

Abstract
Chlorophylls (Chls) are the most important cofactors for capturing solar energy to drive photosynthetic reactions. Five spectral types of Chls have been identified to date, with Chl f having the most red-shifted absorption maximum because of a C21-formyl group substitution of Chl f. However, the biochemical provenance of this formyl group is unknown. Here, we used a stable isotope labeling technique (18O and 2H) to determine the origin of the C21-formyl group of Chl f and to verify whether Chl f is synthesized from Chl a in the cyanobacterial species Halomicronema hongdechloris. In the presence of either H218O or 18O2, the origin of oxygen atoms in the newly synthesized chlorophylls was investigated. The pigments were isolated with HPLC, followed by MS analysis. We found that the oxygen atom of the C21-formyl group originates from molecular oxygen and not from H2O. Moreover, we examined the kinetics of the labeling of Chl a and Chl f from H. hongdechloris grown in 50% D2O-seawater medium under different light conditions. When cells were shifted from white light D2O-seawater medium to far-red light H2O-seawater medium, the observed deuteration in Chl f indicated that Chl(ide) a is the precursor of Chl f. Taken together, our results advance our understanding of the biosynthesis pathway of the chlorophylls and the formation of the formyl group in Chl f.
Keywords: biosynthesis, chlorophyll, cyanobacteria, isotopic tracer, mass spectrometry (MS), photosynthetic pigment, chlorophyll f, deuterated molecules
Introduction
Chlorophylls (Chls)2 are very effective photo-capturing cofactors in oxygenic photosynthetic organisms as they have very strong absorption bands in the visible region of the spectrum, thus enabling them to absorb solar energy efficiently (1). There are five spectral forms of naturally occurring Chls: Chl a, accompanied by its epimer Chl a′, is the most common Chl, present in all oxygenic photosynthetic organisms (2). Chl b is mainly found in green algae and plants as an accessory chlorophyll. The c-type Chls are a group of chlorophylls characterized by a fully oxidized porphyrin macrocycle that differs from the chlorin macrocycle observed in all. Chl d, accompanied with its epimer Chl d′, is the major chlorophyll found in Acaryochloris marina (Loughlin et al. 2013). The most recently identified Chl, Chl f, was reported in 2010, possesses the most red-shifted absorption spectrum of all Chls and extends the photosynthetic active radiation (PAR) compared with that of Chl d in A. marina region even further into the near-infrared region (700–760 nm) of the solar spectrum (3, 4).
Chls are substituted Mg-tetrapyrroles and have a common five-membered macrocyclic ring structure (Fig. 1). The tetrapyrrole macrocycle of all Chls, apart from Chl c, is esterified to a phytol chain. The different Chls possess distinct spectral properties and absorb light energy at different wavelength regions of the solar spectrum, primarily because of their side chain substitutions, excepting the c-type Chls, which differ in the saturation level of the macrocycle, as well as modifications at C-17 (2).
Figure 1.

Chemical structure of chlorophyll a. The substitution site of the formyl group of chlorophyll f is shown as a partial structure. The numbers of carbon are labeled following the International Union of Pure and Applied Chemistry numbering system.
The biosynthetic pathway for Chl a has been well established through a combination of isotopic labeling studies, enzyme biochemistry, and mutant analysis (5–7). It begins with the formation of δ-aminolevulinic acid (ALA), and the final step is the esterification of the hydrophobic phytol side chain, through a total of 17 enzymatic steps (2). Chl b, d, and f have similar chemical structures to that of Chl a with a difference of the formyl group substitution at the C71 methyl, C31 vinyl, or C21 methyl group of Chl a, respectively. Because Chl a is present in all oxygenic photosynthetic organisms, it has been proposed that it is the first synthesized Chl and is subsequently modified during later steps of the biosynthetic pathway to synthesize other Chls, i.e. Chl b, d, and f are synthesized from Chl a. The biosynthetic enzymes identified from plants and cyanobacteria confirmed that Chl b is synthesized from Chl(ide) a through a reaction that converts a methyl to a formyl side group at the C7 position of Chl a (8, 9). The synthesis of Chl b is catalyzed by chlorophyllide a oxygenase, in a two-step reaction that converts the C7 methyl group to a formyl group (9). Chl b can be converted back to Chl a through 7-hydroxymethy chlorophyll (8). The interconversion between Chl a and Chl b helps plants and green algae to adjust pigment composition based on the quality of light.
Stable isotopes have been used successfully to determine the biosynthetic pathways of different molecules. Of these, 13C, 15N, and 18O have been applied to understand the mechanisms of bacteriochlorophyll and Chl biosynthesis (6, 10, 11). The 18O in H218O and 18O2 has been used to investigate the origin of the oxygen atoms in Chl a, b, and d (12–14). There are five oxygen atoms present in the Chl a molecule (C55H72O5N4Mg). The biosynthetic pathway of the Chl a molecule and the origin of its five oxygen atoms are well established (6). The four oxygen atoms at the C133 and C173 carboxyl groups in the Chl a molecule are derived from water during ALA formation (6, 15). Thus, these four oxygen atoms originate from water via a hydratase-type reaction mechanism during synthesis of the precursor ALA. The fifth oxygen atom at the C131-oxo group of Chl a comes from molecular oxygen (O2) through an oxygenase-type reaction in all oxygenic photosynthetic organisms (6). Using 18O-labeling technology, it was further revealed that the sixth oxygen atom at the C7 formyl group of Chl b is derived from O2 and not from water (12). The sixth oxygen atom of the formyl group at the C3 position in Chl d is also derived from O2 through an oxygenase-type reaction mechanism, although the enzyme directly involved in the reaction is yet to be characterized (14).
D2O (2H2O) has been used extensively in biochemical studies to trace the fate of hydrogen atoms and in mechanistic studies on enzyme reactions, as well as for measuring protein synthesis rates (16–18). However, it has been less widely used in the examination of metabolic pathways because of unpredictable toleration of D levels; some of them cannot survive with high levels of D even after acclimation treatments (19–21). Part of the reason for this is that the d-proton can be toxic to an organism at sufficiently high concentrations, possibly because of the stronger hydrogen bond formed with D, which may result in the slowing down or severely limiting metabolic reactions, and in interactions where hydrogen exchange is part of the reaction or binding mechanisms (20, 22, 23). However, a number of studies have reported that a sizable fraction of D2O can be tolerated by some microbes (24). D2O has also been applied to in vivo investigations of the biomembrane structure (25, 26) and in the study of Chl biosynthesis (27, 28).
Because Chl f is a newly discovered chlorophyll, the origin of all of its six oxygen atoms needs to be investigated, especially the one located at the C21-formyl group (Fig. 1). Whether the oxygen at the C21 formyl group in Chl f is incorporated using the same reaction mechanisms as that of Chl b or Chl d is still under debate. However, Chl f has been proposed to have an analogous chemical mechanism for its formyl group substitution (29). Recently, a single enzyme, Chl f synthase, was reported to be responsible for the synthesis of Chl f under far-red light (RL), although the pathway of Chl f biosynthesis still needs to be experimentally validated (30).
Using 18O labeling, we explored whether the origin of the oxygen atoms in Chl f was from water (H218O) or from oxygen (18O2). The kinetics of Chl a and Chl f biosynthesis in response to changed light conditions was investigated using D2O-seawater in the culture medium. By monitoring the changes in the percentage of deuterated chlorophylls induced by the changed light conditions, we aimed to examine whether Chl f is derived from Chl a under far-red light conditions.
Results
H218O experiment
In the Chls extracted from Halomicronema hongdechloris cells grown in the culture having either H218O or 18O2, a 2 Da higher mass above the monoisotopic mass will be observed if one 18O is incorporated. A maximum difference of 10 Da added to the monoisotopic mass is expected for labeled Chl a because it contains five oxygen atoms and a maximum of an additional 12 Da for labeled Chl f as it contains six oxygen atoms.
The maximum incorporation of 18O atoms can be accounted by the heaviest mass peak. For Chl a extracted from H. hongdechloris grown in H218O, the heaviest mass peak observed was 900.5 m/z, which was 8 Da higher than the monoisotopic mass of Chl a (892.5). The results indicate that four oxygen atoms in Chl a are derived from water (Fig. 2A), which is consistent with previous reports (13, 15). Similar to the Chl a, the newly synthesized Chl f was 8 Da heavier with a maximum mass of 914.5 m/z also indicating that four oxygen atoms in Chl f are derived from water (Fig. 2B). The five 18O-labeled Chls (both Chl a and Chl f) were mainly observed after 12 days of incubation in 18H2O-seawater medium to a maximum percentage of ∼10%, which could be due to 18O2 produced from H218O by oxygenic photosynthesis. It was noted that both Chl a and Chl f showed similar profiles of 18O incorporation, with a maximum labeled percentage of ∼80% obtained at 10–12 days and ∼10% for five 18O-labeled chlorophylls at the end of the experimental period (16 days). There were no significant differences observed between Chl a and Chl f for the five 18O atom incorporations. The H218O incubation experiment suggested that there was a complete turnover of Chl a at ∼10 days, and almost all of the extracted Chl a contained 18O atoms. However, the newly synthesized Chl f labeling peaked at ∼12 days, indicating a two-day lag phase between newly synthesized Chl a and Chl f (Fig. 2). The slight delay in the synthesis of Chl f containing 18O may have been caused by the fact that Chl f is synthesized from Chl a (Fig. 2).
Figure 2.

18O incorporated into chlorophyll molecules using H218O-seawater KES medium. 018O atom represents the monoisotopic mass of Chl a (A) or f (B); that is, no any 18O atoms incorporated into chlorophylls. 1–418O atom represents Chl a (A) or f (B) with one to four isotopic 18O incorporations. 518O atom represents Chl a (A) or f (B) may have five isotopic 18O incorporations. Each value represents the average of the three values, and error bars represent the standard deviation.
18O2 gas experiment
The maximum mass of newly synthesized Chl a extracted from the H. hongdechloris cells grown in the presence of 18O2 was 894.5 m/z, which was 2 Da heavier than the monoisotopic mass of Chl a (892.5 m/z) (Fig. 3A). The time course 18O-labeling profile indicated that only one 18O atom was incorporated even after 14 days of incubation time. No Chl a was detected containing two labeled 18O, which was consistent with previous reports of only one oxo-group oxygen atom at the C131 position originating from O2 gas (13). There was no significant increase in the percentage of 18O-labeled Chl a (Fig. 3A) or 18O-labeled Chl f (Fig. 3B) observed after 5 days. This may be due to the relatively small volume of gas phase used in the experimental set-up (an enclosed 6 ml culture vial containing 4 ml gas phase) and that the 18O2 will be equilibrated with 16O2 produced from water by oxygenic photosynthesis activities.
Figure 3.

18O incorporated into chlorophyll molecules by cells grown in the gas phase containing 50% 18O2 gas. 018O atom represents the monoisotopic mass of Chl a (A) or f (B); that is, no 18O atoms were incorporated into chlorophylls. 118O atom represents Chl a (A) or f (B) with one isotopic 18O incorporations. 218O atom and 318O atom represent Chl a (A) and f (B) that may have two or three isotopic 18O incorporations, respectively. Error bars represent standard deviations with n = 6. The points without any error bars are from the repeated experiment that had one replication to check the validity of the data.
The mass spectrometry analysis of purified Chl f from the 18O2 experiment showed a similar profile for 18O labeling, but with a slight delay of 2 days for maximum labeling as compared with the 18O-labeled Chl a (Fig. 3, A and B) However, the maximum mass of the newly synthesized Chl f was 910.5 m/z, which was 4 Da heavier than its monoisotopic peak of 906.5 m/z (Fig. 3B), which suggested that the oxygen atoms at the C21 formyl group and the oxo-group at C131 came from oxygen molecules. The total percentage of 18O-labeled chlorophyll f (including one and two 18O-labeled chlorophyll f) is higher than the total percentage of 18O-labeled Chl a molecules at the same time points. However, the percentage of doubly 18O-labeled Chl f is the same or lower than the percentage of 18O-labeled Chl a, whereas the percentage of singly 18O-labeled Chl f is much higher. No Chl f with three 18O labels was detected, which is in agreement with the H218O results that four oxygen atoms of chlorophylls are derived from water molecules and not from oxygen molecules. The population of the one 18O-labeled Chl f included an 18O substitution either at the C21 formyl group or at the C13 oxo-position. The delay observed in the doubly labeled Chl f was consistent with the profiles of one 18O labeling in Chl a from the same cultures. The percentage of the doubly 18O-labeled Chl f (Fig. 3B) is always the same or lower than the percentage of 18O-labeled Chl a (Fig. 3A), which would be expected if Chl a is the precursor of Chl f. Taken together, Fig. 3 provides direct evidence that the oxygen atom of C21 formyl group in Chl f is derived from molecular oxygen, and the kinetics and percentage of labeling of doubly and singly 18O-labeled Chl f is consistent with it being synthesized from Chl a. However, the data cannot exclude the possibility that chlorophyllide (Chlide) a may be the precursor for Chlide f.
H. hongdechloris in D2O-seawater culture media
H. hongdechloris cells were grown in three different concentrations of D2O-seawater culture medium with final D2O concentrations of 30, 50, or 70% under RL conditions to determine the highest concentration of D2O that could be used without having an adverse effect on growth while still providing sufficient labeling of the chlorophylls to easily discriminate labeled from unlabeled chlorophylls.
There are 72 hydrogen atoms in Chl a and 70 hydrogen atoms in Chl f. The mass spectral profiles of deuterated Chl a obtained from the different percentages of D2O medium are presented, respectively, in Fig. 4. In 30% D2O medium, the peaks of 892–897 m/z comprised of the standard mass distribution of undeuterated Chl a. (i.e. the envelope of deuterated Chl a showed overlaps with undeuterated (unlabeled) Chl a resulting in one mass peak cluster centered at 899 (m/z) in the 30% D2O media (Fig. 4A, insets)). In contrast the Chl a from 50% D2O and 70% D2O cultures has three distinct mass peak clusters or envelopes. In 50% D2O medium, there are undeuterated Chl a molecules at 892–897 m/z, but two distinct deuterated clusters at 898–913 m/z, centered at 907 m/z, and at 914–932 m/z, centered at 920 m/z (Fig. 4B). In 70% D2O medium, the deuterated clusters were at 902–920 m/z, centered at 912 m/z, and at 921–942 m/z, centered at 931 m/z (Fig. 4C). In contrast the chlorophyllide fragment ions of these chlorophylls only have a single deuterated envelope (Fig. 4). The second higher molecular mass clusters correspond to a 36 atom % incorporation of deuterium from the 50% D2O culture and 51 atom % incorporation of deuterium from the 70% D2O culture. The same atom % deuterium is observed in the chlorophyllide fragment ions of these chlorophylls, indicating that the first cluster consists of either undeuterated chlorophyll macrocycle and deuterated phytyl side chain or deuterated chlorophyll macrocycle and undeuterated phytyl side chain (Fig. 4, B and C). MS/MS of 912 m/z ion of the 70% D2O gave both undeuterated and deuterated mass clusters of the chlorophyllide fragment ion, indicating that this ion is indeed a mixture of undeuterated chlorophyll macrocycle and deuterated phytyl tail, as well as deuterated chlorophyll macrocycle and undeuterated phytyl tail.
Figure 4.

Mass spectrum of deuterated chlorophyll a extracted from cells grown in 30% (A), 50% (B), and 70% (C) D2O-seawater KES medium under far-red light conditions for 4 weeks. The envelope formed by the empty bars represents the simulated mass spectra with the predicted deuterated rate of Chl a. If 14% of the hydrogen atoms of chlorophyllide a were deuterated, the mass distribution of Chllide a would have centered at 619 m/z. If 7% of hydrogen atoms of Chl a were deuterated, the mass distribution of Chl a would have centered at 899 m/z. The atom %D is increased following the increased D2O percentage in the KES medium.
Further confirmation of the identities of the different clusters seen in the Chl a from the 50 and 70% D2O cultures was via modeling and MS/MS of the center of each envelope. Fig. 5 shows the MS of Chl a from 50% D2O culture at weeks 1 and 4. In both cases all three clusters are seen, but the quantity of each is different. The modeled intensities shown in Fig. 5C assume one or both parts of the chlorophyll being deuterated at the specified atom %D of the macrocycle ring structure (32 hydrogens) or the phytyl tail (40 hydrogens). In addition MS/MS of the central peak to yield the Chlide a fragment ion confirmed that the first deuterated envelope was a mixture of deuterated and undeuterated macrocycle. The fact that the center of the envelopes does not change significantly between weeks 1and 4 indicates that the lower than expected atom %D incorporation is likely due to H for D isotope discrimination and maybe also some dilution of the D2O during set up of the experiment.
Figure 5.

Mass spectrum of deuterated chlorophyll a extracted from cells grown in 50% D2O-seawater KES medium under far-red light conditions for 1 week (A) and 4 weeks (B) and the comparison with the simulated mass spectra for different incorporation rates of deuteration (C). The mass region between 892 and 897 represents the monoisotopic mass of Chl a without any deuteration. The mass region between 898 and 932 represents deuterated chlorophylls. The envelope formed by the empty bars represents stimulated mass spectra with the predicted deuterated rate of chlorophyll a. If 18% of hydrogen atoms in Chl a were deuterated, the mass distribution of Chl a would be centered at 907 Da. If 36% of hydrogen atoms of Chl a were deuterated, the mass distribution of Chl a would be centered at 921 Da.
In subsequent analysis, the kinetics of deuterated Chl formation was restricted to examining total deuterated compared with undeuterated Chls, with undeuterated chlorophyll being represented by monoisotopic masses in the range 6 mass units higher than the lowest monoisotopic mass and deuterated chlorophylls being >6 mass units above the lowest monoisotopic mass. So undeuterated Chl a was 892–897 m/z, and deuterated Chl a had monoisotopic masses of >897. Using this criterion the rate of deuterated Chl a formation in the cells cultured in 50% D2O and 70% D2O-seawater culture medium was similar (Fig. 6A). 47% of the Chl a molecules from 50% D2O-seawater culture medium were deuterated (labeled) at week 4, which was similar to the 49% of molecules that were deuterated (labeled) from cells grown in 70% D2O-seawater medium. This indicates a similar synthesis and turnover rate for Chl a in both culture conditions. However, the rate of accumulation of deuterated Chl f (Fig. 6B) is lower than the rate of accumulation of Chl a (Fig. 6A). The simplest explanation of this observation is that deuterated Chl f is derived predominantly from the turnover of deuterated Chl a located in the newly synthesized photosynthetic protein complexes. It would be expected that these complexes would contain a higher proportion of unlabeled Chl a than any chlorophylls derived from new synthesis. This is also consistent with the 18O-labeling experiments described above.
Figure 6.

The deuterated chlorophyll percentage recorded from H. hongdechloris grown under far-red light, but in different concentrations of D2O-seawater in the KES medium. A, deuterated percentage of Chl a. B, deuterated percentage of Chl f. The error bars represent standard deviation with n = 6.
Chlorophyll a and f synthesis rates under changing light conditions
Growth in far-red light induces the synthesis of Chl f. Thus, by switching between growth in D2O and/or growth in red or white light and monitoring the changes in the rates of formation of deuterated Chl a and Chl f, we can gain insight into the possible biosynthetic origin of Chl f.
The following D2O-labeling experiments were focused on the dynamic changes of d-labeling ratio of both chlorophylls during changing culture and light conditions.
Treatment 1: white light D2O-labeling experiments
After an initial growth phase of H. hongdechloris cells in 50% D2O-seawater medium under white light (WL)-D2O illumination for 2 weeks, the cells were subjected to three different treatments for another 2 weeks. Chl f was only detected in the cells that were shifted to RL conditions (WR-H2O; Fig. 7A). Because we removed the D2O source at the time when we changed the culture light condition, the deuterated Chl f observed in the WR-H2O treatment at weeks 3 and 4 after incubation will come from either newly synthesized tetrapyrroles and thus have lower percentage labeling than Chl a, or it will have the same labeling percentage as Chl a if it is derived from Chl a. As shown in Fig. 7A, the changing trends of Chl a and Chl f labeling percentage were the same and decreased at the same rate over time, indicating that Chl f is likely to be synthesized from Chl a rather than completely de novo. That is, if Chl f was made de novo, unlabeled or very limited labeling of Chl f might be expected, which is not consistent with what we have observed in Fig. 7. Additionally, the significant decrease in labeling percentages of Chl a in the WW-H2O treatment (p = 0.0027) was the same as that of WR-H2O (p < 0.001), except that cells were kept under WL illumination. This indicates a similar rate of synthesis and turnover of Chl a in the WW-H2O treatment and in the WR-H2O treatments (Fig. 7A).
Figure 7.
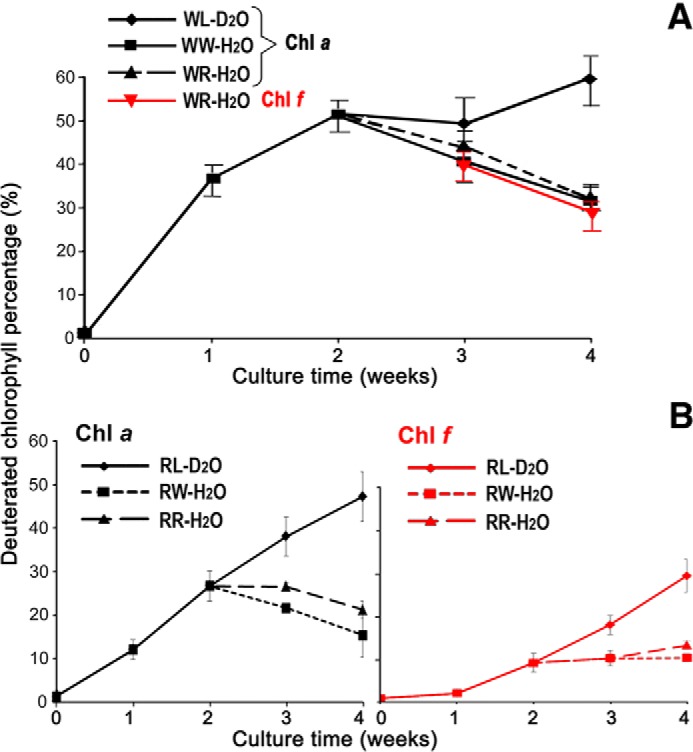
Time course of the percentage of deuterated chlorophylls extracted from the designed 50% D2O treatments (Fig. 8). The percentage of deuterated chlorophylls isolated from D2O treatments. A, cultures are initiated in white light and switched to far-red light. B, cultures are initiated in far-red light and switched to white light. WL, white light and D2O seawater medium; WW, white light with transfer to normal seawater KES medium in weeks 3 and 4; WR, white light switched to far-red light and transfer from D2O seawater to normal seawater medium in weeks 3 and 4; RL, far-red light and D2O seawater medium; RR, far-red light with transfer to normal seawater KES medium in weeks 3 and 4; RW, far-red light switched to white light and transfer from D2O seawater to normal seawater medium in weeks 3 and 4. Chl f data are presented in red. Error bars represent standard variation based on repeated measurements (n = 13, except for RR treatment n = 3).
Treatment 2: far-red light D2O-labeling experiments
After an initial growth phase of H. hongdechloris cells in 50% D2O-seawater medium under RL illumination for 2 weeks, the cells were subjected to three different treatments for another 2 weeks. Both deuterated Chl a and Chl f were present in RL cultures, and the kinetics of d-labeled Chl a and Chl f is shown in Fig. 7B. In week 1, no significant increase of deuterated Chl f was observed, with a clear lag phase in the synthesis of deuterated Chl f indicating that the Chl f may be made from predominantly undeuterated precursors or precursor pools. The percentages of deuterated Chl f in far-red light was always significantly lower than the percentage of deuterated Chl a (p < 0.001), which contrasts with the WL grown cells where the percentage of deuterated Chl a and deuterated Chl f were almost identical (Fig. 7A). In RW-H2O treatment, no new Chl f was synthesized because the RL light source was changed to WL illumination; therefore, the percentage of deuterated Chl f remained the same. In RR-H2O treatment, the cells were still able to synthesize Chl f, but the percentage of deuterated Chl f showed only a very minor increase (p = 0.053), unlike the case for Chl a where a significant decrease in deuterated Chl a was observed (p = 0.012). However, the percentage of deuterated Chl f compared with total Chl f is always lower than the percentage of deuterated Chl a to total Chl a, which is still consistent with Chl a being a precursor of Chl f.
Discussion
Chlorophyll f is known as a “red light–induced” chlorophyll (31) and has a unique modification with a formyl group at the C21 position instead of the methyl group of Chl a (32). The conversion of the C2-methyl group of Chl a to the C2-formyl group of Chl f may have a similar reaction mechanism as that of Chl b synthesis, which has a formyl group at the C7 position (29). Using 18O2 as the isotopic source, two 18O atoms are observed to be incorporated in the newly synthesized Chl f, which was different to only one 18O detected as labeled in Chl a (Fig. 3), indicating that the additional oxygen atom of Chl f originates from molecular oxygen. This clearly demonstrates that the oxygen in the formyl group of Chl f is derived from molecular oxygen and that the formation of the C2-formyl group of Chl f is an oxygenase-type reaction.
The observation in the 18O2 experiment is that doubly 18O-labeled Chl f is always the same or at a lower percentage than the 18O-labeled Chl a in the same experiment. In addition, the singly 18O-labeled Chl f is much higher than that of Chl a. These findings support the conclusion that Chl f is derived from Chl a that comes from both new synthesis as well as from previously synthesized and hence unlabeled Chl a. It also suggests that the substitution of the formyl group at C21 position occurred at a late stage of the chlorophyll biosynthesis pathway.
In our study, H. hongdechloris cells were able to grow in 50% D2O-seawater medium without any significant effects on their growth rate, indicating the tolerances of H. hongdechloris against the toxicity of D. Our results were similar to the other study where strains of Escherichia coli, Bacillus subtilis, and Bacillus thuringiensis showed no significant change in growth rate when grown in the presence of up to 50% D2O; however, a higher percentage of D2O (>50%) reduced the growth rate of the most of these organisms (17). Similarly, Lester et al. (33) evaluated a number of bacterial species including E. coli and found that 50% D2O did not inhibit their growth rate. Thus, D2O up to 50% may provide an ideal measure for labeling studies such as the present study in different bacterial communities. A few studies have also extended the scope of growing algae and other microbes in a high concentration of D2O of ∼99% by either providing them an extended period of adaptation in D2O or by acclimatizing them through a sequential subculturing into increasing concentrations of deuterium (24, 34, 35). In our study, 50% D2O-seawater was used for H. hongdechloris culture without any prior acclimation. This was necessary to perform the experiment where cells were shifted from H2O-seawater to 50% D2O-seawater medium and vice versa.
Our hypothesis that Chl a is the precursor of Chl f was also supported by the D2O-labeling experiments performed under both WL and RL conditions. In the WL-D2O experiment, no Chl f was detected in the first 2 weeks because of being a red light–induced chlorophyll. However, after shifting cells from WL-D2O to WR-H2O for a week, newly synthesized Chl f was as high as ∼39% deuterated percentage, even though the cells were transferred from D2O-seawater to H2O-seawater medium after changing the light conditions (Fig. 7A). The deuterated Chl f observed from the week 3 samples in WL-D2O experiments confirms our hypothesis that Chl f is derived from Chl a. Similar to the WL experiment, the percentage of d-labeling in Chl a and Chl f in RL also suggests that Chl a is the precursor of Chl f. In the RL experiment, a lag phase of incorporation of D in the Chl f was observed after 1 week with only ∼1% deuterated Chl f. This was because deuterated Chl a was unavailable at the beginning of the experiment, and hence mostly unlabeled Chl a was metabolized into unlabeled Chl f, resulting into a very small fraction of the deuterated Chl f. After 1 week, newly synthesized deuterated Chl a was metabolized into the newly synthesized Chl f, and hence ∼10% deuteration labeling was observed in the Chl f. Moreover, a slight increase in the deuterated Chl f at 4 weeks was observed when cells were shifted from culture medium containing D2O at 2 weeks into the medium without D2O (RR-H2O experiment). The observed increase in deuterated Chl f in the RR experiment could be due to the deuterated Chl a metabolizing into Chl f after 1 week of adaptation of the cells into the new medium. Additionally, the delay of 18O-labeling Chl f observed from H218O- and 18O2-labeling experiments (Figs. 2 and 3) indicated that Chl f is synthesized from Chl a or at a late stage of the chlorophyll biosynthesis pathway. The current experimental results cannot exclude the possibility that chlorophyllide a may be the immediate precursor of chlorophyllide f instead of Chl a. We know that the last step of chlorophyll biosynthesis is esterification of the chlorophyllide with a phytol-pyrophosphate to produce chlorophylls. The deuterated chlorophylls can originate from the precursors of the magnesium tetrapyrrole five-membered ring structure (chlorophyllides) and the precursors of the phytol chains. However, we could not distinguish the atom %D in the ring structures from the atom %D in the phytol chains, because of the limitations of mass spectral analysis (> 500 Da) for the small molecular fraction of deuterated phytol chains with a predicted molecular mass of ∼278 Da. The deuterated chlorophyll is the sum of the deuterated ring structure (chlorophyllides) and the deuterated phytol chain. The observed atom %D at the chlorophyllide section, which was higher than that of total chlorophyll atom %D from the same samples, could indicate that the deuterated chlorophyllide percentage was the sum of D labeling at the ring structure, with and without the D labeling at the phytol chain, because of two individual biosynthetic pathways. That Chl f is derived from Chl a further supports the Granick hypothesis of forward direction of evolution for the Chl and heme biosynthesis pathways (36), which states that pathways evolved forward as organisms evolved. However, it is still unclear whether chlorophyllide a or Chl a is the central molecule for Chl f biosynthesis. Moreover, it is also unclear whether the formyl group of Chl f is synthesized through a one-step or two-step reaction from the C21 methyl group of Chl a. Similarly, further work needs to be done to understand whether Chl f degrades via Chl a, similar to Chl b through a hydroxymethyl chlorophyll intermediate (8).
The 18O2 and H218O experiments revealed the origin of all oxygen atoms of Chl f. They also indicates that the extra oxygen at C21 formyl group of Chl f originates through an oxygenase-type of reaction mechanisms. Our D2O and 18O isotope labeling suggests that Chl a is the precursor of Chl f biosynthesis, similar to that of Chl b and Chl d. The results of this study show that D2O can be used as a powerful tool for stable isotope labeling in cyanobacteria to examine biosynthesis pathways. The results reported here have an important implication for understanding the evolution and biosynthesis pathway of chlorophylls and their formyl group incorporations.
Materials and methods
Culture conditions
H. hongdechloris cells were cultured in modified “K” enrichment seawater (KES) medium (37) under either RL with intensity of 10 μE, or under WL at ∼ 20 μE, on a rotating flatbed shaker at ∼100 rpm. Isotopic labeling experiments were performed on cultures grown under RL conditions except where detailed light conditions have been indicated. In each experiment, cultures were homogenized and precultured for 4 days prior to subculturing to the specified conditions, because the doubling time of H. hongdechloris is ∼3–4 days (38).
Sea salt mixture was bought from a local store (Instant Ocean), and seawater medium was made according to the manufacturer's instructions. D2O (2H2O) (99.5% H atom in the 2H form) was obtained from the Australian Nuclear Science and Technology Organization. To make the required percentage of D2O-seawater, the sea salt was dissolved in D2O mixed with H2O to the required percentage of D.
H218O experiment
H. hongdechloris cells were harvested from 1 ml of culture with OD707 nm = 0.2 and transferred into either 1 ml of H218O (97%, 18O; Sigma–Aldrich) or H216O-modified KES artificial seawater medium. The cells were collected for pigment analysis at 4, 8, 12, and 16 days. The experiment was repeated as above, but cells were sampled at 2, 6, 10, and 14 days. Each sampling point had three technical repeats.
18O2 experiment
H. hongdechloris cells harvested from 1 ml of the culture with OD707 nm = 0.2 were transferred to 6-ml vacuum vials (Sarstedt). 4 ml of premixed 50% 18O2 and 50% dinitrogen (N2) gas was injected into the vacuum vials before culture inoculation. The controls were set up by injecting 4 ml of premixed 50% 16O2 and 50% dinitrogen (N2) gas. Two biological replicates for each gas treatment including the control were tested and sampled for pigment isolation up to 16 days. Each purified chlorophyll sample was analyzed with three individual replicates for mass spectral analysis. The whole experiment was repeated, and results of the two experiments were combined to verify the experimental data.
Growth in D2O
Different percentages of D2O-seawater KES medium were used to test the tolerances of H. hongdechloris cell culture to the presence of D2O, and the potential toxicities of D2O of the cultures. H. hongdechloris cells were cultured in three different concentrations of D2O-seawater as 30, 50, and 70% for 4 weeks. The deuterated percentage of isolated chlorophylls compared with undeuterated chlorophyll remaining was indicative of the toxicity of D2O to the culture. We use 24-well culture plates for examining the tolerance of the cells to the presence of D2O and for the following pulse-chase experiments. Each well contains 2 ml of H. hongdochloris cell culture with a Qy value of 0.6 at 665 nm (in 100% methanol) at inoculation time. The samples are collected randomly from individual well following the designed experiments.
Pulse-chase of D2O and H2O
The 50% D2O-seawater treatment was selected as the optimal D2O concentration in terms of growth rate and in being able to easily distinguish labeled from unlabeled Chls, and this treatment was used in all further labeling experiments. The experimental setup relied on the previously reported observations that H. hongdechloris cells growing under WL produce mainly Chl a and no Chl f, whereas when H. hongdechloris is grown under RL light conditions, Chl f accumulates to ∼15% of the Chl a concentration (31, 39). In switching cultures from WL to RL, Chl f becomes detectable within 2 days, and after switching from RL to WL, Chl f starts disappearing within 2 days (38). Comparing the labeling patterns of Chl a and Chl f during a switch of light conditions and at the same time transferring the cultures from D2O to H2O would provide data to determine whether Chl f is synthesized from precursors like Chl a already present in the culture or from de novo synthesis. The cultures were maintained under RL or WL for a minimum of 3 weeks before commencement of the experiments. Two different starting conditions were used in these experiments (Fig. 8): 1) WL-D2O cell cultures were grown under WL in 50% D2O-seawater KES medium for 2 weeks, and 2) RL-D2O cultures were grown under RL in 50% D2O-seawater KES medium for 2 weeks.
Figure 8.
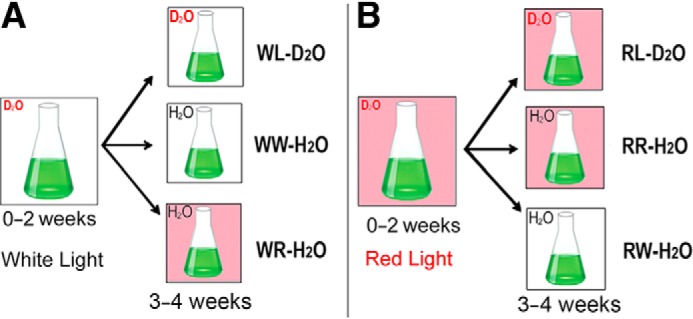
Diagram of the experimental set-up. H. hongdechloris cells were grown in 50% D2O-seawater KES medium either under white light (A) or under far-red light (B). After 2 weeks, the H. hongdechloris cultures were equally divided and transferred to three different conditions. D2O represents the culture grown in D2O-seawater KES medium, and H2O represents the culture grown in normal seawater KES medium. Pink boxes indicate the culture was illuminated under far-red light conditions (730 nm LED light), and white boxes indicate the culture was illuminated by white light (WL). Six treatments are designated as white light and D2O seawater medium (WL-D2O); WW-H2O, white light and changed to normal seawater medium in 3–4 weeks; WR-H2O, switched light to far-red light and changed to normal seawater medium in 3–4 weeks; RL-D2O, far-red light and D2O seawater medium; RR-H2O, far-red light and changed to normal seawater KES medium in 3–4 weeks; RW-H2O, switched light white light and changed to normal seawater medium in 3–4 weeks.
Treatment 1 experiment
WL-D2O cells (Fig. 8A) were equally divided into three subtreatments for another 2 weeks. For condition 1 (WL-D2O), the cells were continuously cultured under the same WL conditions in 50% D2O-seawater KES medium. For condition 2 (WW-H2O), the cells were rinsed with H2O-seawater and cultured in H2O-seawater KES medium but remained under WL conditions. For condition 3 (WR-H2O), the cells were rinsed with H2O-seawater and cultured in H2O-seawater KES medium under RL condition. Cells from each treatment were sampled at weekly intervals for 4 weeks for pigment extraction and further HPLC and mass spectral analysis.
Treatment 2 experiment
RL-D2O cells (Fig. 8B), were equally divided into three subtreatments for another 2 weeks. For condition 1 (RL-D2O), the cells were continuously cultured under the same RL conditions in 50% D2O-seawater KES medium. For condition 2 (RW-H2O), RL-D2O cells were rinsed with H2O-seawater and cultured in H2O-seawater KES medium under WL condition. For condition 3 (RR-H2O), the RL-D2O cells were rinsed with H2O-seawater and cultured in H2O-seawater KES medium but remained under RL conditions. Cells from each treatment were sampled at weekly intervals for 4 weeks for pigment extraction and further HPLC and mass spectral analysis.
Pigment extraction and chlorophyll purification
All pigment extractions and purifications were conducted under dim green light and at 4 °C where possible. The cells were harvested by centrifugation at 13,000 rpm for 5 min, and pigments were extracted using prechilled 100% methanol. The extracted methanolic samples were immediately used for HPLC purification as described previously (40). Chl f and Chl a were separated using a C18 reverse phase column (Kinetex, 100 × 4.6 mm; Phenomenex, Lane Cove, Australia) on a Shimdazu HPLC (model 10A series) using 100% methanol as the mobile phase at a flow rate of 1 ml/min. Purified Chl a and Chl f were collected at their retention times and immediately dried by vacuum centrifugation. Dried Chl a and Chl f were stored in the dark at −80 °C for further mass spectral analysis.
Mass spectrometry
The purified chlorophylls were mixed with 4 μl of the matrix terthiopene (Sigma) (10 mg/ml in acetone) and placed on the sample holder plate. For 18O2 and H218O experiments, pigments were analyzed by MALDI-MS on a Voyager-DE STR (Applied Biosystems) as described before (14). For D2O experiments, mass spectral analyses were performed using an Applied Biosystems Q-STAR Pulsar with a MALDI source (Mass Spectrometry Core Facility, University of Sydney). Data analysis was performed using Data Explorer and Analyst TF 1.7.1 software for the MALDI-MS performed on the Voyager-DE STR and Q-STAR, respectively.
Calculation of isotopic-labeled chlorophylls
Chl a (C55H72N4O5Mg) contains five oxygen atoms and 72 hydrogen atoms and has average molecular mass and monoisotopic mass values of 893.5426 and 892.5353 Da, respectively; Chl f (C55H70N4O6Mg) has six oxygen atoms and 70 hydrogen atoms and has average molecular mass and monoisotopic mass values of 907.5219 and 906.5146 Da, respectively. Non-labeled Chl a has seven mass peaks of 892.5 to 898.5 with the relative ion intensities of 100, 75, 42, 15.6, 3.9, 0.7, and 0.1, associated with the natural abundance of isotopic carbon, oxygen, hydrogen, and magnesium. Non-labeled Chl f has seven mass peaks of 906.5 to 912.5 with the relative intensities of 100, 75, 42.2, 15.8, 4, 0.7, and 0.1. The relative concentrations of each isotopic 18O incorporated into the Chls were calculated as described before (10, 14). The values obtained in the control experiment were deducted from the calculated values of the labeled 18O isotopomers to avoid any noise and artifacts obtained in the mass spectrum.
Chl a molecules ionized by MALDI-TOF analysis have two major ion peak regions at m/z 892.5–898.5 and m/z 614.2–620.5, which correspond to M+ masses of chlorophyll a and chlorophyllide a, respectively. Similarly Chl f molecules ionized during MALDI-TOF analysis have two major ions peak regions at m/z 906.5–912.5 and m/z 628.2–634.5, which correspond to M+ masses of chlorophyll f and chlorophyllide f, respectively. MS data from both chlorophyll and chlorophyllide ions were calculated using the matrix developed previously in both H218O- and 18O-labeling experiments to calculate the abundance of 18O containing chlorophylls (10, 14).
With 50% D2O in the medium, all hydrogen atoms in the chlorophylls could be potentially labeled. The labeling results in an envelope of mass peaks well separated from the unlabeled chlorophyll masses. Unless stated otherwise, the calculation of the ratio of deuterated to undeuterated chlorophylls/chlorophyllides was performed by the sum of peak heights of the first six monoisotopic masses as representing undeuterated chlorophyll (unlabeled chlorophylls), whereas the sum of peak intensities of the monoisotopic masses greater than six represented the deuterated chlorophyll (labeled chlorophylls). The peak heights were only summed for peaks with masses ±0.02 mass units from the expected calculated monoisotopic masses and also only with a signal to noise ratio of >3:1.
The extent of deuterated atom percentage (%D per molecule) in 50% D2O-seawater KES medium was calculated by the simulation described previously (41) and available online at https://www.ncbi.nlm.nih.gov/CBBresearch/Yu/midas/index.html.3 The basic assumption of the program is that the amount of deuterium atoms available for each position in Chl a and Chl f is equal. The expected profile of labeled peaks with different percentages of D out of 72 H atoms in Chl a or 70 H atoms in Chl f were thus calculated and compared with the experimental data to determine the extent of labeling.
Author contributions
H. G. performed the experiments. P. C. L. contributed to the 18O-labeling. H. G., R. D. W., and M. C. designed the experiments, analyzed the data, and wrote the manuscript.
Acknowledgments
We thank Dr. Chris Garvey from the Australian Nuclear Science and Technology Organization for kindly providing D2O (>99%). We also thank Dr. Ben Crossett (Mass Spectrometry Core Facility, University of Sydney) for assisting with the mass spectral analysis.
This work was supported by the Australian Research Council Grant CE140100015. The authors declare that they have no conflicts of interest with the contents of this article.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- Chl
- chlorophyll
- Chlide
- chlorophyllide
- RL
- 730-nm LED red light
- WL
- white light
- ALA
- δ-aminolevulinic acid.
References
- 1. Croce R., and van Amerongen H. (2014) Natural strategies for photosynthetic light harvesting. Nat. Chem. Biol. 10, 492–501 [DOI] [PubMed] [Google Scholar]
- 2. Chen M. (2014) Chlorophyll modifications and their spectral extension in oxygenic photosynthesis. Annu. Rev. Biochem. 83, 317–340 [DOI] [PubMed] [Google Scholar]
- 3. Chen M., Schliep M., Willows R. D., Cai Z.-L., Neilan B. A., and Scheer H. (2010) A red-shifted chlorophyll. Science 329, 1318–1319 [DOI] [PubMed] [Google Scholar]
- 4. Chen M., and Blankenship R. E. (2011) Expanding the solar spectrum used by photosynthesis. Trends Plant Sci. 16, 427–431 [DOI] [PubMed] [Google Scholar]
- 5. Suzuki J. Y., Bollivar D. W., and Bauer C. E. (1997) Genetic analysis of chlorophyll biosynthesis. Annu. Rev. Genet. 31, 61–89 [DOI] [PubMed] [Google Scholar]
- 6. Porra R. J., and Scheer H. (2000) 18O and mass spectrometry in chlorophyll research: derivation and loss of oxygen atoms at the periphery of the chlorophyll macrocycle during biosynthesis, degradation and adaptation. Photosynth. Res. 66, 159–175 [DOI] [PubMed] [Google Scholar]
- 7. Hollingshead S., Bliss S., Baker P. J., and Neil Hunter C. (2017) Conserved residues in Ycf54 are required for protochlorophyllide formation in Synechocystis sp. PCC 6803. Biochem. J. 474, 667–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ito H., Ohtsuka T., and Tanaka A. (1996) Conversion of chlorophyll b to chlorophyll a via 7-hydroxymethyl chlorophyll. J. Biol. Chem. 271, 1475–1479 [DOI] [PubMed] [Google Scholar]
- 9. Tanaka A., Ito H., Tanaka R., Tanaka N. K., Yoshida K., and Okada K. (1998) Chlorophyll a oxygenase (CAO) is involved in chlorophyll b formation from chlorophyll a. Proc. Natl. Acad. Sci. U.S.A. 95, 12719–12723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vavilin D., Brune D. C., and Vermaas W. (2005) 15N-labeling to determine chlorophyll synthesis and degradation in Synechocystis sp. PCC 6803 strains lacking one or both photosystems. Biochim. Biophys. Acta 1708, 91–101 [DOI] [PubMed] [Google Scholar]
- 11. Egorova-Zachernyuk T., van Rossum B., Erkelens C., and de Groot H. (2008) Characterisation of uniformly 13C,15N labelled bacteriochlorophyll a and bacteriopheophytin a in solution and in solid state: complete assignment of the 13C, 1H and 15N chemical shifts. Magn. Reson. Chem. 46, 1074–1083 [DOI] [PubMed] [Google Scholar]
- 12. Porra R. J., Schäfer W., Cmiel E., Katheder I., and Scheer H. (1994) The derivation of the formyl-group oxygen of chlorophyll b in higher plants from molecular oxygen. Eur. J. Biochem. 219, 671–679 [DOI] [PubMed] [Google Scholar]
- 13. Porra R. J., Schäfer W., Katheder I., and Scheer H. (1995) The derivation of the oxygen atoms of the 131-oxo and 3-acetyl groups of bacteriochlorophyll a from water in Rhodobacter sphaeroides cells adapting from respiratory to photosynthetic conditions: evidence for an anaerobic pathway for the formation of isocyclic ring E. FEBS Lett. 371, 21–24 [DOI] [PubMed] [Google Scholar]
- 14. Schliep M., Crossett B., Willows R. D., and Chen M. (2010) 18O labeling of chlorophyll d in Acaryochloris marina reveals that chlorophyll a and molecular oxygen are precursors. J. Biol. Chem. 285, 28450–28456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Porra R. J., Schäfer W., Gad'on N., Katheder I., Drews G., and Scheer H. (1996) Origin of the two carbonyl oxygens of bacteriochlorophyll a. Eur. J. Biochem. 239, 85–92 [DOI] [PubMed] [Google Scholar]
- 16. Busch R., Kim Y. K., Neese R. A., Schade-Serin V., Collins M., Awada M., Gardner J. L., Beysen C., Marino M. E., Misell L. M., and Hellerstein M. K. (2006) Measurement of protein turnover rates by heavy water labeling of nonessential amino acids. Biochim. Biophys. Acta 1760, 730–744 [DOI] [PubMed] [Google Scholar]
- 17. Berry D., Mader E., Lee T. K., Woebken D., Wang Y., Zhu D., Palatinszky M., Schintlmeister A., Schmid M. C., Hanson B. T., Shterzer N., Mizrahi I., Rauch I., Decker T., Bocklitz T., et al. (2015) Tracking heavy water (D2O) incorporation for identifying and sorting active microbial cells. Proc. Natl. Acad. Sci. U.S.A. 112, E194–E203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kasumov T., Willard B., Li L., Sadygov R. G., and Previs S. (2015) Proteome dynamics with heavy water: instrumentations, data analysis, and biological applications. In Recent Advances in Proteomics Research (Magdeldin S., ed) Chapter 2, InTech, Rijeka, Croatia [Google Scholar]
- 19. Pengelly W. L., and Bandurski R. S. (1983) Analysis of indole-3-acetic acid aetabolism in Zea mays using deuterium oxide as a tracer. Plant Physiol. 73, 445–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hochuli M., Szyperski T., and Wüthrich K. (2000) Deuterium isotope effects on the central carbon metabolism of Escherichia coli cells grown on a D2O-containing minimal medium. J. Biomol. NMR 17, 33–42 [DOI] [PubMed] [Google Scholar]
- 21. Tcherkez G., Mahé A., Gauthier P., Mauve C., Gout E., Bligny R., Cornic G., and Hodges M. (2009) In folio respiratory fluxomics revealed by 13C isotopic labeling and H/D isotope effects highlight the noncyclic nature of the tricarboxylic acid “cycle” in illuminated leaves. Plant Physiol. 151, 620–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mosin O. V., and Ignatov I. (2014) Biological influence of deuterium on procariotic and eukaryotic cells. Eur. J. Mol. Biotechnol. 3, 11–24 [Google Scholar]
- 23. Mosin O., and Ignatov I. (2015) Isotopic effects of deuterium in various biological objects as the cells of methylotrophic, chemoheterotrophic, photoorganotrophic microorganisms and green algae. J. Health Med. Nursing 11, 2422–8419 [Google Scholar]
- 24. Mosin O., and Ignatov I. (2012) Isotope effects of deuterium in bacterial and microalgae cells at growth on heavy water (D2O). Water Chem. Ecol. 3, 83–94 [Google Scholar]
- 25. Garvey C. J., Lenné T., Koster K. L., Kent B., and Bryant G. (2013) Phospholipid membrane protection by sugar molecules during dehydration: insights into molecular mechanisms using scattering techniques. Int. J. Mol. Sci. 14, 8148–8163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Y., Lin Y., Garvey C. J., Birch D., Corkery R. W., Loughlin P. C., Scheer H., Willows R. D., and Chen M. (2016) Characterization of red-shifted phycobilisomes isolated from the chlorophyll f-containing cyanobacterium Halomicronema hongdechloris. Biochim. Biophys. Acta 1857, 107–114 [DOI] [PubMed] [Google Scholar]
- 27. Katz J. J., Dougherty R. C., Svec W. A., and Strain H. H. (1964) Exchangable H in chlorophyll and the path of H in photosynthesis. J. Am. Chem. Soc. 86, 4220–4221 [Google Scholar]
- 28. Folly P., and Engel N. (1999) Chlorophyll b to chlorophyll a conversion precedes chlorophyll degradation in Hordeum vulgare L. J. Biol. Chem. 274, 21811–21816 [DOI] [PubMed] [Google Scholar]
- 29. Kräutler B. (2011) A new factor in life's quest for energy. Angew. Chem. Int. Ed. 50, 2439–2441 [DOI] [PubMed] [Google Scholar]
- 30. Ho M.-Y., Shen G., Canniffe D. P., Zhao C., and Bryant D. A. (2016) Light-dependent chlorophyll f synthase is a highly divergent paralog of PsbA of photosystem II. Science 353, aaf9178. [DOI] [PubMed] [Google Scholar]
- 31. Chen M., Li Y., Birch D., and Willows R. D. (2012) A cyanobacterium that contains chlorophyll f: a red-absorbing photopigment. FEBS Lett. 586, 3249–3254 [DOI] [PubMed] [Google Scholar]
- 32. Willows R. D., Li Y., Scheer H., and Chen M. (2013) Structure of chlorophyll f. Org. Lett. 15, 1588–1590 [DOI] [PubMed] [Google Scholar]
- 33. Lester W. Jr., Sun S. H., and Seber A. (1960) Observations on the influence of deuterium on bacterial growth. Ann. N.Y. Acad. Sci. 84, 667–677 [DOI] [PubMed] [Google Scholar]
- 34. Chorney W., Scully N. J., Crespi H. L., and Katz J. J. (1960) The growth of algae in deuterium oxide. Biochim. Biophys. Acta 37, 280–287 [DOI] [PubMed] [Google Scholar]
- 35. Lu M. F., Zhang Y. J., and Zhang H. Y. (2013) Isotope effects on cell growth and sporulation, and spore heat resistance, survival and spontaneous mutation of Bacillus cereus by deuterium oxide culture. Afr. J. Microbiol. Res. 7, 604–611 [Google Scholar]
- 36. Granick S. (1965) Evolution of heme and chlorophyll. In Evolving genes and proteins (Bryson V., and Vogel H., eds) pp. 67–88, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 37. Keller M. D., Selvin R. C., Claus W., and Guillard R. R. R. (1987) Media for the culture of oceanic ultraphytoplankton. J. Phycol. 23, 633–638 [Google Scholar]
- 38. Li Y., Lin Y., Loughlin P. C., and Chen M. (2014) Optimization and effects of different culture conditions on growth of Halomicronema hongdechloris: a filamentous cyanobacterium containing chlorophyll f. Front. Plant Sci. 5, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Akutsu S., Fujinuma D., Furukawa H., Watanabe T., Ohnishi-Kameyama M., Ono H., Ohkubo S., Miyashita H., and Kobayashi M. (2011) Pigment analysis of a chlorophyll f-containing cyanobacterium strain KC1isolated from Lake Biwa. Photomed. Photobiol. 33, 35–40 [Google Scholar]
- 40. Loughlin P. C., Willows R. D., and Chen M. (2014) In vitro conversion of vinyl to formyl groups in naturally occurring chlorophylls. Sci. Rep. 4, 6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Willows R., Netting A., and Milborrow B. (1994) Endogenous biosynthetic precursors of (+)-abscisic acid: I. incorporation of isotopes from 2H2O, 18O2 and [5-18O]mevalonic acid. Funct. Plant Biol. 21, 327–343 [Google Scholar]