

Abstract
Tumors depend on their microenvironment for sustained growth, invasion, and metastasis. In this environment, endothelial cells (ECs) are an important stromal cell type interacting with malignant cells to facilitate tumor angiogenesis and cancer cell extravasation. Of note, lysosomal acid lipase (LAL) deficiency facilitates melanoma growth and metastasis. ECs from LAL-deficient (lal−/−) mice possess enhanced proliferation, migration, and permeability of inflammatory cells by activating the mammalian target of rapamycin (mTOR) pathway. Here we report that lal−/− ECs facilitated in vivo tumor angiogenesis, growth, and metastasis, largely by stimulating tumor cell proliferation, migration, adhesion, and transendothelial migration via increased expression of IL-6 and monocyte chemoattractant protein 1 (MCP-1). This prompted us to look for lysosomal proteins that are involved in lal−/− EC dysfunctions. We found that lal−/− ECs displayed increased expression of Rab7, a late endosome/lysosome-associated small GTPase. Moreover, Rab7 and mTOR were co-increased and co-localized to lysosomes and physically interacted in lal−/− ECs. Rab7 inhibition reversed lal−/− EC dysfunctions, including decreasing their enhanced migration and permeability of tumor-stimulatory myeloid cells, and suppressed EC-mediated stimulation of in vitro tumor cell transmigration, proliferation, and migration and in vivo tumor growth and metastasis. Finally, Rab7 inhibition reduced overproduction of reactive oxygen species and increased IL-6 and MCP-1 secretion in lal−/− ECs. Our results indicate that metabolic reprogramming resulting from LAL deficiency enhances the ability of ECs to stimulate tumor cell proliferation and metastasis through stimulation of lysosome-anchored Rab7 activity.
Keywords: cancer therapy, endothelial cell, lysosome, tumor metastasis, tumor microenvironment, Rab7 GTPase, lysosomal acid lipase
Introduction
Tumorigenesis is a complex process during which tumors depend on the microenvironment for sustained growth, invasion, and metastasis (1). Endothelial cells (ECs)3 are one of the important stromal cell types within the tumor microenvironment that have a strong influence on tumor cells and tumor-associated immune cells. ECs regulate tumor proliferation and progression in several ways. First, ECs participate in tumor angiogenesis by forming tumor vasculature, which provides tumor cells with oxygen and nutrients to satisfy their growing need (2). It has already been reported that cross-talk between tumors and ECs promotes tumor angiogenesis (3). Second, ECs interact with cancer cells, affecting the efficiency of cancer cell migration and extravasation (4). Third, ECs influence tumor proliferation and progression by regulating immune cells (5). For example, ECs isolated from tumor tissue are able to inhibit T cell functions (6). To effectively limit the contributions of ECs to tumor proliferation and progression, it is necessary to block the pathogenic influence of ECs on tumor cells.
The angiogenic switch of ECs in tumor proliferation and progression is metabolically demanding and mediated by adaptations in EC metabolism (7). Emerging evidence indicates that the metabolic change determines not only EC differentiation and angiogenesis but also the pathogenic functions of ECs in cancer (8). We have demonstrated that metabolic reprogramming during deficiency of lysosomal acid lipase (LAL) increases EC proliferation, migration, and the permeability of tumor-promoting myeloid-derived suppressor cells (MDSCs) and suppression of T cell proliferation and functions (9). LAL is a key enzyme in the metabolic pathway of neutral lipids that hydrolyzes cholesteryl esters and triglycerides in the lysosome of cells to generate free fatty acids and cholesterol (10, 11). Metabolic switch of LAL deficiency in ECs induces overactivation of the mammalian target of rapamycin (mTOR) pathway, which is a lysosome-anchored metabolic regulator and mediates lal−/− EC dysfunction (9). LAL is a lysosome-localized enzyme that influences endomembrane trafficking and mTOR shuttling to lysosomes (12, 13). While searching for lysosomal proteins that are involved in LAL and mTOR activities, Rab7 GTPase was found to be up-regulated in lal−/− ECs, similar to what was observed in lal−/− MDSCs (14). Rab7 GTPase has been reported to be a key regulatory protein for proper aggregation and fusion of late endocytic structures in the perinuclear region and consequently for the biogenesis and maintenance of the lysosomal compartment in cells (15, 16). We further identified that Rab7 GTPase was co-localized with mTOR and lysosomes in lal−/− ECs and physically interacted with mTOR to influence its downstream signaling. In this study, we focused on how Rab7 GTPase in lal−/− ECs affects their stimulation of tumor proliferation, growth, and metastasis. These findings provide mechanistic insight into LAL deficiency facilitating metastasis and indicate a potential target on ECs for anti-tumor therapy.
Results
ECs with LAL deficiency stimulated tumor angiogenesis, growth, and metastasis
Angiogenesis is essential for tumor growth and metastasis, during which ECs and tumor cells interact to facilitate the formation of new blood vessels. To see whether LAL deficiency plays a role in EC stimulation of tumor angiogenesis, an in vivo Matrigel plug assay was performed with wild-type (lal+/+) or lal−/− ECs mixed with tumor cells. The mixture was subcutaneously injected into lal+/+ recipient mice for a tumor angiogenesis and growth study. Ten days post-injection, the plugs were harvested, sectioned, and stained with H&E. Plugs mixed with lal−/− ECs and B16 melanoma cells had a larger size and heavier weight than those mixed with lal+/+ ECs and B16 melanoma cells (Fig. 1, A–C). Consistently, H&E staining revealed robust melanoma cell proliferation in plugs containing lal−/− ECs (Fig. 1D) and more newly formed microvessels in plugs containing lal−/− ECs than in those with lal+/+ ECs (Fig. 1D, arrows). Similar results were observed when ECs were mixed with Lewis lung carcinoma (LLC) cells (Fig. 1, A–D).
Figure 1.
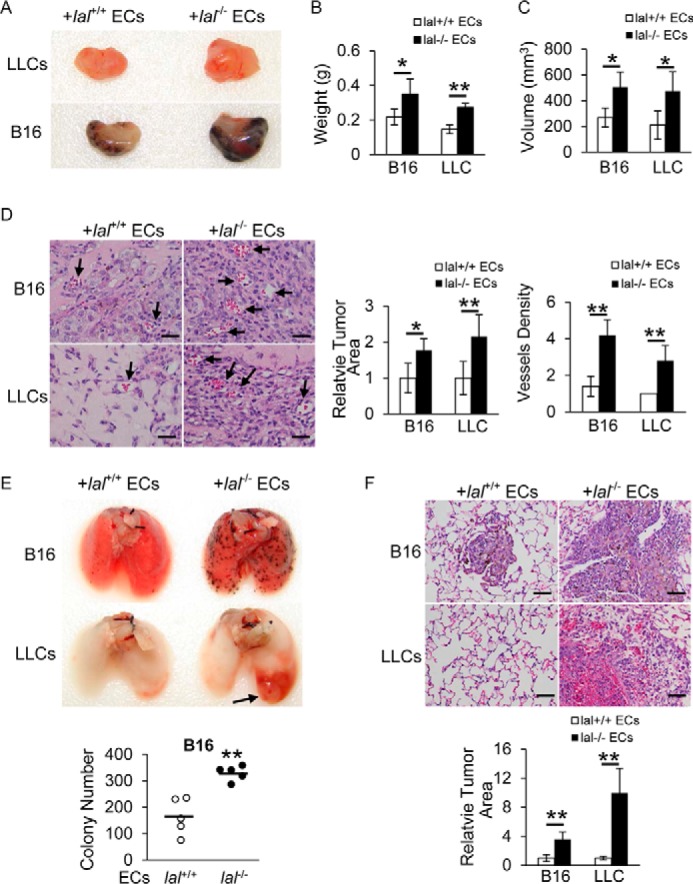
lal−/− ECs stimulated tumor angiogenesis, growth, and metastasis in vivo. A, B16 melanoma or LLC cells (1 × 105) were mixed with lal+/+ or lal−/− ECs (2 × 105) in Matrigel and implanted subcutaneously into lal+/+ recipient mice for 10 days. A representative gross picture of tumor growth is shown (n = 5). B, statistical analysis of tumor plug weight. C, statistical analysis of tumor plug volume. D, left panel, representative H&E staining of tumor plug sections. Arrows indicate newly formed microvessels. Original magnification, ×400. Scale bars = 100 μm. Right panel, statistical analysis of relative tumor areas and quantification of vessel density by counting the number of vessels per field. E, B16 melanoma or LLC cells (4 × 105) were mixed with lal+/+ or lal−/− ECs (4 × 105) and injected intravenously into lal+/+ mice for 2 weeks. Representative lungs and quantitative analysis of B16 melanoma colonies in the lungs are shown (n = 5). F, representative H&E staining of the lungs in E, including statistical analysis of relative tumor areas. Original magnification, ×200. Scale bars = 200 μm. For all experiments, data are expressed as mean ± S.D. n = 4∼5. *, p < 0.05; **, p < 0.01.
Next, lal+/+ or lal−/− ECs mixed with B16 melanoma cells were co-injected into the tail veins of lal+/+ recipient mice to detect the potential effect of ECs on tumor cell invasion. Two weeks after injection, compared with mice that received lal+/+ ECs and melanoma cells, mice co-injected with lal−/− ECs and melanoma cells developed more melanoma colonies in the lungs (Fig. 1E). H&E staining revealed more neoplastic melanoma cells in lungs that received lal−/− ECs than in those that received lal+/+ ECs (Fig. 1F). A similar stimulation of metastatic invasion was observed when lal−/− ECs were co-injected with LLC cells into lal+/+ recipient mice, with more neoplastic cells in lungs that received lal−/− ECs (Fig. 1, E and F). Taken together, ECs with LAL deficiency stimulated tumor angiogenesis, growth, and metastasis.
ECs with LAL deficiency stimulated tumor cell proliferation, migration, adhesion, and transendothelial migration
The stimulatory effects of lal−/− ECs on tumor growth and invasion were further examined by in vitro experiments. To evaluate the influence of lal−/− ECs on tumor cell proliferation, tumor cells were treated with lal+/+ or lal−/− EC-conditioned medium (CM) for 72 h. As shown in Fig. 2A, compared with lal+/+ EC-CM treatment, lal−/− EC-CM treatment significantly increased the proliferation of B16 melanoma and LLC cells. Attachment of cancer cells to ECs is the first step of the extravasation process (4). Therefore, whether LAL deficiency in ECs affects tumor cell attachment was examined. CellTrackerTM Green 5-chloromethylfluorescein diacetate (CMFDA)–labeled tumor cells (B16 melanoma or LLC cells) were added to the EC monolayer and incubated for 3 h. As Fig. 2B shows, both B16 melanoma and LLC cells showed significantly more adhesion to lal−/− ECs compared with lal+/+ ECs. After attachment to ECs, tumor cells transmigrate through EC junctions. A Transwell assay was performed to determine tumor cell transmigration across the endothelial monolayers formed by lal+/+ or lal−/− ECs. ECs were seeded into Transwell upper chambers and grown to confluence. CMFDA-labeled LLC cells were loaded on the EC monolayers. Fifteen hours later, the LLC cells that had migrated to the lower chamber were counted. Consistent with their higher adhesion to tumor cells, lal−/− ECs showed increased permeability, with more LLC cells migrating to the lower chamber than lal+/+ ECs (Fig. 2C). Our pioneer study showed that transmigrated B16 cells across the endothelial monolayer tightly attached to the membrane of the inserts and did not migrate to the bottom of the chamber. In addition, an in vitro tumor cell migration assay was performed to determine whether lal−/− ECs influence tumor cell migration. Tumor cells were treated with mitomycin C to eliminate the potential effects of cell proliferation in these assays. As shown in Fig. 2D, 24 h after co-culture with lal−/− EC-CM, LLC cells migrated more efficiently into the area of an artificial wound compared with those treated with lal+/+ EC-CM. However, no significant difference was observed when B16 melanoma cells were treated with lal−/− EC-CM. The discrepancy between LLC and B16 cells means that cancer cells from different origins have different features. Taken together, these results suggested that lal−/− ECs stimulated tumor cell proliferation, migration, adhesion, and transendothelial migration in vitro.
Figure 2.
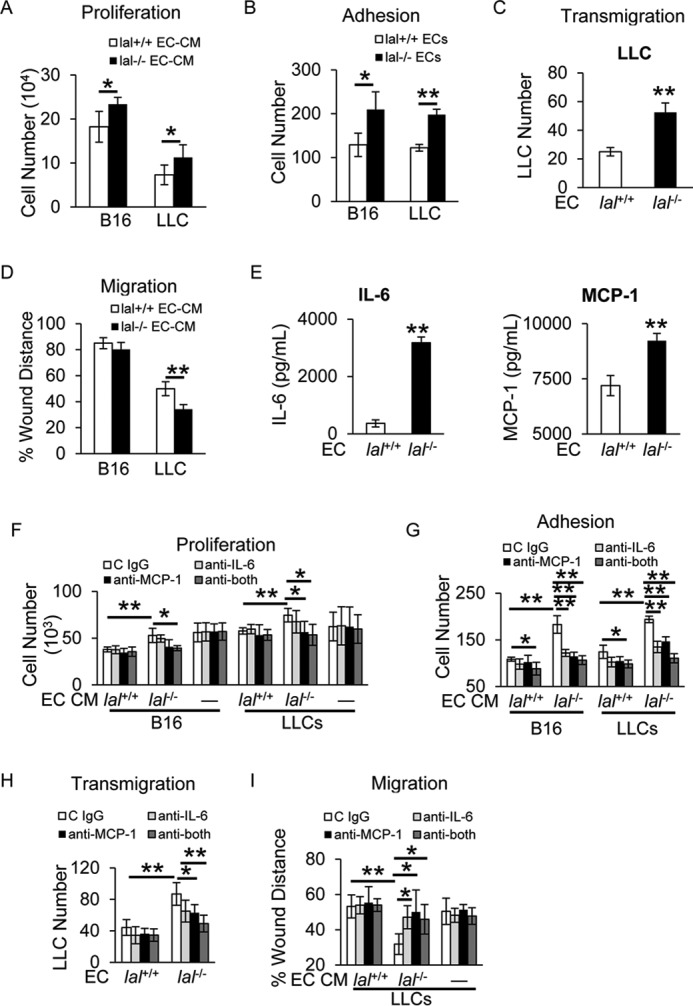
lal−/− ECs stimulated tumor cell proliferation, migration, adhesion, and transendothelial migration in vitro. A, B16 melanoma or LLC cells (2 × 104) were co-cultured with CM of lal+/+ or lal−/− ECs in vitro for 72 h for tumor cell enumeration. B, CMFDA-labeled tumor cell (B16 melanoma or LLC cells, 1 × 104) adhesion to a lal+/+ or lal−/− EC monolayer. Total cell number of five fields in each well was quantified using a fluorescence microscope. C, CMFDA-labeled LLC transendothelial migration across the lal+/+ or lal−/− EC monolayers in the Transwell assay. LLC cells migrated to the lower chamber were counted. D, tumor cell migration after lal+/+ or lal−/− EC-CM treatment by in vitro wound healing assay at 15 h in the presence of mitomycin C. E, secretion of IL-6 and MCP-1 by ECs in the culture medium was measured by ELISA. F, antibody neutralization against IL-6 and MCP-1 individually or in combination in ECs; IgG was the control. The CM was harvested and used to treat B16 melanoma or LLC cells (5 × 103) in vitro for 72 h for tumor cell enumeration. To exclude the potential effects of neutralizing antibody on tumor cells, neutralizing antibodies or control (C) IgG were added to tumor cell culture medium directly. G, CMFDA-labeled tumor cell (B16 melanoma or LLC cells, 1 × 104) adhesion to lal+/+ or lal−/− EC monolayers that were pretreated with neutralizing antibody against IL-6, MCP-1 individually or in combination or control IgG. H, CMFDA-labeled LLC transendothelial migration across the lal+/+ or lal−/− ECs monolayers that were pretreated with neutralizing antibodies or control IgG. I, tumor cell migration after neutralizing antibody–pretreated EC-CM treatment was assessed by in vitro wound healing assay in the presence of mitomycin C. For all experiments, data are expressed as mean ± S.D. n = 3∼4. *, p < 0.05; **, p < 0.01.
We previously reported that chemokines and cytokines secreted by lal−/− ECs are responsible for mediating myeloid cell transendothelial migration (9). Here we further investigated whether these cytokines/chemokines play a role in the tumor-stimulating abilities of lal−/− ECs. An ELISA confirmed the elevated secretion of IL-6 and monocyte chemoattractant protein 1 (MCP-1) by lal−/− ECs (Fig. 2E), consistent with their up-regulated mRNA levels, as we reported previously (9). To examine whether IL-6 and MCP-1 secreted by lal−/− ECs facilitated tumor cell proliferation, EC were treated with anti-IL-6 or anti-MCP-1 neutralizing antibodies. CM harvested afterward was used to treat tumor cells for 72 h. As shown in Fig. 2F, compared with control IgG-treated EC-CM, LLC cell proliferation was decreased with anti-MCP-1 treated lal−/− EC-CM, whereas anti-IL-6–treated EC-CM had no effect on cancer cell proliferation (B16 showed a decrease with no statistical difference). Combination of anti-IL-6 and anti-MCP-1 antibody–treated lal−/− EC-CM did not further block LLC cell proliferation. Therefore, MCP-1 was a primary factor responsible for stimulating LLC cell proliferation. To exclude the potential effect of neutralizing antibodies on tumor cell proliferation, antibodies were directly added to tumor cell culture medium, which had no effect on tumor cell proliferation.
To study whether IL-6 and MCP-1 secreted by lal−/− ECs facilitated tumor cell adhesion and transmigration, ECs were treated with anti-IL-6 or anti-MCP-1 neutralizing antibodies. As Fig. 2G shows, both B16 melanoma and LLC cells showed decreased adhesion to lal−/− ECs that were pretreated with anti-IL-6 or anti-MCP-1 antibody or both. The results in Fig. 2H show that fewer LLC cells transmigrated through ECs that were pretreated with anti-MCP-1 antibody or both antibodies than through those treated with control IgG. Finally, their effects on tumor cell migration were examined. Because there was no significant difference in B16 melanoma cell migration between lal+/+ and lal−/− EC-CM (Fig. 2D), only LLC cells were tested here. After co-culture with lal−/− EC-CM that was pretreated with anti-IL-6 or anti-MCP-1 antibodies or both, LLC cells migrated less efficiently into the area of an artificial wound compared with those co-cultured with control IgG. Neutralizing antibodies alone had no direct effect on LLC cell migration (Fig. 2I). Taken together, increased secretion of IL-6 and MCP-1 by lal−/− ECs contributed to enhanced tumor cell proliferation, migration, adhesion, and transendothelial migration.
LAL deficiency increased Rab7 GTPase expression in ECs
Rab7 GTPase is a key regulatory protein controlling biogenesis and trafficking of lysosomes. In lal−/− mice, increased Rab7 GTPase expression has been observed in MDSCs (14). Similarly, Western blot and immunocytochemical staining analyses detected an increased level of Rab7 GTPase in lal−/− ECs (Fig. 3, A and B). Furthermore, immunofluorescence co-staining demonstrated co-localization of the lysosomal marker LAMP1 and Rab7 GTPase; lal−/− ECs showed many more LAMP1-positive lysosomes around the perinuclear region (Fig. 3C). The metabolic master regulator mTOR plays a key role in controlling lal−/− EC metabolic reprogramming (9). Increased expression and co-localization of Rab7 GTPase and mTOR on the lysosomes of lal−/− ECs were detected (Fig. 3C). These results imply that Rab7 GTPase actively participates in lal−/− EC tumorigenesis, perhaps through regulation of the mTOR pathway.
Figure 3.

LAL deficiency increased Rab7 GTPase expression and co-localization with LAMP1 and mTOR in ECs. A, left panel, representative Western blot analysis of the Rab7 GTPase protein level in lal−/− versus lal+/+ ECs. Right panel, statistical analyses of three independent Western blot analyses. *, p < 0.05. B, immunocytochemical staining of Rab7 GTPase in lal−/− versus lal+/+ ECs. Original magnification, ×400. Scale bars = 100 μm. C, immunofluorescence co-staining of the lysosomal marker LAMP1, Rab7 GTPase, and mTOR in lal−/− versus lal+/+ ECs. Original magnification, ×600. Scale bars = 20 μm.
Rab7 GTPase interacted with mTOR and influenced its downstream signaling
We have recently reported that the mTOR signaling pathway is regulated by Rab7 GTPase in myeloid cells (17). To investigate whether the same regulation occurs in lal−/− ECs, siRNA (a pool of three different sets) was used to effectively knock down Rab7 GTPase expression in lal−/− ECs, as confirmed by immunofluorescence staining (Fig. 4A). Western blot analysis showed that Rab7 GTPase siRNA knockdown not only effectively reduced Rab7 GTPase protein expression by around 70% but also down-regulated mTOR and downstream pS6 (phospho-S6 ribosomal protein) levels in lal−/− ECs (Fig. 4B). Three individual Rab7 GTPase siRNAs were also tested separately, which achieved the same results (supplemental Fig. 1). An immunoprecipitation assay using anti-Rab7 GTPase antibody showed increased mTOR immunoprecipitation intensity of lal−/− EC lysate compared with lal+/+ EC lysate (Fig. 4C). These findings were further confirmed by GST pulldown assay using purified recombinant Rab7 proteins (Fig. 4D, left panel). After incubation with EC protein extracts, GST-Rab7 GTPase fusion protein pulled down mTOR protein in both lal+/+ and lal−/− ECs, with stronger mTOR intensity in lal−/− ECs (Fig. 4D, center and right panels). As a control, GST showed no pulldown signal in both ECs. Therefore, Rab7 GTPase is physically associated with mTOR with enhanced interaction in lal−/− ECs. We have recently reported that, in myeloid cells, Rab7 GTPase interacts with the N-terminal heat repeat domain of mTOR (17). This was confirmed in ECs by a GST–mTOR N-terminal fusion fragment (M1A). A GST–mTOR C-terminal fusion fragment (M4) was used as a negative control. The GST–M1A mTOR fusion fragment showed a stronger interaction with endogenous Rab7 GTPase protein in lal−/− EC cell extract than that of lal+/+ ECs, whereas the GST–M4 mTOR fusion fragment showed no pulldown (Fig. 4F, right panel).
Figure 4.

Rab7 GTPase interacted with mTOR and influenced mTOR downstream signaling. A, immunofluorescence staining of Rab7 GTPase in ECs after Rab7 GTPase siRNA knockdown. Original magnification, ×600. Scale bars = 10 μm. C, control. B, Rab7 GTPase siRNA knockdown reduced mTOR, phosphorylated S6 and Rab7 GTPase protein levels in ECs by Western blot analysis. C, immunoprecipitation (IP) assay of Rab7 GTPase and mTOR interaction. EC lysates were immunoprecipitated by control IgG (IgG) or anti-Rab7 GTPase antibody (Rab7) and detected by anti-mTOR antibody in Western blot analysis. Left panel, a representative immunoprecipitation detected by Western blot analysis. Right panel, statistical analyses of three independent immunoprecipitation assays. IB, immunoblot. D, GST pulldown assay of Rab7 GTPase and mTOR interaction. Left panel, the purity of recombinant GST and Rab7–GST fusion protein was visualized by Coomassie Blue staining on SDS-PAGE. Center panel, lal+/+ and lal−/− EC lysates were incubated with purified recombinant GST or Rab7–GST fusion protein, pulled down with GST beads, and detected by anti-mTOR antibody in Western blot analysis. Right panel, statistical analyses of three independent Rab7-GST pulldown assays. MW, molecular weight. E, schematic of the mTOR domain structure (top) and PCR-generated GST–mTOR M1A and M4 fusion fragments (bottom). F, left panel, expression and purification of two recombinant mTOR fragment GSTs. Top right panel, pulldown of endogenous Rab7 GTPase in lal+/+ or lal−/− ECs by recombinant mTOR M1A–GST and mTOR M4–GST fusion fragments. Bottom right panel, statistical analyses of three independent mTOR fragment GST pulldown assays. One microgram of input was loaded on the gels for the pulldown and immunoprecipitation experiments. *, p < 0.05; **, p < 0.01.
Inhibition of Rab7 GTPase impaired lal−/− EC permeability and migration and decreased reactive oxygen species overproduction
To investigate whether increased Rab7 GTPase expression is responsible for lal−/− EC dysfunction, siRNAs were used to knock down Rab7 GTPase expression in lal−/− ECs. An in vitro wound healing assay was performed to determine lal−/− EC migrating abilities. The results showed that migration toward the scratch was delayed in lal−/− ECs with Rab7 GTPase siRNA transfection at 15 h after creating the scratch (Fig. 5A), indicating that Rab7 GTPase inhibition impaired lal−/− EC increased migration.
Figure 5.
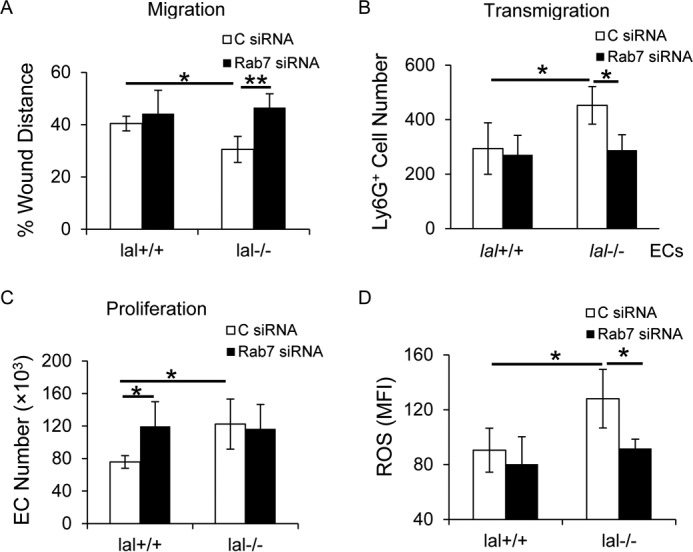
Inhibition of Rab7 GTPase impaired lal−/− EC permeability and migration and decreased ROS overproduction. A, EC migration after Rab7 GTPase siRNA knockdown was assessed by an in vitro wound healing assay in the presence of mitomycin C. C, control. B, MDSC transendothelial migration was determined after Rab7 GTPase siRNA knockdown in ECs for 48 h. CMFDA-labeled bone marrow–derived MDSCs were added to the siRNA-transfected EC monolayers. Four hours later, MDSCs in the lower chamber were counted. C, EC proliferation after Rab7 GTPase siRNA knockdown. D, statistical analysis of mean fluorescent intensity (MFI) of the ROS level in ECs after Rab7 GTPase siRNA knockdown by flow cytometry. For all experiments, scrambled siRNA transfection served as a control, and data are expressed as mean ± S.D. n = 4∼5. *, p < 0.05; **p < 0.01.
ECs control transmigration of leukocytes or tumor cells from the vasculature to inflammatory or metastatic sites. Next, EC permeability was analyzed by Transwell assay. After ECs were transfected with Rab7 GTPase or control siRNA for 48 h, CMFDA-labeled lal+/+ bone marrow–derived Ly6G+ cells were loaded on the EC monolayer. Four hours later, the number of Ly6G+ cells in the lower chamber was significantly reduced across lal−/− ECs transfected with Rab7 GTPase siRNA compared with those transfected with control siRNA (Fig. 5B), suggesting that Rab7 GTPase inhibition reduced the permeability of lal−/− ECs. Rab7 GTPase inhibition did not affect lal−/− EC proliferation (Fig. 5C).
Reactive oxygen species (ROS) overproduction has been observed in lal−/− ECs and is a mechanism underlying lal−/− EC dysfunctions that are mediated by mTOR overactivation (9). To test whether Rab7 GTPase regulates ROS production in lal−/− ECs, the ROS level was measured by flow cytometry analysis in ECs transfected with Rab7 GTPase or control siRNA. As Fig. 5D shows, knocking down Rab7 GTPase expression significantly reduced ROS production in lal−/− ECs.
Inhibition of Rab7 GTPase impaired lal−/− EC stimulation of tumor growth and metastasis in vivo and transendothelial migration, proliferation, and migration in vitro
Because lal−/− ECs stimulated tumor growth and metastasis, it would be intriguing to know the role of Rab7 GTPase in these pathogenic functions. To study B16 melanoma growth, ECs after Rab7 GTPase or control siRNA transfection were subcutaneously injected with B16 melanoma cells into the flank region of lal+/+ recipient mice. Ten days post-injection, mice injected with Rab7 GTPase siRNA–transfected lal−/− ECs developed less tumor burden than those with control siRNA–transfected lal−/− ECs (Fig. 6A). We made similar observations at other time points (7, 14, 17, 21, and 24 days; data not shown). To study B16 melanoma invasion, ECs after Rab7 GTPase or control siRNA transfection were co-injected with B16 melanoma cells into lal+/+ recipient mice intravenously. Two weeks later, mice injected with Rab7 GTPase siRNA–transfected lal−/− ECs developed fewer melanoma metastatic lesions in their lungs (Fig. 6B, top panel). Statistical analysis revealed fewer melanoma colonies in the lungs of mice that received Rab7 GTPase siRNA-transfected ECs than in those that received control siRNA–transfected ECs (Fig. 6B, bottom panel). This was further examined by an in vitro transendothelial migration study. A Transwell assay was performed with ECs transfected with Rab7 GTPase or control siRNA and cultured in the upper chamber for 48 h. CMFDA-labeled LLC cells were loaded on the EC monolayer. Fifteen hours later, LLC cells in the lower chamber were significantly fewer across lal−/− ECs transfected with Rab7 GTPase siRNA than those across lal−/− ECs transfected with control siRNA (Fig. 6C). These results suggest that Rab7 GTPase inhibition reduced lal−/− EC permeability, reducing tumor cell transendothelial migration and invasion. In addition, the effects of Rab7 GTPase knockdown in ECs on tumor cell proliferation and migration were investigated. ECs were transfected with Rab7 GTPase or control siRNA for 24 h, and the transfection medium was then replaced with DMEM with 5% FBS. Two days later, CM was harvested to treat tumor cells. As shown in Fig. 6D, compared with CM harvested from control siRNA–transfected lal−/− ECs, both B16 melanoma and LLC cell proliferation was significantly inhibited with CM from Rab7 GTPase siRNA–transfected lal−/− ECs. An in vitro tumor cell migration assay showed that LLC cells migrated less efficiently into the wound area after co-culture with CM from Rab7 GTPase siRNA–transfected lal−/− ECs than those co-cultured with CM from control siRNA–transfected lal−/− ECs (Fig. 6E).
Figure 6.
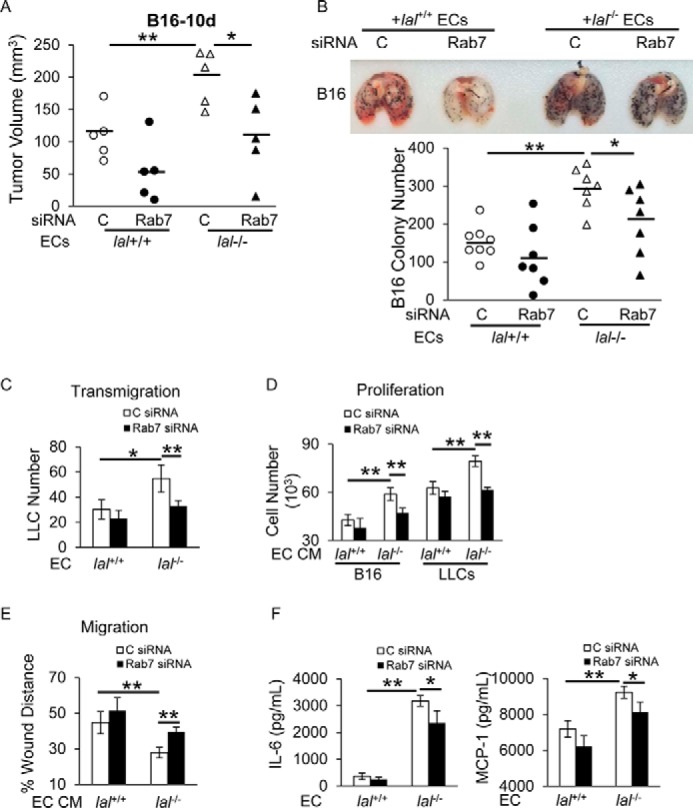
Inhibition of Rab7 GTPase impaired lal−/− EC stimulation of tumor growth and metastasis in vivo and transendothelial migration, proliferation, and migration in vitro. A, ECs (2 × 105) with Rab7 GTPase or control siRNA knockdown were mixed with B16 melanoma cells (1 × 105) and injected subcutaneously into the flank region of lal+/+ mice. Quantitative analysis of tumor volume 10 days post-injection was statistically analyzed. B, ECs (4 × 105) with Rab7 GTPase or control siRNA knockdown were mixed with B16 melanoma cells (4 × 105) and co-injected intravenously into lal+/+ mice for 2 weeks. Representative lungs are shown. Quantitative analysis of melanoma colonies in the lungs of lal+/+ recipient mice was statistically analyzed. C, control. C, LLC cell transendothelial migration was determined after Rab7 GTPase siRNA knockdown in ECs. D, B16 melanoma or LLC cell (5 × 103) proliferation was examined with CM of ECs transfected with Rab7 GTPase or control siRNA. E, tumor cell migration was assessed by in vitro wound healing assay after treatment with CM of ECs transfected with Rab7 GTPase or control siRNA. F, ELISA of secreted IL-6 and MCP-1 by ECs with Rab7 GTPase or control siRNA knockdown. In all experiments, scrambled siRNA transfection served as a control, and data are expressed as mean ± S.D. n = 4∼8. *, p < 0.05; **, p < 0.01.
We have shown that increased secretion of IL-6 and MCP-1 by lal−/− ECs contributed to enhanced tumor cell proliferation, migration, and transendothelial migration (Fig. 2, E–I). To investigate whether Rab7 GTPase affects lal−/− EC tumor-stimulating abilities by regulating secretion of chemokines/cytokines, the levels of IL-6 and MCP-1 were examined after Rab7 GTPase knockdown by ELISA. The results showed that knockdown of Rab7 GTPase reversed the elevated secretion of IL-6 and MCP-1 in lal−/− ECs (Fig. 6F). Taken together, Rab7 GTPase knockdown reduced lal−/− EC stimulation of tumor cell transendothelial migration, proliferation, and migration through down-regulating cytokine/chemokine secretion.
Inhibition of Rab7 GTPase increased tube formation and tumor binding to ECs
Rab7 GTPase did not always overlap with mTOR in regulating lal−/− EC functions. Flow cytometry analysis showed that Rab7 GTPase knockdown by siRNA transfection increased the expression level of VEGFR2 in lal−/− ECs (Fig. 7A). As reported before, VEGFR2 mediates impairment of lal−/− EC tube-forming capability (9). Indeed, Rab7 GTPase knockdown reversed the tube-forming impairment of lal−/− ECs and formed more complete tube networks than control siRNA (Fig. 7B). In a separate study, Rab7 GTPase knockdown in lal−/− ECs significantly increased B16 melanoma and LLC cell binding to lal−/− ECs than those transfected with control siRNA (Fig. 7C), which was associated with elevated expression of E-selectin and P-selectin in lal−/− ECs with Rab7 GTPase siRNA transfection compared with those with control siRNA transfection (Fig. 7D).
Figure 7.
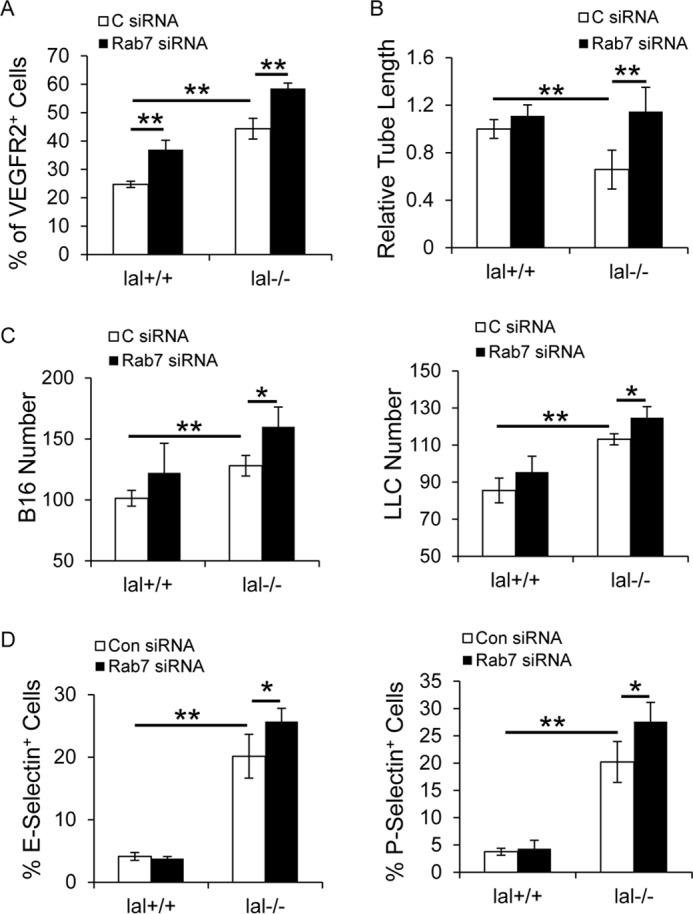
Inhibition of Rab7 GTPase increased tube formation and tumor binding to lal−/− ECs. A, flow cytometry analysis of VEGFR2 expression in ECs after Rab7 GTPase siRNA knockdown. C, control. B, EC in vitro Matrigel tube formation was assessed after Rab7 GTPase siRNA knockdown. Statistical analysis of cumulative tube lengths 6 h after EC seeding on Matrigel is shown. C, CMFDA-labeled tumor cell (B16 melanoma or LLC cells, 1 × 104) adhesion to ECs after Rab7 GTPase siRNA knockdown. Five fields in each well were quantified using a fluorescence microscope. D, flow cytometry analysis of expression of E-selectin and P-selectin in ECs after Rab7 GTPase siRNA knockdown. In all experiments, scrambled siRNA transfection served as a control, and data are expressed as mean ± S.D. n = 4∼5. *, p < 0.05; **, p < 0.01.
Discussion
The tumor environment contains various stromal cells that nurture tumor initiation, growth, and metastasis. ECs are a very important component of stromal cells in the tumor environment (1) and serve as a barrier to control penetration of tumor cells and tumor-stimulating inflammatory cells into organs (9). ECs not only regulate anti-tumor immunity (i.e. myeloid and T cell functions) but also directly influence tumor proliferation, growth, and metastasis through paracrine and juxtacrine mechanisms (18, 19). To control tumorigenesis, ECs are a critical target for cancer therapy. Understanding the molecular mechanisms and new pathways that govern EC functions can greatly facilitate new drug discovery.
A dysregulated metabolism has been reported to lead to EC dysfunction (7, 8). We have strong evidence showing that a neutral lipid metabolism controlled by LAL plays a critical role in EC anti-tumor functions (9). LAL deficiency significantly changes EC functions toward tumor promotion. In this study, lal−/− ECs facilitated in vivo tumor angiogenesis, growth, and metastasis (Fig. 1) and directly stimulated in vitro tumor proliferation, migration, adhesion, and transendothelial migration in co-culture with B16 melanoma or LLC cells (Fig. 2). Lal−/− EC tumorigenic functions are largely mediated by the lysosome-anchored metabolic regulator mTOR, as we reported previously (9). In lal−/− myeloid cells, in the absence of fatty acid metabolism, cells have to overactivate the glycolytic pathway to produce ATP through oxidative phosphorylation (14, 20), which, however, can cause mitochondrial impairment and lead to increased ROS production (21). ROS overproduction was observed in lal−/− ECs, and neutralization of ROS by antioxidant N-acetyl-l-cysteine reversed lal−/− EC dysfunction (9). Inhibition of the mTOR pathway reversed ROS overproduction in lal−/− ECs and rescued EC dysfunction (9).
Because membrane trafficking causes mTOR shuttling to lysosomes and regulates mTOR signaling, identification of factors that regulate lysosomal genesis and distribution will greatly benefit our understanding to control unwanted metabolic reprogramming in lal−/− ECs and, therefore, to control tumor angiogenesis, migration, and penetration. In this report, we have identified that Rab7 GTPase, a lysosomal protein controlling lysosomal genesis and trafficking, was up-regulated in lal−/− ECs by Western blot assay (Fig. 3A) and immunocytochemical staining (Fig. 3B). Immunofluorescence co-staining showed enhanced lysosomal genesis in association with increased expression of Rab7 GTPase and mTOR in lal−/− ECs. Interestingly and importantly, co-localization of Rab7 GTPase with mTOR on lysosomes has been observed (Fig. 3C), suggesting that the lysosome serves as a major warehouse for mTOR and Rab7 GTPase to interact. In this study, Rab7 GTPase knockdown resulted in down-regulation of mTOR downstream signaling in ECs (Fig. 4B), indicating that lysosome-anchored Rab7 GTPase regulates mTOR signaling in ECs. Physical interaction between Rab7 GTPase and mTOR in EC extract has been identified. Rab7 GTPase interacted with mTOR through its N-terminal heat repeat domain, and LAL deficiency leads to enhanced interaction between these two proteins in ECs (Fig. 4, C–F). This observation is consistent with our recent observation in myeloid cells (17). Rab7 GTPase is a late endosome/lysosome-associated small GTPase, participating in multiple regulatory mechanisms in endosomal sorting, biogenesis of lysosomes, and phagocytosis. Its mutations or dysfunctions result in traffic disorders that cause various diseases, such as cancer, lipid metabolism disease, and neuropathy (16, 22, 23). The Rab7 small GTPase is an early-induced melanoma driver whose levels can be up-regulated in melanoma as part of a lysosome-associated signature (24).
Because Rab7 GTPase and mTOR were co-localized on lysosomes of lal−/− ECs and Rab7 physically interacts with mTOR, we speculate that Rab7 GTPase is involved in mTOR-mediated cellular and pathogenic phenotypes, as we reported previously (9). At the cellular level, Rab7 GTPase inhibition decreased ROS overproduction in lal−/− ECs (Fig. 5D), which was increased by mTOR overactivation. Several other mTOR-mediated cellular phenotypes were investigated by blocking Rab7 GTPase in lal−/− ECs. We have shown that LAL deficiency increases ECs migration (9). Knocking down Rab7 GTPase in lal−/− ECs impaired enhanced EC migration (Fig. 5A). ECs serve as a barrier controlling the penetration of leukocytes into organs. An inflammatory status caused by LAL deficiency increases EC permeability to allow tumor cells and tumor-stimulating immune cells transmigrating through the EC monolayer and accumulating in distal organs of lal−/− mice (9, 25). Rab7 GTPase knockdown decreased LLC cell and Ly6G+ cell transmigration across the lal−/− EC monolayer (Figs. 5B and 6C). At the pathogenic level, lal−/− ECs stimulated in vivo tumor angiogenesis, growth, and metastasis (Fig. 1). Rab7 GTPase inhibition reduced not only in vivo tumor growth and metastasis (Fig. 6, A and B) but also in vitro tumor cell proliferation and migration (Fig. 6, D and E). For the in vivo experiments, the difference in tumor growth could be due to the initial differences in ECs transfected with Rab7 or control siRNAs. In addition, transfected Rab7 siRNAs can cause profound biological effects, including irreversible genetic and epigenetic changes. Rab7 siRNA transfection also decreased transendothelial migration of Ly6G+ myeloid cells (Fig. 5B), which plays an important role in tumor progression. Therefore, although cells could not retain siRNAs for a long time, a difference in tumor growth has been observed. Taken together, these observations collectively indicate that the lysosomal trafficking-controlling Rab7 GTPase regulates the LAL–mTOR axis in ECs, which contributes greatly to tumorigenesis.
Besides mTOR-mediated cellular and pathogenic phenotypes, the paracrine mechanism may contribute to lal−/− EC tumorigenic effects, as evidenced by increased secretion of IL-6 and MCP-1 by lal−/− ECs, and these tumorigenic effects were reversed by anti-IL-6 and/or anti-MCP-1 neutralizing antibodies (Fig. 2, E–I). Inhibition of Rab7 GTPase reversed the elevated secretion of IL-6 and MCP-1 in lal−/− ECs (Fig. 6F), suggesting that Rab7 GTPase could regulate lal−/− EC tumorigenic effects through modulating cytokine/chemokine secretion.
Although most functions of Rab7 overlap with mTOR, there are a few cellular events promoted by mTOR that were enhanced further by Rab7 GTPase inhibition in lal−/− ECs. mTOR inhibition has been reported to decrease EC tube-forming capability (26). Rab7 GTPase inhibition increased lal−/− EC tube-forming capability in vitro (Fig. 7B), in association with up-regulated expression of VEGFR2 and adhesion molecules (E-selectin and P-selectin) (Fig. 7, A and D). Rab7 GTPase inhibition also increased B16 melanoma and LLC cell binding ability to lal−/− ECs (Fig. 7C). These discrepancies suggest that, in addition to interaction with mTOR, Rab7 GTPase may influence EC functions by interacting with mTOR-independent pathways. Nevertheless, the overall outcome demonstrated that tumor growth and metastasis were reduced with Rab7 GTPase inhibition of lal−/− ECs, as shown in Fig. 6, A and B. In conclusion, Rab7 GTPase is a crucial lysosomal component that regulates LAL-controlled neutral lipid metabolism in ECs, actively participates in the regulation of tumor proliferation, progression, and metastasis, and serves as a therapeutic target for cancer treatment.
Experimental procedures
Animals and cell lines
lal+/+ and lal−/− mice of the FVB/N background were bred in-house. All scientific protocols involving the use of animals were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine and followed guidelines established by the Panel on Euthanasia of the American Veterinary Medical Association. Animals were housed under Institutional Animal Care and Use Committee–approved conditions in a secured animal facility at Indiana University School of Medicine. The murine B16 melanoma cell line and LLC cell line (ATCC, Manassas, VA) were cultured in DMEM supplemented with 10% FBS (Gibco).
Isolation and in vitro culture of pulmonary ECs
ECs were isolated from lungs and cultured in vitro as we described previously (9).
In vivo Matrigel plug assay with ECs and tumor cells
This assay was performed according to established methods with minor modifications (9, 27). ECs and tumor cells were collected separately. After washing with PBS, 2 × 105 ECs were mixed with 1 × 105 B16 melanoma or LLC cells in 500 μl of Matrigel Basement Membrane Matrix (BD Biosciences). The cell–Matrigel mixture was then injected subcutaneously into the abdomen of 3-month-old lal+/+ mice. After 10 days, the mice were sacrificed, and plugs were harvested from underneath the skin. The plugs were fixed, embedded, sectioned, stained with H&E, and examined using microscopy.
Mouse tumor growth and metastasis models
The tumor growth and metastasis models have been described recently (27, 28). A pilot study has been performed to determine the best ratio between ECs and tumor cells. To test the tumor growth potential, 4 × 105 ECs and 2 × 105 B16 melanoma or LLC cells were mixed, centrifuged, resuspended in 100 μl of PBS, and then injected subcutaneously into the flank region of 3-month-old lal+/+ recipient mice. Tumor volume (length × width2 × π/6) was monitored twice every week for 4 weeks. To test the metastasis potential, 4 × 105 ECs and 4 × 105 B16 melanoma or LLC cells were mixed, centrifuged, resuspended in 200 μl of PBS, and then injected intravenously into 3-month-old lal+/+ mice. Two weeks after the injection, the mice were sacrificed, and the lungs were inflated with 4% paraformaldehyde for examination of metastasis.
In vitro conditioned medium treatment
To collect the CM of ECs, cells were cultured until 90% confluent. The culture medium was replaced with 2 ml of DMEM with 5% FBS in each well of 6-well plates. After 24 h, the medium was collected and centrifuged to remove cell debris. For the neutralization study, before harvesting the medium, ECs were pretreated with 10 μg/ml neutralizing antibody against IL-6 or MCP-1 or control IgG for 1 h, and CM was collected afterward. For the siRNA knockdown study, ECs were transfected with Rab7 GTPase or control siRNA for 24 h. The transfection medium was then replaced with DMEM with 5% FBS. Two days later, CM was harvested for further study.
To determine the effect of EC-CM on tumor cell proliferation, B16 melanoma or LLC cells were seeded at a density of 2 × 104 cells/well into a 24-well plate. After cells attached to the plates, 500 μl of CM harvested from lal+/+ or lal−/− ECs was added to each well. Seventy-two hours later, B16 melanoma and LLC cells were counted. For the neutralization and siRNA knockdown study, 5 × 103 B16 melanoma or LLC cells were seeded into a 96-well plate, and 200 μl of CM was added to each well. To exclude the potential effect of neutralizing antibodies on tumor cell proliferation, antibodies were directly added to tumor cell culture medium as a control.
To analyze the effect of EC-CM on tumor cell migration, in vitro wound healing assays were performed as described previously (29). Briefly, B16 melanoma or LLC cells were seeded at a density of 1.5 × 105 cells/well into a 24-well plate and incubated overnight to form a confluent monolayer. A scratch was created by scraping the cell monolayer in a straight line with a p200 pipette tip. After washing three times with PBS, the medium was changed to 500 μl of CM harvested from lal+/+ or lal−/− ECs and 5 μg/ml mitomycin C (Sigma-Aldrich). Cells were kept in culture at 37 °C, 5% CO2. Images were taken 0 and 15 h after creating the scratch. Migration was estimated by measuring the distances from one side of the scratch to the other side using NIS Elements imaging software (Nikon, Melville, NY). To exclude the potential effect of neutralizing antibodies on tumor cell migration, antibodies were directly added to tumor cell culture medium as a control.
Tumor cell binding assay
The tumor cell binding assay was performed as described previously with minor modifications (30). ECs were plated in 24-well plates and incubated until they formed a uniform monolayer. Tumor cells (B16 melanoma or LLC cells) were prelabeled with CellTrackerTM Green CMFDA (Invitrogen) for 30 min at 37 °C. Labeled tumor cells (1 × 104) were then added to the EC monolayer and incubated for 3 h at 37 °C. At the end of incubation, cells were gently washed, and the binding of tumor cells to ECs was quantified under five fields in each well using a fluorescence microscope (Nikon). For the neutralization study, ECs were pretreated with 10 μg/ml neutralizing antibody against IL-6 or MCP-1 or control IgG for 1 h.
Transwell assay
Tumor cell or MDSC transendothelial migration was determined by Transwell assay as we reported previously, with minor modifications (9, 31). Briefly, ECs were grown on Transwells until they were confluent. CMFDA-labeled LLC cells (2 × 104 cells in 250 μl of medium) or MDSCs (1 × 104 cells in 250 μl of medium) were added to the EC monolayer. For tumor cell transmigration, hepatocyte growth factor was added to the lower chamber as a chemoattractant. Tumor cells were allowed to transmigrate for 15 h. The LLC cells that crossed the ECs were counted under five random microscopic fields (4, 32). For MDSCs, after 4 h, their transendothelial migration was determined by counting their numbers in the lower chamber. For the neutralization study, ECs were pretreated with 10 μg/ml neutralizing antibody against IL-6 or MCP-1 or control IgG for 1 h.
ELISA
The expression levels of IL-6 and MCP-1 in EC-CM were measured using ELISA kits (BD Biosciences) according to the instructions of the manufacturer.
Western blot analysis
Western blot analysis was performed as described previously (9, 27) with antibodies against Rab7 GTPase (rabbit monoclonal antibodies, 1:1000, Cell Signaling Technology, Beverly, MA).
Immunofluorescence staining
ECs were fixed for 15 min in 4% paraformaldehyde and permeabilized for 10 min in 0.02% Triton X-100. After washing, ECs were blocked with 1% BSA in PBST (PBS and 0.1% Tween 20) for 1 h. Cells were incubated with rabbit anti-Rab7 GTPase antibodies (1:500) (Cell Signaling Technology), rat anti-lysosomal associate membrane protein 1 (LAMP1) antibodies (1:500, Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-mTOR antibodies (1:500), or rabbit anti-mTOR antibodies (1:500, Cell Signaling Technology) at 4 °C overnight. The following day, cells were incubated with fluorescence-conjugated secondary antibody for 1 h. Images were examined under a Nikon fluorescence microscope.
Small interfering RNA transfection
Before transfection, ECs were seeded into 6-well plates at a density of 2.5 × 105 cells/well and incubated overnight. For siRNA-mediated gene knockdown, 50 nmol/liter Rab7 siRNA SMARTpool or control siRNA (Dharmacon, Chicago, IL) was transfected into cells with DharmaFECT Transfection Reagent IV (Dharmacon) according to the protocol of the manufacturer. After 72 h of transfection, cells were harvested for further analysis.
Three sets of Rab7 siRNAs were designed and synthesized by Integrated DNA Technologies using Custom Dicer-Substrate siRNA software: Rab7 siRNA1, 5′rArCrArGrGrArArArCrArGrArArGrUrGrGrArArCrUrGrUAC (sense) and 5′rGrUrArCrArGrUrUrCrCrArCrUrUrCrUrGrUrUrUrCrCrUrGrUrUrU (antisense); Rab7 siRNA2, 5′rGrUrUrGrUrGrUrUrGrGrGrArArArCrArArGrArUrUrGrACmC (sense) and 5′rGrGrUrCrArArUrCrUrUrGrUrUrUrCrCrCrArArCrArCrArArCrArA (antisense); Rab7 siRNA3, 5′rGrGrArArGrArArArGrUrGrUrUrGrCrUrGrArArGrGrUrCAT (sense) and 5′rArUrGrArCrCrUrUrCrArGrCrArArCrArCrUrUrUrCrUrUrCrCrUrA (antisense).
Purification of recombinant Rab7 GTPase and mTOR truncated fusion proteins
Recombinant Rab7 GTPase and mTOR truncated fusion proteins were purified as we described previously (17).
Immunoprecipitation assay
The immunoprecipitation assay was performed as we described previously (17). Proteins associated with Rab7 GTPase protein were detected by Western blot analysis using anti-mTOR antibody (Cell Signaling Technology). One microgram of “input” was loaded on the gels. Quantification of protein levels was determined by measuring band intensity using Image Lab 4.1 software (Bio-Rad) and calculating the ratio of the protein of interest to an internal loading control.
GST pulldown assay
The pulldown assay was performed as we described previously to evaluate the interaction between Rab7 GTPase and mTOR truncated fragments (GST–mTOR–M1A and GST–mTOR–M4) (17). Proteins associated with the mTOR truncated fragment fusion protein were detected by Western blot analysis using anti-Rab7 GTPase antibody (Cell Signaling Technology). One microgram of input was loaded on the gels.
In vitro wound healing assay
The in vitro wound healing assay was performed to analyze EC migration as described previously (9, 29).
Tube formation assay
The in vitro angiogenic activity of ECs was determined by Matrigel tube formation assay, as we described previously (9, 33). Tube formation was defined as a tube-like structure exhibiting a length four times its width (34).
Flow cytometry analysis
ECs after siRNA transfection were harvested and washed with PBS. To detect the VEGFR-2 expression level, cells were incubated with allophycocyanin (APC)-conjugated anti-mouse VEGFR-2 antibody (eBioscience, San Diego, CA). To examine the effects of Rab7 siRNA knockdown on the expression of adhesion molecules, cells were incubated with phycoerythrin-conjugated anti-mouse E-selectin and phycoerythrin-conjugated anti-mouse P-selectin antibodies (eBioscience), respectively. For flow cytometry analysis, ≥10,000 cells were acquired and scored using a LSRII machine (BD Biosciences). Data were processed using the CellQuest software program (BD Biosciences).
ROS measurement
The ROS level in ECs was measured by flow cytometry as we described previously (9).
Statistics
Data were expressed as mean ± S.D. Differences between two treatment groups were compared by Student's t test. When more than two groups were compared, one-way analysis of variance with post hoc Newman–Keul multiple comparisons test was used. Results were considered statistically significant when p < 0.05. All analyses were performed with GraphPad Prism 5.0 (GraphPad, San Diego, CA).
Author contributions
T. Z. designed and performed the experiments, analyzed and interpreted the data, and wrote the manuscript. X. D. performed the experiments and analyzed and interpreted the data. C. Y. and H. D. designed the experiments, analyzed and interpreted the data, and revised the manuscript.
Supplementary Material
Acknowledgments
We thank Michele Klunk for technical support and animal maintenance.
This work was supported by National Institutes of Health Grants HL087001 (to H. D.) and CA138759 and CA152099 (to C. Y.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Fig. 1 and Table 1.
- EC
- endothelial cell
- LAL
- lysosomal acid lipase
- MDSC
- myeloid-derived suppressor cell
- mTOR
- mammalian target of rapamycin
- LLC
- Lewis lung carcinoma
- CM
- conditioned medium
- CMFDA
- 5-chloromethylfluorescein diacetate
- ROS
- reactive oxygen species.
References
- 1. Quail D. F., and Joyce J. A. (2013) Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weis S. M., and Cheresh D. A. (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med. 17, 1359–1370 [DOI] [PubMed] [Google Scholar]
- 3. Zeng Q., Li S., Chepeha D. B., Giordano T. J., Li J., Zhang H., Polverini P. J., Nor J., Kitajewski J., and Wang C.-Y. (2005) Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell 8, 13–23 [DOI] [PubMed] [Google Scholar]
- 4. Reymond N., d'Água B. B., and Ridley A. J. (2013) Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 13, 858–870 [DOI] [PubMed] [Google Scholar]
- 5. Young M. R. (2012) Endothelial cells in the eyes of an immunologist. Cancer Immunol. Immunother. 61, 1609–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mulligan J. K., Rosenzweig S. A., and Young M. R. (2010) Tumor secretion of VEGF induces endothelial cells to suppress T cell functions through the production of PGE(2). J. Immunother. 33, 126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goveia J., Stapor P., and Carmeliet P. (2014) Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol. Med. 6, 1105–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghesquière B., Wong B. W., Kuchnio A., and Carmeliet P. (2014) Metabolism of stromal and immune cells in health and disease. Nature 511, 167–176 [DOI] [PubMed] [Google Scholar]
- 9. Zhao T., Ding X., Du H., and Yan C. (2014) Myeloid-derived suppressor cells are involved in lysosomal acid lipase deficiency-induced endothelial cell dysfunctions. J. Immunol. 193, 1942–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yan C., and Du H. (2014) Lysosomal acid lipase is critical for myeloid-derived suppressive cell differentiation, development, and homeostasis. World J. Immunol. 4, 42–51 [Google Scholar]
- 11. Grabowski G. A., Du H., and Charnas L. (2014) Chapter 142: Lysosomal acid lipase deficiencies: The Wolman disease/cholesteryl ester storage disease spectrum. In The Online Metabolic and Molecular Bases of Inherited Disease (Beaudet A. L., Vogelstein B., Kinzler K. W., Antonarakis S. E., Ballabio A., Gibson K. M., and Mitchell G. eds.) pp. The McGraw-Hill Companies, Inc., New York [Google Scholar]
- 12. Korolchuk V. I., Saiki S., Lichtenberg M., Siddiqi F. H., Roberts E. A., Imarisio S., Jahreiss L., Sarkar S., Futter M., Menzies F. M., O'Kane C. J., Deretic V., and Rubinsztein D. C. (2011) Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 13, 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zoncu R., Efeyan A., and Sabatini D. M. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yan C., Ding X., Dasgupta N., Wu L., and Du H. (2012) Gene profile of myeloid-derived suppressive cells from the bone marrow of lysosomal acid lipase knock-out mice. PLoS ONE 7, e30701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bucci C., Thomsen P., Nicoziani P., McCarthy J., and van Deurs B. (2000) Rab7: a key to lysosome biogenesis. Mol. Biol. Cell 11, 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guerra F., and Bucci C. (2016) Multiple roles of the small GTPase Rab7. Cells 5, E34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ding X., Zhang W., Zhao T., Yan C., and Du H. (2017) Rab7 GTPase controls lipid metabolic signaling in myeloid-derived suppressor cells. Oncotarget 8, 30123–30137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krishnamurthy S., Dong Z., Vodopyanov D., Imai A., Helman J. I., Prince M. E., Wicha M. S., and Nör J. E. (2010) Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer Res. 70, 9969–9978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee E., Pandey N. B., and Popel A. S. (2014) Lymphatic endothelial cells support tumor growth in breast cancer. Sci. Rep. 4, 5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ding X., Wu L., Yan C., and Du H. (2015) Establishment of lal−/− myeloid lineage cell line that resembles myeloid-derived suppressive cells. PLoS ONE 10, e0121001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ding X., Du H., Yoder M. C., and Yan C. (2014) Critical role of the mTOR pathway in development and function of myeloid-derived suppressor cells in lal−/− mice. Am. J. Pathol. 184, 397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang M., Chen L., Wang S., and Wang T. (2009) Rab7: roles in membrane trafficking and disease. Biosci. Rep. 29, 193–209 [DOI] [PubMed] [Google Scholar]
- 23. Numrich J., and Ungermann C. (2014) Endocytic Rabs in membrane trafficking and signaling. Biol. Chem. 395, 327–333 [DOI] [PubMed] [Google Scholar]
- 24. Alonso-Curbelo D., Riveiro-Falkenbach E., Pérez-Guijarro E., Cifdaloz M., Karras P., Osterloh L., Megías D., Cañón E., Calvo T. G., Olmeda D., Gómez-López G., Graña O., Sánchez-Arévalo Lobo V. J., Pisano D. G., Wang H. W., et al. (2014) RAB7 controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell 26, 61–76 [DOI] [PubMed] [Google Scholar]
- 25. Qu P., Shelley W. C., Yoder M. C., Wu L., Du H., and Yan C. (2010) Critical roles of lysosomal acid lipase in myelopoiesis. Am. J. Pathol. 176, 2394–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Farhan M. A., Carmine-Simmen K., Lewis J. D., Moore R. B., and Murray A. G. (2015) Endothelial cell mTOR complex-2 regulates sprouting angiogenesis. PLoS ONE 10, e0135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao T., Du H., Ding X., Walls K., and Yan C. (2015) Activation of mTOR pathway in myeloid-derived suppressor cells stimulates cancer cell proliferation and metastasis in lal−/− mice. Oncogene 34, 1938–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao T., Yan C., and Du H. (2016) Lysosomal acid lipase in mesenchymal stem cell stimulation of tumor growth and metastasis. Oncotarget 7, 61121–61135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liang C.-C., Park A. Y., and Guan J.-L. (2007) In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329–333 [DOI] [PubMed] [Google Scholar]
- 30. Yadav A., Kumar B., Yu J.-G., Old M., Teknos T. N., and Kumar P. (2015) Tumor-associated endothelial cells promote tumor metastasis by chaperoning circulating tumor cells and protecting them from anoikis. PLoS ONE 10, e0141602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao T., Du H., Blum J. S., and Yan C. (2016) Critical role of PPARγ in myeloid-derived suppressor cell-stimulated cancer cell proliferation and metastasis. Oncotarget 7, 1529–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reymond N., Riou P., and Ridley A. J. (2012) in Rho GTPases: Methods and Protocols (Rivero F., ed.) pp 123–142, Springer New York, New York [Google Scholar]
- 33. Zhao T., Li J., and Chen A. F. (2010) MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am. J. Physiol. Endocrinol. Metab. 299, E110–E116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamada H., Kim M. K., Iwakura A., Ii M., Thorne T., Qin G., Asai J., Tsutsumi Y., Sekiguchi H., Silver M., Wecker A., Bord E., Zhu Y., Kishore R., and Losordo D. W. (2006) Estrogen receptors α and β mediate contribution of bone marrow–derived endothelial progenitor cells to functional recovery after myocardial infarction. Circulation 114, 2261–2270 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.