

Abstract
Protein tyrosine phosphatases (PTPs) have been challenging targets for inhibitor design, because all PTPs share a highly conserved active site structure, which is positively charged and requires negatively charged moieties for tight binding. in this study, we developed cell-permeable bicyclic peptidyl inhibitors against T-cell PTP (TCPTP), which feature a cell-penetrating motif in one ring and a target-binding sequence in the second ring.
Graphical abstract
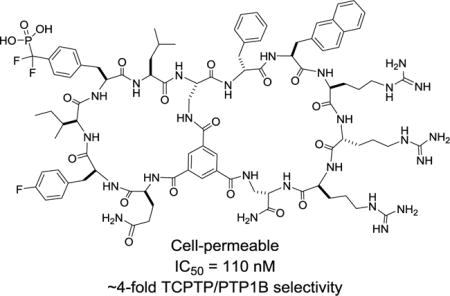
Cell-permeable, biologically active bicyclic peptidyl inhibitors against T-cell protein-tyrosine phosphatase were directly isolated from a combinatorial library.
The human genome encode >100 proteins with protein tyrosine phosphatase (PTP) activity.1 Together with protein kinases, the PTPs mediate the execution and regulation of many cellular processes such as signal transduction. Modulation of the activities of PTPs is expected to have therapeutic benefits for a variety of human diseases and conditions.2 For example, PTP1B has been pursued as a target for treatment of type II diabetes and potentially obesity,3 whereas SHP-2 is a target for anticancer drug design.4 T-cell PTP (TCPTP) is involved in haematopoiesis and cytokine response and is linked to the development of several inflammatory disorders including type 1 diabetes, Crohn’s disease, and rheumatoid arthritis.5 Selective inhibitors against PTPs would provide novel therapeutic agents as well as valuable chemical probes for investigating their physiological and pathological roles. However, designing isoform-specific PTP inhibitors has been challenging, because all of the PTPs share a highly conserved active site structure, which is positively charged. High-affinity binding to the PTP active site requires a negatively charged species which is, however, generally impermeable to the cell membrane. To overcome this problem, we recently developed a bicyclic peptide approach, featuring a short cell-penetrating peptide (CPP; e.g., FΦRRRR where Φ is L-2-naphthylalanine) in one ring for endocytic cellular uptake and a target-binding motif in the second ring.6 Application of this method to PTP1B resulted in a cell-permeable, potent, and selective inhibitor for PTP1B (KD = 37 nM). Importantly, the bicyclic peptidyl inhibitor displayed a 17-fold selectivity over TCPTP, which shares 90% sequence identity with PTP1B within the active site. Presumably, the peptidyl inhibitor achieves isoform selectivity by engaging in additional interactions with the less conserved protein surfaces beyond the active site. The bicyclic approach was subsequently shown to be effective for delivering a wide variety of peptide sequences including negatively charged phosphopeptides into the cytosol of mammalian cells.7 In this study, we set out to test whether the bicyclic peptide approach might be applied to generate isoform-specific inhibitors against other members of the PTP superfamily.
We chose TCPTP as the target, because it has been difficult to develop inhibitors with selectivity for TCPTP over PTP1B.8 To this end, we designed a bicyclic peptide library featuring a degenerate peptide sequence, X1-X2-X3-F2Pmp-X4 [where F2Pmp is L-(phosphonodifluoromethyl)phenylalanine and X1-X4 are any of the 24 amino acid building blocks], in the first (or N-terminal) ring and 12 different amphipathic CPP motifs in the second (or C-terminal) ring (Fig. 1). F2Pmp is a non-hydrolysable analogue of phosphotyrosine (pY), which binds to the active site of PTPs with modest affinity and selectivity.9 The 24 building blocks included 10 proteinogenic L-amino acids (Ala, Ser, Pro, Ile, Asp, Gln, His, Tyr, Trp, and Gly), 5 unnatural α-L-amino acids [norleucine (Nle), 2-aminobutyruc acid (Abu), 4-fluorophenylalanine (Fpa), phenylglycine (Phg), and pipecolic acid (Pip)], and 9 α-D-amino acids [D-Ala, D-Pro, D-Val, D-Thr, D-Leu, D-Asn, D-Glu, D-Phe, D-2-naphthylalanine (D-Nal)]. The 12 CPP sequences consisted of different combinations of two or three aromatic hydrophobic residues (e.g., Phe and Nal) and three or four L- or D-arginine residues (Table S1).10 The bicyclic peptide library has a theoretical diversity of 3.98 × 106 and was synthesized on 2 g of TentaGel microbeads (~90 μm; ~2.8 × 106 beads/g), as detailed in Supporting Information. Briefly, each library bead was topologically segregated into two different layers, with a unique bicyclic peptide synthesized in the surface layer and a linear peptide of identical sequence prepared in the inner core as an encoding tag.6,11 To increase the stringency of library screening, the ligand loading density of the surface layer (but not the inner layer) was reduced by 10-fold.12 In addition, during the coupling of F2Pmp, a 9:1 (mol/mol) ratio of Fmoc-Tyr/Fmoc-F2Pmp was used, resulting in another 10-fold reduction in the F2Pmp content. Altogether, the loading density of F2Pmp-containing bicyclic peptides on the bead surface was decreased by 100-fold, relative to that of the linear encoding peptides inside the beads. We have previously demonstrated that reduction of the surface ligand density greatly reduces nonspecific binding caused by simultaneous interaction of a single protein molecule with multiple ligand molecules on the solid support.12
Fig. 1.
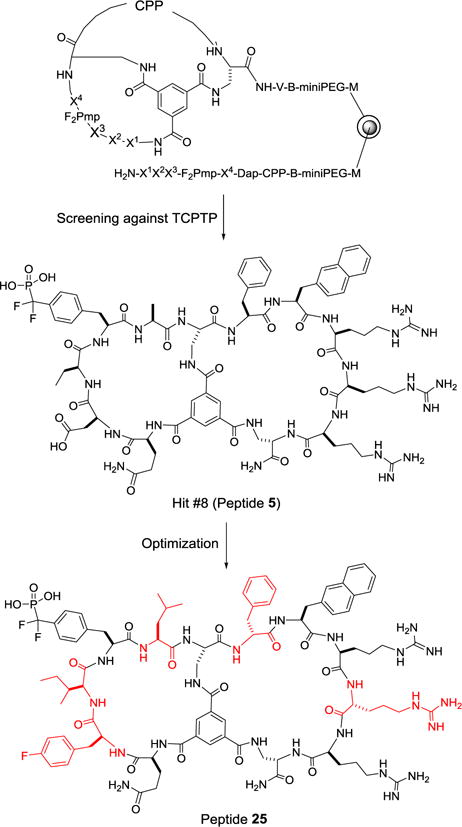
Structures of the bicyclic peptide library, the initial hit selected for optimization, and the optimization product (peptide 25). Residues modified during optimization are highlighted in red.
Approximately 400 mg of the bicyclic peptide library (~1.1 × 106 compounds) was subjected to two rounds of screening for binding to TCPTP. During the first round, the library was incubated with biotinylated TCPTP (300 nM) and subsequently a streptavidin-alkaline phosphatase conjugate. After removal of any unbound proteins, the protein-bound beads were stained by incubating with a solution of 5-bromo-4-chloro-3-indolyl phosphate (BCIP).13 Turquoise colored beads (189 beads) were separated into intensely colored (49 beads) and medium colored categories (140 beads) and exhaustively washed to remove the bound proteins and the indigo dye. The two pools of beads were next incubated with 20 nM Texas red-labeled TCPTP and the fluorescent beads (total 137 positive beads) were separated into 5 groups on the basis of the screening results: group A (intensely colored during both rounds; 4 beads), group B (intensely colored in BCIP screening but medium colored during Texas-red screening; 14 beads), group C (intensely colored in BCIP screening but lightly colored during Texas-red screening; 26 beads), group D (medium colored in both screenings; 23 beads), and group E (medium colored in BCIP screening and lightly colored during Texas-red screening; 70 beads). The 137 positive beads were subjected to partial Edman degradation-mass spectrometry (PED-MS) analysis,14 resulting in 60 unambiguous, complete sequences (Table S2).
The two group A hits and three of the group B hits (Table 1, peptides 1–5), which should represent the most potent binders of TCPTP, were individually resynthesized and tested for inhibition of TCPTP and PTP1B activities in the solution phase. Surprisingly, the two group A peptides (peptides 1 and 2) and peptide 3 from group B did not show significant inhibition of TCPTP. Peptides 4 and 5 (both from group B) were relatively potent inhibitors of TCPTP, with IC50 values of 550 and 660 nM, respectively. Peptide 5 showed slightly higher TCPTP/PTP1B selectivity (2.6-fold) than peptide 4 (2-fold) and was selected for further study. The reason for lack of TCPTP inhibition by peptides 1–3 was not investigated; it is possible that the peptides bind to TCPTP at a site(s) other than the active site.
Table 1.
Sequences of bicyclic peptides and their inhibitory activities against TCPTP and PTP1B
| Peptide No. | Sequencea | CPP | IC50 (nM) | |
|---|---|---|---|---|
| TCPTP | PTP1B | |||
| 1 | Pro-Tyr-Ala-F2Pmp-Pip | I | ND | ND |
| 2 | Pro-Ile-Asp-F2Pmp-Pip | J | ND | ND |
| 3 | Asp-Asp-thr-F2Pmp-Pip | J | ND | ND |
| 4 | Asp-glu-Asp-F2Pmp-Ile | J | 550 ± 140 | 1200 ± 700 |
| 5 | Gln-Asp-Abu-F2Pmp-Ala | C | 660 ± 280 | 1750 ± 260 |
| 6 | Gln-Asp-Phe-F2Pmp-Ala | J | 470 ± 70 | 1190 ± 30 |
| 7 | Gln-Asp-Ile-F2Pmp-Ala | J | 340 ± 50 | 1150 ± 490 |
| 8 | Gln-Asp-Val-F2Pmp-Ala | J | 380 ± 110 | 580 ± 70 |
| 9 | Gln-Asp-Ala-F2Pmp-Ala | J | 1110 ± 70 | 1430 ± 40 |
| 10 | Gln-Asp-Ser-F2Pmp-Ala | J | 530 ± 40 | 840 ± 10 |
| 11 | Gln-Asp-Nva-F2Pmp-Ala | J | 610 ± 280 | 1320 ± 250 |
| 12 | Gln-Asp-Nle-F2Pmp-Ala | J | 580 ± 100 | 1100 ± 60 |
| 13 | Gln-Asp-Ile-F2Pmp-Abu | I | 400 | 1600 |
| 14 | Gln-Asp-Ile-F2Pmp-Ile | J | 240 | 500 |
| 15 | Gln-Asp-Ile-F2Pmp-Val | J | 420 | 730 |
| 16 | Gln-Asp-Ile-F2Pmp-thr | J | >2000 | >2000 |
| 17 | Gln-Asp-Ile-F2Pmp-Gln | J | 210 ± 50 | 590 ± 260 |
| 18 | Gln-Asp-Ile-F2Pmp-Asn | J | 380 ± 5 | 910 ± 410 |
| 19 | Gln-Asp-Ile-F2Pmp-Nle | J | 290 ± 120 | 840 ± 150 |
| 20 | Gln-Asp-Ile-F2Pmp-Leu | J | 140 ± 40 | 260 ± 60 |
| 21 | Gln-Asp-Ile-F2Pmp-Tle | J | 680 ± 140 | 560 ± 90 |
| 22 | Gln-glu-Ile-F2Pmp-Leu | J | 1120 | 1270 |
| 23 | Gln-asp-Ile-F2Pmp-Leu | J | 1090 | 1340 |
| 24 | Gln-phe-Ile-F2Pmp-Leu | J | 250 | 530 |
| 25 | Gln-Fpa-Ile-F2Pmp-Leu | J | 110 ± 16 | 420 ± 200 |
| 26 | Gln-Glu-Ile-F2Pmp-Leu | J | 250 | 410 |
| 27 | Gln-Tyr-Ile-F2Pmp-Leu | J | 140 | 210 |
| 28 | Ala-Fpa-Ile-F2Pmp-Leu | J | 140 ± 10 | 280 ± 2 |
| 29 | Pro-Fpa-Ile-F2Pmp-Leu | J | 140 ± 20 | 290 ± 70 |
| 30 | glu-Fpa-Ile-F2Pmp-Leu | J | 47 ± 3 | 46 ± 17 |
| 31 | Phg-Fpa-Ile-F2Pmp-Leu | J | 310 ± 230 | 350 ± 240 |
| 32 | ser-Fpa-Ile-F2Pmp-Leu | J | 140 ± 40 | 100 ± 20 |
| 33 | Gln-Fpa-Ile-Tyr-Leu | J | ND | ND |
Only the randomized region is shown. Three-letter codes in all lowercase letters indicate D-amino acids. ND, not determined due to no significant inhibition.
To improve the potency and selectivity of peptide 5, we first replaced its CPP motif C (Phe-Nal-Arg-Arg-Arg) with CPP motif J (D-Phe-Nal-Arg-D-Arg-Arg), because the latter was the most frequently selected among the 12 different CPP sequences, especially among the intensely colored hits (42% of group A, B, and C hits). In addition, alternating stereochemistry in the CPP sequence had previously been shown to improve the cellular uptake efficiency.10 We also replaced the L-2-aminobutyic acid (Abu) at the pY-1 position (relative to F2Pmp, which is designated as position 0) with serine and other amino acids containing hydrophobic side chains (Table 1, peptides 6–12) and found that isoleucine gave both the highest potency against TCPTP and the best TCPTP/PTP1B selectivity (~3.4-fold). We therefore kept isoleucine as the pY-1 residue and replaced the Ala residue at the pY+1 position with Abu, Ile, Val, D-Thr, Gln, Asn, norleucine (Nle), Leu, or tert-leucine (Tle) (Table 1, peptides 13–21). The results showed that a leucine at the pY+1 position gave the highest potency against TCPTP (IC50 = 140 nM), although the TCPTP/PTP1B selectivity was slightly reduced (~1.8-fold). Next, the Ile-F2Pmp-Leu motif was kept constant and the pY-2 residue (Asp) was replaced with D-glutamic acid, D-aspartic acid, D-phenylalanine, Fpa, Glu, or Tyr (Table 1, peptides 22–27). We found that Fpa at this position gives a good balance of potency and selectivity (IC50 = 110 nM against TCPTP and a ~4-fold TCPTP/PTP1B selectivity for peptide 25). Additionally, substitution of Fpa for the negatively charged Asp is expected to improve the cell-permeability of the bicyclic peptide inhibitor.10 Further modification at the pY-3 position (Table 1, peptides 28–32) did not improve the TCPTP/PTP1B selectivity, although substitution of D-Glu for Gln produced the most potent inhibitor of the series (IC50 = 47 nM against TCPTP for peptide 30). Importantly, peptide 25 did not significantly inhibit any of the other PTPs tested, including CD45, PTPRD, SHP-1, SHP-2, and VHR (<20% inhibition at 1.5 μM; Fig. 2A). A control peptide (peptide 33), which contains a Tyr in place of F2Pmp but is otherwise identical to peptide 25, was also prepared. Peptide 33 had no inhibitory activity against TCPTP (Figure 2B).
Fig. 2.
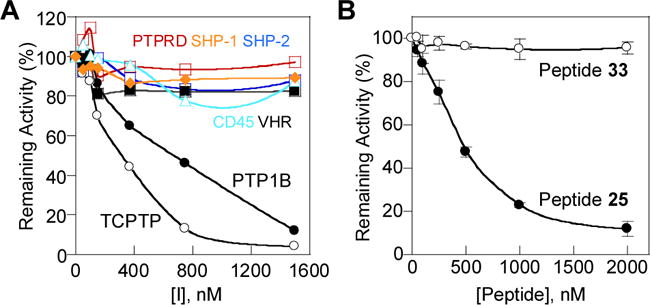
Selectivity of peptide 25 for TCPTP. (A) Plot of remaining activity of TCPTP, PTP1B, CD45, PTPRD, SHP-1, SHP-2, and VHR as a function of peptide 25 concentration. (B) Comparison of peptides 25 and 33 for their inhibitory activity toward TCPTP.
Peptides 25 and 33 were next labeled with fluorescein isothiocyanate (FITC) and their entry into HeLa cells was monitored by live-cell confocal microscopy. Both peptides entered HeLa cells efficiently (Fig. 3A,B). The predominantly punctate fluorescence pattern suggested that at least a fraction of the endocytosed peptides was retained inside the endosomal/lysosomal compartments, although binding of cytosolic peptides to the ER localized TCPTP would also produce a punctate fluorescence pattern. To determine whether a fraction of the internalized peptides reached the cytosol, we labeled peptides 25 and 33 with naphthofluorescein (NF) and quantitated their cellular entry by flow cytometry. Because NF has a pKa value of 7.8 and is non-fluorescent inside the acidic endosomes and lysosomes (pH ≤6), it has been used to effectively quantitate the cytosolic entry of CPPs.10,15 The flow cytometry data indicate that peptides 25 and 33 enter the cytosol and nucleus of HeLa cells with 35% and 180% efficiencies, respectively (an efficiency of 100% is defined as equal peptide concentration in the extracellular growth medium and the cytosol) (Fig. 3C). The lower cellular entry efficiency of peptide 25 than 33 is expected, as the negatively charged F2Pmp would partially neutralize the arginine positive charges and reduce the CPP efficiency.7,10
Fig. 3.
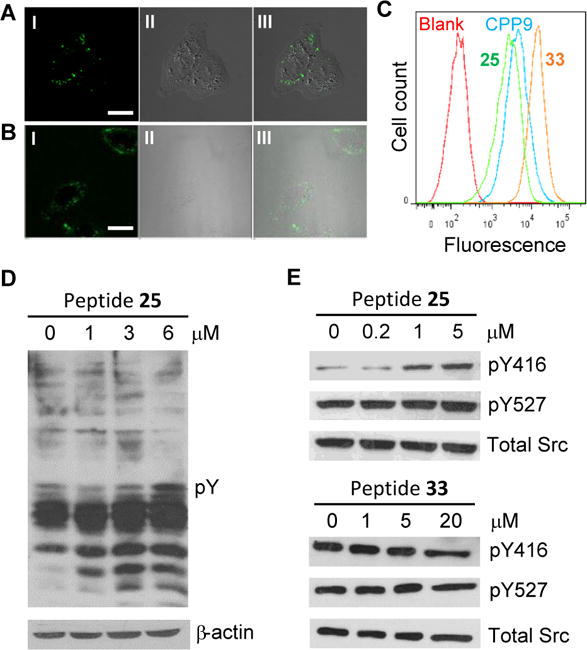
Cellular activity of peptide 25. (A,B) Confocal microscopic images of HeLa cells after treatment with 5 μM FITC-labelled peptide 25 (A) or 33 (B) for 2 h. I, GFP channel; II, DIC; and III, overlap of I and II. Scale bar, 20 μm. (C) Flow cytometry analysis of the relative cytosolic entry efficiency of cyclic CPP9 and peptides 25 and 33. (D) Anti-pY western blot showing the effect of peptide 25 on the global pY level in HeLa cells. (E) Western blot analysis showing the effect of peptide 25 (control peptide 33) on the phosphorylation levels of Tyr-416 and Tyr-527 of Src kinase in C6 cells.
The ability of peptide 25 to modulate the intracellular TCPTP activity was assessed by incubating HeLa cells with increasing concentrations of peptide 25 (0–6 μM) for 24 h, separating the whole cell lysate by SDS-PAGE, and western blotting with an anti-pY antibody. Treatment with peptide 25 caused dramatic, dose-dependent increase in the global pY levels of intracellular proteins, especially in the lower molecular-weight range (Fig. 3D). The increase in the pY level was already apparent at 1 μM and appeared to plateau at 3 μM peptide 25. To determine whether the observed increase in the pY levels was at least partially caused by inhibition of TCPTP, we examined the effect of peptide 25 on the tyrosine phosphorylation of the Src kinase. It has previously been reported that the pY-416 site in Src is a specific substrate of TCPTP, whereas pY-527 is dephosphorylated by PTP1B.8,16 C6 glioma cells were incubated for 24 h with peptide 25 (0–5 μM) and then stimulated with epidermal growth factor (EGF; 50 ng/mL) for 10 min. The cells were lysed and the lysate was separated by SDS-PAGE. The proteins were transferred to a nitrocellulose membrane and blotted with antibodies specific for the pY-416 and pY-527 sites of Src. In agreement with the previous studies,8,16 treatment with peptide 25 dose-dependently increased the phosphorylation level of the Tyr-416 site, but had no significant effect on the Tyr-527 site (Fig. 3E). Again, the maximal enhancement was reached at ~1 μM peptide 25. In contrast, despite its 5-fold more efficient cellular entry, peptide 33 did not change the phosphorylation level at either the Tyr-416 site or the Tyr-527 site at up to 20 μM concentration (Fig. 3E). These results strongly argue that peptide 25 was able to efficiently enter the mammalian cells and inhibit the intracellular TCPTP.
In conclusion, we have obtained a relatively potent and cell-permeable bicyclic peptidyl inhibitor against TCPTP, a challenging intracellular target for inhibitor discovery. This inhibitor may serve as a useful tool for investigating the biological function of TCPTP in signalling pathways. Together with our previously reported examples,6,7 this work demonstrates that bicyclic peptides featuring both cell-penetrating and target-binding units may provide a general strategy for developing cell-permeable ligands against intracellular proteins.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (GM110208 and GM122459).
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustellin T. Cell. 2004;117:699. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 2.Zhang ZY. Acc Chem Res. 2017;50:122. doi: 10.1021/acs.accounts.6b00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elchelby M, Payette P, Michaliszyn E, Cromlish W, Collins S, Lee Loy A, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramanchandran C, Gresser MJ, Tremblay ML, Kennedy BP. Science. 1999;283:1544. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 4.Chan G, Kalaitzidis D, Neel BG. Cancer Metastasis Rev. 2008;27:179. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 5.Chistiakova DA, Chistiakova EI. Int J Diabetes Mellitus. 2010;2:114. [Google Scholar]
- 6.Lian W, Jiang B, Qian Z, Pei D. J Am Chem Soc. 2014;136:9830. doi: 10.1021/ja503710n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian Z, LaRochelle JR, Jiang B, Lian W, Hard RL, Selner N, Luechapanickhul R, Barrios AM, Pei D. Biochemistry. 2014;53:4034. doi: 10.1021/bi5004102. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jiang B, Pei D. J Med Chem. 2015;58:6306. doi: 10.1021/acs.jmedchem.5b00411. [DOI] [PMC free article] [PubMed] [Google Scholar]; Trinh TB, Upadhyaya P, Qian Z, Pei D. ACS Comb Sci. 2016;18:75. doi: 10.1021/acscombsci.5b00164. [DOI] [PMC free article] [PubMed] [Google Scholar]; Qian Z, Rhode CA, McCroskey LC, Wen J, Appiah-Kubi G, Wang DJ, Guttridge DC, Pei D. Angew Chem Int Ed. 2017;56:1525. doi: 10.1002/anie.201610888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang S, Chen L, Luo Y, Gunawan A, Lawrence DS, Zhang ZY. J Am Chem Soc. 2009;131:13072. doi: 10.1021/ja903733z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Wu L, Otaka A, Smyth MS, Roller PP, Burke TR, den Hertog J, Zhang ZY. Biochem Biophys Res Commun. 1995;216:976. doi: 10.1006/bbrc.1995.2716. [DOI] [PubMed] [Google Scholar]
- 10.Qian Z, Martyna A, Hard RL, Wang J, Appiah-Kubi G, Coss C, Phelps MA, Rossman JS, Pei D. Biochemistry. 2016;55:2601. doi: 10.1021/acs.biochem.6b00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joo SH, Xiao Q, Ling Y, Gopishetty B, Pei D. J Am Chem Soc. 2006;128:13000. doi: 10.1021/ja063722k. [DOI] [PubMed] [Google Scholar]
- 12.Chen X, Tan PH, Zhang Y, Pei D. J Comb Chem. 2009;11:604. doi: 10.1021/cc9000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wavreille AS, Garaud M, Zhang Y, Pei D. Methods. 2007;42:207. doi: 10.1016/j.ymeth.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thakkar A, Wavreille AS, Pei D. Anal Chem. 2006;78:5935. doi: 10.1021/ac0607414. [DOI] [PubMed] [Google Scholar]
- 15.Qian Z, Dougherty PG, Pei D. Chem Commun. 2015;51:2162. doi: 10.1039/c4cc09441g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan C, Zhu M, Wang Q, Lu L, Xing S, Fu X, Jiang Z, Zhang S, Li Z, Li Z, Zhu R, Ma L, Xu L. Chem Commun. 2012;48:1153. doi: 10.1039/c2cc16818a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.