

Abstract
The acylation of lysine residues in superoxide dismutase-1 (SOD1) has been previously shown to decrease its rate of nucleation and elongation into amyloid-like fibrils linked to amyotrophic lateral sclerosis. The chemical mechanism underlying this effect is unclear, i.e. hydrophobic/steric effects versus electrostatic effects. Moreover, the degree to which the acylation might alter the prion-like seeding of SOD1 in vivo has not been addressed. Here, we acylated a fraction of lysine residues in SOD1 with groups of variable hydrophobicity, charge, and conformational entropy. The effect of each acyl group on the rate of SOD1 fibril nucleation and elongation were quantified in vitro with thioflavin-T (ThT) fluorescence, and we performed 594 iterate aggregation assays to obtain statistically significant rates. The effect of the lysine acylation on the prion-like seeding of SOD1 was assayed in spinal cord extracts of transgenic mice expressing a G85R SOD1–yellow fluorescent protein construct. Acyl groups with >2 carboxylic acids diminished self-assembly into ThT-positive fibrils and instead promoted the self-assembly of ThT-negative fibrils and amorphous complexes. The addition of ThT-negative, acylated SOD1 fibrils to organotypic spinal cord failed to produce the SOD1 inclusion pathology that typically results from the addition of ThT-positive SOD1 fibrils. These results suggest that chemically increasing the net negative surface charge of SOD1 via acylation can block the prion-like propagation of oligomeric SOD1 in spinal cord.
Keywords: amyloid, amyotrophic lateral sclerosis (ALS) (Lou Gehrig disease), fibril, oligomer, protein aggregation, lysine acetylation, motor neuron disease, succinylation
Introduction
The role that long-range Coulombic interactions play in the nucleation, propagation, and toxicity of amyloid-like oligomers is generally overlooked. These electrostatic forces scale with the net charge (Z) of the self-assembling proteins, unlike short-range Coulombic interactions that scale with local patterns of surface charge (1–5). In the hydrophobic core of a protein or protein aggregate, these long-range Coulombic forces can extend dozens of angstroms further than through aqueous solution, where the Debye radius is only ∼10 Å at pH 7.4, I = 0.1 m (6, 7).
Can long-range electrostatic forces be medicinally targeted, for example, to increase electrostatic repulsion between protein subunits that self-assemble into neurotoxic complexes? Can Z be viewed as a druggable parameter and chemically permuted like other classic parameters in biochemistry such as an enzyme's Vmax or Km? To date, there have been only a handful of studies to determine whether such electrostatic forces can be chemically (medicinally) amplified to inhibit self-assembly (1, 8, 9). This potentially new subfield of medicinal chemistry is obscured by the general difficulty in quantifying the net charge of a folded protein in solution and the lack of tools available for measuring the net charge of a folded protein in solution (6). For example, the magnitude by which Z is affected by a missense mutation, post-translational modification, fluctuation in subcellular pH, or co-factor binding has only been measured for a few proteins, using “protein charge ladders” (6, 10, 11). In these cases, measured values of net charge can differ in magnitude (and sign/polarity) from predicted values (6, 10, 11).
In this paper, we hypothesize that lysine acylation (lysine-ϵ-NH3+ → lysine-ϵ-NHCOR) represents a plausible mechanism by which protein self-assembly might be controlled electrostatically. Although this electrostatic hypothesis is unconventional, it is not unreasonable. Pharmacological agents such as aspirin, the simplest aryl ester, can transfer acetyl groups directly to lysine residues of ∼100 different proteins (12–15). In general, the neutralization of a single lysine-ϵ-NH3+ in a protein is expected to have a large effect on its overall net charge because proteins are predicted to have low values of net charge to begin with (“low” defined as Z between −10 and +10) (16). Could acyl groups function as potent electrostatic inhibitors of undesired interactions between proteins? We conducted this study to begin to answer this question, within the context of the amyloidogenesis of SOD1 (17, 18).2 The self-assembly of SOD1, a net negatively charged protein at pH 7.4, into amyloid-like complexes is regarded as one of many causes of a subset of ALS (19–21).
The toolbox of lysine acyl modifications utilized by biological systems is broader than once assumed and includes methylation, formylation, propionylation, butyrylation, crotonylation, malonylation, succinylation, glutarylation, and myristoylation (22). Previous studies have shown that acetylation of lysine residues (and other types of chemical modifications) in neurotoxic proteins can affect processes linked to neurodegeneration (23–25), but the biochemical mechanisms are unclear. For example, acetylation of lysine residues in TDP-43 is enhanced by oxidative stress, and disrupts RNA- and DNA-binding sites and triggers TDP-43 aggregation (24). In mice, SOD1 has been reported to undergo succinylation and acetylation at multiple lysine residues; however, any role in ALS is unclear (26). The acetylation of Tau can either promote or decrease aggregation depending on the site of acetylation (25). In the case of SOD1, palmitoylation of cysteine has been shown to cause partitioning of SOD1 to cell membranes, which might increase its aggregation propensity by increasing the probability of surface-catalyzed aggregation (27).
We have recently shown that acetylation of multiple lysine residues in apo-SOD1 (up to ∼6 of 11 lysines in the SOD1 chain) with aspirin impedes the (in vitro) nucleation and elongation of amyloid fibrils as measured by microplate-based ThT assays (1). This protective effect was rationalized to be electrostatic in nature and originate from the increased electrostatic repulsion of acetylated SOD1 via neutralization of lysine-ϵ-NH3+ to lysine-ϵ-NHCOCH3. For example, apo-SOD1 has a net negative charge of −6.92 ± 0.14 per subunit (11), which is increased by approximately −0.9 units per acetyl group. Although our previous study proved that post-translational modifications can inhibit SOD1 fibrillization via transferring of acetyl groups, it did not examine: (i) whether acetylation promoted alternate (amorphous) pathways of SOD1 aggregation (28); (ii) if natural and unnatural acyl modifications of greater negative charge (i.e. ΔZ > 1) could inhibit SOD1 fibrillization into amyloid by a greater magnitude than acetyl modifications; and (iii) whether acylation of lysine residues in SOD1 could inhibit the prion-like seeding of SOD1 complexes in living systems. In the present study, we answer these three questions.
In this study, we quantified how acylation of a fraction of lysine in purified, recombinant WT apo-SOD1 with natural and unnatural groups of variable negative charge and hydrophobicity (Figs. 1 and 2) affected: (i) the net surface charge of SOD1 at pH 7.4; (ii) the kinetics and morphology of amyloid formation during microplate-based assays; and (iii) the ability of WT SOD1 fibrils to seed the aggregation of ALS-variant SOD1 in organotypic spinal cord derived from transgenic G85R-SOD1 mice.
Figure 1.

Increasing the net negative charge (Z) of apo-SOD1 via acylation of lysine-ϵ-NH3+. Acylation of SOD1 by PM is shown as an example. The net charge of WT apo-SOD1 is −12.13 ± 0.08 (per dimer) with pH 7.4 10 mm potassium phosphate buffer (11). The formal change in charge upon attachment of PM group to lysine is ΔZformal = −4.0. The actual change in charge upon acylation of lysine in SOD1 (ΔZCE; determined in this paper) is −3.33 ± 0.04, because of “charge regulation” (29, 30). The values of ΔZformal and ΔZCE are listed for each acyl group studied in this paper in Fig. 2.
Figure 2.

Natural (N) and unnatural (U) acyl modifications examined in this study. ΔZformal refers to the formal difference in net charge upon acylation of a single lysine-ϵ-NH3+. ΔZCE refers to the measured difference in net charge upon acylation of a single lysine-ϵ-NH3+ that reflects charge regulation (29, 30) (measured in this study using CE). Errors represent the standard deviation (n = 3). Log P represents the calculated hydrophobicity for each acyl group. Errors represent the standard deviation of hydrophobicity calculations from Chemdraw and ChemAxon programs.
Results and discussion
The natural and unnatural acyl modifications examined in this study increase the magnitude of net negative charge of SOD1 by −1 to −4 formal units; hydrophobicity of the lysine-ϵ-NH3+ is increased from log P ≈ +1.0 to log P ≈ −2.0 (Fig. 2). Purified, recombinant WT apo-SOD1 were acylated with different anhydrides at various stoichiometric ratios that resulted, through trial and error, in the desired average stoichiometry of ∼1 acyl group per SOD1 monomer for each type of acyl group (Table 1). The average number of acylated lysine (Table 1) was determined by integration of mass spectra and weighted averaging of each “rung” (Fig. 3, a and b, and supplemental Figs. S1 and S2). Stoichiometries ranged from 0.70 acyl groups per monomer for tricarboxybiphenylated SOD1 to 1.45 for tricarboxycyclobutylated SOD1; however, stoichiometries for all other groups ranged from 0.89 to 1.22 acyl groups per monomer (Table 1). The percentage of SOD1 that remained unacylated after addition of anhydrides ranged from 10.7% in tricarboxycyclobutylated samples to 40.9% in tricarboxybiphenylated samples; however, unacetylated SOD1 ranged from 20.8 to 36.2% for all other types of acyl groups (Table 1). These stoichiometries were also confirmed with capillary electrophoresis (CE), which detects proteins by absorbance at 214 nm (Fig. 3, c and d). Thus, this study examined how partially acylated SOD1 proteins—mixtures of regioisomers that contain subpopulations of unmodified SOD1—affect aggregation.
Table 1.
Kinetic parameters of in vitro fibrilization of WT apo-SOD1 (100 mm TCEP, 37 °C, pH 7.4) before and after acylation
The data are shown as means ± S.D.
| Modification Ac(N)a | Lag time ratiob | Propagation rate ratiob | Maximal ThT fluorescence ratiob | Hazard ratioc | Unmodified SOD1d | ΔTme |
|---|---|---|---|---|---|---|
| % | °C | |||||
| CB Ac(1.45) | 1.37 ± 0.17 (p = 0.09) | 0.94 ± 0.30 (p = 0.8) | 0.45 ± 0.35 (p = 0.08) | 0.48 (p = 0.15) | 10.7 | −3.2 |
| PM Ac(1.22) | 1.88 ± 0.08 (p < 0.0001) | 0.94 ± 0.07 (p = 0.4) | 0.36 ± 0.20 (p < 0.0001) | 0.25 (p < 0.0001) | 23.2 | −3.6 |
| BT Ac(0.98) | 0.87 ± 0.08 (p = 0.09) | 0.84 ± 0.24 (p = 0.5) | 0.66 ± 0.18 (p = 0.02) | 0.21 (p < 0.0001) | 20.8 | −4.5 |
| BP Ac(0.70) | 1.46 ± 0.09 (p = 0.0004) | 1.48 ± 0.28 (p = 0.2) | 0.35 ± 0.15 (p < 0.0001) | 0.47 (p = 0.005) | 40.9 | −4.0 |
| SA Ac(0.90) | 0.98 ± 0.06 (p = 0.8) | 1.03 ± 0.14 (p = 0.8) | 1.25 ± 0.16 (p = 0.2) | 2.95 (p = 0.001) | 36.2 | −2.1 |
| CA Ac(1.10) | 0.92 ± 0.09 (p = 0.3) | 2.06 ± 0.06 (p < 0.0001) | 1.37 ± 0.15 (p = 0.04) | 3.83 (p < 0.0001) | 27.9 | −5.3 |
| GA Ac(0.89) | 0.99 ± 0.09 (p = 0.9) | 4.42 ± 0.24 (p = 0.2) | 0.61 ± 0.36 (p = 0.2) | 1.18 (p = 0.61) | 34.3 | −3.8 |
| PhA Ac(1.07) | 1.01 ± 0.13 (p = 0.9) | 0.87 ± 0.16 (p = 0.4) | 1.18 ± 0.17 (p = 0.3) | 1.11 (p = 0.69) | 23.9 | −4.0 |
| AA Ac(1.05) | 1.00 ± 0.06 (p = 0.9) | 1.04 ± 0.07 (p = 0.6) | 1.29 ± 0.15 (p = 0.07) | 1.09 (p = 0.72) | 29.7 | −2.2 |
a Average stoichiometry of the acylated protein was calculated as a weighted average of integrated mass spectra.
b Ratios for all kinetic parameters were calculated as τmod/τ/unmod, kmod/kunmod, and ThTmod/ThTunmod, with p values calculated with respect to unmodified SOD1 using an unpaired Student's t test at 95% confidence interval (p < 0.05).
c Hazard ratios and their corresponding p values were calculated from the statistical comparison between Kaplan–Meier plots of each modified SOD1 and its corresponding unmodified protein, using the log-rank (Mantel–Cox) algorithm at 95% confidence interval (p < 0.05).
d Calculated via integration of deconvoluted mass spectra.
e The values of ΔTm are calculated as Tm(unmod) − Tm(acylated).
Figure 3.

Quantifying charge regulation associated with lysine acylation using protein charge ladders and CE. a and b, electrospray ionization mass spectra of unmodified (a) and pyromellitylated (b) WT apo-SOD1 proteins (denoted as WT-PM(1.22)) used in amyloid assays. Mass spectra for all other acyl modifications are shown in supplemental Figs. S1 and S2. c and d, CE electropherograms of protein charge ladders of WT apo-SOD1 with acyl groups with ΔZformal = −4 (c) and acyl groups with ΔZformal = −2 (d) for the purpose of measuring ΔZ of acylation. e and f, average values of measured ΔZ (ΔZCE) for all modifications with ΔZformal = −4 (e) and all modifications with ΔZformal = −2 (f). The number of acyl modifications in CE electropherograms refers to the number of modifications per SOD1 dimer, whereas the number of acyl modifications in mass spectra refers to modifications per monomer (electrospray ionization leads to dissociation of SOD1 dimer). ΔZCE values in e and f are average values for each type of modification. The values of ΔZCE for each acyl group are listed in Fig. 2.
Trypsin digestion and tandem mass spectrometry resulted in between 98 and 100% sequence coverage and suggested that lysine residues were semirandomly acylated by each anhydride (supplemental Table S1 and Figs. S3–S7). Generally, the acylation of surface lysine-ϵ-NH3+ in proteins is not purely random per se and is not necessarily controlled by the solvent accessibility of a particular lysine (but rather the pKa of lysine-ϵ-NH3+) (6). The CA and BT modifications did not result in as many measureable regioisomers as other modifications, according to MS/MS (supplemental Table S1). For example, only Lys-36 and Lys-128 were found to be acylated in SOD1-BT solutions, and only Lys-9 was found to be acylated in SOD1-CA. We do not know whether these results are artifacts of altered trypsinization or reflect selective acylation. We also detected sporadic acylation at His, Thr, Ser, Cys, and Arg residues with certain modifications (supplemental Table S1). The N-terminal α-NH3+ group was uniformly acetylated in all protein solutions prior to acylation with anhydrides as the N-terminal α-NH3+ group of human SOD1 is properly acetylated when expressed in yeast.
The formal (predicted) change in net charge of SOD1 imparted by each modification (ΔZformal) does not accurately reflect the actual change in the net surface charge of SOD1 because of “charge regulation” (the adjustment of pKa values of functional groups in response to the neutralization of lysine or addition of charged groups) (29, 30). To determine the actual ΔZ for each type of acylation, we performed CE on both unmodified and modified proteins and calculated the ΔZ associated with each acylation using the protein charge ladder method (10, 11) (Fig. 3, c–f). To do so, we plotted the electrophoretic mobility of each acyl peak (each “rung”) versus the number of acylations (N) associated with that rung (Fig. 3, e and f). The actual ΔZ (denoted ΔZCE) was calculated by obtaining the x intercept, which is equal to the quotient of the net charge of unmodified apo-SOD1 (previously determined to be ZCE = −12.13 ± 0.08, per dimeric protein (31)) and the ΔZ imparted by each modification (Fig. 3, e and f). In the case of tricarboxylic acylating groups (ΔZformal = −4), we determined that ΔZCE values ranged from −3.06 ± 0.05 to −3.33 ± 0.04, with the average ΔZCE = −3.19 ± 0.10 (Table 1 and Fig. 3e).
The ΔZCE of acylating groups with ΔZformal = −2 ranged from ΔZCE = −1.64 ± 0.04 to −1.82 ± 0.03, with the average ΔZCE = −1.72 ± 0.07 (Fig. 3f). In these charge ladders, we also observed a secondary “charge ladder,” that is, a distinct population of rungs in this same ladder. The rungs of this minor ladder are designated with asterisks in Fig. 3d. This distribution of minor rungs exhibited a ΔZCE = −0.87 ± 0.02 per modification (supplemental Fig. S8), which might be due to acylation of partially charged residues (e.g. serine or histidine; supplemental Table S2).
To determine the effect of acylating agents on the thermostability of WT apo-SOD1, we performed differential scanning calorimetry (DSC) on unmodified and variably acylated SOD1 proteins (Fig. 4). All acyl modifications caused a modest decrease in melting temperature (Tm) of WT apo-SOD1, up to a ΔTm value of approximately −5 °C (Fig. 4 and Table 1), which is similar to previous analyses of acylated SOD1 (1, 31) and bovine carbonic anhydrase II (32) (acylation does not uniformly destabilize proteins, e.g. acetylation increases the thermostability of α-amylase (33)).
Figure 4.
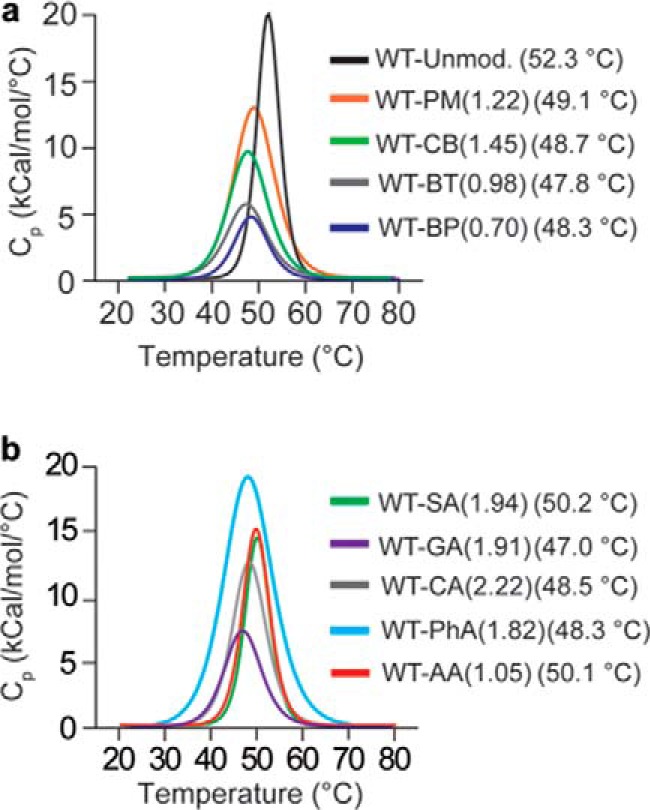
Effect of lysine acylation on the thermostability of WT apo-SOD1. a and b, DSC thermograms of unmodified and acylated WT apo-SOD1, modified with acyl groups with ΔZformal = −4 (a) and ΔZformal ≤ −2 (b). The number of acyl modifications are listed per monomeric SOD1. The values in the parentheses next to each SOD1 protein represent the corresponding melting temperature.
Effect of surface charge and hydrophobicity on fibril nucleation and elongation of WT apo-SOD1 in microplate assays
Similar to our previous studies (1, 28, 31), we utilized a 96-well microplate-based ThT fluorescence assay (ntotal = 594) to investigate the effect of acylation on the kinetics of apo-SOD1 fibrillization. Because the aggregation of SOD1 is stochastic (34), we performed ≥10 replicate assays for each type of acylated protein (i.e. analyzed ≥10 iterate wells) and calculated mean rates of fibril nucleation and elongation (Figs. 5 and 6). For each type of acylated protein, we also performed a control ThT assay on unacylated SOD1 that was exposed to the same concentration of organic solvent as the acylated protein. Trace organic solvent (0.01–0.1% v/v) is initially required to dissolve each anhydride into SOD1 solutions but is washed out during centrifugal filtration and buffer transfer; nevertheless, we performed separate controls for each acyl group.
Figure 5.

Effect of fractional acylation of WT apo-SOD1 with highly charged groups (ΔZformal = −4) on SOD1 fibrillization into ThT-positive species. a and b, replicate ThT fluorescence assays of unmodified (a) and singly acylated (b) WT apo-SOD1. Replicates that resulted in zero fluorescence (denoted ThT(−)) are also shown. Regions of lag phase are magnified to illustrate stochasticity. Insets show TEM micrographs of fibrillar and amorphous complexes of unmodified and pyromellitylated WT apo-SOD1 after the completion of ThT fibrillization assay (images are representative and include analysis of ThT-negative and ThT-positive wells). The TEM micrographs for all other modified and unmodified SOD1 fibrils are shown in supplemental Fig. S9. c, average plots of ThT fluorescence for all acylated WT apo-SOD1 and their corresponding unmodified proteins (averaged from a and b). Replicates that resulted in zero fluorescence (i.e. ThT(−)) were included in average plots. d, SDS-PAGE of SOD1 solutions before starting (upper gel) and after termination of ThT assay (lower gel).
Figure 6.

Effect of fractional acylation of WT apo-SOD1 with weakly charged groups (ΔZformal ≤ −2) on SOD1 fibrillization into ThT-positive species. a and b, replicate ThT fluorescence assays of unmodified (a) and singly acylated (b) WT apo-SOD1. Replicates that resulted in zero fluorescence (denoted ThT(−)) are also shown. c, plots of average ThT fluorescence for all unmodified and acylated WT apo-SOD1 (averaged from a and b). Replicates that resulted in zero fluorescence (i.e. ThT(−)) were included in average plots. d, SDS-PAGE of all apo-SOD1 solutions before starting (left gel) and after termination of ThT assay (right gel). The TEM micrographs for all modified and unmodified SOD1 proteins are shown in supplemental Fig. S11.
Iterate longitudinal plots of ThT fluorescence are shown in Figs. 5 and 6 for unmodified and singly acylated SOD1 proteins. Generally, all modifications with ΔZformal = −4 diminished the rate of apo-SOD1 fibrillization into ThT-positive fibrils and/or diminished the maximal fluorescence of fibrils (Fig. 5, a–c). Pyromellitylated SOD1 proteins showed a ∼64% decrease in the intensity of ThT fluorescence compared with unmodified proteins (Fig. 5c). Moreover, pyromellitylated and tricarboxybiphenylated apo-SOD1 underwent amyloid nucleation (into ThT-positive fibrils) at slower rates than unmodified SOD1, whereas tricarboxybenzophenonylated and tricarboxycyclobutylated apo-SOD1 proteins showed no significant difference in lag time compared with unmodified apo-SOD1 (p = 0.09; Fig. 5c and Table 1). Pyromellitylation also increased the number of wells that exhibited no apparent increase in ThT fluorescence. The absence of (or decrease in) ThT fluorescence is not definitive proof that a modification inhibited the fibrillization of SOD1. Amyloid fibrils of apo-SOD1 with minimal ThT fluorescence have been reported (34).
To determine the morphology of acylated SOD1 aggregates, we performed transmission electron microscopy (TEM) on wells that contain both acylated and unmodified SOD1 aggregates (insets in Fig. 5, a and b, and supplemental Fig. S9). All wells that exhibited increases in ThT fluorescence were found with TEM to contain a mixture of fibrillar and amorphous aggregates. The fraction of wells that exhibited no significant increase in ThT fluorescence also contained fibrillar and amorphous complexes; however, a few ThT-negative wells contained only amorphous aggregates (insets in Fig. 5, a and b).
The low level—or complete absence—of ThT fluorescence exhibited by many wells is not caused by incomplete aggregation or variation in concentration of SOD1 proteins. Analysis of the ±concentration of soluble SOD1 with SDS-PAGE, Bradford assay, and bicinchoninic acid (BCA) assay confirmed that proteins existed at similar concentrations prior to initiating the assay (Fig. 5d). For example, SDS-PAGE bands for unmodified and singly tricarboxybiphenylated apo-SOD1 proteins had statistically similar intensities (2.8 × 103 and 3.1 × 103 a.u., respectively; Fig. 5d, upper panel). Moreover, analysis of sample supernatants at the end of each aggregation assay (with SDS-PAGE) showed that SOD1 completely aggregated into a sedimentable species (Fig. 5d, lower panel). We reiterate that the analysis of ThT-negative wells with TEM also showed the presence of fibrillar SOD1 species (Fig. 5, a and b). Thus, the SOD1 protein self-assembles into ThT-negative fibrils and amorphous complexes, and acylation can increase the probability that SOD1 forms these ThT-negative assemblies.
The observation that polycarboxylic acyl groups promote the formation of fibrils with low (or zero) ThT fluorescence suggests that these acylated fibrils have different structures or gross morphologies than unmodified fibrils. We found that, in general, acylation of apo-SOD1 with modifications possessing ΔZformal = −4 produced slightly shorter fibrils than unmodified protein (supplemental Fig. S10). For example, singly pyromellitylated SOD1 fibrils were (on average) 17.8 ± 7.5 nm shorter than unmodified fibrils. Tricarboxycyclobutylation had the most pronounced effect on the length of SOD1 fibrils; these fibrils were shorter than unmodified fibrils by 38.7 ± 5.4 nm (supplemental Fig. S10).
In summary, acyl groups that change the net charge of SOD1 by ΔZformal = −4 generally inhibit the aggregation of SOD1 into ThT-positive fibrils but promote the formation of shorter fibrils with lower or zero ThT fluorescence. The ability of polycarboxylic modifications such as PM to inhibit fibrillization into ThT-positive species could arise from: (i) increased electrostatic repulsions between SOD1 polypeptides (1) and/or (ii) aromatic rings that might alter the packing of SOD1 polypeptides into ThT-positive fibrils (35).
To discern between electrostatic and hydrophobic effects, we modified WT apo-SOD1 with a series of aromatic and non-aromatic moieties that are rotational, aromatic, and electrostatic analogs of one another (GA, SA, PhA, CA, and AA; Fig. 2). ThT fluorescence aggregation assays were then performed on all modified proteins (Fig. 6). These groups add either one or two formal units of net negative charge (ΔZformal ≤ −2) to apo-SOD1 upon lysine acylation (Fig. 2 and supplemental Fig. S2). Four of these acylating groups add a linear chain of 2–5 carbons (both saturated and unsaturated) to the protein sequence (Fig. 2). One acyl group (from phthalic anhydride, PhA) possesses a benzene ring with a single carboxylic acid, thus acting as the steric analog of the pyromellityl group, but with a ΔZformal = −2 (Fig. 2). Another group, CA, represents a hydrophobic, rotationally locked (unsaturated) analog of SA.
In general, acyl groups that imparted at most −2 units of charge resulted in weaker inhibitory effects on SOD1 fibrillization (into ThT-positive species) compared with acyl groups with ΔZformal = −4 (Fig. 6, a–c). Despite their structural similarity, phthalylation had a smaller kinetic effect on SOD1 aggregation than pyromellitylation. For example, the difference in fibrillization lag time (Δτ) = 11.94 ± 1.54 h for PM-SOD1 (Δτ is calculated as τPM-SOD1 − τunmod-SOD1), whereas PhA-SOD1 showed only a minor difference in lag time compared with unmodified SOD1 (Δτ = 0.07 ± 0.96 h; Fig. 6, a–c). The greater inhibitory effect of pyromellitylation compared with phthalylation (ΔΔτ = 10.33 ± 2.49 h) suggests that the two additional units of negative charge imparted to SOD1 by the PM modification is the source of the inhibitory effect of PM, not the steric bulk added by the benzene ring.
The succinylation of ∼0.90 lysines in SOD1 did not significantly alter the nucleation or propagation of ThT-positive fibrils (Fig. 6, a–c, and Table 1). The citraconylation of ∼1.10 lysines in SOD1, on the other hand, accelerated the propagation of SOD1 by ∼2-fold (Fig. 6, a–c, and Table 1). The citraconyl group, the unsaturated, methylated analog of succinyl moiety, is both rigid and hydrophobic (it is the only modification in this study with a non-rotameric secondary methyl group; Fig. 2). The isoelectric nature of citraconyl and succinyl groups (Fig. 2) suggests that the acceleration of aggregation by CA groups is caused by their more rigid, hydrophobic groups, which we presume will drive burial and promote London dispersion interactions.
In addition to extracting standard metrics of aggregation (e.g. lag time, propagation rate) from ThT fluorescence assays, we also analyzed ThT fluorescence data with Kaplan–Meier estimators (36) to determine the probability that acylated SOD1 fibrillized into ThT-positive complexes versus ThT-negative complexes (Fig. 7). We have recently shown that Kaplan–Meier estimators can be used to determine the probability that ALS-mutant SOD1 will fibrillize (in vitro) into ThT-positive species, relative to the probability that WT SOD1 will fibrillize into ThT-positive species. These probabilistic metrics scale better with patient phenotypes (i.e. survival time) than classical aggregation metrics and might be more clinically relevant (36). Each downward step in the Kaplan–Meier plot (Fig. 7) indicates the time at which an individual microplate well reached half of its maximum ThT fluorescence. This inflection point (X0) therefore expresses both the nucleation rate and elongation (secondary nucleation) rate for fibrillizing proteins in each well (37, 38), according to the formula X0 = τ + 2/k. In the present study, Kaplan–Meier estimators express the probability of fibrillization of acylated SOD1 into ThT-positive species relative to unacylated SOD1. The principal readout in this analysis is a hazard ratio (HR) of fibrillization (34). Hazard ratios > 1 indicate that acylation promoted fibrillization into ThT-positive species; ratios < 1 demonstrate that modifications inhibited fibrillization into ThT-positive species; and ratios = 1 demonstrate no effect.
Figure 7.

Effect of acylation on the probability of SOD1 aggregation studied with Kaplan–Meier estimators. Kaplan–Meier plots of SOD1 aggregation in microplate wells, extracted from ThT fluorescence data in Figs. 5 and 6. Each down-step represents aggregation of a single well into a ThT-positive species. The red trace in each plot represents acylated protein, and the black trace represents the unacylated control. Values next to each plot indicate the percentage of wells that showed no significant increase in ThT fluorescence over 120 h (i.e. ThT-negative wells). In all plots, p values were calculated with respect to unmodified SOD1 using log rank (Mantel–Cox) algorithm at 95% confidence interval (p < 0.05).
The Kaplan–Meier plots of SOD1 fibrillization (Fig. 7) provide a convenient bird's-eye view of the fibrillization of SOD1 in all individual wells of the microplate, especially wells that did not fibrillize into ThT-positive species. At high TCEP concentrations (i.e. 100 mm), SOD1 does not always form ThT-positive species, that is, iterate wells do not exhibit fluorescence above baseline (31). These negative effects have not been observed at 10 mm TCEP (1, 34) and appear because of increasing ionic strength (39). Kaplan–Meier analyses can account for (and quantify) the iterate assays that showed baseline (zero) ThT fluorescence (i.e. ThT-negative wells) (31).
Fibrillization of individual wells containing apo-SOD1 proteins with acyl modifications that imparted ΔZCE > −3.0 occurred at a lower probability compared with wells with unmodified protein (Fig. 7). Hazard ratios were calculated to be < 0.5 for SOD1 proteins modified with all tricarboxylic acyl groups (Fig. 7). According to Kaplan–Meier analysis, modifications that imparted ≤2 formal units of charge led to minimal or no reduction in probability of fibrillization into ThT-positive species and in some cases increased probability of fibrillization (Fig. 7). Citraconylation of SOD1 resulted in a significantly higher probability of aggregation into ThT-positive fibrils than unmodified SOD1 protein, with a hazard ratio of 3.83 (Fig. 7 and Table 1); succinylation also increased the probability of fibrillization into ThT-positive species but to a lesser extent (HR = 2.95; Fig. 7 and Table 1). Acetylation had a negligible effect on fibrillization (HR = 1.09; Fig. 7 and Table 1).
We point out that our previous report on the inhibitory effects of acetylation on SOD1 fibrillization does not necessarily conflict with the results here, because the proteins in the current study contain ∼1 modification (on average; Table 1) per subunit, as well as a substantial population of unmodified SOD1, whereas our previous study analyzed SOD1 proteins with up to ∼6 acetyl modifications per subunit (and an absence of unacylated SOD1) (1).
The number of wells that do not aggregate into ThT-positive species in these microplate assays is, itself, an important metric. For unmodified SOD1 proteins, the percentage of wells that failed to form ThT-positive fibrils varied from 0 to 13% of all wells (Fig. 7). Several different acyl groups increased the percentage of wells that failed to form ThT-positive complexes. Pyromellitylation exhibited the largest effect and increased the probability that SOD1 formed ThT-negative complexes from 12% of wells to 34% of wells (Fig. 7).
Effect of lysine acylation on the pathway of SOD1 aggregation
The aggregation of SOD1 has been observed to occur stochastically and proceed along amorphous and fibrillar pathways that are in competition (have similar ΔEa) at pH 7.4 and 37 °C (28). We sought to determine whether acylation alters these pathways of aggregation, e.g. favors non-fibrillar pathways over fibrillar or vice versa. Non-fibrillar aggregates cannot be detected with ThT fluorescence but are detectable using 1-anilino-8-naphthalenesulfonate (ANS) fluorescence (28, 39, 40); ANS produces a fluorescent readout when bound to either fibrillar or amorphous aggregates (40). Microplate-based iterate ANS assays of WT and mutant apo-SOD1 aggregation yield a mixture of sigmoidal and exponential aggregation processes; that is, a minor fraction of iterate microplate wells rapidly form amorphous aggregates (exponential regime), whereas the majority of wells produce fibrillar aggregates (sigmoidal regime) (28).
Iterate assays of ANS fluorescence were performed on a subset of acylated SOD1 proteins (glutarylated, pyromellitylated, and tricarboxybenzophenonylated apo-SOD1 proteins; Fig. 8a). Acylated and unacylated apo-SOD1 proteins each exhibited sigmoidal and exponential increases in ANS fluorescence (Fig. 8a). Wells that exhibit sigmoidal kinetics were found with TEM to contain both fibrillar and amorphous species (supplemental Fig. S12), whereas wells exhibiting exponential kinetics contained only amorphous aggregates (supplemental Fig. S12), consistent with the results from our previous study on unmodified SOD1 (28).
Figure 8.

Effect of apo-SOD1 acylation on the rate of formation of ANS-positive complexes. a, longitudinal plots of ANS fluorescence for WT apo-SOD1 modified with GA(0.89), PM(1.22), and BT(0.98). b, comparison plots of average lag time, propagation rate, and ThT fluorescence intensity for the aggregation of all modified SOD1 proteins and their corresponding unmodified proteins. The data are extracted from sigmoidal regimes only. c, Kaplan–Meier plots for the aggregation of acylated and unmodified SOD1 proteins extracted from data in a. Upper and lower plots in c show the Kaplan–Meier analysis for sigmoidal (upper panels) and exponential (lowers panels) regimes. In all plots, p values were calculated with respect to unmodified SOD1 using log rank (Mantel–Cox) algorithm at 95% confidence interval (p < 0.05).
The absolute nucleation rates (lag times) and propagation rates in ANS assays (i.e. the sigmoidal regime) were slower than in ThT assays (Fig. 8), as we previously observed for unacylated SOD1 (28). This difference might be caused by the inhibitory effects that ANS can have on protein aggregation (41). Despite these absolute differences in rates as measured by the two assays, the shift in the rate of SOD1 nucleation and propagation upon acylation were statistically similar in both ThT and ANS assays (Figs. 5, 6, and 8). For example, single pyromellitylation delayed the nucleation of apo-SOD1 according to ANS and ThT fluorescence assays (ΔτANS = 9.34 ± 2.05 h and ΔτThT = 11.94 ± 1.54 h) but did not alter SOD1 propagation rate (ΔkANS = 0.01 ± 0.003 h−1 and ΔkThT = 0.02 ± 0.02 h−1). Glutarylation and tricarboxybenzophenonylation did not significantly change the rate of nucleation or oligomer propagation of SOD1 in either assay (Fig. 8, a and b).
ANS fluorescence suggests that acylation can decrease the probability of fibrillar aggregation and increase the probability of amorphous aggregation. For example, ∼41% of wells containing unmodified apo-SOD1 aggregated in the exponential regime, whereas ∼60% of wells with glutarylated SOD1 aggregated exponentially (insets in Fig. 8a). Similar trends were observed in the case of PM- and BT-modified SOD1 (insets in Fig. 8a). Together, these results suggest that acylation (both natural and unnatural) can weakly favor (in our opinion) one specific SOD1 aggregation pathway over others (in this case amorphous pathways over fibrillar ones). Pyromellitylation imparts at least a fraction of its inhibitory effects on fibrillization via altering the mechanism (pathway) of SOD1 aggregation.
We also analyzed the sigmoidal and exponential aggregation of individual wells with Kaplan–Meier estimators (Fig. 8c). Pyromellitylation diminished the probability of sigmoidal aggregation (hazard ratio = 0.11, p < 0.001; Fig. 8c) but had a statistically insignificant effect on exponential aggregation (hazard ratio = 0.52, p = 0.51; Fig. 8c). The fibrillization rate of BT-modified apo-SOD1 (i.e. the sigmoidal regime) was statistically similar to unmodified SOD1 (hazard ratio = 0.74, p = 0.49; Fig. 8c), as was the amorphous aggregation (i.e. exponential regime) of unmodified and BT-modified SOD1 (hazard ratio = 0.26, p = 0.13; Fig. 8c).
Effect of WT SOD1 acylation on seeding the aggregation of G85R SOD1 in organotypic mouse spinal cord
In transgenic mice, the co-expression of WT SOD1 protein increases the toxicity of the ALS-mutant SOD1 (42–44). The cause of this synergy is unknown; that is, it is not known whether WT SOD1 directly interacts with mutant SOD1 in ways that promote the aggregation of mutant SOD1 or whether WT SOD1 competes with mutant SOD1 for protective factors (45). Additionally, recent studies have demonstrated the ability for ThT-positive fibrils of recombinant WT SOD1 to induce SOD1 pathology and motor neuron disease in an in vivo model for SOD1 prion-like seeding (46).
To determine how acylation of SOD1 affected the ability of its aggregates to seed the aggregation of ALS-variant SOD1 in living systems, we added acylated and unacylated aggregates (that formed in microplate wells during ThT assays) to organotypic spinal cord cultures prepared from the G85R-SOD1·YFP transgenic mouse model (Fig. 9). This model expresses the ALS-linked G85R SOD1 transgene below the threshold level required to develop motor neuron disease or inclusion pathology and has been used extensively to study the prion-like seeding of oligomeric SOD1 (to seed aggregation in pools of soluble SOD1) (46). These cultures can be kept alive for months (47) and provide a convenient platform for monitoring (using fluorescence microscopy) the ability of exogenously added solutions of aggregated SOD1 to induce formation of fluorescent inclusions from SOD1·YFP, which is endogenously expressed by the mice (46). The readout in these qualitative assays are large, spatially localized inclusions of G85R-SOD1·YFP (46). There is a positive correlation between the formation of these SOD1·YFP inclusions and the onset of ALS symptoms in mice (48).
Figure 9.

Seeding properties of unmodified (a–c) and singly pyromellitylated fibrils (d–f) of WT apo-SOD1 after 4-week incubation with organotypic spinal cord cultures from transgenic ALS mice. Representative confocal micrographs of inclusion pathology induced in spinal cord slices from transgenic G85R-SOD1·YFP mice (harvested at ∼7 days old). Fibrils were grown in microplate wells and added in a well-specific manner to organotypic spinal cord as a function of ThT fluorescence: high fluorescence (a and f), low fluorescence (b and e), and zero fluorescence (c and d). The value on the top right of each confocal micrograph represents the percentage of replicate spinal cord slices that showed SOD1 inclusions (n = 5). The plots below the confocal micrographs show ThT fluorescence traces (left) and TEM micrographs (right) corresponding to the particular microplate well/fibril solution that was added to spinal cord.
Spinal cord cultures were prepared from mice at ∼7 days of age and treated with SOD1 fibrils from wells that produced fibrils with high, low, and zero ThT fluorescence (Fig. 9). We found that the addition of WT SOD1 fibrils with high ThT fluorescence induced the formation of large, visible inclusions of G85R-SOD1·YFP, regardless of whether the aggregates were composed of acylated (pyromellitylated for these experiments) or unmodified apo-SOD1 (Fig. 9, a, b, e, and f). In contrast, the addition of ThT-negative SOD1 fibrils did not induce the formation of G85R-SOD1·YFP inclusions, regardless of whether the aggregates were composed of acylated or unmodified apo-SOD1 (Fig. 9, c and d). When considering that acylation of SOD1 increases the probability that the protein forms a ThT-negative fibril or an amorphous oligomer (Figs. 5, 7, and 8), the tissue culture assays suggest that the acylation of WT apo-SOD1 will inhibit its ability to seed further aggregation by driving the acylated protein into a ThT-negative assembly.
The ability of ThT-positive WT SOD1 fibrils to induce inclusions that we observed in this study is in agreement with our recent study (46) (Fig. 9, a, b, e, and f). In this paper, however, we also show that ThT-negative fibrils, produced simultaneously from the same batch of SOD1 in the same microplate, do not induce inclusions of ALS-variant SOD1 (Fig. 9, c and d). In the context of ALS, the question of whether acylation of SOD1 would increase or decrease the toxicity of SOD1 (i.e. accelerate or decelerate the onset or progression of neurodegeneration) depends on whether these large assemblies are protective or toxic.
Because pyromellitylation had the greatest inhibitory effect on SOD1 fibrillization, we became interested in determining whether the pyromellitylation of a larger fraction of lysine residues in WT apo-SOD1 protein would completely abolish its ability to form ThT-positive fibrils. To this end, we acylated an average of 4.16 lysines in apo-SOD1 (per monomeric protein) with pyromellitic dianhydride (Fig. 10a). This higher degree of lysine pyromellitylation did not abolish the tertiary structure of native apo-SOD1 but did destabilize it by ΔTm = −8.9 °C (Fig. 10b). After performing nine replicate ThT fluorescence aggregation assays, we observed that none of the iterate wells containing apo-SOD1-PM(4.16) fibrillized into ThT-positive species (Fig. 10c). The addition of these ThT-negative fibrils to spinal cord cultures did not seed aggregation (Fig. 10c), in accordance with our results from singly pyromellitylated SOD1 fibrils that were ThT-negative. Thus, the pyromellitylation of approximately four stoichiometric equivalents of lysine in SOD1 abolishes its ability to seed ALS-variant SOD1 aggregation in organotypic spinal cord cultures of transgenic mice.
Figure 10.

Pyromellitylation of 4. 16 lysine residues (37% of all lysines in SOD1) abolishes the ability of SOD1 to form ThT-positive fibrils in microplate assays and inhibits seeding in organotypic spinal cord cultures. a, electrospray ionization mass spectrum. b, DSC thermogram of pyromellitylated WT apo-SOD1 (4.16 acylated lysine per monomer). c, representative confocal micrograph of spinal cord slices from transgenic G85R-SOD1·YFP mice that were exposed with apo-SOD1-PM(4.16) fibril homogenates. The value on the top right of the confocal image represents the percentage of discrete spinal cord slices that showed SOD1 inclusions (n = 5). The plots next to the confocal image show TEM micrograph (upper panel) and ThT fluorescence traces (lower panel) corresponding to the microplate well/fibril solution that was added to spinal cord.
We also compared the in vitro seeding activity of singly pyromellitylated SOD1 fibrils that were ThT-negative and ThT-positive. These experiments were performed on soluble, unacylated “native” apo-SOD1, by addition of 5 μl of acylated fibril seeds (formed from in vitro microplate assays; supplemental Fig. S13a). The rate of unseeded nucleation of SOD1 fibrils were statistically similar to rate of nucleation of SOD1 fibrils that were seeded with ThT-negative, acylated fibrils (p = 0.5; supplemental Fig. S13b). In contrast, SOD1 fibrils nucleated faster (by ∼5 h) when seeded with ThT-positive, acylated fibrils compared with unseeded controls (p = 0.005; supplemental Fig. S13b). The rate of fibril propagation was faster in the presence of ThT-negative and ThT-positive seeds than in the absence of seeds; however, the increase in propagation rate was more significant for SOD1 oligomers seeded with ThT-positive fibrils (p = 0.02 for ThT-negative versus p = 0.007 for ThT-positive; supplemental Fig. S13c). ThT-positive seeds also increased the probability of SOD1 fibrillization according to Kaplan–Meier analysis (p = 0.04; Fig. S13d), whereas no significant changes in probability of fibrillization were observed in the presence of ThT-negative seeds (p = 0.21; supplemental Fig. S13d). In conclusion, the results from our in vitro seeding assays support our seeding assays in organotypic spinal cord: ThT-negative SOD1 fibrils lack the ability to seed the fibrillization of soluble apo-SOD1.
Conclusion
This paper demonstrates that attachment of negatively charged acyl groups to a fraction of lysine residues in SOD1 can inhibit its aggregation into ThT-positive fibrils that lack seeding activity in organotypic spinal cord, while promoting the formation of ThT-negative fibrils and amorphous complexes that lack seeding activity in organotypic spinal cord. These protective effects appear to be electrostatic in nature, that is, they are not caused by increases in native state thermostability and are not caused by steric disruptions to polypeptide packing in fibrils and amorphous complexes. We are hesitant to refer to the seeding activity of ThT-positive SOD1 fibrils as “prion-like activity.” For example, we do not know whether ThT-positive aggregates crossed cell membranes in cultured spinal cord and directly catalyzed the aggregation of intracellular SOD1 proteins or whether the ThT-positive aggregates merely lurked in extracellular space and aggregated with soluble SOD1 that existed in extracellular space (SOD1 exists in cerebrospinal fluid at nanomolar concentrations (49)).
One limitation of this study is that it did not test the toxicity of acylated SOD1 fibrils, because it is difficult to comprehensively assay the cytotoxicity of SOD1 aggregates among the entire population of motor neurons in organotypic spinal cord slabs. Recently, Eisenberg and co-workers (50) have cultured primary motor neurons (2D cultures derived from embryonic stem cells) to assay the toxicity of ThT-positive SOD1 fibrils and oligomers (ThT-negative aggregates were not reported or studied). The primary motor neurons in this study were assayed 12 h after incubation with aggregates, which is shorter than the lag phase of the seeded fibrillization processes we measure in vitro. These cultured cells and others (e.g. neuroblastoma cells) are viable for only a few days to a maximum of 2 weeks. Our organotypic spinal cord—a 3D culture, per se—is incubated with fibrils for 4 weeks, and the only conventional assay that might be appropriate is the ethidium homodimer-1 assay. This assay requires manual counting of cells on the surface of the tissue slab (51), i.e. spinal cord cross-sections used in our current study are ∼300-μm-thick, and the bulk of cells are not observed with microscopy. We do note that in our experience with organotypic spinal cord slabs, substantial toxicity would be directly observed by a change in the spinal cord morphology and a shrinking of the sections over time. None of the fibril preparations, even those that induced SOD1 inclusions, were observed to result in shrinking, suggesting either a low level and/or a slow rate of toxicity. Future studies will be needed to address the issue of fibril toxicity.
Another caveat—perhaps a perceived weakness—of this study is that it generated and analyzed mixtures of acylated and unacylated apo-SOD1 proteins. For example, 10.7–40.9% of apo-SOD1 remained unacylated in samples used in this study (Table 1), and the fraction that was acylated included multiple regioisomers (supplemental Tables S1 and S2). Thus, this study analyzed the aggregation of mixtures of unmodified and quasi-randomly modified SOD1 proteins. This statistical feature is, however, an asset not a liability. Our results suggest that subpopulations of SOD1 bearing randomly acylated lysine residues can be potent inhibitors of SOD1 self-assembly into ThT-positive fibrils; that is, the acylation of almost any lysine residue in SOD1 can impede its fibrillization into ThT-positive fibrils. Our explanation for this nonspecific effect is that acylation increases long-range Coulombic repulsions between SOD1 polypeptides; that is, it does not matter which lysine is acylated because acylation of any of the 11 lysine in SOD1 will increase its net negative charge by ∼0.9 units and increase electrostatic repulsion of subunits.
An interesting finding of this study deals with the polydispersity of SOD1 and with how polydispersity affects its rate and probability of fibrillization. When a single lysine is randomly acylated in SOD1, a theoretical maximum of 11 regioisomers are generated; when two lysines are acylated, a maximum of 55 regioisomers can be generated (i.e. r = 11!/[N!(11 − N)!], where N is the number of acylated lysines for a particular CE rung) (52). Acylation of lysines is of course not entirely random but is at least partially random according to tandem mass spectrometry (supplemental Tables S1 and S2). Thus, the sequence homology (monodispersity) of SOD1 chains is diminished to less than 100% by fractional acylation. How might a decrease in monodispersity affect the rate of formation or free energy of fibrillar SOD1? The threshold level of polydispersity/sequence homology required for two chains to self-assemble into amyloid has not been quantified. We presume that an SOD1 protein with an acetylated Lys-91 (for example) will pack better with another SOD1 protein that possesses an acetylated Lys-91, compared with an SOD1 chain with unacetylated Lys-91. This study suggests that the increase in polydispersity associated with fractional lysine acylation is not a major factor in disfavoring SOD1 fibrillization. For example, pyromellitylated SOD1 proteins have the same approximate polydispersity as tricarboxycyclobutylated SOD1 proteins (according to MS/MS; supplemental Table S1) yet have divergent effects on fibrillization.
This study represents the first systematic determination of how acylation of a protein affects its net surface charge and rate of fibril nucleation and propagation (53, 54). These data will assist us (and others) in the design of small molecules that electrostatically control the self-assembly of amyloid-like oligomers via acylation (1, 55). For example, recent studies have shown that the simplest aryl ester, aspirin, can impede the aggregation of multiple proteins linked to neurodegeneration, presumably via acetylation of proteins (other than aspirin's primary target, cyclooxygenase) (8). Can more complex pharmacons be designed to transfer more electrostatically potent groups than the acetyl group of aspirin (ΔZ = −1)? Our results suggest that single acyl groups can electrostatically disrupt the formation of classical prion-like amyloid assemblies if the acyl group is sufficiently charged. Toward this goal of “drugging Z,” we have recently imagined and are now synthesizing “ball and chain flail” compounds that contain amyloid binding motifs (the “handle”) attached to flexible linkers (the “chain”) that are capped with an acylating head group (the “ball”). We hypothesize that these small molecules will selectively acylate amyloid-like oligomers or fibrils and electrostatically disrupt their propagation.
Experimental procedures
Expression, purification, and demetallation of human WT SOD1
Human WT SOD1 was recombinantly expressed in Saccharomyces cerevisiae, purified, demetallated, and characterized as previously described (1). Briefly, S. cerevisiae cells were shaken at 30 °C for ∼7 days postinoculation (to obtain an average optical density of 1.8 a.u.) and were centrifuged at 4500 rpm. Cell pellets were lysed using 0.5-mm glass beads, followed by ammonium sulfate precipitation and three successive chromatographic separations: hydrophobic interaction chromatography, ion-exchange chromatography, and size-exclusion chromatography. Upon the completion of size-exclusion chromatography, purified SOD1 proteins were demetallated via dialysis in three demetallation buffers (∼6 days), which are as follows: (i) 100 mm sodium acetate, 10 mm EDTA, pH 3.8; (ii) 100 mm sodium acetate, 10 mm NaCl, pH 3.8; and (iii) 100 mm sodium acetate, pH 5.5. All demetallated proteins were flash frozen with liquid nitrogen and kept at −80 °C.
Inductively coupled plasma mass spectrometry
The absence of metal ions in purified WT SOD1 proteins was confirmed using a 7900 inductively coupled plasma MS instrument (Agilent Technologies, Santa Clara, CA). All WT apo-SOD1 proteins contained < 0.02 molar equivalents of zinc or copper.
Acylation of WT apo-SOD1 with different acylating groups
Acylating agents that were used to modify WT apo-SOD1 were purchased from Sigma–Aldrich and include: cyclobutane-1,2,3,4-tetracarboxylic dianhydride (CB), PM, BT, 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BP), SA, CA, GA, PhA, and AA. To perform acylation reactions, WT apo-SOD1 proteins were first transferred to a reaction buffer containing 100 mm N-(2-hydroxyethyl)piperazine-N′-(4-butanesulfonic acid), pH 9.0. Stock solutions of different acylating agents were prepared by dissolution in either DMSO, 1,4-dioxane, or dimethylformamide (DMF) to a final concentration of 1 m (except for CB and BP, which were dissolved to a final stock concentration of 0.5 m). Different degrees of WT apo-SOD1 acylation were obtained by adding different molar equivalents of acylating agents to the reaction solution (to a final concentration of up to 1 mm; [SOD1] = 10 μm), followed by constant and gentle stirring for 2 h at 4 °C. Identical volumes of DMSO, DMF, or dioxane were added to a solution of unmodified WT apo-SOD1 (i.e. the control solution) to account for any possible effects of organic solvents on the structure or stability of native SOD1. The final concentration of organic solvents in the aggregation assay (per modification) are as follows: 0.01% DMSO (PM); 0.1% DMF (CB); 0.04% DMSO (BP); 0.01% DMSO (BT); 0.02% DMSO (SA); 0.01% DMSO (CA); 0.01% DMSO (GA); 0.01% DMSO (PhA); and 0.04% dioxane (AA). Acylation reactions were stopped after 2 h, and 10-μl aliquots were taken for further characterization, as described below.
Mass spectrometry (MS)
Acylated WT apo-SOD1 proteins were diluted in 0.1% formic acid solution to a final concentration of 1 μm and analyzed using a Synapt G2 ion-mobility spectrometry mass spectrometry instrument under positive ion mode. The proteins were desalted for 10 min on a C-18 trapping column (98% water, 2% acetonitrile) prior to MS. Mass spectra of proteins were then deconvoluted using MaxEnt1 module in MassLynx software (Waters Corporation). The number of modifications per SOD1 subunit was calculated from deconvoluted spectra. In particular, we expressed the number of modifications as the weighted average of each modification per SOD1 subunit.
Trypsin digestion, tandem mass spectrometry (MS/MS), and proteomics analysis
Partially acylated apo-SOD1 protein solutions were transferred to 50 mm Tris-HCl buffer, pH 8.8 (to achieve optimal trypsin activity), using centrifugal filtration. Transferred protein solutions were digested with trypsin Gold (Promega®) at a ratio of 1:20 (trypsin:SOD1; [SOD1] = 150 μm). DTT was added to SOD1–trypsin mixture at a final concentration of 2 mm to ensure complete disulfide reduction. The SOD1 tryptic digest was incubated at 37 °C for 24 h, prior to mass spectrometry analysis. A LTQ LX/Orbitrap Discovery LC/MS instrument (Thermo Fisher Scientific) was used to acquire mass spectra for apo-SOD1 tryptic peptides. SOD1 tryptic digests were separated on a reversed-phase C-18 column using a gradient mixture of water and acetonitrile as mobile phase. The proteomics analysis was performed using the SEQUEST protein identification software (Thermo Fisher Scientific). Human SOD1 amino acid sequence was used as the template for protein identification. The sequence coverage among all acylated SOD1 proteins varied from 98 to 100%. Only peptides with Xcorr ≥ 4.0 and ppm ≤ 0.05 were considered as positive hits.
Capillary electrophoresis
The degree to which each modification altered the net charge of WT apo-SOD1 was quantified using CE, as previously described (11). Briefly, acylated and unmodified apo-SOD1 proteins were transferred to CE running buffer (10 mm KPO4, pH 7.4) and analyzed using a Beckman P/ACE instrument, equipped with a bare fused-silica capillary. All CE experiments were performed at 22 °C.
Differential scanning calorimetry
The impact of all acyl modifications on the thermostability of WT apo-SOD1 was measured using DSC. Unmodified and acylated proteins were transferred to 10 mm KPO4 buffer (pH 7.4) and heated from 20 to 80 °C at a scan rate of 1 °C/min, using a MicroCal VP-DSC calorimeter (Malvern Instruments). Normalization of protein concentration and baseline correction were performed on DSC thermograms prior to fitting with a two-state non-binding model, from which the values of melting temperature (Tm) were extracted.
ThT and ANS aggregation assays
High-throughput ThT and ANS aggregation assays were performed, as previously described (1, 28). Briefly, acylated and unmodified WT apo-SOD1 proteins were transferred to aggregation buffer (10 mm KPO4, 5 mm EDTA, pH 7.4) via centrifugal filtration (four or five cycles of 1:10 dilution, followed by 10-fold concentration). Solutions were then filtered using a 200-μm GHP Acrodisc® syringe filter.
Concentration of all apo-SOD1 solutions (modified and unmodified) were measured using Bradford assay and BCA assay (instead of UV-visible spectrophotometry), because of the high UV absorption of aromatic moieties in PM, BT, BP, and PhA modifications. Both Bradford and BCA assays were performed according to well established, standard procedures (56, 57). BSA was used as standard protein for both Bradford and BCA assays.
After quantifying the concentration of all modified and unmodified SOD1 proteins, TCEP was added to each protein solution ([TCEP] = 100 mm, [SOD1] = 30 μm per dimer), and solutions were gently shaken at room temperature for 6 h to achieve ∼90% disulfide reduction (28). Solutions were then filtered with a 0.2-μm GHP Acrodisc® syringe filter to remove any preformed oligomers that might possibly seed apo-SOD1 aggregation. ThT and/or ANS were added to WT apo-SOD1 solutions at a final concentration of 20 μm, thoroughly mixed, and solutions were aliquoted (200 μl each) into the wells of a 96-well, non-binding black polystyrene plate. Each well contained a clean and dust-free 1/8-inch Teflon bead (McMaster-Carr®). The plate was firmly sealed with a polypropylene seal to prevent solvent evaporation and TCEP oxidation. The assays were performed using an Ascent® Fluoroskan FL microplate reader (Thermo Fisher Scientific) with a built-in temperature controller (37 °C). Fluorescence was recorded every 15 min for up to ∼168 h, with intermittent orbital gyration at 360 rpm (gyration occurred every 15 s, for 15 s; gyration diameter = 3 mm). Analysis of SOD1 aggregation data, including fitting kinetic traces, extraction of mean kinetic parameters (i.e. lag time, propagation rate, ThT/ANS fluorescence intensity), construction of Kaplan–Meier plots, and calculation of Hazard ratios, were performed as previously described (1, 34).
Transmission electron microscopy
To determine the morphology of SOD1 aggregates after the termination of each aggregation assay, we performed TEM microscopy using a JEOL JEM-1010 transmission electron microscope (JEOL Ltd.). TEM samples for SOD1 aggregates were prepared, as previously described (34).
Mice
Studies that involved transgenic mice were approved by the Institutional Animal Care and Use Committee at the University of Florida in accordance with the National Institutes of Health guidelines. All animals were housed (between one to five to a cage) and nourished on ad libitum food and water. A 14-h light and 10-h dark cycle was used. The construction and pathology of transgenic G85R-SOD1·YFP mice have been previously described (58) and are maintained on the FVB/NJ background.
Organotypic spinal cord cultures
The organotypic spinal cord slice cultures were prepared as previously described (46). Briefly, G85R-SOD1·YFP mouse pups were euthanized at 7 days old by CO2 asphyxiation and were immediately decapitated. Spinal cords were dissected and cut to a length of 350 μm using a McIlwain Tissue Chopper. Spinal cord slices were cultured on 0.4-μm Millipore Millicell-CM membrane inserts; incubated in medium containing 50% (v/v) minimal essential medium, 25 mm HEPES, 25% (v/v) heat-inactivated horse serum, 25% (v/v) Hanks' balanced salt solution; and supplemented with 6.4 mg/ml glucose and 2 mm glutamine. Cultures were incubated at 37 °C in a 5% CO2/95% air humidified environment.
To perform the seeding experiments, fibril homogenates from in vitro microplate preparations were vigorously shaken, and 1 μl of homogenate was micropipetted directly to the top of each spinal cord section (after the cross-section was incubated for ∼1 week). The sections were incubated for 1 month after the addition of aggregated SOD1 from in vitro microplate assays. To image sections, samples were fixed with 4% paraformaldehyde for 4 h, rinsed in PBS, and put directly onto slides. The slides were then coverslipped in Vectastain mounting medium containing DAPI. The images were visualized using an Olympus DSU-IX81 spinning disc confocal microscope.
Author contributions
S. R., and A. A. performed the experiments and data analysis. C. M. C. performed TEM experiments. C. M. C., D. L. P., and Y. S. purified the SOD1 proteins. J. I. A. performed seeding experiments in organotypic spinal cord. S. R., A. A., and B. F. S. wrote the manuscript. All authors approved the final version of the manuscript.
Supplementary Material
This work was supported by National Science Foundation Grant CHE 1352122 (to B. F. S.), Welch Foundation Grant AA-1854 (to B. F. S.), and NINDS, National Institutes of Health Grant 1R01NA092788-01 (to J. I. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1 and S2 and Figs. S1–S13.
- SOD1
- superoxide dismutase-1
- ALS
- amyotrophic lateral sclerosis
- DSC
- differential scanning calorimetry
- CE
- capillary electrophoresis
- TEM
- transmission electron microscopy
- TCEP
- tris(2-carboxyethyl)phosphine
- ANS
- 1-anilino-8-naphthalenesulfonate
- ThT
- thioflavin-T
- CB
- cyclobutane-1,2,3,4-tetracarboxylic dianhydride
- PM
- pyromellitic dianhydride
- BT
- benzophenone-3,3′,4,4′-tetracarboxylic dianhydride
- BP
- 3,3′,4,4′-biphenyltetracarboxylic dianhydride
- SA
- succinic anhydride
- CA
- citraconic anhydride
- GA
- glutaric anhydride
- PhA
- phthalic anhydride
- AA
- acetic anhydride
- BCA
- bicinchoninic acid
- HR
- hazard ratio
- DMF
- dimethylformamide.
References
- 1. Abdolvahabi A., Shi Y., Rhodes N. R., Cook N. P., Martí A. A., and Shaw B. F. (2015) Arresting amyloid with Coulomb's law: acetylation of ALS-linked SOD1 by aspirin impedes aggregation. Biophys. J. 108, 1199–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mompeán M., Chakrabartty A., Buratti E., and Laurents D. V. (2016) Electrostatic repulsion governs TDP-43 C-terminal domain aggregation. PLoS Biol. 14, e1002447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taylor J. D., Hawthorne W. J., Lo J., Dear A., Jain N., Meisl G., Andreasen M., Fletcher C., Koch M., Darvill N., Scull N., Escalera-Maurer A., Sefer L., Wenman R., Lambert S., et al. (2016) Electrostatically-guided inhibition of Curli amyloid nucleation by the CsgC-like family of chaperones. Sci. Rep. 6, 24656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiti F., Stefani M., Taddei N., Ramponi G., and Dobson C. M. (2003) Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 424, 805–808 [DOI] [PubMed] [Google Scholar]
- 5. Lawrence M. S., Phillips K. J., and Liu D. R. (2007) Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 129, 10110–10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gitlin I., Carbeck J. D., and Whitesides G. M. (2006) Why are proteins charged? Networks of charge-charge interactions in proteins measured by charge ladders and capillary electrophoresis. Angew. Chem. Int. Ed. Engl. 45, 3022–3060 [DOI] [PubMed] [Google Scholar]
- 7. Shi Y., Acerson M. J., Shuford K. L., and Shaw B. F. (2015) Voltage-induced misfolding of zinc-replete ALS mutant superoxide dismutase-1. ACS Chem. Neurosci. 6, 1696–1707 [DOI] [PubMed] [Google Scholar]
- 8. Ayyadevara S., Balasubramaniam M., Alla R., Mehta J. L., and Shmookler Reis R. J. (2017) Aspirin-mediated acetylation protects against multiple neurodegenerative pathologies by impeding protein aggregation. Antioxid. Redox. Signal., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Profit A. A., Vedad J., and Desamero R. Z. (2017) Peptide conjugates of benzene carboxylic acids as agonists and antagonists of amylin aggregation. Bioconjug. Chem. 28, 666–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shi Y., Mowery R. A., and Shaw B. F. (2013) Effect of metal loading and subcellular pH on net charge of superoxide dismutase-1. J. Mol. Biol. 425, 4388–4404 [DOI] [PubMed] [Google Scholar]
- 11. Shi Y., Abdolvahabi A., and Shaw B. F. (2014) Protein charge ladders reveal that the net charge of ALS-linked superoxide dismutase can be different in sign and magnitude from predicted values. Protein Sci. 23, 1417–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alfonso L. F., Srivenugopal K. S., and Bhat G. J. (2009) Does aspirin acetylate multiple cellular proteins? Mol. Med. Rep. 2, 533–537 [DOI] [PubMed] [Google Scholar]
- 13. Tatham M. H., Cole C., Scullion P., Wilkie R., Westwood N. J., Stark L. A., and Hay R. T. (2017) A proteomic approach to analyze the aspirin-mediated lysine acetylome. Mol. Cell. Proteomics 16, 310–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bateman L. A., Zaro B. W., Miller S. M., and Pratt M. R. (2013) An alkyne-aspirin chemical reporter for the detection of aspirin-dependent protein modification in living cells. J. Am. Chem. Soc. 135, 14568–14573 [DOI] [PubMed] [Google Scholar]
- 15. Schmitt N. D., and Agar J. N. (2017) Parsing disease-relevant protein modifications from epiphenomena: perspective on the structural basis of SOD1-mediated ALS. J. Mass Spectrom. 52, 480–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Felder C. E., Prilusky J., Silman I., and Sussman J. L. (2007) A server and database for dipole moments of proteins. Nucleic Acids Res. 35, W512–W521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chattopadhyay M., Durazo A., Sohn S. H., Strong C. D., Gralla E. B., Whitelegge J. P., and Valentine J. S. (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. U.S.A. 105, 18663–18668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oztug Durer Z. A., Cohlberg J. A., Dinh P., Padua S., Ehrenclou K., Downes S., Tan J. K., Nakano Y., Bowman C. J., Hoskins J. L., Kwon C., Mason A. Z., Rodriguez J. A., Doucette P. A., Shaw B. F., et al. (2009) Loss of metal ions, disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase. PLoS One 4, e5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Renton A. E., Chiò A., and Traynor B. J. (2014) State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rotunno M. S., and Bosco D. A. (2013) An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front. Cell Neurosci. 7, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Furukawa Y., Anzai I., Akiyama S., Imai M., Cruz F. J., Saio T., Nagasawa K., Nomura T., and Ishimori K. (2016) Conformational disorder of the most immature Cu, Zn-superoxide dismutase leading to amyotrophic lateral sclerosis. J. Biol. Chem. 291, 4144–4155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choudhary C., Weinert B. T., Nishida Y., Verdin E., and Mann M. (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550 [DOI] [PubMed] [Google Scholar]
- 23. Oueslati A., Fournier M., and Lashuel H. A. (2010) Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: implications for Parkinson's disease pathogenesis and therapies. Prog. Brain Res. 183, 115–145 [DOI] [PubMed] [Google Scholar]
- 24. Cohen T. J., Hwang A. W., Restrepo C. R., Yuan C. X., Trojanowski J. Q., and Lee V. M. (2015) An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 6, 5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cook C., Stankowski J. N., Carlomagno Y., Stetler C., and Petrucelli L. (2014) Acetylation: a new key to unlock tau's role in neurodegeneration. Alzheimers Res. Ther. 6, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Banks C. J., Rodriguez N. W., Gashler K. R., Pandya R. R., Mortenson J. B., Whited M. D., Soderblom E. J., Thompson J. W., Moseley M. A., Reddi A. R., Tessem J. S., Torres M. P., Bikman B. T., and Andersen J. L. (2017) Acylation of superoxide dismutase 1 (SOD1) at K122 governs SOD1-mediated inhibition of mitochondrial respiration. Mol. Cell Biol. 37, e00354–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Antinone S. E., Ghadge G. D., Lam T. T., Wang L., Roos R. P., and Green W. N. (2013) Palmitoylation of superoxide dismutase 1 (SOD1) is increased for familial amyotrophic lateral sclerosis-linked SOD1 mutants. J. Biol. Chem. 288, 21606–21617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abdolvahabi A., Shi Y., Chuprin A., Rasouli S., and Shaw B. F. (2016) Stochastic formation of fibrillar and amorphous superoxide dismutase oligomers linked to amyotrophic lateral sclerosis. ACS Chem. Neurosci. 7, 799–810 [DOI] [PubMed] [Google Scholar]
- 29. Menon M. K., and Zydney A. L. (2000) Determination of effective protein charge by capillary electrophoresis: effects of charge regulation in the analysis of charge ladders. Anal. Chem. 72, 5714–5717 [DOI] [PubMed] [Google Scholar]
- 30. Gitlin I., Mayer M., and Whitesides G. M. (2003) Significance of charge regulation in the analysis of protein charge ladders. J. Phys. Chem. B 107, 1466–1472 [Google Scholar]
- 31. Shi Y., Rhodes N. R., Abdolvahabi A., Kohn T., Cook N. P., Marti A. A., and Shaw B. F. (2013) Deamidation of asparagine to aspartate destabilizes Cu,Zn superoxide dismutase, accelerates fibrillization, and mirrors ALS-linked mutations. J. Am. Chem. Soc. 135, 15897–15908 [DOI] [PubMed] [Google Scholar]
- 32. Shaw B. F., Arthanari H., Narovlyansky M., Durazo A., Frueh D. P., Pollastri M. P., Lee A., Bilgicer B., Gygi S. P., Wagner G., and Whitesides G. M. (2010) Neutralizing positive charges at the surface of a protein lowers its rate of amide hydrogen exchange without altering its structure or increasing its thermostability. J. Am. Chem. Soc. 132, 17411–17425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shaw B. F., Schneider G. F., Bilgiçer B., Kaufman G. K., Neveu J. M., Lane W. S., Whitelegge J. P., and Whitesides G. M. (2008) Lysine acetylation can generate highly charged enzymes with increased resistance toward irreversible inactivation. Protein Sci. 17, 1446–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abdolvahabi A., Shi Y., Rasouli S., Croom C. M., Aliyan A., Martí A. A., and Shaw B. F. (2017) Kaplan–Meier meets chemical kinetics: intrinsic rate of SOD1 amyloidogenesis decreased by subset of ALS mutations and cannot fully explain age of disease onset. ACS Chem. Neurosci. 8, 1378–1389 [DOI] [PubMed] [Google Scholar]
- 35. Ivanova M. I., Sievers S. A., Guenther E. L., Johnson L. M., Winkler D. D., Galaleldeen A., Sawaya M. R., Hart P. J., and Eisenberg D. S. (2014) Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc. Natl. Acad. Sci. U.S.A. 111, 197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rich J. T., Neely J. G., Paniello R. C., Voelker C. C. J., Nussenbaum B., and Wang E. W. (2010) A practical guide to understanding Kaplan–Meier curves. Otolaryng. Head Neck Surg. 143, 331–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferrone F. (1999) Analysis of protein aggregation kinetics. Methods Enzymol. 309, 256–274 [DOI] [PubMed] [Google Scholar]
- 38. Knowles T. P., Waudby C. A., Devlin G. L., Cohen S. I., Aguzzi A., Vendruscolo M., Terentjev E. M., Welland M. E., and Dobson C. M. (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537 [DOI] [PubMed] [Google Scholar]
- 39. Yoshimura Y., Lin Y., Yagi H., Lee Y. H., Kitayama H., Sakurai K., So M., Ogi H., Naiki H., and Goto Y. (2012) Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation. Proc. Natl. Acad. Sci. U.S.A. 109, 14446–14451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Younan N. D., and Viles J. H. (2015) A comparison of three fluorophores for the detection of amyloid fibers and prefibrillar oligomeric assemblies. ThT (thioflavin T); ANS (1-anilinonaphthalene-8-sulfonic acid); and bisANS (4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid). Biochemistry 54, 4297–4306 [DOI] [PubMed] [Google Scholar]
- 41. Kundu B., and Guptasarma P. (1999) Hydrophobic dye inhibits aggregation of molten carbonic anhydrase during thermal unfolding and refolding. Proteins 37, 321–324 [DOI] [PubMed] [Google Scholar]
- 42. Deng H. X., Shi Y., Furukawa Y., Zhai H., Fu R., Liu E., Gorrie G. H., Khan M. S., Hung W. Y., Bigio E. H., Lukas T., Dal Canto M. C., O'Halloran T. V., and Siddique T. (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7142–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fukada K., Nagano S., Satoh M., Tohyama C., Nakanishi T., Shimizu A., Yanagihara T., and Sakoda S. (2001) Stabilization of mutant Cu/Zn superoxide dismutase (SOD1) protein by coexpressed wild SOD1 protein accelerates the disease progression in familial amyotrophic lateral sclerosis mice. Eur. J. Neurosci. 14, 2032–2036 [DOI] [PubMed] [Google Scholar]
- 44. Wang L., Deng H. X., Grisotti G., Zhai H., Siddique T., and Roos R. P. (2009) Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Hum. Mol. Genet. 18, 1642–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu G., Ayers J. I., Roberts B. L., Brown H., Fromholt S., Green C., and Borchelt D. R. (2015) Direct and indirect mechanisms for wild-type SOD1 to enhance the toxicity of mutant SOD1 in bigenic transgenic mice. Hum. Mol. Genet. 24, 1019–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ayers J. I., Diamond J., Sari A., Fromholt S., Galaleldeen A., Ostrow L. W., Glass J. D., Hart P. J., and Borchelt D. R. (2016) Distinct conformers of transmissible misfolded SOD1 distinguish human SOD1-FALS from other forms of familial and sporadic ALS. Acta Neuropathol. 132, 827–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rothstein J. D., Jin L., Dykes-Hoberg M., and Kuncl R. W. (1993) Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 90, 6591–6595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nagy M., Fenton W. A., Li D., Furtak K., and Horwich A. L. (2016) Extended survival of misfolded G85R SOD1-linked ALS mice by transgenic expression of chaperone Hsp110. Proc. Natl. Acad. Sci. U.S.A. 113, 5424–5428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Frutiger K., Lukas T. J., Gorrie G., Ajroud-Driss S., and Siddique T. (2008) Gender difference in levels of Cu/Zn superoxide dismutase (SOD1) in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 9, 184–187 [DOI] [PubMed] [Google Scholar]
- 50. Sangwan S., Zhao A., Adams K. L., Jayson C. K., Sawaya M. R., Guenther E. L., Pan A. C., Ngo J., Moore D. M., Soriaga A. B., Do T. D., Goldschmidt L., Nelson R., Bowers M. T., Koehler C. M., et al. (2017) Atomic structure of a toxic, oligomeric segment of SOD1 linked to amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. U.S.A. 114, 8770–8775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. de Lavor M. S., Binda N. S., Fukushima F. B., Caldeira F. M., da Silva J. F., Silva C. M., de Oliveira K. M., Martins B. D., Torres B. B., Rosado I. R., Gomez R. S., Gomez M. V., and de Melo E. G. (2015) Ischemia-reperfusion model in rat spinal cord: cell viability and apoptosis signaling study. Int. J. Clin. Exp. Pathol. 8, 9941–9949 [PMC free article] [PubMed] [Google Scholar]
- 52. Carbeck J. D., Colton I. J., Gao J. M., and Whitesides G. M. (1998) Protein charge ladders, capillary electrophoresis, and the role of electrostatics in biomolecular recognition. Acc. Chem. Res. 31, 343–350 [Google Scholar]
- 53. Tanaka M., and Komi Y. (2015) Layers of structure and function in protein aggregation. Nat. Chem. Biol. 11, 373–377 [DOI] [PubMed] [Google Scholar]
- 54. Iyer A., Roeters S. J., Schilderink N., Hommersom B., Heeren R. M., Woutersen S., Claessens M. M., and Subramaniam V. (2016) The impact of N-terminal acetylation of α-synuclein on phospholipid membrane binding and fibril structure. J. Biol. Chem. 291, 21110–21122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eisele Y. S., Monteiro C., Fearns C., Encalada S. E., Wiseman R. L., Powers E. T., and Kelly J. W. (2015) Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 14, 759–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Walker J. M. (2009) The bicinchoninic acid (BCA) assay for protein quantitation. The Protein Protocols Handbook, pp. 11–15, Humana Press, Totowa, NJ [Google Scholar]
- 57. Kruger N. J. (2009) The Bradford method for protein quantitation. The Protein Protocols Handbook, pp. 17–24, Humana Press, Totowa, NJ [Google Scholar]
- 58. Wang J., Farr G. W., Zeiss C. J., Rodriguez-Gil D. J., Wilson J. H., Furtak K., Rutkowski D. T., Kaufman R. J., Ruse C. I., Yates J. R. 3rd, Perrin S., Feany M. B., and Horwich A. L. (2009) Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc. Natl. Acad. Sci. U.S.A. 106, 1392–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.