room temperature. HCl (2 M, 15 mL) was added and the reaction was cooled to 4 °C and allowed to react overnight. The solution was then taken up in 40 mL of ethyl acetate, and washed with H2O (5 × 20 mL) and LiCl(aq) (3 × 20 mL), then dried with NaSO4. The solvent was removed under reduced pressure and then purified by column chromatography (1:1 ethyl acetate: hexane) to give a product in 52% yield (0.035 g). 1H NMR (400 MHz; CDCl3): δ = 1.49–1.51 (6H, m, 3×CH2), 1.53–1.80 (6H, m, 3×CH2), 1.87 (6H, s, 2×CH3), 1.92 (2H, q, CH2), 2.74 (6H, s, 2×CH3), 3.14 (2H, t, CH2), 3.19 (2H, t, CH2), 4.26 (2H, m, 2×CH), 4.28 (2H, d, CH2), 7.2 (1H, s, CH), 7.99 (3H, m, 3×CH), 8.09 (2H, m, 2×CH), 8.27, (3H, m, 3×CH).
Tumor Cell Culture
The carcinoma cell lines MCF-7, MDA-MD-231, A-549, and PC-3 were obtained from the Tissue Culture Shared Resource (Georgetown University, Lombardi Cancer Center, Washington, DC). The cells were grown in Dulbecco’s modified Eagle medium (DMEM). All media was supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin–glutamine. Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Immunohistochemistry Assay
Immunofluorescence protocols from Cell Signaling and ABCam were followed in this experiment. The MCF-7, MDA-MD-231, A-549, and PC-3 carcinoma cell lines were grown in six-well plates containing glass slides to 40% confluence. The media was removed and the cells were covered with a depth of 2–3 mm with 4% formaldehyde diluted in warm PBS. The cells were allowed to fix for 15 min at room temperature. The fixative was aspirated, and the cells were rinsed three times with PBS for 5 min each. The specimens were blocked with TBST (Tris-buffered saline/0.3% Tween-20) containing 20% FBS for 60 min and the blocking solution was aspirated. The specimens were incubated with a 1/1000 dilution of anti-APaseP antibody (Santa Cruz Biotechnology), and incubated overnight at 4 °C. The specimens were rinsed three times in 1× PBS for 5 min each. The fluorochrome-conjugated secondary antibody, Alexafluor 488 labeled anti-IgG secondary, was diluted in antibody dilution buffer and treated for 1–2 h at room temperature in the dark. The specimens were rinsed three times in 1× PBS for 5 min each. The coverslip was mounted and cured overnight at room temperature. The slides were imaged on a Zeiss LSM 510 confocal microscope 1.4-numerical aperture plan-apochromat oil-immersion objective at 63× magnification. Fluorescent and differential interference contrast (DIC) images of the specimens were obtained. The fluorescence images were overlaid with DIC images in order to locate the APaseP signal.
Binding Affinity Assay
Human recombinant XPNPEP1, APaseP (Abnova, Tapei City, Taiwan), was immobilized on a C1 (28 RU) sensor chip. Immobilization was performed by amine coupling of XPNPEP1, in pH 4.0 and 10% acetate buffer. Samples were prepared in HBS-P (10 mM Hepes + 150 mM NaCl + 0.05% P-20). All kinetic measurements were conducted at 25 °C at a flow rate of 100 μL/min with a contact time of 60 s and dissociation time of 120 s. Double-referencing was performed to account for bulk effects caused by changes in the buffer composition or nonspecific binding.31,32 Data were evaluated with BIAevaluation software, and the best fit (lowest χ2) was obtained using a 1:1 binding model.31,32 The sensorgram was fitted globally over the association and dissociation phases. Equilibrium dissociation constants (affinity) were calculated from the rate constants (KD = koff/kon).
Cell Viability Assay
The MCF-7 breast adenocarcinoma cells were maintained in a humidified CO2 (5%) incubator at 37 °C in media. Compounds 1B, 2B, and 3B were freshly dissolved in media at 25 μM and subsequently serially diluted in prewarmed culture medium. Methylthisazolyldiphenyltetrazolium bromide (MTT) was used as described by the manufacturer. Cells were plated out in 96-well plates at a density of 7850 cells/well followed by addition of compounds the next day. Cell culture medium was replaced with 50 μL prewarmed medium containing the different concentrations of the compounds from 500 nm to 25 μM.33,36 After 96 h incubation, cell proliferation was assessed by MTT assay. Briefly, MTT was dissolved in prewarmed medium at 5 mg/mL, and 50 μL was added to each well. Following an incubation period of 120 min, the medium was gently aspirated. The formed formazan crystals were dissolved in 100 μL DMSO, and the absorbance was measured with a plate reader (BMG labtech) at 570 nm.
Fluorescence Imaging Study
MCF-7, MB-MDA-231, A-549, and PC-3 carcinoma cells were cultured in media on a fluorodish with glass bottom (World Precision Instruments, Sarasota, FL) and incubated in a humidified atmosphere of 5% CO2 at 37 °C overnight. After removing the media, the cells were incubated with 10 μM of compounds 1A, 2A, 3A, 4, and 5 in media for 60 min and then washed with media (3×). The cells were then bathed in fresh media, and microscopic examination was conducted on a Zeiss LSM 510 Meta Confocal Microscope (Carl Zeiss, Inc., Jena, German). The microscope was set to a TAMRA filter with an excitation wavelength of 565–615 nm and an emission wavelength of LP560 nm.37–39 ImageJ software was used for image processing and analysis.40–42 The image magnification was 63×. The pseudocolor of TAMRA is green. Z-stack analysis was performed on MCF-7 and A-549 cells after 10 μM treatment with compound 3B and sections of 0.1 μm were acquired in a Z-stack from top to bottom of the cell.37
Flow Cytometry
Cellular studies were acquired using a BD Biosciences Fortessa cell analyzer (San Jose, CA). A 488 nm wavelength laser was used to excite MCF-7 cells treated with titrated concentrations from 0, 0.1, 1, and 10 μM of 3B in media for 60 min. After dose dependent treatment of the cells, analysis was performed to determine the extent of fluorescent cellular uptake of 3B. The TAMRA fluorophore was excited using an argon ion laser (15 mW, 488 nm) and probed with fluorescence detection using a 550 nm long-pass filter.38,43
Tumor Xenografts
The animal procedures were performed according to a protocol approved by the Animal Care and Use Committee (GUACUC) at Georgetown University Medical Center. Female athymic nude mice (nu/nu), obtained from National Cancer Institute (Bethesda, MD) at 4 to 6 weeks of age, were subcutaneously inoculated on the left side of the animal with 2.5 × 106 MCF-7 cells suspended in a mixture of 50 μL of PBS and 50 μL of matrixgel basement membrane (BD Biosciences, San Jose, CA). When the tumor implants reached ~80 mm3, the tumor-bearing mice were subjected to in vivo studies.35,44,45 After 1 week of acclimatization while being fed a basal diet, mice used in the study were implanted with a pellet of 17β-estradiol subcutaneously (1.7 mg over 60 day release, and producing a 3–4 nM E2 blood level) at least 2 days before tumor cell inoculation.45,46
In Vivo Tumor Imaging
In vivo fluorescence imaging was performed with a Maestro 2.6 In Vivo Fluorescent Imaging System (Woburn, MA). A M-MSI-FLTR-GREEN filter was used, with excitation 503 to 555 nm, and emission filter set to 580 nm long-pass. The acquisition settings were set to 550 to 800 in 10 nm steps. The pseudocolor image represents the spatial distribution of photon counts within the animal. Background fluorescence was measured and subtracted by setting up a background measurement. Images were acquired and analyzed using Cri software (Cambridge Research & Instrumentation Inc., Woburn, MA). Mice bearing MCF-7 tumor xenograft were anesthesized by inhalation of isoflurane (1–3%) in oxygen (Abbott Laboratories, Chicago, IL), as generated by a non-rebreathing nose-cone system with an exhaust evacuator and F-Air canister built into the Maestro imaging system. Mice were then injected via the tail vein with 10 mg/kg of the breast tissue specific drug delivery conjugate in PBS. One mouse was not injected and was used as a blank control.37 Images were obtained immediately after the injection of breast tissue specific conjugate. The mice were imaged for 20 min after administration of the peptide. The dorsal and ventral sides of each mouse were imaged. The tumor area, liver, and bladder area were designated as regions of interest.37
Acknowledgments
This work was supported by a DOD Breast Cancer Research Program Concept Award to M. A. P., Georgetown University Medical Center Tumor Biology fellowship to A.C., Lombardi Comprehensive Cancer Center grant (P30CA051008), and the Georgetown University Center of Drug Discovery. We would like to thank Alexandra Gadiano, Paige Morgan, Andy Ji, and Tabari Baker Ph.D. for their support and commitment to science. Peter Johnson from the Microscopy and Imagery Shared Resource, and Aykut Uren M.D., and Yasemin Saygideger Kont M.D. from the Biacore Molecular Interaction Service (BMISR) for technical assistance and discussion.
Footnotes
Notes
The authors declare the following competing financial interest(s): A patent application has been filed by Georgetown University on behalf of the inventors that are listed as authors in this article.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconj-chem.5b00481.
Florescence detection, binding affinity, and spectroscopic characterization (PDF)
References
- 1.Vu T, Claret FX. Trastuzumab: Updated Mechanisms of Action and Resistance in Breast Cancer. Front Oncol. 2012;2:62. doi: 10.3389/fonc.2012.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–33. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rapaka RS. Membranes and Barriers: Targeted Drug Delivery. NIDA Research Monograph. 1995;154:1–254. [PubMed] [Google Scholar]
- 4.Essler M, Ruoslahti E. Molecular specialization of breast vasculature: a breast-homing phage-displayed peptide binds to aminopeptidase P in breast vasculature. Proc Natl Acad Sci U S A. 2002;99:2252–7. doi: 10.1073/pnas.251687998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myrberg H, Zhang L, Mäe M, Langel U. Design of a tumor-homing cell-penetrating peptide. Bioconjugate Chem. 2008;19:70–5. doi: 10.1021/bc0701139. [DOI] [PubMed] [Google Scholar]
- 6.Vanhoof G, et al. Kininase activity in human platelets: Cleavage of the Arg1-Pro2 bond of bradykinin by aminopeptidase P. Biochem Pharmacol. 1992;44:479–487. doi: 10.1016/0006-2952(92)90439-p. [DOI] [PubMed] [Google Scholar]
- 7.Cottrell GS, Hyde RJ, Lim J, Parsons MR, Hooper NM, Turner AJ. Identification of critical residues in the active site of porcine membrane-bound aminopeptidase P. Biochemistry. 2000;39:15129–35. doi: 10.1021/bi0015865. [DOI] [PubMed] [Google Scholar]
- 8.Polaina J, MacCabe AP. Industrial enzymes: structure, function and applications. Vol. 45. Springer; Dordrecht, The Netherlands: 2007. pp. 245–254. [Google Scholar]
- 9.Hahnenkamp A, Schafers M, Bremer C, Holtke C. Design and synthesis of small-molecule fluorescent photoprobes targeted to aminopeptdase N (APN/CD13) for optical imaging of angiogenesis. Bioconjugate Chem. 2013;24:1027–38. doi: 10.1021/bc400074w. [DOI] [PubMed] [Google Scholar]
- 10.Pathuri G, Hedrick AF, Disch BC, Doan JT, Ihnat MA, Awasthi V, Gali H. Synthesis and evaluation of novel Tc-99m labeled probestin conjugates for imaging APN/CD13 expression in vivo. Bioconjugate Chem. 2012;23:115–24. doi: 10.1021/bc200546b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukhopadhyay S, Barnes CM, Haskel A, Short SM, Barnes KR, Lippard SJ. Conjugated platinum(IV)-peptide complexes for targeting angiogenic tumor vasculature. Bioconjugate Chem. 2008;19:39–49. doi: 10.1021/bc070031k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakanishi M, Moriyama A, Narita Y, Sasaki M. Aminopeptidase-M from Human-Liver. 1 Solubilization, Purification, and Some Properties of the Enzyme. J Biochem. 1989;106:818–825. doi: 10.1093/oxfordjournals.jbchem.a122937. [DOI] [PubMed] [Google Scholar]
- 13.Warder SE, Tucker LA, McLoughlin SM, Strelitzer TJ, Meuth JL, Zhang Q, Sheppard GS, Richardson PL, Lesniewski R, et al. Discovery, identification, and characterization of candidate pharmacodynamic markers of methionine aminopeptidase-2 inhibition. J Proteome Res. 2008;7:4807–20. doi: 10.1021/pr800388p. [DOI] [PubMed] [Google Scholar]
- 14.Kao YJ, Starnes WL, Behal FJ. Human kidney alanine aminopeptidase: physical and kinetic properties of a sialic acid containing glycoprotein. Biochemistry. 1978;17:2990–4. doi: 10.1021/bi00608a008. [DOI] [PubMed] [Google Scholar]
- 15.Sakabe M, Asanuma D, Kamiya M, Iwatate RJ, Hanaoka K, Terai T, Nagano T, Urano Y. Rational design of highly sensitive fluorescence probes for protease and glycosidase based on precisely controlled spirocyclization. J Am Chem Soc. 2013;135:409–14. doi: 10.1021/ja309688m. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y, Zhu J, Xiang K, Li Y, Sun R, Ma J, Sun H, Liu Y. Synthesis and immunomodulatory activity of [60]fullerene-tuftsin conjugates. Biomaterials. 2011;32:9940–9. doi: 10.1016/j.biomaterials.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 17.Schoenen FJ, Whipple D, Baillargeon P, Brown CL, Chase P, Ferguson J, Fernandez-Vega V, Hodder P, Mathew R, Neuenswander B, Porubsky P, Rogers S, Skinner-Adams T, Sosa M, Spicer T, To J, Tower NA, Trenholme KR, Wang J, Weiner WS, Aube J, Rosen H, White EL, Gardiner DL, Dalton JP. Inhibitors of the Plasmodium falciparum M17 Leucine Aminopeptidase. Probe Reports from the NIH Molecular Libraries Program. 2010:1–13. [PubMed] [Google Scholar]
- 18.Zorko M, Langel U. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv Drug Delivery Rev. 2005;57:529–545. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 19.Prochiantz A. Messenger proteins: homeoproteins, TAT and others. Curr Opin Cell Biol. 2000;12:400–406. doi: 10.1016/s0955-0674(00)00108-3. [DOI] [PubMed] [Google Scholar]
- 20.Joliot AH, Triller A, Volovitch M, Pernelle C, Prochiantz A. alpha-2,8-Polysialic acid is the neuronal surface receptor of antennapedia homeobox peptide. New Biol. 1991;3:1121–34. [PubMed] [Google Scholar]
- 21.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–50. [PubMed] [Google Scholar]
- 22.Persson D, Isakson P, Goks M, et al. Uptake of analogs of penetratin, Tat(48–60) and oligoarginine in live cells. Biochem Biophys Res Commun. 2003;307:100–107. doi: 10.1016/s0006-291x(03)01135-5. [DOI] [PubMed] [Google Scholar]
- 23.Akdag IO, Ozkirimli E. The Uptake Mechanism of the Cell-Penetrating pVEC Peptide. J Chem. 2013;2013:1–9. [Google Scholar]
- 24.Saar K, Lindgren M, Hansen M, Eiriksdottir E, Jiang Y, Rosenthal-Aizman K, Sassian M, Langel U. Cell-penetrating peptides: a comparative membrane toxicity study. Anal Biochem. 2005;345:55–65. doi: 10.1016/j.ab.2005.07.033. [DOI] [PubMed] [Google Scholar]
- 25.Säälik P, Elmquist A, Hansen M, Padari K, Saar K, Viht K, Langel Ü, Pooga M. Protein cargo delivery properties of cell-penetrating peptides. A comparative study. Bioconjugate Chem. 2004;15:1246–1253. doi: 10.1021/bc049938y. [DOI] [PubMed] [Google Scholar]
- 26.Vives E. Present and future of cell-penetrating peptide mediated delivery systems: “Is the Trojan horse too wild to go only to Troy? J Controlled Release. 2005;109:77–85. doi: 10.1016/j.jconrel.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 27.Liu C, Chang L, Wang H, Bai J, Ren W, Li Z. Upconversion Nanophosphor: An efficient phosphopeptides- recognizing matrix and luminescence resonance energy transfer donor for robust detection of protein kinase activity. Anal Chem. 2014;86:6095–6102. doi: 10.1021/ac501247t. [DOI] [PubMed] [Google Scholar]
- 28.Amblard M, Fehrentz JA, Martinez J, Subra G. Methods and protocols of modern solid phase peptide synthesis. Mol Biotechnol. 2006;33:239–54. doi: 10.1385/MB:33:3:239. [DOI] [PubMed] [Google Scholar]
- 29.Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. A comparative study of bioorthogonal reactions with azides. ACS Chem Biol. 2006;1:644–648. doi: 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]
- 30.Shao C, Wang X, Xu J, Zhao J, Zhang Q, Hu Y. Carboxylic acid-promoted copper(I)-catalyzed azide-alkyne cycloaddition. J Org Chem. 2010;75:7002–5. doi: 10.1021/jo101495k. [DOI] [PubMed] [Google Scholar]
- 31.Myszka DG. Improving biosensor analysis. J Mol Recognit. 1999;12:279–284. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 32.Khalili H, Godwin A, Choi J-w, Lever R, Brocchini S. Comparative binding of disulfide-bridged PEG-Fabs. Bioconjugate Chem. 2012;23:2262–2277. doi: 10.1021/bc300372r. [DOI] [PubMed] [Google Scholar]
- 33.Brakel RV, Vulders RCM, Bokdam RJ, Grüll H, Robillard MS. A doxorubicin prodrug activated by the Staudinger reaction. Bioconjugate Chem. 2008;19:714–718. doi: 10.1021/bc700394s. [DOI] [PubMed] [Google Scholar]
- 34.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2002;278:585–90. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 35.Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, de Plater L, Guyader C, et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res. 2007;13:3989–98. doi: 10.1158/1078-0432.CCR-07-0078. [DOI] [PubMed] [Google Scholar]
- 36.Noronha S, Bernardo W, Barros AJ, Nakaie CR, Shimuta SI, Silva I, Noronha S. Effects on cell viability and on apoptosis in tumoral (MCF-7) and in normal (MCF10A) epithelial breast cells after human chorionic gonadotropin and derivated-angiotensin peptides treatments. J Cancer Ther. 2013;04:65–69. [Google Scholar]
- 37.Zou P, Xu S, Povoski SP, Wang A, Johnson MA, Martin EW, Subramaniam V, Xu R, Sun D. Near-Infrared fluorescence labeled anti-TAG-72 monoclonal antibodies for tumor imaging in colorectal cancer xenograft mice. Mol Pharmaceutics. 2009;6:428–440. doi: 10.1021/mp9000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christian N, Milone MC, Ranka SS, Li G, Frail PR, Davis KP, Bates FS, Therien MJ, et al. Tat-functionalized near-infrared emissive polymersomes for dendritic cell labeling. Bioconjugate Chem. 2007;18:31–40. doi: 10.1021/bc0601267. [DOI] [PubMed] [Google Scholar]
- 39.Meerovich I, Koshkaryev A, Thekkedath R, Torchilin V. Screening and optimization of ligand conjugates for lysosomal targeting. Bioconjugate Chem. 2011;22:2271–2282. doi: 10.1021/bc200336j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burgess A, Vigneron S, Brioudes E, Labbé JC, Lorca T, Castro A. Loss of human greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A. 2010;107:12564–9. doi: 10.1073/pnas.0914191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18:533–543. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Potapova TA, Sivakumar S, Flynn JN, Li R, Gorbsky GJ. Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited. Mol Biol Cell. 2011;22:1191–1206. doi: 10.1091/mbc.E10-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Summers HD, Brown MR, Holton MD, Tonkin JA, Hondow N, Brown AP, Brydson R, Rees P. Quantification of nanoparticle dose and vesicular inheritance in proliferating cells. ACS Nano. 2013;7:6129–6137. doi: 10.1021/nn4019619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar A, Hao G, Liu L, Ramezani S, Hsieh JT, Öz OK, Sun X. Click-Chemistry strategy for labeling antibodies with copper-64 via a cross-bridged tetraazamacrocyclic chelator scaffold. Bioconjugate Chem. 2015;26:782–9. doi: 10.1021/acs.bioconjchem.5b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhelev Z, Bakalova R, Aoki I, Matsumoto KI, Gadjeva V, Anzai K, Kanno I. Nitroxyl radicals for labeling of conventional therapeutics and noninvasive magnetic resonance imaging of their permeability for blood-brain barrier: relationship between structure, blood clearance, and MRI signal dynamic in the brain. Mol Pharmaceutics. 2009;6:504–12. doi: 10.1021/mp800175k. [DOI] [PubMed] [Google Scholar]
- 46.Chew GL, Huang D, Huo CW, Blick T, Hill P, Cawson J, Frazer H, et al. Dynamic changes in high and low mammographic density human breast tissues maintained in murine tissue engineering chambers during various murine peripartum states and over time. Breast Cancer Res Treat. 2013;140:285–297. doi: 10.1007/s10549-013-2642-7. [DOI] [PubMed] [Google Scholar]