

Abstract
Insulin resistance is a major risk factor for many diseases. However, its underlying mechanism remains unclear in part because it is triggered by a complex relationship between multiple factors, including genes and the environment. Here, we used metabolomics combined with computational methods to identify factors that classified insulin resistance across individual mice derived from three different mouse strains fed two different diets. Three inbred ILSXISS strains were fed high-fat or chow diets and subjected to metabolic phenotyping and metabolomics analysis of skeletal muscle. There was significant metabolic heterogeneity between strains, diets, and individual animals. Distinct metabolites were changed with insulin resistance, diet, and between strains. Computational analysis revealed 113 metabolites that were correlated with metabolic phenotypes. Using these 113 metabolites, combined with machine learning to segregate mice based on insulin sensitivity, we identified C22:1-CoA, C2-carnitine, and C16-ceramide as the best classifiers. Strikingly, when these three metabolites were combined into one signature, they classified mice based on insulin sensitivity more accurately than each metabolite on its own or other published metabolic signatures. Furthermore, C22:1-CoA was 2.3-fold higher in insulin-resistant mice and correlated significantly with insulin resistance. We have identified a metabolomic signature composed of three functionally unrelated metabolites that accurately predicts whole-body insulin sensitivity across three mouse strains. These data indicate the power of simultaneous analysis of individual, genetic, and environmental variance in mice for identifying novel factors that accurately predict metabolic phenotypes like whole-body insulin sensitivity.
Keywords: ceramide, glucose metabolism, insulin resistance, metabolomics, skeletal muscle metabolism, genetic diversity, metabolite signature, strain differences
Introduction
The development of disease in humans often results from a complex interplay between genetic, epigenetic, and environmental factors. Although widely held that the environment plays a major role in metabolic disease, defining the heritable contribution has proven difficult. Genome-wide association studies have revealed a number of SNPs associated with metabolic disease, but collectively these do not account for the majority of disease in the population (1, 2). One difficulty likely involves the complex interaction between genes and environmental factors such as diet and life-style choices (e.g. exercise) (3). Several human trials have demonstrated significant modification (positive and negative) of disease risk associated with specific SNPs in response to the intake of specific or total fats (4–9). These data suggest that dietary recommendations for fat and other macronutrients at a population level may be ill-advised and that more work is needed to establish how the environment and genetics interact. Certain genotypes might be better suited to specific combinations of macronutrients in the diet and a mismatch may be detrimental to health.
Insulin resistance is a major risk factor for many metabolic diseases (10). A number of defects have been linked to insulin resistance, including impaired insulin signaling, accumulation of specific lipid species (11–13), oxidative stress (14, 15), or inflammation (16). Many of these studies have focused on the use of a single mouse strain, notably the C57Bl/6J mouse. However, it has emerged that genetic background has a significant impact on metabolism (17–23), so there is a growing need for a more expansive analysis of insulin resistance in mice of different genetic backgrounds. Indeed, a number of panels comprising many different recombinant inbred mouse strains have been developed such as ILSXISS (24, 25), HMDP (23), BXD (26), and collaborative cross (27), and several pivotal studies of dietary manipulation in these panels have provided novel insights into metabolism. For example, although calorie restriction is widely thought to mediate a prolonged life span in mice (28), when this was examined across 41 different inbred ILSXISS mouse strains there was profound variation in the response. Although calorie restriction was found to extend life span in a few strains, ∼10 strains underwent life-shortening in response to the same manipulation (29). Similarly, marked variation in the metabolic response to high-fat, high-sucrose feeding was reported across more than 100 inbred HMDP mouse strains (23), and diversity has also been reported among commonly used mouse strains (22).
In addition to genetic diversity in metabolism, it has also been reported that individual mice from exactly the same genetic background also display considerable variability in their metabolic response to diet, further adding to the complexity of metabolic studies in mice. For example, a 4-fold difference in adiposity was observed in C57Bl/6J mice fed a high fat diet (30), and others have reported similar variability in body weight and glucose tolerance in this strain (31). Despite these reports, individual variability has been largely ignored as most studies of insulin resistance consider averaged data from groups of mice.
These present findings highlight the key role of genetics and possibly epigenetics in metabolic homeostasis and the complex interplay between genes and the environment in determining the risk of metabolic disease. Here, we encapsulate environmental, genetic, as well as individual variability and focus on the metabolome as a read-out of the integrated contribution of these parameters to a range of phenotypes, most notably insulin resistance. We studied three inbred ILSXISS strains on two different diets and subjected them to metabolic phenotyping and metabolomics analysis of skeletal muscle. Using machine learning, we have identified a novel insulin sensitivity signature in muscle, comprising C22:1-CoA, C2-carnitine and C16:0-ceramide.
Results
Metabolic heterogeneity between strains
We have studied the metabolic response to a diet high in fat and sucrose (HFD)5 in three distinct inbred mouse strains from the ILSXISS cross (24, 25, 29). Specific strains were selected based on their divergent responses to calorie restriction (29), with strain #89 showing extended life span, #97 reduced life span, and #50 showed no change in life span in response to calorie restriction. The three strains differed in body weight on a chow diet (Fig. 1A), and HFD increased body weight in two of the three strains (Fig. 1B). Body composition measured after 4 weeks on the diet revealed HFD increased adiposity in all strains (Fig. 1C). Although strain #89 appeared resistant to HFD-induced weight gain, this strain showed a HFD-induced increase in adiposity accompanied by a decrease in lean mass (Fig. 1D). Oxygen consumption, metabolic rate, energy intake, and ambulatory activity were similar between strains (Fig. 1E, data not shown).
Figure 1.
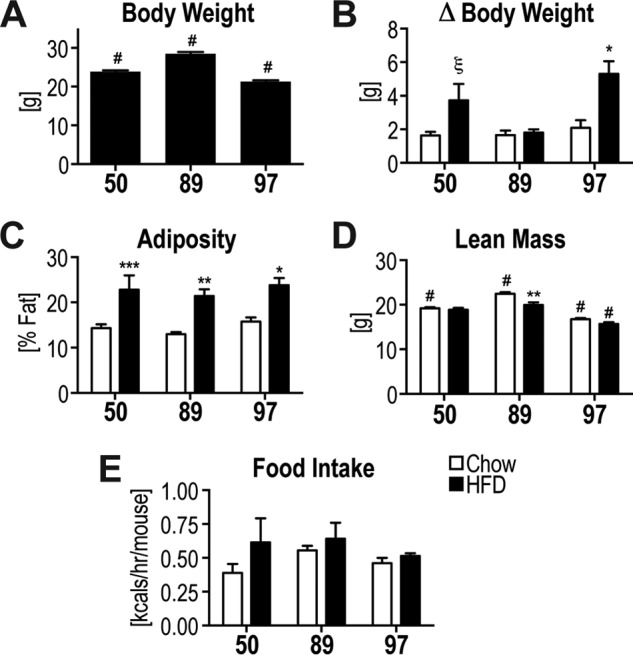
Variability in body composition in response to HFD. A, starting body weight. B, change in body weight during 6-week diet intervention. C–E, body composition analysis determined adiposity (C) and lean mass (D) at week 4 of the diet intervention. E, energy intake of mice is shown. Data are mean ± S.E., n = 5–11 (A–D) and n = 3–5 (E); * different from corresponding chow group: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ξ, p < 0.05 (one-tailed t test); # different from all other strains p < 0.05.
Marked variability was evident in glucose tolerance between the strains (Fig. 2A). Strain #50, and to a lesser degree #89, exhibited HFD-induced glucose intolerance as apparent from the incremental area under the curve (iAUC) of the glucose tolerance test (GTT) (Fig. 2B). Strain #89 was significantly more glucose-tolerant than the other strains both under chow and HFD-fed conditions (Fig. 2B). In fact, the glucose tolerance observed in strain #89 fed a HFD was indistinguishable from that observed in the other strains when fed a chow diet (Fig. 2B). Strains #50 and #97 developed hyperinsulinemia upon HFD feeding, both under fasting conditions and during the GTT (Fig. 2, D and E). The hyperinsulinemia in strain #97 was much higher than in other strains, and this probably accounted for the normal glucose tolerance upon HFD feeding (Fig. 2, A, B, D, and E). Only strain #50 developed fasting hyperglycemia on HFD (Fig. 2C), and strain #97 displayed significantly lower fasting glucose on HFD compared with the other strains on HFD (Fig. 2C). Despite the widespread variability in glucose tolerance/hyperinsulinemia, insulin action assessed ex vivo in white adipose tissue (WAT) explants was impaired in all HFD-fed strains at 10 nm insulin (Fig. 2F). Interestingly, only strain #97 displayed reduced 2-deoxyglucose (2DG) uptake in the presence of the more physiological 0.5 nm insulin dose. This strain also showed a significantly higher insulin-stimulated 2DG uptake on chow diet than any other strain (Fig. 2F).
Figure 2.
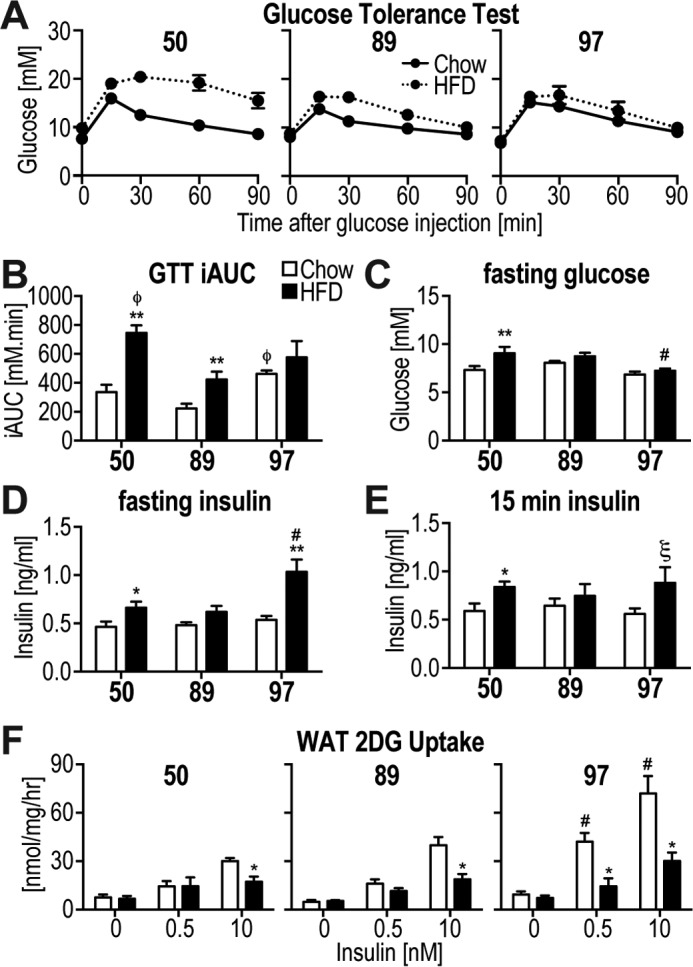
Variability in metabolic phenotypes in response to HFD. A and B, intraperitoneal GTT at a glucose dose of 2 g/kg lean mass was performed on the mice at week 5 of the diet intervention. A, blood glucose was measured during the GTT. B, iAUC analysis from data in A. C, fasting blood glucose was assessed. D and E, blood insulin was assessed after fasting (D) and at 15 min following glucose injection (E). F, 2-[3H]deoxyglucose uptake was assessed in response to indicated doses of insulin in WAT explants. Data are mean ± S.E., n = 5–10 (A–E), n = 5–6 (F); *, different from corresponding chow group: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ξ, p < 0.05 (one-tailed t test); #, different from all other strains p < 0.05; φ, different from strain #89 p < 0.05.
113 metabolites significantly correlated with metabolic parameters
The large variability in metabolic phenotypes observed in these mice upon HFD feeding, ranging from marked glucose intolerance (strain #50), hyperinsulinemia with normal glucose tolerance (strain #97), to mild glucose intolerance that was indistinguishable from other strains on chow diet (strain #89), provided an ideal cohort to identify metabolites associated with insulin resistance across different strains and diets. Here, we studied skeletal muscle due to its prominent role in whole-body insulin sensitivity. We subjected skeletal muscle (quadriceps) from the three strains fed two diets to metabolomics assessment using mass spectrometry and measured 165 metabolites in 29 mice. For further analyses, we also included sums and ratios of some of the metabolites, resulting in a total of 218 metabolite parameters (supplemental Table S1). We performed correlation analyses whereby each metabolite was correlated with each metabolic phenotype, including GTT iAUC, fasting glucose, fasting insulin, adiposity, HOMA-IR, WAT 2DG uptake, and a factor referred to here as the “insulin resistance index” (IRI), calculated as GTT iAUC × fasting insulin. To take into account the genetic and in particular the individual variability, rather than grouping mice and using averaged data, each mouse was used as an individual data point (n = 29). We identified 113 metabolite parameters that correlated with one or more of the metabolic phenotypes (Fig. 3A and supplemental Table S1). Of these, 44 metabolites correlated with IRI, including metabolites previously implicated in insulin resistance such as long-chain (LC) and very-long-chain (VLC) CoAs and carnitines (13). Nine of these showed a correlation coefficient of ≥0.5 or ≤ −0.5 (Fig. 3B).
Figure 3.
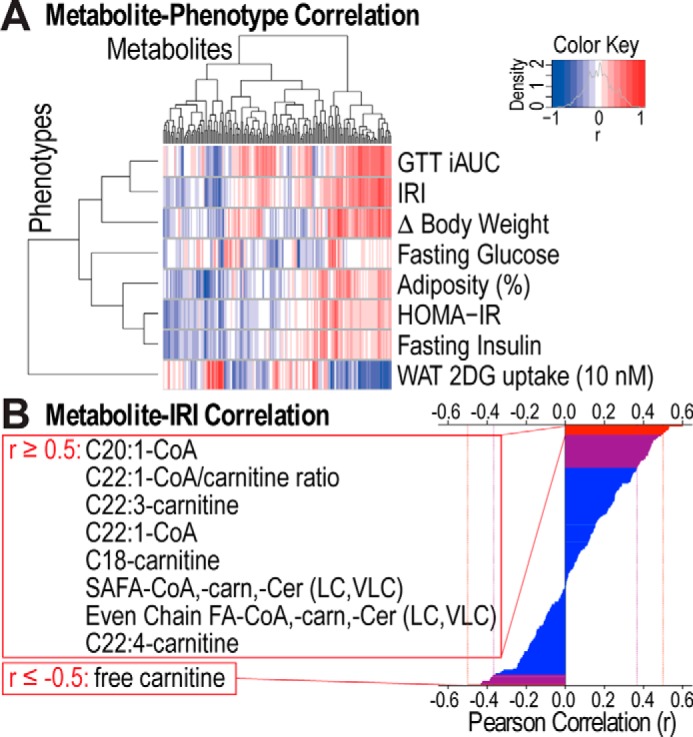
113 metabolites significantly correlate with metabolic phenotypes. A, hierarchical clustering and heat map of Pearson correlation coefficients of correlations between metabolites and metabolic phenotypes are displayed. Blue and red corresponds to negative or positive correlation, respectively. B, Pearson correlation coefficients of correlations between metabolites and IRI are shown. 44 metabolites significantly correlated with IRI (purple and red) and 9 of these had a correlation coefficient ≥0.5 or ≤ −0.5 (red). FA, fatty acyl; SAFA, saturated FA; carn, carnitine; Cer, ceramide; LC, long-chain; VLC, very-long-chain.
Metabolites changed with insulin resistance, diet, and between strains
We next wanted to assess the relationship between individual metabolites and various features of our cohort, including insulin sensitivity, diet, or strain of origin. To assess insulin sensitivity, we employed an insulin resistance index or IRI that relied on both glucose tolerance and insulin levels. This was necessary as use of either of these parameters alone would have mis-represented animals with normal glucose tolerance but substantial hyperinsulinemia or vice versa. As shown in Fig. 4, there was a 12-fold range in IRI across all animals. An upper tertile cutoff was used to separate insulin-resistant from insulin-sensitive mice. Intriguingly, although most insulin-resistant mice were fed a HFD, it is noteworthy that there were several HFD-fed mice among the insulin-sensitive mice (Fig. 4, arrowheads). Notably, most of the latter mice were from strain #89. This highlights the importance of including this strain in the analysis.
Figure 4.
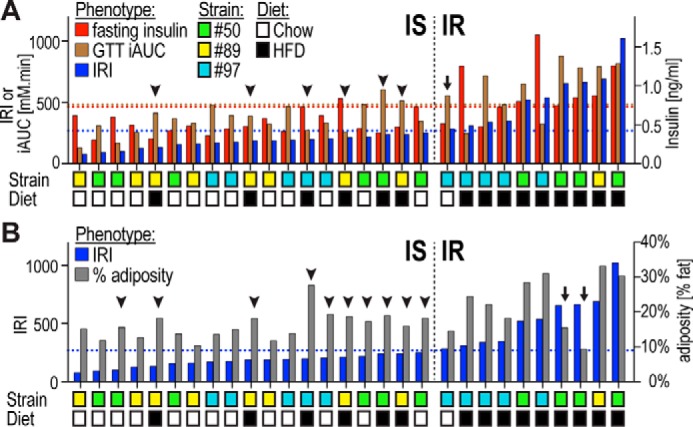
Metabolic phenotypes in individual mice. A and B, indicated metabolic phenotypes are shown in 29 individual mice ranked for insulin resistance as assessed by the IRI. A, insulin-resistant (IR) chow-fed mice (arrow) and insulin-sensitive (IS) HFD-fed mice (arrowheads) are indicated. B, two HFD-fed IR mice (arrows) displayed lower adiposity than any HFD-fed IS mice or even some chow-fed IS mice (arrowheads). Upper tertile cutoffs of metabolic phenotypes are indicated with dotted lines.
Table 1 summarizes significant differences in metabolic phenotypes in the 29 mice when grouped based on insulin resistance, diet, or strains. As expected, adiposity, GTT AUC and iAUC, fasting insulin and glucose, and IRI and HOMA-IR were all significantly changed with insulin resistance and with diet (Table 1). Although adiposity was significantly increased in insulin-resistant mice (Table 1), it appears that adiposity was not driving the classification of some HFD-fed mice into the insulin-sensitive group: two HFD-fed insulin-resistant mice displayed lower adiposity than any of the HFD-fed insulin-sensitive mice and even some of the chow-fed insulin-sensitive mice (Fig. 4B, arrows and arrowheads). Somewhat unexpected were the changes between strains as they were grouped independently from diet. Notably, strain #97 showed significantly lower body weight compared with both other strains, and strain #89 displayed significantly higher fasting glucose than the other strains. The GTT AUC was significantly different between strains #50 and #97.
Table 1.
Metabolic phenotypes of mouse groups based on IR, diet, or strain
Significantly different phenotypes (p < 0.05) are indicated in bold.
| Phenotypes | IR |
Diet |
Strain |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low IRI | High IRI | p value | Chow | HFD | p value | #50 | #89 | #97 | p value 50 vs. 89 | p value 50 vs. 97 | p value 89 vs. 97 | |
| Body weight (g) | 24.7 | 25.0 | 0.8123 | 24.5 | 25.1 | 0.5759 | 25.3 | 27.2 | 22.2 | 0.1061 | 0.0022 | 0.0002 |
| Adiposity (%) | 16.1 | 22.7 | 0.0057 | 14.5 | 22.0 | 0.0007 | 17.9 | 17.1 | 20.0 | 0.7985 | 0.4815 | 0.3455 |
| GTT AUC | 1058 | 1345 | 0.0041 | 1018 | 1286 | 0.0050 | 1321 | 1104 | 1039 | 0.1212 | 0.0314 | 0.3980 |
| GTT iAUC | 360 | 624 | 0.0002 | 354 | 542 | 0.0085 | 541 | 379 | 427 | 0.1251 | 0.2140 | 0.5400 |
| Fasting glucose (mm) | 7.6 | 7.2 | 0.3816 | 7.4 | 7.6 | 0.6647 | 7.2 | 8.6 | 6.7 | 0.0080 | 0.2447 | 0.0014 |
| Fasting insulin (ng/ml) | 0.52 | 0.91 | 0.0003 | 0.51 | 0.79 | 0.0093 | 0.66 | 0.58 | 0.72 | 0.4673 | 0.6900 | 0.3449 |
| IRI (1000×) | 177 | 537 | 0.0000 | 173 | 420 | 0.0013 | 392 | 225 | 277 | 0.1736 | 0.2825 | 0.4572 |
| HOMA-IR | 4.3 | 7.5 | 0.0029 | 4.1 | 6.7 | 0.0153 | 5.4 | 5.4 | 5.5 | 0.9811 | 0.9795 | 0.9604 |
We next examined metabolite changes between insulin-resistant and insulin-sensitive mice. Of the 44 metabolites that significantly correlated with IRI (Fig. 3B), C22:1-CoA was the only metabolite that was significantly changed (q <0.05) with a 2.3-fold increase in insulin-resistant mice (Fig. 5).
Figure 5.
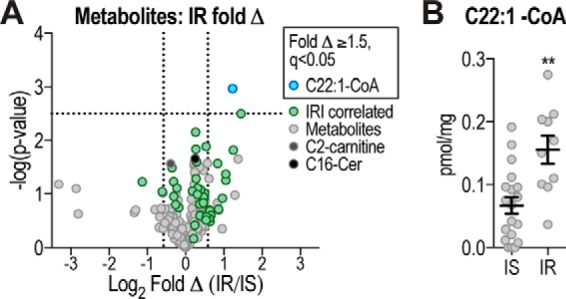
Metabolites changed with insulin resistance. A, volcano plot shows metabolite log2 fold change (Δ) of IR compared with IS mice plotted against −log10 p value, indicating C22:1-CoA as significantly changed (q <0.05, above horizontal line) metabolite. Vertical lines indicate ±1.5-fold change (±0.58 log2 fold change). C16:0-ceramide and C2-carnitine are indicated. B, C22:1-CoA levels in IR and IS mice are shown. Data are mean ± S.E., n = 10–19; **, p < 0.01.
Analysis of metabolite changes between HFD and chow-fed mice identified seven metabolites that were significantly (q <0.05) changed ≥1.5-fold (Fig. 6A). Notably, C22:1-CoA was not among these metabolites. C19:3-CoA was the most significantly increased metabolite in HFD mice, whereas other metabolites (free carnitine and C2-carnitine) were decreased in HFD mice (Fig. 6, A and B). C19:3-CoA as well as other polyunsaturated acyl-CoAs likely originated from the HFD itself. Notably, there was no overlap between metabolite changes with insulin resistance or diet, which is likely due to the fact that some mice fed a HFD were quite insulin-sensitive and vice versa (Fig. 4).
Figure 6.
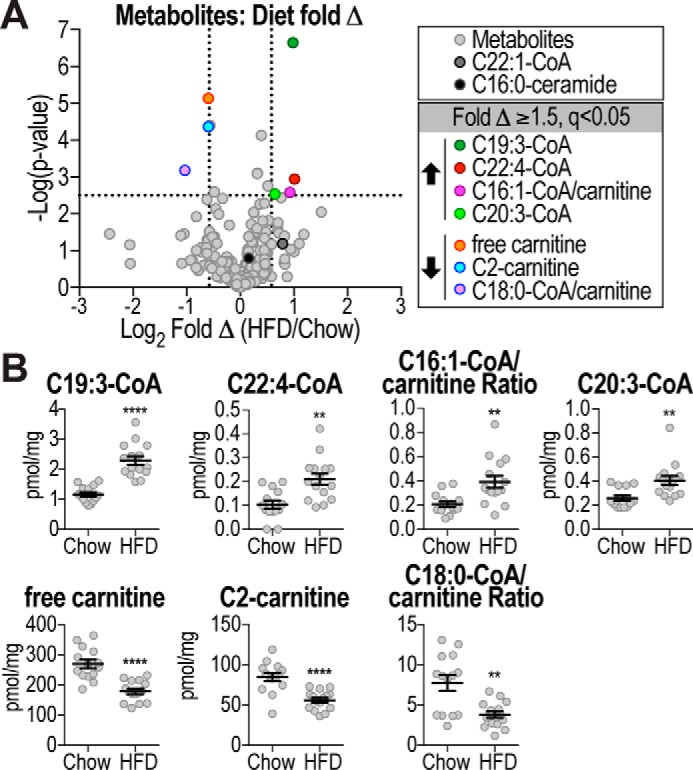
Metabolites changed with diet. A, volcano plot shows metabolite log2 fold change (Δ) of HFD-fed mice compared with chow-fed mice plotted against −log10 p value, indicating significantly changed (q <0.05, above horizontal line) metabolites. Vertical lines indicate ±1.5-fold change (±0.58 log2 fold change). C22:1-CoA and C16:0-ceramide are indicated. B, metabolite levels or ratios of significantly changed metabolites (fold Δ ≥1.5, q<0.05) in chow and HFD-fed mice are shown. Data are mean ± S.E., n = 14–15; **, p < 0.01; ****, p < 0.0001.
Interestingly, a number of metabolites were significantly changed between the different strains (Fig. 7). Arg and ratios comprising Arg, were significantly increased in strain #50 compared with strains #89 and #97 (Fig. 7, A and B). Other metabolites that were different in strain #50 compared with other strains included C14:0-ceramide and malate/citrate ratio. Only two metabolites were significantly altered between strain #97 and #89, Tyr and C16:2-CoA (Fig. 7, A and B).
Figure 7.

Metabolites changed between strains. A, volcano plots show metabolite log2 fold changes (Δ) between different strains plotted against −log10 p value, indicating significantly changed (q <0.05, above horizontal line) metabolites. Dotted lines indicate ±1.5-fold change (±0.58 log2 fold change). C22:1-CoA, C2-carnitine, and C16:0-ceramide are indicated. B, metabolite levels or ratios of significantly changed metabolites (fold Δ ≥1.5, q <0.05) between strains are shown. Data are mean ± S.E., n = 9–10; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Orn, ornithine; Cit, citrulline; Asx, Asp/Asn.
In summary, distinct and non-overlapping metabolites were changed with insulin resistance or diet or between strains. This outcome emphasizes the usefulness of including different strains in the analysis that are less susceptible to HFD-induced insulin resistance (such as the #89 strain), thereby making it feasible to differentiate between HFD feeding and insulin resistance per se.
Insulin resistance classification analysis and novel signature
Given that the metabolite changes with HFD or insulin resistance were distinct and the HFD-fed mice were distributed across a range of insulin sensitivities, we reasoned that these 29 mice present an ideal cohort to perform unbiased classification analyses. We used machine learning to perform classification analyses to determine the classification accuracy (CA) of metabolites in distinguishing mice based on insulin resistance, diet, or strain, using the ensemble learner, random forest (32, 33). Similar to that described above, this was performed using data from individual mice, whereby each mouse's metabolite(s) were utilized to classify that mouse into a certain group, and the accuracy reflects “observed” versus “true” classification. In this analysis, a ratio of 2 represents 100% CA, whereas a ratio of 1 reflects random chance (34). We assessed the CA ratio of the significant 113 metabolites (Cor Met) to classify mice based on insulin resistance (1.37 CA ratio), diet (1.85), and strain (1.72), and the Cor Met signature classified mice significantly better than random chance in all three cases (Fig. 8A). The classification analysis allows determination of the contribution of each individual metabolite to the overall classification accuracy of the signature by using “leave-1-out” analysis. In addition to C22:1-CoA, which was also found to be elevated in insulin-resistant mice (Fig. 5), three additional metabolites were identified that classified the mice according to insulin resistance: C19:3-CoA, C2-carnitine, and C16:0-ceramide (Table 2). This is interesting as C19:3-CoA and C2-carnitine were significantly changed with diet (Fig. 6) and were also among the best diet classifiers (Table 2). This is not surprising as the insulin-resistant group is over-represented with HFD-fed mice (Fig. 4). In addition, ceramide has been identified as a major driver of insulin resistance in other studies (35, 36). Arg and Arg-based metabolites were among the best strain classifiers (Table 2).
Figure 8.
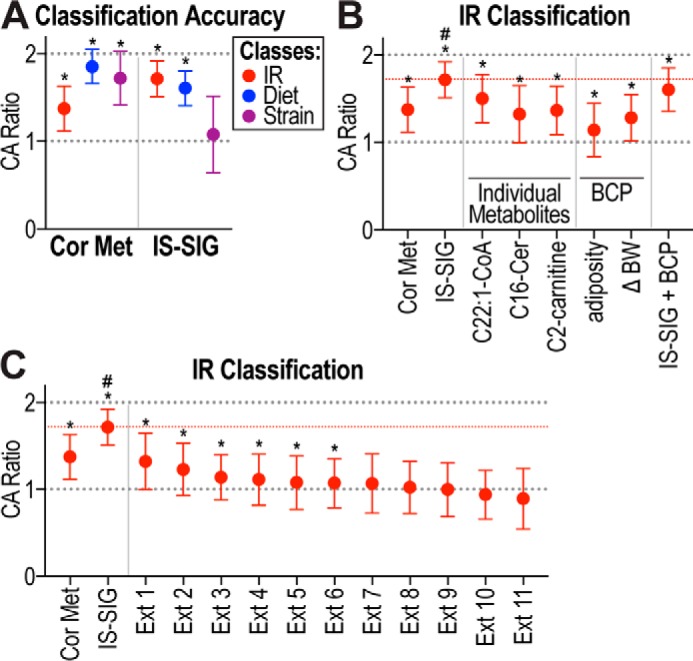
Classification analysis and novel IS-SIG. A, CA ratios are shown of Cor Met and IS-SIG to classify mice based on IR, diet, or strain. B and C, CA ratios are shown of Cor Met, IS-SIG, individual metabolites, body composition parameters (BCP), including adiposity and body weight change (Δ BW), or external signatures (Ext) to classify mice based on IR. CA ratio of 2 represents 100% accuracy, and CA ratio of 1 represents random chance (dotted lines). Median CA ratio of IS-SIG is indicated with a red dotted line. Data are shown as median CA ratio ± confidence limits calculated over 500–1000 bootstrapping runs; *, p < 0.001 compared with random (dotted line at 1); #, p < 0.0001 compared with all other signatures. Cer, ceramide.
Table 2.
Best classifiers for classification based on IR, diet, and strain
The four best classifiers are listed for classification based on IR, diet, and strain. Metabolite levels (pmol/mg) or ratios are shown of mice grouped based on IR, diet, or strain. Significantly changed metabolites (q <0.05) are indicated in bold. Orn, ornithine; Cit, citrulline; SC, short-chain; IR, insulin resistance.
| IR |
Diet |
Strain |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low IRI | High IRI | q value | Chow | HFD | q value | #50 | #89 | #97 | q value 50 vs. 89 | q value 50 vs. 97 | q value 89 vs. 97 | |
| IR classifiers | ||||||||||||
| C22:1-CoA | 0.066 | 0.156 | 0.0484 | 0.071 | 0.122 | 0.2529 | 0.106 | 0.081 | 0.103 | 0.7102 | 0.9632 | 0.7709 |
| C19:3-CoA | 1.51 | 2.19 | 0.0976 | 1.16 | 2.29 | 0.0001 | 1.99 | 1.73 | 1.52 | 0.7102 | 0.4138 | 0.7559 |
| C2-carnitine | 76 | 58 | 0.1232 | 85 | 56 | 0.0024 | 75 | 70 | 64 | 0.8266 | 0.3178 | 0.8785 |
| C16-Ceramide | 0.875 | 1.041 | 0.4172 | 0.880 | 0.980 | 0.4450 | 0.872 | 0.891 | 1.03 | 0.8833 | 0.2381 | 0.5277 |
| Diet classifiers | ||||||||||||
| C19:3-CoA | 1.514 | 2.193 | 0.0976 | 1.163 | 2.294 | 0.0001 | 1.99 | 1.73 | 1.52 | 0.7102 | 0.4138 | 0.7559 |
| Free carnitine | 239 | 193 | 0.1673 | 270 | 179 | 0.0008 | 202 | 194 | 271 | 0.8710 | 0.1459 | 0.1697 |
| Sum of SC-carnitines | 84 | 65 | 0.1325 | 93 | 63 | 0.0024 | 84 | 76 | 71 | 0.7102 | 0.2727 | 0.9323 |
| C2-carnitine | 76 | 58 | 0.1232 | 85 | 56 | 0.0024 | 75 | 70 | 64 | 0.8266 | 0.3178 | 0.8785 |
| Strain classifiers | ||||||||||||
| Arg | 92 | 125 | 0.6844 | 98 | 108 | 0.9001 | 191 | 58 | 56 | 0.0171 | 0.0061 | 0.9567 |
| Arg/(Orn + Cit) ratio | 0.660 | 0.770 | 0.8058 | 0.695 | 0.698 | 0.9812 | 1.206 | 0.465 | 0.395 | 0.0016 | 0.0002 | 0.1939 |
| C12-OH/C10-DC-carnitine | 0.055 | 0.081 | 0.4172 | 0.060 | 0.068 | 0.7724 | 0.065 | 0.042 | 0.083 | 0.1848 | 0.4498 | 0.1892 |
| Arg/Orn ratio | 2.698 | 3.719 | 0.6217 | 2.791 | 3.292 | 0.7473 | 4.878 | 1.928 | 2.232 | 0.0171 | 0.0183 | 0.6126 |
We next tested whether a signature composed of the best insulin resistance classifiers could improve the CA observed with the Cor Met signature. C19:3-CoA was not included in this signature as, while being a top insulin resistance classifier, it was the most significantly increased metabolite in HFD-fed mice and likely originated directly from the diet. The novel signature, here referred to as IS-SIG, comprises C22:1-CoA, C2-carnitine, and C16:0-ceramide. IS-SIG classified mice based on insulin resistance (1.72 CA ratio) more accurately than Cor Met (1.37) and better than based on diet (1.61), while performing poorly in classifying mice according to their strain of origin (1.08) (Fig. 8A). We next tested the contribution of individual metabolites within this signature to the classification performance/accuracy of IS-SIG and determined the classification accuracy of these three metabolites individually. Although all three metabolites individually performed significantly better than random chance, they did not perform as well as IS-SIG (C22:1-CoA, 1.5; C2-carnitine, 1.36; and C16:0-ceramide, 1.32) in classifying mice based on insulin resistance (Fig. 8B).
Increases in body weight or adiposity have been associated with insulin resistance, and these parameters were significantly correlated with IRI in our cohort (Table 1 and supplemental Table S1). We therefore determined whether these body composition parameters were able to classify mice based on insulin resistance on their own or whether they improved the IS-SIG classification accuracy when combined with this metabolite signature (Fig. 8B). Intriguingly, body weight change (ΔBW, 1.28 CA ratio) or adiposity (1.14) performed much worse than IS-SIG (1.72) and did not improve the classification accuracy of IS-SIG when combined with IS-SIG, but rather reduced its classification accuracy (1.60). These data show that insulin resistance is independent of body weight gain or adiposity in our cohort, which is consistent with the observed diversity of adiposity in both insulin-sensitive and -resistant mice (Fig. 4B).
We next compared the classification accuracy of Cor Met and IS-SIG to distinguish mice based on insulin resistance with 11 metabolite signatures (Table 3) that were previously shown to correlate with insulin resistance (35–46). Only 6 of the 11 published signatures performed significantly better than random chance (Fig. 8C). A number of these signatures contained ceramides (Table 3, Ext 1, 3, 6, 9, and 11), and their performance depended on the combination and nature of the ceramide. For example, Ext 1 was C16-ceramide (35, 36) that was also part of IS-SIG (Fig. 8B) and performed the best out of all external signatures, whereas two signatures did not perform well (e.g. C14- (Ext 9) or C18-ceramide (Ext 11)). Branched chain amino acids were also included in a number of these signatures (Table 3, Ext 2, 4, 5, 8, and 10), and also their classification accuracy varied and relied on inclusion of additional metabolites (Table 3). In summary, IS-SIG performed significantly better than any of the tested external signatures in distinguishing mice based on insulin resistance (Fig. 8C).
Table 3.
External signatures
Metabolites, species, sample origin, and references are listed for 11 external signatures (Ext 1–11). carn, carnitine; Cer, ceramide; Glx, glutamate/glutamine.
| Signature | Ext 1 | Ext 2 | Ext 3 | Ext 4 | Ext 5 | Ext 6 | Ext 7 | Ext 8 | Ext 9 | Ext 10 | Ext 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Refs. | 35, 36 | 44 | 38 | 45 | 37 | 41 | 42 | 46 | 39 | 43 | 40 |
| Species | Mouse | Human | Human | Human | Human | Human | Human | Human | Human | Human | Human |
| Sample | Adipose/Liver | Plasma | Plasma | Plasma | Serum | Muscle | Serum | Plasma | Plasma | Serum | Muscle |
| Metabolites | C16-Cer | Val | Sum Cer | Val | Val | Sum Cer | Gly | Val | Sum Cer | Val | C18-Cer |
| Leu/Ile | C18-Cer | Leu/Ile | Leu/Ile | C14-Cer | Leu/Ile | C14-Cer | Leu/Ile | ||||
| Gly | C20-Cer | C3-carn | C3-carn | C16-Cer | Ala | C3-carn | |||||
| Phe | C24-Cer | C5-carn | C5-carn | C24:1-Cer | Glx | Phe | |||||
| Glx | C24:1-Cer | Ala | Phe | Tyr | |||||||
| Glx | Tyr | ||||||||||
| Met | Pyruvate | ||||||||||
| Phe | Malonyl/hydroxy-butyryl-CoA | ||||||||||
| Tyr |
Discussion
This study adds to the growing literature on the significant contribution of genetic diversity to environmental influences on metabolism. Here, we studied metabolism of several inbred mouse strains in response to HFD feeding, and we observed a diverse response ranging from robust β-cell compensatory response with no glucose intolerance to markedly impaired glucose tolerance in response to HFD. Conversely, impaired adipocyte insulin action was observed in all strains. Hence, as is also emerging from human studies (2), this study shows that the metabolic response to the environment, such as diet, is highly divergent in large part due to genetic diversity but also due to individual variance. By embracing this diversity and studying muscle metabolomics in a cohort of mice across three different strains and two diets, we identified a novel muscle metabolic signature that is diagnostic of insulin resistance.
Wide ranging phenotypic diversity was observed across the strains in response to HFD particularly in glucose tolerance (Fig. 2). Strain differences in diet-induced glucose intolerance have previously been documented (17, 20, 22). Of the three ILSXISS lines tested in this study, strain #97 resisted changes in glucose tolerance following HFD similar to reports in BALB/c mice (22). However, unlike BALB/c mice, protection from glucose intolerance in strain #97 appeared to be driven by hyperinsulinemia (Fig. 2). Moreover, this strain became obese on a HFD despite normal glucose tolerance (Figs. 1 and 2). Strain #89 developed mild glucose intolerance in response to a HFD with no changes in body weight or fasting/fed insulin (Figs. 1 and 2), and four of the five HFD-fed mice of this strain were classified insulin-sensitive (Fig. 4). Together with previous studies (17, 20, 22, 23), these findings confirm that genetic diversity governs the metabolic response to calorie-excess diets and show that this response is highly variable between strains involving a complex combinatorial pattern of changes in β-cells, liver, muscle, and adipocyte function.
Three metabolites were identified as the strongest classifiers when mice were distinguished based on insulin sensitivity, C22:1-CoA, C2-carnitine, and C16-ceramide (Table 2), and when combined into a signature (IS-SIG) these metabolites classified mice based on insulin resistance more accurately than each of the metabolites individually, Cor Met, any previously published external signature, or body composition parameters (Fig. 8, B and C). The IS-SIG metabolite C22:1-CoA/erucoyl-CoA was significantly correlated with insulin resistance (Fig. 3B) and was the only metabolite that was significantly elevated in insulin-resistant mice (Fig. 5). Most notably, changes in this metabolite did not correspond to diet or mouse background (Figs. 6 and 7). In support of a role in insulin sensitivity, serum levels of erucic acid were reported to be significantly correlated with insulin resistance in humans with type 2 diabetes (47). Mice are capable of de novo synthesis of erucic acid (48), and the elongase required for its synthesis (Elovl3) is highly induced in brown adipose tissue upon cold exposure, linking it to metabolism (49). Whole-body deletion of Elovl3 in mice results in reduced barrier function of the skin, reduced lipid accumulation in brown adipose tissue, and resistance to diet-induced obesity (50–52). Notably, Elovl3 also synthesizes C20:1-CoA, the metabolite that was most correlated with insulin sensitivity (Fig. 3), suggesting that Elovl3 and its products might play a role in insulin resistance. Furthermore, erucic acid has a low oxidation rate, and it inhibits mitochondrial oxidation (53–55). This is of interest as mitochondrial dysfunction has been implicated in insulin resistance (56). Future studies will be required to explore the mechanistic link between this metabolite and insulin resistance in muscle. The second IS-SIG metabolite, C2-carnitine or acetyl-carnitine, was significantly reduced upon HFD feeding (Fig. 6) and was a strong diet classifier (Table 2). The reduction in C2-carnitine levels in muscle might reflect inefficient fatty acid breakdown and has been linked to diminished activity of carnitine acetyltransferase and/or reduced glucose flux through the pyruvate dehydrogenase complex (57–59). The third IS-SIG metabolite, C16:0-ceramide, has previously been implicated in insulin resistance (35, 36). C16:0-ceramide was elevated in WAT from obese humans and mice (36) and genetic manipulations that modulated C16:0-ceramide globally, in brown adipose tissue, or in liver affected whole-body insulin sensitivity (35, 36). Although C16:0-ceramide was not changed in insulin-resistant mice (Fig. 5), it was identified as a strong insulin resistance classifier using classification analysis (Table 2), and it was capable of distinguishing mice based on insulin resistance better than other external signatures (Fig. 8C, Ext 1). Although other studies specifically focused on the role of C16:0-ceramide (35, 36), here we identified this metabolite out of 218 metabolites as an insulin resistance diagnostic in a totally unbiased manner using machine learning.
The fact that the three metabolites combined (IS-SIG) were a better diagnostic of insulin resistance than each metabolite individually (Fig. 8B) could suggest that they are characteristic of distinct modes of insulin resistance raising the possibility that different strains exhibit different mechanisms of insulin resistance, a fact that has been largely overlooked in studies using single mouse strains. In support of this, C2-carnitine and C22:1-CoA, both of which might be indicative of mitochondrial dysfunction, were not correlated with each other (data not shown). This suggests that although these metabolites might be diagnostic of a common defect, their mechanism is likely different. Notably, the inclusion of body composition parameters worsened IS-SIG's performance in distinguishing mice based on insulin resistance, indicating that the signature acts independently of adiposity or body weight change (Fig. 8B).
A number of metabolites were significantly changed with diet or between strains (Figs. 6 and 7). Polyunsaturated LC and VLC acyl-CoAs were increased with HFD, suggesting these are likely acquired via the diet with lard and safflower oil as the sources of fat. Two metabolites that were decreased in skeletal muscle from HFD-fed mice were free carnitine and C2-carnitine (mentioned above). The diminution in free carnitine could stem from increased production of the LC acyl-carnitines, due to a rise in fatty acid supply (57). Although the HFD-mediated increased metabolites are likely diet-specific, reduced free carnitine and/or C2-carnitine might be useful biomarkers to determine food intake or, in particular, fat intake in studies involving humans.
Surprisingly, a number of metabolites were significantly different between strains, including Arg, Tyr, and C14-ceramide (Fig. 7). These metabolites did not change with either diet or insulin sensitivity. Notably, some of the external signatures contained Tyr (Ext 5, 8, and 10) or C14-ceramide (Ext 6 and 9), and this possibly negatively affected their performance in classifying mice based on insulin resistance (Fig. 8C). Hence, these metabolites are likely tightly controlled by the genetic variance between these strains but in a manner unrelated to insulin sensitivity.
The ILSXISS lines were originally derived from an 8-way-cross of distinct inbred mouse lines (24, 25). Given that some of these parental strains differ in their susceptibility to develop diet-induced metabolic disease (17, 20, 22, 23), the observed variance in metabolic responses to HFD observed herein was somewhat predictable. The novel utility of the present findings lies in the unique combinations of phenotypes present within the ILSXISS strains with respect to various hallmark indicators of the metabolic syndrome as well as the use of individual animals in the analysis and machine-learning-mediated classifications. The phenotypic diversity makes the ILSXISS cross (24, 29), as well as similar murine crosses (23, 26, 27), powerful tools in the search for mechanistic understanding related to nutrient-induced disease. Future studies incorporating larger numbers of strains and other metabolic tissues will allow for the capture of even greater phenotypic and metabolomic diversity, more closely resembling that observed in human studies, and this will subsequently lend itself to identification of underlying genetic drivers.
In conclusion, in this study we have exploited the variance in insulin sensitivity between different inbred mouse strains, between individual mice within an inbred strain, and between mice fed different diets to identify metabolites in skeletal muscle that best classify mice according to this variance in insulin sensitivity. Machine learning identified an insulin sensitivity signature that comprises three metabolites, C22:1-CoA, C2-carnitine, and C16:0-ceramide. Previous studies have implicated both ceramides and acylcarnitines as major contributors to insulin resistance in muscle highlighting the significance of this approach. C22:1-CoA, a novel metabolite implicated in insulin resistance in this study, is of major interest as this metabolite inhibits mitochondrial oxidative capacity, and serum C22:1 fatty acid levels are correlated with human insulin resistance. Strikingly, the contribution of each of these metabolites on their own to insulin sensitivity was considerably less than their combined contribution, suggesting that there are likely multiple pathways that contribute to insulin resistance and providing major justification for the development of multiparametric signatures that classify insulin sensitivity at a population level. These studies provide an important proof-of-principle for exploiting omics platforms such as metabolomics that represent an integrated output of multiple variables, including genetic, environmental, and possibly epigenetic variables for identifying the full repertoire of pathways and factors that contribute to insulin resistance at a population level.
Experimental procedures
Animals and metabolic phenotypes
All experiments were approved by the Garvan Institute and University of Sydney Animal Ethics Committees. Male mice were group-housed on a 12-h light/dark cycle with free access to chow diet (13% calories from fat, 65% carbohydrate, and 22% protein) or HFD (47% fat (7:1 lard-to-safflower oil ratio), 32% carbohydrate, and 21% protein) and water. Body composition, indirect calorimetry, GTTs, and insulin measurements were performed as described previously (60, 61). After 6 weeks on the diet and a 4-h fast, mice were anesthetized at ∼11 a.m. with isofluorane, and tissues were removed and freeze-clamped, immediately submerged into liquid N2, and stored at −80 °C. 2-[3H]Deoxyglucose uptake into epidydimal fat explants was performed as described previously (60). The IRI was calculated as fasting insulin (ng/liter) multiplied by the iAUC of the GTT (mm·min). HOMA-IR was calculated as fasting insulin (microunits/ml) multiplied by fasting glucose (mm) and divided by 22.5.
Metabolomics assessment
Whole-quadriceps skeletal muscle was powdered under liquid N2, aliquoted, lysed in appropriate buffer (50% acetonitrile, 0.3% formic acid; except for acyl-CoAs: 50% 2-propanol, 50% 0.1 m KH2PO4, pH 4.45) using a Tissue Lyzer II (Qiagen), and subjected to metabolomics analysis using stable isotope dilution techniques. Amino acids and acylcarnitine were measured as described previously (62, 63) using a Waters Acquity UPLC system equipped with a TQD and MassLynx 4.1 operating system. Organic acids were quantified as described previously (64) employing Trace Ultra GC coupled to ISQ MS operating under Xcalibur 2.2 (Thermo Fisher Scientific). Ceramides and acyl-CoA esters were extracted, purified, and analyzed as described previously (65–67). Acyl-CoAs were analyzed by flow injection analysis using positive electrospray ionization on a Waters Xevo TQS, employing methanol/water (80:20%, v/v) containing 30 mm NH4OH as the mobile phase. Spectra were acquired in the multichannel acquisition mode monitoring the neutral loss of 507 atomic mass units (phosphoadenosine diphosphate) and scanning from m/z 750 to 1060. Heptadecanoyl-CoA was employed as an internal standard for LC and VLC CoA esters. CoAs were quantified using authentic saturated (C0-C18) and unsaturated (C16:1, C18:2, C18:1, and C20:4) acyl-CoA calibrators. All reported CoAs, including low abundant C22:1-CoA, were within detection limits of the assay. Corrections for heavy isotope effects, mainly 13C, to the adjacent m + 2 spectral peaks were made empirically by referring to the observed spectra for the analytical standards.
Computational analyses, correlation and hierarchical clustering
All analyses were performed in the R programming environment (68). Pairwise Pearson's correlation coefficient (r) was calculated for all parameters, and the resulting r values were plotted as heat maps in R. Hierarchical clustering was performed using complete linkage for agglomeration.
Computational analyses, classification studies
Random forest (32, 33) was used to build three classification models using diet-, strain-, and IRI-based stratifications as the response variables. Binary classes were assigned to samples for IRI (>270,000 = 1; <270,000 = 0) or diet (HFD = 1; chow = 0), or strains (1 = strain #50; 2 = strain #89; and 3 = strain #97). Two IRI cutoffs were considered, upper tertile and median. The upper tertile cutoff achieved a greater fold difference between insulin-sensitive and insulin-resistant groups than the median cutoff. Training and testing data sets were obtained through a 3/4 to 1/4 split, bootstrapped over 500 times. Sampling was done with replacement. The class imbalance (IRI, 10 and 19 mice) was balanced using the SMOTE algorithm (69, 70) oversampling technique (with k = 5) while training the models. ConfusionMatrix from the caret package (71) was used to calculate associated error and accuracy statistics. CAs are observed versus true classification of the testing set. For each model, at least 1000 trees were generated (ntree = 1000) to ensure robustness, and mtry ranged from 1 to 55 depending on the number of features tested. Importance of each feature was assessed via a mean decrease in accuracy. To obtain the ratio, the CA was divided by the random CA where the mouse identities were randomized with confidence limits calculated by the delta method (72).
Statistical analysis
Data are presented as mean ± S.E., unless otherwise indicated. Statistical analyses were performed using t test, analysis of variance (ANOVA), or multiple t test analysis using GraphPad Prism software. Significance was set at p < 0.05 or q <0.05 (multiple t test analysis) and p values are indicated.
Author contributions
K. H. F. W., J. S., and D. E. J. designed the study. J. S. and D. E. J. wrote the manuscript. K. H. F. W., J. S., X. Y. Z., D. J. F., C. C. M., K. C. T., N. J. H., S. P. M., C. E. X., C. H. Y., S. A., and G. J. C. performed mouse experiments. K. H. F. W., O. I., K. W., and D. M. M. performed metabolomics analysis. J. S. performed data analysis. R. C. performed classification analysis. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Drs. Kyle Hoehn and Nigel Turner (University of New South Wales) for critically reading the manuscript and Dr. Jean Yang (University of Sydney) for bioinformatics advice.
This work was supported in part by National Health and Medical Research Council (NHMRC) Project Grants GNT1061122, GNT1086851, and GNT1086850 (to D. E. J.) and National Institutes of Health Grants 2R01DK089312 and 2P01-DK058398 (to D. M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or NHMRC.
This article contains supplemental Table S1.
- HFD
- diet high in fat and sucrose
- CA
- classification accuracy
- IRI
- insulin resistance index
- GTT
- glucose tolerance test
- 2DG
- 2-deoxyglucose
- VLC
- very-long chain
- LC
- long chain
- AUC
- area under the curve
- iAUC
- incremental area under the curve
- WAT
- white adipose tissue
- BW
- body weight
- Ext
- external
- IR
- insulin-resistant
- IS
- insulin-sensitive
- IS-SIG
- insulin sensitivity signature
- Cor Met
- 113 correlated metabolites.
References
- 1. Imamura M., and Maeda S. (2011) Genetics of type 2 diabetes: the GWAS era and future perspectives (Review). Endocr. J. 58, 723–739 [DOI] [PubMed] [Google Scholar]
- 2. Lotta L. A., Gulati P., Day F. R., Payne F., Ongen H., van de Bunt M., Gaulton K. J., Eicher J. D., Sharp S. J., Luan J., De Lucia Rolfe E., Stewart I. D., Wheeler E., Willems S. M., Adams C., et al. (2017) Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet. 49, 17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Phillips C. M. (2013) Nutrigenetics and metabolic disease: current status and implications for personalised nutrition. Nutrients 5, 32–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Phillips C. M., Goumidi L., Bertrais S., Field M. R., Cupples L. A., Ordovas J. M., Defoort C., Lovegrove J. A., Drevon C. A., Gibney M. J., Blaak E. E., Kiec-Wilk B., Karlstrom B., Lopez-Miranda J., McManus R., et al. (2010) Gene-nutrient interactions with dietary fat modulate the association between genetic variation of the ACSL1 gene and metabolic syndrome. J. Lipid Res. 51, 1793–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Phillips C. M., Goumidi L., Bertrais S., Field M. R., Cupples L. A., Ordovas J. M., McMonagle J., Defoort C., Lovegrove J. A., Drevon C. A., Blaak E. E., Kiec-Wilk B., Riserus U., Lopez-Miranda J., McManus R., et al. (2010) ACC2 gene polymorphisms, metabolic syndrome, and gene-nutrient interactions with dietary fat. J. Lipid Res. 51, 3500–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Phillips C. M., Goumidi L., Bertrais S., Field M. R., McManus R., Hercberg S., Lairon D., Planells R., and Roche H. M. (2011) Gene-nutrient interactions and gender may modulate the association between ApoA1 and ApoB gene polymorphisms and metabolic syndrome risk. Atherosclerosis 214, 408–414 [DOI] [PubMed] [Google Scholar]
- 7. Shen J., Arnett D. K., Pérez-Martínez P., Parnell L. D., Lai C. Q., Peacock J. M., Hixson J. E., Tsai M. Y., Straka R. J., Hopkins P. N., and Ordovás J. M. (2008) The effect of IL6–174C/G polymorphism on postprandial triglyceride metabolism in the GOLDN studyboxs. J. Lipid Res. 49, 1839–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Robitaille J., Gaudet D., Perusse L., and Vohl M. C. (2007) Features of the metabolic syndrome are modulated by an interaction between the peroxisome proliferator-activated receptor-δ−87T>C polymorphism and dietary fat in French-Canadians. Int. J. Obes. 31, 411–417 [DOI] [PubMed] [Google Scholar]
- 9. Scacchi R., Pinto A., Rickards O., Pacella A., De Stefano G. F., Cannella C., and Corbo R. M. (2007) An analysis of peroxisome proliferator-activated receptor γ (PPAR-γ 2) Pro12Ala polymorphism distribution and prevalence of type 2 diabetes mellitus (T2DM) in world populations in relation to dietary habits. Nutr. Metab. Cardiovasc. Dis. 17, 632–641 [DOI] [PubMed] [Google Scholar]
- 10. Samuel V. T., and Shulman G. I. (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148, 852–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chavez J. A., Siddique M. M., Wang S. T., Ching J., Shayman J. A., and Summers S. A. (2014) Ceramides and glucosylceramides are independent antagonists of insulin signaling. J. Biol. Chem. 289, 723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Erion D. M., and Shulman G. I. (2010) Diacylglycerol-mediated insulin resistance. Nat. Med. 16, 400–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koves T. R., Ussher J. R., Noland R. C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J. R., Newgard C. B., Lopaschuk G. D., and Muoio D. M. (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56 [DOI] [PubMed] [Google Scholar]
- 14. Anderson E. J., Lustig M. E., Boyle K. E., Woodlief T. L., Kane D. A., Lin C. T., Price J. W. 3rd., Kang L., Rabinovitch P. S., Szeto H. H., Houmard J. A., Cortright R. N., Wasserman D. H., and Neufer P. D. (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 119, 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoehn K. L., Salmon A. B., Hohnen-Behrens C., Turner N., Hoy A. J., Maghzal G. J., Stocker R., Van Remmen H., Kraegen E. W., Cooney G. J., Richardson A. R., and James D. E. (2009) Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. U.S.A. 106, 17787–17792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee B. C., and Lee J. (2014) Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 1842, 446–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrikopoulos S., Massa C. M., Aston-Mourney K., Funkat A., Fam B. C., Hull R. L., Kahn S. E., and Proietto J. (2005) Differential effect of inbred mouse strain (C57BL/6, DBA/2, 129T2) on insulin secretory function in response to a high fat diet. J. Endocrinol. 187, 45–53 [DOI] [PubMed] [Google Scholar]
- 18. Rossmeisl M., Rim J. S., Koza R. A., and Kozak L. P. (2003) Variation in type 2 diabetes-related traits in mouse strains susceptible to diet-induced obesity. Diabetes 52, 1958–1966 [DOI] [PubMed] [Google Scholar]
- 19. Almind K., and Kahn C. R. (2004) Genetic determinants of energy expenditure and insulin resistance in diet-induced obesity in mice. Diabetes 53, 3274–3285 [DOI] [PubMed] [Google Scholar]
- 20. Gallou-Kabani C., Vigé A., Gross M. S., Rabès J. P., Boileau C., Larue-Achagiotis C., Tomé D., Jais J. P., and Junien C. (2007) C57BL/6J and A/J mice fed a high-fat diet delineate components of metabolic syndrome. Obesity 15, 1996–2005 [DOI] [PubMed] [Google Scholar]
- 21. Ferrara C. T., Wang P., Neto E. C., Stevens R. D., Bain J. R., Wenner B. R., Ilkayeva O. R., Keller M. P., Blasiole D. A., Kendziorski C., Yandell B. S., Newgard C. B., and Attie A. D. (2008) Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS Genet. 4, e1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Montgomery M. K., Hallahan N. L., Brown S. H., Liu M., Mitchell T. W., Cooney G. J., and Turner N. (2013) Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 56, 1129–1139 [DOI] [PubMed] [Google Scholar]
- 23. Parks B. W., Sallam T., Mehrabian M., Psychogios N., Hui S. T., Norheim F., Castellani L. W., Rau C. D., Pan C., Phun J., Zhou Z., Yang W. P., Neuhaus I., Gargalovic P. S., Kirchgessner T. G., et al. (2015) Genetic architecture of insulin resistance in the mouse. Cell Metab. 21, 334–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Williams R. W., Bennett B., Lu L., Gu J., DeFries J. C., Carosone-Link P. J., Rikke B. A., Belknap J. K., and Johnson T. E. (2004) Genetic structure of the LXS panel of recombinant inbred mouse strains: a powerful resource for complex trait analysis. Mamm. Genome 15, 637–647 [DOI] [PubMed] [Google Scholar]
- 25. Bennett B., Carosone-Link P., Zahniser N. R., and Johnson T. E. (2006) Confirmation and fine mapping of ethanol sensitivity quantitative trait loci, and candidate gene testing in the LXS recombinant inbred mice. J. Pharmacol. Exp. Ther. 319, 299–307 [DOI] [PubMed] [Google Scholar]
- 26. Wang X., Pandey A. K., Mulligan M. K., Williams E. G., Mozhui K., Li Z., Jovaisaite V., Quarles L. D., Xiao Z., Huang J., Capra J. A., Chen Z., Taylor W. L., Bastarache L., Niu X., Pollard K. S., et al. (2016) Joint mouse-human phenome-wide association to test gene function and disease risk. Nat. Commun. 7, 10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Churchill G. A., Airey D. C., Allayee H., Angel J. M., Attie A. D., Beatty J., Beavis W. D., Belknap J. K., Bennett B., Berrettini W., Bleich A., Bogue M., Broman K. W., Buck K. J., Buckler E., et al. (2004) The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 36, 1133–1137 [DOI] [PubMed] [Google Scholar]
- 28. Weindruch R. (1996) The retardation of aging by caloric restriction: studies in rodents and primates. Toxicol. Pathol. 24, 742–745 [DOI] [PubMed] [Google Scholar]
- 29. Liao C. Y., Rikke B. A., Johnson T. E., Diaz V., and Nelson J. F. (2010) Genetic variation in the murine life span response to dietary restriction: from life extension to life shortening. Aging Cell 9, 92–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koza R. A., Nikonova L., Hogan J., Rim J. S., Mendoza T., Faulk C., Skaf J., and Kozak L. P. (2006) Changes in gene expression foreshadow diet-induced obesity in genetically identical mice. PLoS Genet. 2, e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burcelin R., Crivelli V., Dacosta A., Roy-Tirelli A., and Thorens B. (2002) Heterogeneous metabolic adaptation of C57BL/6J mice to high-fat diet. Am. J. Physiol. Endocrinol. Metab. 282, E834–E842 [DOI] [PubMed] [Google Scholar]
- 32. Breiman L. (2001) Random forests. Machine Learn. 45, 5–32 [Google Scholar]
- 33. Liaw A., and Wiener M. (2002) Classification and regression by randomForest. R News 2, 18–22 [Google Scholar]
- 34. Chaudhuri R., Khoo P. S., Tonks K., Junutula J. R., Kolumam G., Modrusan Z., Samocha-Bonet D., Meoli C. C., Hocking S., Fazakerley D. J., Stöckli J., Hoehn K. L., Greenfield J. R., Yang J. Y. H., and James D. E. (2015) Cross-species gene expression analysis identifies a novel set of genes implicated in human insulin sensitivity. NPJ Syst. Biol. Appl. 1, 15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raichur S., Wang S. T., Chan P. W., Li Y., Ching J., Chaurasia B., Chaurasia B., Dogra S., Öhman M. K., Takeda K., Sugii S., Pewzner-Jung Y., Futerman A. H., and Summers S. A. (2014) CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 20, 687–695 [DOI] [PubMed] [Google Scholar]
- 36. Turpin S. M., Nicholls H. T., Willmes D. M., Mourier A., Brodesser S., Wunderlich C. M., Mauer J., Xu E., Hammerschmidt P., Brönneke H. S., Trifunovic A., LoSasso G., Wunderlich F. T., Kornfeld J. W., Blüher M., Krönke M., and Brüning J. C. (2014) Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 20, 678–686 [DOI] [PubMed] [Google Scholar]
- 37. Newgard C. B., An J., Bain J. R., Muehlbauer M. J., Stevens R. D., Lien L. F., Haqq A. M., Shah S. H., Arlotto M., Slentz C. A., Rochon J., Gallup D., Ilkayeva O., Wenner B. R., Yancy W. S. Jr., et al. (2009) A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 9, 311–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haus J. M., Kashyap S. R., Kasumov T., Zhang R., Kelly K. R., Defronzo R. A., and Kirwan J. P. (2009) Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kasumov T., Solomon T. P., Hwang C., Huang H., Haus J. M., Zhang R., and Kirwan J. P. (2015) Improved insulin sensitivity after exercise training is linked to reduced plasma C14:0 ceramide in obesity and type 2 diabetes. Obesity 23, 1414–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bergman B. C., Brozinick J. T., Strauss A., Bacon S., Kerege A., Bui H. H., Sanders P., Siddall P., Wei T., Thomas M. K., Kuo M. S., and Perreault L. (2016) Muscle sphingolipids during rest and exercise: a C18:0 signature for insulin resistance in humans. Diabetologia 59, 785–798 [DOI] [PubMed] [Google Scholar]
- 41. Straczkowski M., Kowalska I., Nikolajuk A., Dzienis-Straczkowska S., Kinalska I., Baranowski M., Zendzian-Piotrowska M., Brzezinska Z., and Gorski J. (2004) Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes 53, 1215–1221 [DOI] [PubMed] [Google Scholar]
- 42. Thalacker-Mercer A. E., Ingram K. H., Guo F., Ilkayeva O., Newgard C. B., and Garvey W. T. (2014) BMI, RQ, diabetes, and sex affect the relationships between amino acids and clamp measures of insulin action in humans. Diabetes 63, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Floegel A., Stefan N., Yu Z., Mühlenbruch K., Drogan D., Joost H. G., Fritsche A., Häring H. U., Hrabě de Angelis M., Peters A., Roden M., Prehn C., Wang-Sattler R., Illig T., Schulze M. B., Adamski J., Boeing H., and Pischon T. (2013) Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 62, 639–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Palmer N. D., Stevens R. D., Antinozzi P. A., Anderson A., Bergman R. N., Wagenknecht L. E., Newgard C. B., and Bowden D. W. (2015) Metabolomic profile associated with insulin resistance and conversion to diabetes in the Insulin Resistance Atherosclerosis Study. J. Clin. Endocrinol. Metab. 100, E463–E468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Magkos F., Bradley D., Schweitzer G. G., Finck B. N., Eagon J. C., Ilkayeva O., Newgard C. B., and Klein S. (2013) Effect of Roux-en-Y gastric bypass and laparoscopic adjustable gastric banding on branched-chain amino acid metabolism. Diabetes 62, 2757–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Würtz P., Mäkinen V. P., Soininen P., Kangas A. J., Tukiainen T., Kettunen J., Savolainen M. J., Tammelin T., Viikari J. S., Rönnemaa T., Kähönen M., Lehtimäki T., Ripatti S., Raitakari O. T., Järvelin M. R., and Ala-Korpela M. (2012) Metabolic signatures of insulin resistance in 7,098 young adults. Diabetes 61, 1372–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kusunoki M., Tsutsumi K., Nakayama M., Kurokawa T., Nakamura T., Ogawa H., Fukuzawa Y., Morishita M., Koide T., and Miyata T. (2007) Relationship between serum concentrations of saturated fatty acids and unsaturated fatty acids and the homeostasis model insulin resistance index in Japanese patients with type 2 diabetes mellitus. J. Med. Invest. 54, 243–247 [DOI] [PubMed] [Google Scholar]
- 48. Tucci S., Behringer S., and Spiekerkoetter U. (2015) De novo fatty acid biosynthesis and elongation in very-long chain acyl-CoA dehydrogenase-deficient mice supplemented with odd or even medium-chain fatty acids. FEBS J. 282, 4242–4253 [DOI] [PubMed] [Google Scholar]
- 49. Tvrdik P., Asadi A., Kozak L. P., Nedergaard J., Cannon B., and Jacobsson A. (1997) Cig30, a mouse member of a novel membrane protein gene family, is involved in the recruitment of brown adipose tissue. J. Biol. Chem. 272, 31738–31746 [DOI] [PubMed] [Google Scholar]
- 50. Zadravec D., Brolinson A., Fisher R. M., Carneheim C., Csikasz R. I., Bertrand-Michel J., Borén J., Guillou H., Rudling M., and Jacobsson A. (2010) Ablation of the very-long-chain fatty acid elongase ELOVL3 in mice leads to constrained lipid storage and resistance to diet-induced obesity. FASEB J. 24, 4366–4377 [DOI] [PubMed] [Google Scholar]
- 51. Westerberg R., Tvrdik P., Undén A. B., Månsson J. E., Norlén L., Jakobsson A., Holleran W. H., Elias P. M., Asadi A., Flodby P., Toftgård R., Capecchi M. R., and Jacobsson A. (2004) Role for ELOVL3 and fatty acid chain length in development of hair and skin function. J. Biol. Chem. 279, 5621–5629 [DOI] [PubMed] [Google Scholar]
- 52. Westerberg R., Månsson J. E., Golozoubova V., Shabalina I. G., Backlund E. C., Tvrdik P., Retterstøl K., Capecchi M. R., and Jacobsson A. (2006) ELOVL3 is an important component for early onset of lipid recruitment in brown adipose tissue. J. Biol. Chem. 281, 4958–4968 [DOI] [PubMed] [Google Scholar]
- 53. Christiansen R. Z., Christophersen B. O., and Bremer J. (1977) Monoethlenic C20 and C22 fatty acids in marine oil and rapeseed oil. Studies on their oxidation and on their relative ability to inhibit palmitate oxidation in heart and liver mitochondria. Biochim. Biophys. Acta 487, 28–36 [DOI] [PubMed] [Google Scholar]
- 54. Reubsaet F. A., Veerkamp J. H., Trijbels J. M., and Monnens L. A. (1989) Total and peroxisomal oxidation of various saturated and unsaturated fatty acids in rat liver, heart and m. quadriceps. Lipids 24, 945–950 [DOI] [PubMed] [Google Scholar]
- 55. Bremer J., and Norum K. R. (1982) Metabolism of very-long chain monounsaturated fatty acids (22:1) and the adaptation to their presence in the diet. J. Lipid Res. 23, 243–256 [PubMed] [Google Scholar]
- 56. Muoio D. M., and Neufer P. D. (2012) Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metab. 15, 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Seiler S. E., Martin O. J., Noland R. C., Slentz D. H., DeBalsi K. L., Ilkayeva O. R., An J., Newgard C. B., Koves T. R., and Muoio D. M. (2014) Obesity and lipid stress inhibit carnitine acetyltransferase activity. J. Lipid Res. 55, 635–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Muoio D. M., Noland R. C., Kovalik J. P., Seiler S. E., Davies M. N., DeBalsi K. L., Ilkayeva O. R., Stevens R. D., Kheterpal I., Zhang J., Covington J. D., Bajpeyi S., Ravussin E., Kraus W., Koves T. R., and Mynatt R. L. (2012) Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 15, 764–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li Q., Deng S., Ibarra R. A., Anderson V. E., Brunengraber H., and Zhang G. F. (2015) Multiple mass isotopomer tracing of acetyl-CoA metabolism in Langendorff-perfused rat hearts: channeling of acetyl-CoA from pyruvate dehydrogenase to carnitine acetyltransferase. J. Biol. Chem. 290, 8121–8132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tan S. X., Fisher-Wellman K. H., Fazakerley D. J., Ng Y., Pant H., Li J., Meoli C. C., Coster A. C., Stöckli J., and James D. E. (2015) Selective insulin resistance in adipocytes. J. Biol. Chem. 290, 11337–11348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hoehn K. L., Turner N., Swarbrick M. M., Wilks D., Preston E., Phua Y., Joshi H., Furler S. M., Larance M., Hegarty B. D., Leslie S. J., Pickford R., Hoy A. J., Kraegen E. W., James D. E., and Cooney G. J. (2010) Acute or chronic upregulation of mitochondrial fatty acid oxidation has no net effect on whole-body energy expenditure or adiposity. Cell Metab. 11, 70–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. An J., Muoio D. M., Shiota M., Fujimoto Y., Cline G. W., Shulman G. I., Koves T. R., Stevens R., Millington D., and Newgard C. B. (2004) Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat. Med. 10, 268–274 [DOI] [PubMed] [Google Scholar]
- 63. Wu J. Y., Kao H. J., Li S. C., Stevens R., Hillman S., Millington D., and Chen Y. T. (2004) ENU mutagenesis identifies mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease. J. Clin. Invest. 113, 434–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jensen M. V., Joseph J. W., Ilkayeva O., Burgess S., Lu D., Ronnebaum S. M., Odegaard M., Becker T. C., Sherry A. D., and Newgard C. B. (2006) Compensatory responses to pyruvate carboxylase suppression in islet β-cells. Preservation of glucose-stimulated insulin secretion. J. Biol. Chem. 281, 22342–22351 [DOI] [PubMed] [Google Scholar]
- 65. Merrill A. H. Jr, Sullards M. C., Allegood J. C., Kelly S., and Wang E. (2005) Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods 36, 207–224 [DOI] [PubMed] [Google Scholar]
- 66. Magnes C., Sinner F. M., Regittnig W., and Pieber T. R. (2005) LC/MS/MS method for quantitative determination of long-chain fatty acyl-CoAs. Anal. Chem. 77, 2889–2894 [DOI] [PubMed] [Google Scholar]
- 67. Minkler P. E., Kerner J., Ingalls S. T., and Hoppel C. L. (2008) Novel isolation procedure for short-, medium-, and long-chain acyl-coenzyme A esters from tissue. Anal. Biochem. 376, 275–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. R Development Core Team (2015) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 69. Chawla N. V., Bowyer K. W., Hall L. O., and Kegelmeyer W. P. (2002) SMOTE: Synthetic Minority Over-sampling Technique. JAIR 16, 321–357 [Google Scholar]
- 70. Torgo L. (2010) Data Mining with R, Learning with Case Studies, Chapman and Hall/CRC, CRC Press, Boca Raton, FL [Google Scholar]
- 71. Kuhn M., Wing J., Weston S., Williams A., Keefer C., Engelhardt A., Cooper T., Mayer Z., Kenkel B., the R Core Team, Benesty M., Lescarbeau R., Ziem A., Scrucca L., Tang Y., Candan C., and Hunt C. (2016) caret: Classification and Regression Training. R package version 6.0 [Google Scholar]
- 72. Oehlert G. W. (1992) A note on the delta method. American Statistician 46, 27–29 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.