

Abstract
With the recent advent of several generations of targeted DNA nucleases, most recently CRISPR/Cas9, genome editing has become broadly accessible across the biomedical community. Importantly, the capacity of these nucleases to modify specific genomic loci associated with human disease could render new classes of genetic disease, including autosomal dominant or even idiopathic disease, accessible to gene therapy. In parallel, the emergence of adeno-associated virus (AAV) as a clinically important vector raises the clear possibility of integrating these two technologies towards the development of gene editing therapies. Though clear challenges exist, numerous proof of concept studies in preclinical models offer exciting promise for the future of gene therapy.
Keywords: AAV, gene therapy, gene editing, CRISPR/Cas9, zinc-finger nuclease
Introduction
Gene therapy, the treatment of disease via the delivery of genetic material to cells, has enabled incurable diseases to now be considered as therapeutic targets, including both monogenic diseases with well-defined underlying genetic etiology as well as idiopathic diseases with candidate gene targets. Throughout most of its history, the major barrier to gene therapy has been delivery. A major advance has been the development of safe and effective delivery vectors, and the most prominent for in vivo gene therapy have been based on adeno-associated viruses (AAV). Natural AAVs offer reasonable infectivity, a lack of pathogenicity, numerous variants with different tissue tropisms, and negligible genomic integration. As a result, vectors based on AAV have begun to show increasing clinical promise, primarily in studies involving gene augmentation where additional copies of genes are delivered to replace the functionality of null alleles in recessive diseases or to overexpress a potentially therapeutic factor. In particular, AAV has been successful in trials for monogenic recessive disorders including Leber’s congenital amaurosis type 2 (LCA2) [1, 2], hemophilia B [3, 4], spinal muscular atrophy [5], and lipoprotein lipase deficiency [6, 7]. The last of these is the basis for a clinically approved gene therapy product in the European Union, and it is anticipated that a gene therapy for LCA2 may be approved in the US in 2017. In addition, early-stage clinical trials have demonstrated some positive signs in harnessing AAV to treat more complex disorders, such as overexpressing SERC2A in heart failure patients [8] and expressing the VEGF inhibitor sFLT-1 in patients with age-related macular degeneration [9].
While gene therapy is thus gathering increasing momentum, particularly for monogenic disease, a number of disorders are not amenable to gene augmentation therapy. For instance, autosomal dominant genetic diseases require the elimination or modification of the disease-causing allele. In addition, AAV has a limited carrying capacity of <5 kb [10], and mutated genes whose cDNAs exceed this threshold require alternate approaches. Furthermore, while non-integrating vectors like AAV might be safer than integrating vectors, they can insufficient to treat disease requiring gene delivery in actively mitotic cells due to progressive dilution of the delivered extrachromosomal genetic cargo with each cell division [11].
Gene editing technology in the form of targeted nucleases, with the capacity to directly and permanently edit and modify the cellular genome, can potentially address such challenges. For example, these nucleases may offer the capability to specifically eliminate dominant disease alleles, correct endogenous genes, or integrate exogenous genes at safe harbors, resulting in permanent changes that are heritable in mitotic cells. These approaches could be applied for direct in vivo therapy, and AAV’s potential for high delivery efficiency coupled with the enhanced efficacy of AAV genomes as DNA donors for homology directed repair also offers the capacity for ex vivo modification of cells for subsequent engraftment. Coupling the potential of gene editing technology with the increasingly well-established, safe, and effective gene delivery capabilities of AAV may thus render new classes of genetic diseases accessible to gene therapy.
Therapeutic Gene Editing
Targeted gene editing has two primary goals – disrupting a sequence or introducing a precisely defined modification to a sequence – and both strategies begin with generating a DNA break at the locus of interest. For disruption, the non-homologous end joining (NHEJ) cellular repair mechanism directly rejoins the two ends and typically introduces small insertions or deletions (indels) at the cut site [12]. When placed near the 5’ end of a coding sequence, such indels generally disrupt the reading frame and thereby effectively knocks out the target gene. For precise modification, a DNA template containing both the desired modification and flanking regions of DNA homologous to the target area, i.e. homology arms, is co-delivered with the nuclease. The homology-directed repair (HDR) pathway can then splice the template in place of the damaged DNA within the region between the homology arms, thereby mediating specific gene modification (Fig. 1) [13, 14].
Figure 1. Cellular mechanisms of DNA repair following DNA double-strand break.
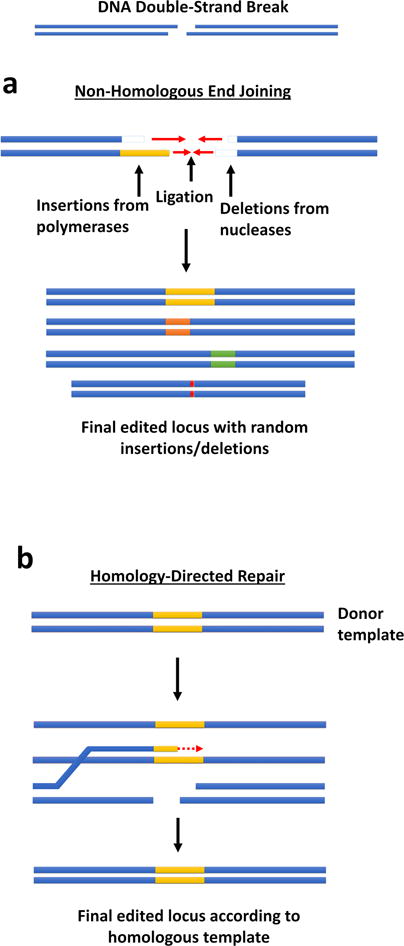
When a double-strand DNA break occurs, one of two cellular mechanisms repairs the damage. (a) In non-homologous end joining (NHEJ), polymerases and nucleases clean up the damaged ends by adding or deleting small numbers of nucleotides until they can be rejoined by ligases. The final ligated product contains small insertions or deletions (indels) at the damage site, often resulting in a frameshift. (b) In homology-directed repair (HDR), the 3’ overhang strand at a site of DNA damage can displace a strand in a separate DNA duplex with homology to that strand (a donor template). Polymerases extend the damaged end according to the homologous template DNA duplex, and the strand either returns to its original complementary strand, annealing to the other original damaged end; or the donor template strand and the previously damaged strand can undergo complete crossover and recombination. Either option results in a repaired strand.
Both strategies require a means to generate targeted DNA strand breaks, and the first such engineerable tool was zinc-finger nucleases (ZFNs). An “alphabet” of individual zinc finger (ZF) DNA binding domains that bind to specific three-nucleotide targets was identified, and these ZFs could then be modularly assembled to target new desired genetic loci and fused with Fok I nuclease domains to yield custom nucleases [15, 16]. This advance opened the door not only to a broad range of important basic research applications but also to the potential capacity to treat disorders including HIV infection [17], hemophilia [18], sickle cell anemia [19], and others. For instance, the use of ZFNs to knock out the HIV co-receptor CCR5 within T cells, and thus render them resistant to HIV infection, is currently in clinical trials [17]. While estalished ZFNs are indeed quite effective enzymes, generating new nucleases is difficult since the target specificity of each individual ZF domain can depend on the context of its neighboring domains [20], requiring a high level of expertise, ZFN library selection methods, and thus a time-consuming process to generate specific ZFNs for new targets.
In 2009, the DNA binding domains of the transcription activator-like effector (TALE) class of bacterial transcription factors was found to consist of modular elements [21, 22]. Excitingly, these individual TALE domains were found bound single nucleotides with strong specificity and importantly with minimal context dependence, unlike ZFNs. Thus, TALE DNA binding domains could be linked together with near-ideal modularity to target virtually any desired DNA sequence, and fusion to a FokI nuclease domain yielded TALE nucleases (TALENs) [23]. Simple assembly kits made the generation of new functional TALENs rapid and accessible to researchers. That said, the resulting TALEN constructs were very large and thus challenged the carrying capacity of delivery vectors like AAV, and the repetitive nature of the TALEN coding sequence led to concerns with recombination in the context of these ssDNA viral vectors.
While the simplicity of TALENs seemed unlikely to be surpassed, in 2012, it was demonstrated that the bacterial anti-viral adaptive immune mechanism known as the CRISPR/Cas9 system [24–26] could be re-engineered for targeted gene editing [27], a finding that was subsequently applied for genome editing in human cells [28–30]. Three components of the system from Streptococcus pyogenes are necessary and sufficient for enzymatic activity: the CRISPR-associated protein 9 (Cas9) nuclease; the CRISPR targeting RNA (crRNA) that is complementary to a target DNA sequence; and the trans-activating crRNA (tracrRNA) that hybridizes with a crRNA, enables it to bind to Cas9, and helps direct cleavage activity to the encoded locus. Fusing the two RNA components into a single guide RNA strand (sgRNA) further simplified the system such that virtually any desired target strand of DNA could be targeted and cleaved by simply changing the targeting RNA sequencing, limited only by the requirement for a small adjacent sequence known as the protospacer-adjacent motif. With this discovery, DNA cleavage and editing no longer required even simple modular protein assembly but merely modification of ~20 nucleotides of the targeting sgRNA. The simplicity and efficacy of the resulting CRISPR/Cas9 system make effective gene editing broadly accessible.
With the potential to effectively modify virtually any locus, CRISPR/Cas9 gene editing offers promise for both in vitro and in vivo genome editing. Successful application of this work for therapeutic purposes, however, will hinge upon an effective and reliable method for delivering the CRISPR/Cas9 machinery to affected cells.
AAV
Adeno-associated virus (AAV) is the most clinically successful in vivo gene therapy vector to date. AAVs are a family of non-enveloped, single-stranded DNA viruses that naturally require the presence of a helper virus, such as adenovirus, to replicate. The 4.7 kb AAV genome contains two short (~150 nucleotide) viral inverted terminal repeat sequences (ITRs) flanking two genes, rep and cap, encoding proteins for replication and capsid formation, respectively. Because these genes can function in trans, the virus can be engineered for assembly of virus particles with recombinant genomes containing only the desired genetic cargo flanked by the ITRs. AAV has numerous natural serotypes with somewhat different viral capsid sequences and tissue tropisms, indicating that differences in the viral capsid proteins can lead to differences in infectivity. As a non-integrating virus with a strong safety record, AAV has strong promise as a clinical gene delivery vehicle, and as mentioned above there are numerous examples of strong proof of concept in clinical trials as well as a regulatory approval in the European Union [1–9].
In addition to delivering DNA sequences for direct expression, the single stranded nature of the AAV genome can serve as an effective template that inherently stimulates the homologous recombination pathway to mediate gene targeting [31]. Specifically, viral delivery of a HDR construct with homology to a chromosomal locus, even without a nuclease component, can result in recombination into the target locus at a rate of up to 1% [32], a rate >1,000-fold higher than conventional plasmid donors [33] or other viral vectors [34]. Successful AAV-mediated gene targeting has been achieved in neural stem cells [35], human pluripotent stem cells [36], and hematopoietic stem cells (HSCs) [37], among other cell types. In addition, as discussed below, combining AAV with a nuclease offers even stronger potential.
Despite its success, AAV has significant challenges as a gene therapy delivery system. Natural serotypes AAV are inefficient on many target cells and tissues, do not have the capacity for targeted delivery to specific cells, and can be neutralized by antibodies that are prevalent within the human population due to prior natural AAV exposure. Additionally, the packaging capacity of AAV is approximately the size of its wild type genome (4.7 kb), and cargos that are greater than 10% beyond this size are not possible to package [10]. Finally, while a non-integrating virus offers the potential for lower genotoxicity and thus higher safer than an integrating one, the lack of a specific mechanism for vector integration means that the cargo will be diluted over time in mitotic target cells, and treating diseases with a cargo that must persist for efficacy is more difficult.
Different approaches have been taken to address these challenges. For example, directed evolution – or the generation of large AAV variant libraries and iterative selection in vitro or in vivo for enhanced gene delivery properties – has generated novel AAV variants with greatly improved delivery efficiencies for a range of applications and targets [38]. These include enhanced delivery to lung epithelium in human organotypic culture tissue [39] and a pig model of cystic fibrosis [40], enhanced retrograde transport for targeting specific neuronal populations in vivo [41], greatly improved biodistribution to target tissues such as outer retinal photoreceptors upon simple administration to the vitreous [42], and other applications. In addition, a large cargo can be delivered in AAV by packaging fragments of a gene in two separate vectors, and the full product can then be reconstituted in vivo via trans-splicing or homologous recombination of the two separate vectors [43], though with a significant decrease in overall efficiency. At any rate, the potential for highly efficient natural and in particular engineered AAV delivery to therapeutically relevant targets makes it a strong choice for gene therapy, including for therapeutic applications of CRISPR/Cas9.
Nucleases and AAV for Therapeutic Gene Editing
One major focus of gene editing has been ex vivo engineering of cellular therapies, in which a specific patient cells are harvested, edited, and re-engrafted. Compared to a direct in vivo therapy, more in vitro delivery options are available. As a prominent example, CD4+ T cells harvested from HIV-infected patients were edited to disrupt the CCR5 locus and thereby confer resistance to HIV infection, followed by reintroducion into patients. This approach has been implemented with both ZFNs and TALENs, and the ZFN-based approach – in which the nuclease was delivered with an adenoviral vector – is currently in clinical trials in which the engineered cells were shown to persist following administration [17]. In addition to CCR5 disruption, this CCR5 locus has been edited within HSCs via AAV donor template delivery and the ZFNs transiently expression through mRNA electroporation [44].
In addition to CCR5 disruption for HIV [45, 46], therapeutic treatment of β-globinopathies [47] such as sickle cell disease and β-thalassemia [48, 49] has been explored. Ex vivo cell therapy thus has strong potential to address unmet medical need, though efforts are currently focused predominantly on the hematopoietic system since its cells can readily be harvested and cultured. In vivo delivery will be needed to address most other tissue targets.
For in vivo editing, AAV’s packaging capacity posed initial challenges for CRISPR/Cas9 delivery, as the combined size of the initially best-characterized Streptococcus pyogenes Cas9 (SpCas9), the sgRNA, and promoters for each was simply too large to fit into a single AAV vector. However, two primary approaches for utilizing AAV as a CRISPR/Cas9 delivery vector have since emerged. Since the initial discovery and characterization of SpCas9, thousands of CRISPR/Cas9 proteins have been identified [50], many of which are significantly smaller than SpCas9. The best-characterized alternative Cas9 protein, derived from Staphylococcus aureus (SaCas9), is nearly 1 kb shorter than SpCas9 and can thus be accommodated along with its sgRNA in AAV [51]. Other non-Cas9 CRISPR proteins, such as Cpf1 [52], offer new binding and cleavage characteristics in addition to being more compact. With these smaller CRISPR/Cas9 proteins, the entire system can fit comfortably in a single AAV vector, though there are still inflexible limitations on the maximal size of promoters. As an alternative, some studies have packaged SpCas9 and the sgRNA in separate vectors for co-administration [53]. This approach is particularly useful for HDR-modification applications, where for example one vector could be used to deliver the nuclease and sgRNA and a second vector the HDR template.
Both the use of smaller Cas9s and the dual vector approach have been successfully implemented in vivo for an increasing number of applications, both to disrupt endogenous gene expression as well as to precisely correct disease alleles. In early 2015, SaCas9 and its sgRNA were combined in a single AAV8 vector to disrupt and thereby knock out expression of a cholesterol regulatory gene, proprotein convertase subtilisin/kinexin type 9 (PCSK9), in the adult mouse liver [51]. The result was reduced circulating cholesterol levels.
Later in 2015, another group demonstrated the ability to correct the ornithine transcarbamylase (OTC) locus, a gene responsible for a potentially life-threatening metabolic disease, in the liver using two vectors [54]. One AAV8 vector contained SaCas9, and the second harbored the sgRNA along with the HDR repair template. Co-delivery successfully corrected a mutation in ~10% of the cells within neonatal mouse liver, leading to significantly greater survival of affected mice.
Two-vector systems have also been successfully used for targeted gene disruption. In 2016, three research groups used either SaCas9 or SpCas9 in a two-vector system to disrupt an exon within the dystrophin gene that harbored a disease-causing mutation within a mouse model of Duchenne’s muscular dystrophy (DMD) [53, 55, 56]. Loss of dystrophin expression in the corresponding human monogenic recessive disorder leads to progressive muscle degeneration. To restore expression of this essential protein, they targeted sites within the splice sites flanking the mutation-containing exon 23, and the resulting successful elimination of this non-essential exon from the mRNA led to a functional protein product. AAV was administered via several routes – including direct intramuscular injection, intravenous injection, retro-orbital injection, and neonatal intraperitoneal injection – which to varying levels of functional dystrophin production in muscle tissue. While the fraction of muscle cells corrected was low, as in the OTC liver study, it was sufficient in these models of DMD to restore significant levels of muscle function.
Challenges
While gene editing therapy offers considerable promise, numerous challenges still must be overcome. First, there is a risk of engineered nucleases cutting unintended sites with imperfect but very close homology to the nuclease target site. Such off-target editing is well known to occur within in vitro contexts [57], and this risk can be further amplified by viral delivery methods such as AAV that can lead to persistent Cas9/gRNA expression in non-dividing cells for durations far longer than needed for the genome editing, an undesirable condition since the likelihood of undesired off-target cutting increases with nuclease residence time [58, 59]. This can been mitigated ex vivo though delivery mRNA encoding the nuclease or even recombinant nuclease proteins [44], but these methods do not translate well to in vivo contexts.
Numerous approaches have thus been developed to reduce such off-target effects, and one strategy is Cas9 protein engineering. For example, mutant form of Cas9 capable of only nicking one strand of DNA, rather than cleaving both, was combined with two sgRNAs targeting opposite strands near the desired locus. The resulting paired nicks yielded double-strand breaks that could be harnessed to generate indels or achieve HDR, but single nicks (such as at an off-target site that matches one sgRNA but not the other) instead lead to high-fidelity repair through the base excision repair pathway. The result is reduced off-target editing [60]. In another approach, rational modifications were introduced into Cas9 to reduce non-specific DNA contacts and thereby decrease binding affinity to non-specific targets without substantially affecting on-target editing rates [61, 62].
A third approach, based on the correlation between residence time and off-target activity, has been controlling the activity of Cas9 after delivery to minimize the total duration of its activity. One approach introduces inteins into the structure of Cas9 that only splice themselves out and generate active Cas9 in the presence of a small molecule ligand. By providing the small molecule for only a short duration, the activity window for editing can be reduced, thereby limiting off-target editing [63]. Another approach is the use of self-inactivating Cas9 vectors, where sgRNA target sites are engineered into the delivered viral genome itself to target the Cas9 expression cassette for destruction at the same time as targeting the desired genomic locus. The result is reduced residence time and off-target editing [64, 65].
While it would clearly be preferable to use a system with reduced off-target cutting, assessing the actual clinical risks of off-target modifications is challenging. In vitro assays that detect off-target cutting can be highly sensitive, such that only a subset of at-risk sites are actually cut in vivo. Furthermore, off-target modifications can be highly variable in location and sequence, and understanding how sequence changes they translate to functional risk of an adverse event is very difficult. Future work may focus increasingly on functional assays of off-target cutting impact, such as cell transformation.
Persistent expression also raises the risk of an immune response to the expression of a bacterial protein in a human cell, which can result in immune elimination of therapeutically corrected cells. For example, expression of AAV-delivered SpCas9 in a mouse has elicited clear immune recognition, though the subsequent cellular damage in this animal model was minimal [66]. Methods of effective transient delivery, such as self-inactivating vectors, may reduce immune responses by limiting the time of exposure.
Efficacy in vivo is an additional challenge. While successes in the highly accessible liver bode well for future work, low editing rates in other tissues, while therapeutically sufficient for the strong work in the DMD model, raise concerns for diseases that may require greater levels of correction or larger animals (or humans) that tend to be more difficult targets for gene delivery compared to mouse models. Delivery therefore remains a major challenge, particularly in vivo, and improved delivery systems including novel AAV variants engineered by directed evolution or rational design are therefore needed for human gene therapy. Improving delivery efficiency to target tissues will increase the efficacy of AAV-mediated genome editing, and improving vector selectivity or targeting to these tissues can enhance the safety profile by reducing potential side effects in tissues unaffected by the disease.
Conclusion
The era of gene editing has transformed virtually every area of biology, and clinical gene therapy is among the most exciting. The advent of CRISPR/Cas9 has enabled readily engineerable, accessible, and effective gene editing, and this technology is positioned to combine with AAV vectors to assist with in vitro editing and to make in vivo clinical gene editing therapy a reality. Addressing additional challenges in the field – including Cas9 target fidelity, Cas9 immunogenicity, and AAVs engineered for optimal gene delivery in the clinic – will enable next generation gene and genome editing therapeutics.
Table 1.
Therapeutic applications of gene editing via AAV combined with engineered nucleases.
| Disease | Target gene | Target cell/tissue | Nuclease used | AAV serotype used | Modification type | Reference | Note |
|---|---|---|---|---|---|---|---|
| HIV infection | CCR5 | CD4+ T cells | ZFN | rAAV6 | Disruption | [44] | Donor template only in AAV; nuclease mRNA |
| Hemophilia B | FIX | Hepatocytes | ZFN | rAAV8 | Correction | [67] | |
| Cardiovascular/ cholesterol homeostasis | PCSK9 | Liver/hepatocytes | SaCas9 | rAAV8 | Disruption | [51] | |
| DMD | Dmd | Muscle | SpCas9 | rAAV9 | Exon excision | [53] | |
| Muscle | SaCas9 | rAAV9 | Exon excision | [55] | |||
| Muscle | SaCas9 | rAAV8 | Exon excision | [56] | |||
| Metabolic disease | OTC | Liver/hepatocytes | SaCas9 | rAAV8 | Correction | [54] | |
| Hepatitis B infection | HBV polymerase | HepAD38 cells | ZFN | rAAV2 | Disruption | [68] | |
| Tyrosinaemia | Fah | Hepatocytes/liver | SaCas9 | rAAV8 | Correction | [69] | Donor template only in AAV; nuclease mRNA |
Abbreviations list
- AAV
adeno-associated virus
- NHEJ
non-homologous end joining
- HDR
homology-directed repair
- ZF
zinc finger
- ZFN
zinc-finger nuclease
- TALE
transcription activator-like effector
- TALEN
transcription activator-like effector nuclease
- CRISPR
clustered regularly interspaced short palindromic repeats
- crRNA
CRISPR targeting RNA
- tracrRNA
trans-activating crRNA
- sgRNA
single guide RNA
- HSC
hematopoietic stem cell
- SpCas9
Streptococcus pyogenes Cas9
- SaCas9
Staphylococcus aureus Cas9
- PCSK9
proprotein convertase subtilisin/kinexin type 9
- OTC
ornithine transcarbamylase
- DMD
Duchenne’s muscular dystrophy
- CCR5
C–C chemokine receptor type 5
- FIX
coagulation factor IX
- Dmd
dystrophin
- HBV
Hepatitis B virus
- Fah
Fumarylacetoacetate hydrolase
References
- 1.Bainbridge JWB, Smith AJ, Barker SS, et al. Effect of Gene Therapy on Visual Function in Leber’s Congenital Amaurosis. N Engl J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 2.Bennett J, Ashtari M, Wellman J, et al. AAV2 Gene Therapy Readministration in Three Adults with Congenital Blindness. Sci Transl Med. 2012;4:120ra15. doi: 10.1126/scitranslmed.3002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nathwani AC, Tuddenham EGD, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–65. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathwani AC, Reiss UM, Tuddenham EGD, et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N Engl J Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.d’Ydewalle C, Sumner CJ. Spinal Muscular Atrophy Therapeutics: Where do we Stand? Neurotherapeutics. 2015;12:303–316. doi: 10.1007/s13311-015-0337-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stroes ES, Nierman MC, Meulenberg JJ, et al. Intramuscular Administration of AAV1-Lipoprotein Lipase S447X Lowers Triglycerides in Lipoprotein Lipase – Deficient Patients. Heart. 2008;28:2303–2304. doi: 10.1161/ATVBAHA.108.175620. [DOI] [PubMed] [Google Scholar]
- 7.Carpentier AC, Frisch F, Labbé SM, Gagnon R, de Wal J, Greentree S, Petry H, Twisk J, Brisson D, Gaudet D. Effect of Alipogene Tiparvovec (AAV1-LPL S447X) on Postprandial Chylomicron Metabolism in Lipoprotein Lipase-Deficient Patients. J Clin Endocrinol Metab. 2012;97:1635–1644. doi: 10.1210/jc.2011-3002. [DOI] [PubMed] [Google Scholar]
- 8.Greenberg B, Butler J, Felker GM, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387:1178–1186. doi: 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- 9.Constable IJ, Pierce CM, Lai C-M, et al. Phase 2a Randomized Clinical Trial: Safety and Post Hoc Analysis of Subretinal rAAV.sFLT-1 for Wet Age-related Macular Degeneration. EBioMedicine. 2016;14:168–175. doi: 10.1016/j.ebiom.2016.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong J-Y, Fan P-D, Frizzell RA. Quantitative Analysis of the Packaging Capacity of Recombinant Adeno-Associated Virus. Hum Gene Ther. 1996;7:2101–2112. doi: 10.1089/hum.1996.7.17-2101. [DOI] [PubMed] [Google Scholar]
- 11.Kymäläinen H, Appelt JU, Giordano FA, et al. Long-term episomal transgene expression from mitotically stable integration-deficient lentiviral vectors. Hum Gene Ther. 2014;25:428–42. doi: 10.1089/hum.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.San Filippo J, Sung P, Klein H. Mechanism of Eukaryotic Homologous Recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 14.Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- 15.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93:1156–60. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bibikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim YG, Chandrasegaran S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol. 2001;21:289–97. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tebas P, Stein D, Tang WW, et al. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. N Engl J Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anguela XM, Sharma R, Doyon Y, et al. Robust ZFN-mediated genome editing in adult hemophilic mice. Blood. 2013;122:3283–7. doi: 10.1182/blood-2013-04-497354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoban MD, Cost GJ, Mendel MC, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125 doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramirez CL, Foley JE, Wright DA, et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods. 2008;5:374–375. doi: 10.1038/nmeth0508-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–12. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 22.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 23.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient Design and Assembly of Custom TALEN and Other TAL Effector-Based Constructs for DNA Targeting. Nucleic Acids Res. 2011;39:e82–e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terns MP, Terns RM. CRISPR-based adaptive immune systems. Curr Opin Microbiol. 2011;14:321–327. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhaya D, Davison M, Barrangou R. CRISPR-Cas Systems in Bacteria and Archaea: Versatile Small RNAs for Adaptive Defense and Regulation. Annu Rev Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 26.Wiedenheft B, Sternberg SH, Doudna Ja. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–8. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 27.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA – Guided DNA Endonuclease in Adaptice Bacterial Immunity. Science. 2012;337:816–822. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science (80-) 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vasileva A, Linden RM, Jessberger R. Homologous recombination is required for AAV-mediated gene targeting. Nucleic Acids Res. 2006;34:3345–3360. doi: 10.1093/nar/gkl455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirata R, Chamberlain J, Dong R, Russell DW. Targeted transgene insertion into human chromosomes by adeno-associated virus vectors. Nat Biotechnol. 2002;20:735–738. doi: 10.1038/nbt0702-735. [DOI] [PubMed] [Google Scholar]
- 33.Russell DW, Hirata RK. Human gene targeting by viral vectors. Nat Genet. 1998;18:325–330. doi: 10.1038/ng0498-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellis J, Bernstein A. Gene targeting with retroviral vectors: recombination by gene conversion into regions of nonhomology. Mol Cell Biol. 1989;9:1621–7. doi: 10.1128/mcb.9.4.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotterman MA, Vazin T, Schaffer DV. Enhanced selective gene delivery to neural stem cells in vivo by an adeno-associated viral variant. Development. 2015;142:1885–92. doi: 10.1242/dev.115253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asuri P, Bartel MA, Vazin T, Jang J-H, Wong TB, Schaffer DV. Directed evolution of adeno-associated virus for enhanced gene delivery and gene targeting in human pluripotent stem cells. Mol Ther. 2012;20:329–38. doi: 10.1038/mt.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paiboonsukwong K, Ohbayashi F, Shiiba H, Aizawa E, Yamashita T, Mitani K. Correction of mutant Fanconi anemia gene by homologous recombination in human hematopoietic cells using adeno-associated virus vector. J Gene Med. 2009;11:1012–1019. doi: 10.1002/jgm.1382. [DOI] [PubMed] [Google Scholar]
- 38.Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet. 2014;15:445–51. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Excoffon KJDA, Koerber JT, Dickey DD, Murtha M, Keshavjee S, Kaspar BK, Zabner J, Schaffer DV. Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc Natl Acad Sci U S A. 2009;106:3865–70. doi: 10.1073/pnas.0813365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steines B, Dickey DD, Bergen J, et al. CFTR gene transfer with AAV improves early cystic fibrosis pig phenotypes. JCI insight. 2016;1:e88728. doi: 10.1172/jci.insight.88728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tervo DGR, Hwang B-Y, Viswanathan S, et al. A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron. 2016;92:372–382. doi: 10.1016/j.neuron.2016.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, Flannery JG, Schaffer DV. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5:189ra76. doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 43.Trapani I, Colella P, Sommella A, et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol Med. 2013;6 doi: 10.1002/emmm.201302948. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Exline CM, DeClercq JJ, et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol. 2015;33:1256–1263. doi: 10.1038/nbt.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang CX, Cannon PM. Clinical Applications of Genome Editing to HIV Cure. AIDS Patient Care STDS. 2016;30:539–544. doi: 10.1089/apc.2016.0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DiGiusto DL, Cannon PM, Holmes MC, et al. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol Ther Methods Clin Dev. 2016;3:16067. doi: 10.1038/mtm.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie F, Ye L, Chang JC, Beyer AI, Wang J, Muench MO, Kan YW. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014;24:1526–33. doi: 10.1101/gr.173427.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu P, Tong Y, Liu X, et al. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2–654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci Rep. 2015;5:12065. doi: 10.1038/srep12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shmakov S, Smargon A, Scott D, et al. Diversity and evolution of class 2 CRISPR–Cas systems. Nat Rev Microbiol. 2017 doi: 10.1038/nrmicro.2016.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–71. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–3. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang Y, Wang L, Bell P, et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34:334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tabebordbar M, Zhu K, Cheng JKW, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–11. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nelson CE, Hakim CH, Ousterout DG, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science (80-) 2016;351 doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsai SQ, Joung JK. Defining and improving the genome-wide specificities of CRISPR–Cas9 nucleases. Nat Rev Genet. 2016;17:300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pruett-Miller SM, Reading DW, Porter SN, Porteus MH. Attenuation of Zinc Finger Nuclease Toxicity by Small-Molecule Regulation of Protein Levels. PLoS Genet. 2009;5:e1000376. doi: 10.1371/journal.pgen.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaj T, Guo J, Kato Y, Sirk SJ, Barbas CF. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods. 2012;9:805–7. doi: 10.1038/nmeth.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ran FA, Hsu PD, Lin C-Y, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–9. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science (80-) 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davis KM, Pattanayak V, Thompson DB, Zuris JA, Liu DR. Small molecule–triggered Cas9 protein with improved genome-editing specificity. Nat Chem Biol. 2015;11:316–318. doi: 10.1038/nchembio.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Epstein BE, Schaffer DV. 119. Engineering a Self-Inactivating CRISPR System for AAV Vectors. Mol Ther. 2016;24:S50. [Google Scholar]
- 65.Ruan G-X, Barry E, Yu D, et al. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol Ther. 2017;0:1–25. doi: 10.1016/j.ymthe.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chew WL, Tabebordbar M, Cheng JKW, Mali P, Wu EY, Ng AHM, Zhu K, Wagers AJ, Church GM. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H, Haurigot V, Doyon Y, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–21. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weber ND, Stone D, Sedlak RH, De Silva Feelixge HS, Roychoudhury P, Schiffer JT, Aubert M, Jerome KR. AAV-mediated delivery of zinc finger nucleases targeting hepatitis B virus inhibits active replication. PLoS One. 2014;9:e97579. doi: 10.1371/journal.pone.0097579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yin H, Song C-Q, Dorkin JR, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]