

Abstract
Apicomplexan parasites such as Toxoplasma gondii rely on a unique form of locomotion known as gliding motility. Generating the mechanical forces to support motility are divergent class XIV myosins (MyoA) coordinated by accessory proteins known as light chains. Although the importance of the MyoA–light chain complex is well-established, the detailed mechanisms governing its assembly and regulation are relatively unknown. To establish a molecular blueprint of this dynamic complex, we first mapped the adjacent binding sites of light chains MLC1 and ELC1 on the MyoA neck (residues 775–818) using a combination of hydrogen–deuterium exchange mass spectrometry and isothermal titration calorimetry. We then determined the 1.85 Å resolution crystal structure of MLC1 in complex with its cognate MyoA peptide. Structural analysis revealed a bilobed architecture with MLC1 clamping tightly around the helical MyoA peptide, consistent with the stable 10 nm Kd measured by isothermal titration calorimetry. We next showed that coordination of calcium by an EF-hand in ELC1 and prebinding of MLC1 to the MyoA neck enhanced the affinity of ELC1 for the MyoA neck 7- and 8-fold, respectively. When combined, these factors enhanced ELC1 binding 49-fold (to a Kd of 12 nm). Using the full-length MyoA motor (residues 1–831), we then showed that, in addition to coordinating the neck region, ELC1 appears to engage the MyoA converter subdomain, which couples the motor domain to the neck. These data support an assembly model where staged binding events cooperate to yield high-affinity complexes that are able to maximize force transduction.
Keywords: cell motility, crystal structure, isothermal titration calorimetry (ITC), myosin, protein structure, Toxoplasma gondii, X-ray crystallography, host cell invasion
Introduction
Protozoan parasites of phylum Apicomplexa cause significant disease in humans and animals. Of particular relevance to human health are Plasmodium spp., the causative agents of malaria (1); Cryptosporidium parvum, an opportunistic pathogen affecting cancer and AIDS patients as well as young children (2, 3); and Toxoplasma gondii, which infects ∼30% of all humans on earth and causes serious disease in immunocompromised individuals and neonatally infected fetuses (4–6).
Central to the pathogenesis of apicomplexans is a unique form of substrate-dependent locomotion termed “gliding motility,” which is essential for traversing the environment and invading host cells (7). Driving motility is a class XIV unconventional myosin motor (MyoA), which is notably divergent from canonical myosins in that it lacks a “tail” and conventional sequence motifs in both the neck and motor regions (8–10). Thus, the mechanisms that enable MyoA to function with a step size and velocity similar to canonical fast muscle myosins are not well-understood (11).
Genetic studies in the model apicomplexan T. gondii have led to a functional model of MyoA in complex with its accessory proteins that form part of the “glideosome” complex located between the outer plasma membrane and the inner membrane complex (Fig. 1) (11–14). Although the MyoA motor domain generates the force for motility, the interactions between the MyoA neck, essential light chain 1 (ELC1),2 and myosin light chain 1 (MLC1) support force transduction from the motor to the gliding-associated protein complex, which provides the crucial link between the glideosome and the parasite cytoskeleton (7). MLC1 also supports localization of the motor complex, because of its unique N-terminal anchoring function (15), and the presence of ELC1 enhances the in vitro velocity of the motor (16). Thus, defining the molecular mechanisms underlying apicomplexan motility necessitates a detailed characterization of the MyoA–MLC1–ELC1 complex.
Figure 1.
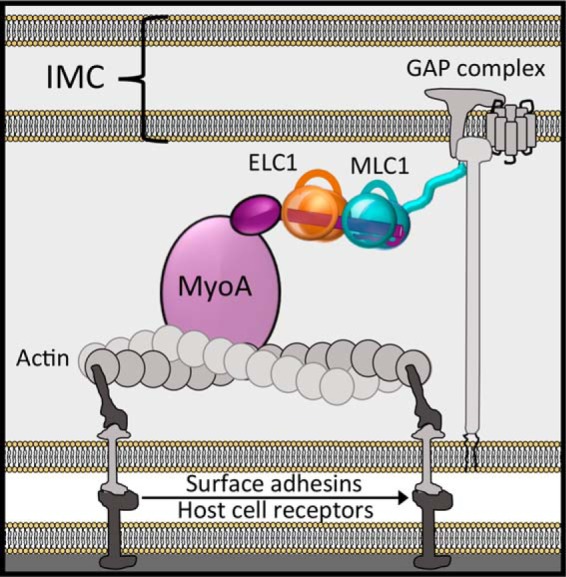
Leading model describing the general architecture of MyoA (motor domain, light purple circle; converter domain, dark purple oval), with ELC1 (orange) and MLC1 (teal) bound to the neck region (dark purple cylinder), and accessory proteins comprising the glideosome macromolecular complex.
Here we use a combination of solution-binding and structural studies to establish a molecular blueprint of the MyoA–MLC1–ELC1 complex from the model organism T. gondii. Our data support a model that relies on staged and cooperative binding events to generate high-affinity complexes that are optimal for efficient force transduction. These data, combined with the divergence of MyoA from canonical myosins, may prove valuable for ongoing research in targeted therapeutic development.
Results
MLC1 forms a bimolecular complex with the MyoA neck
To map the assembly of MLC1 and ELC1 on MyoA, we first synthesized a MyoA neck peptide incorporating residues 775–818, which was designed based on secondary structure predictions and previous studies of MyoA from T. gondii and Plasmodium falciparum (17, 18). A C775S mutation was included to prevent spurious disulfide bond formation with free cysteines in ELC1. We recombinantly produced full-length ELC1 and an N-terminal deletion construct of MLC1 (Ala66–Glu210; hereafter referred to simply as MLC1). Notably, the N-terminal tail was also removed in related plasmodium experiments (18), and actin-based motility assays show that its removal does not affect in vitro motility, because MyoA bound with either full-length MLC1 or the Ala66–Glu210 truncation moved actin filaments with no significant difference in velocity (2.58 ± 0.25 or 2.62 ± 0.02 μm/s, respectively). Solution-binding studies were then performed using hydrogen-deuterium exchange mass spectrometry (HDX-MS), which quantifies exchange of amide hydrogens with deuterium to reveal differences in secondary structure upon complex formation. HDX-MS studies were carried out using a short pulse of deuterium exposure (3 s at 0 and 23 °C) with MyoA neck peptide (775–818) in the presence of MLC1 and ELC1 individually and collectively (Fig. 2a). The apo MyoA neck peptide was fully deuterated at every time point of exchange, indicating a complete lack of secondary structure. Decreased amide exchange was observed in residues 775–795 upon ELC1 binding, consistent with a recent study that identified a binding region of 770–800 (17). MLC1 binding induced large decreases in exchange in residues 801–818. These decreases in HDX suggest that there is a disorder-order transition in the MyoA neck peptide upon either MLC1 or ELC1 binding. MLC1 binding alone also induced a small decrease in exchange in residues 779–784, which might reflect either an additional MLC1 protein binding with low affinity in the absence of ELC1 or MLC1 causing an allosteric conformational change at the ELC1 binding site. We used isothermal titration calorimetry (ITC) to address this possibility and definitively measure stoichiometry. These data show that MLC1 bound to MyoA neck constructs 775–818 and 801–818 with a 1:1 stoichiometry (Fig. 2b) (Table 1), consistent with a discrete binary complex. Notably, a larger entropic penalty was associated with MLC1 binding the full MyoA neck (775–818) (Table 1), consistent with the HDX data that showed a general order imparted by MLC1 binding.
Figure 2.

MLC1-binding region on MyoA neck analyzed by HDX, ITC, and X-ray crystallography reveals several key interactions crucial to complex formation. a, HDX heat maps showing difference in percentage of backbone amide deuterium incorporation of MyoA neck induced by binding of light chains at two time points of exchange. Putative directly ordered regions are marked with solid lines, and putative indirectly ordered regions are marked with dashed lines. Sequence of the MyoA peptide is shown. Red coloring of terminal residues indicates N-terminal acetylation or C-terminal amidation. Refer to supplemental Table S1 for the full HDX-MS data set relating to this figure. b, representative ITC binding isotherms following the titration of MLC1 into a solution of MyoA (775–818) or MyoA (801–818). c, left panel, 1.85 Å resolution crystal structure of MLC1 in complex with MyoA (801–831) with close-ups of the apical salt bridge (inset 1) formed between MLC1 Asp145 and MyoA Arg808 and MLC1 “clamp” (inset 2) formed via two hydrogen bonds between Glu179 and backbone amides of Tyr114 and Ala115. Right panel, electrostatic surface maps of MLC1 and the MyoA neck peptide. In this view, the MyoA neck peptide has been extracted from the surrounding MLC1 and rotated to reveal the basic surface along its helical longitudinal axis.
Table 1.
Summary of ITC thermodynamic parameters
All experiments were performed in triplicate. Thermodynamic parameters were fitted using Origin software (Microcal).
| Light chain | MyoA/light chain | CaCl2 | Molar ratio | Kd | ΔH | −TΔS |
|---|---|---|---|---|---|---|
| nm | kcal/mol | kcal/mol | ||||
| MLC1 | 801–818 | − | 1.05 ± 0.01 | 10.0 ± 1.4 | −26.9 ± 0.2 | 16.0 ± 0.2 |
| 775–818 | − | 1.00 ± 0.01 | 43.1 ± 6.5 | −40.7 ± 0.6 | 30.6 ± 0.6 | |
| ELC1 | 775–818 | − | 0.99 ± 0.03 | 590.6 ± 46.5 | −5.7 ± 0.2 | −3.0 ± 0.1 |
| + | 1.02 ± 0.03 | 82.8 ± 17.7 | −10.4 ± 0.1 | 0.8 ± 0.2 | ||
| 775–818/MLC1 | − | 1.02 ± 0.04 | 71.8 ± 5.0 | −9.9 ± 0.1 | 0.1 ± 0.1 | |
| + | 1.02 ± 0.04 | 11.5 ± 5.2 | −13.2 ± 0.4 | 2.3 ± 0.5 | ||
| FL/MLC1 | − | 1.02 ± 0.01 | 1.9 ± 1.5 | −15.8 ± 0.3 | 3.7 ± 0.8 | |
| ELC1 WT | + | 0.97 ± 0.38 | 2.3 E4 ± 0.9 E4 | 1.3 ± 0.3 | −7.6 ± 0.1 | |
| MgCl2 | 0.34 ± 0.09 | 14.4 E4 ± 2.1 E4 | 9.0 ± 2.5 | −14.3 ± 2.4 | ||
| ELC1 D80A | + | 1.43 ± 0.05 | 1.8 E4 ± 0.3 E4 | 0.9 ± 0.1 | −7.3 ± 0.1 |
To complement the solution-binding studies and further dissect the molecular interface, we next determined the 1.85 Å resolution crystal structure of MLC1 in complex with a truncated MyoA neck peptide (Fig. 2c). Several MyoA constructs were tested in combination with MLC1, although only the complex with the MyoA neck peptide (801–831) yielded diffraction quality crystals. Structural analysis revealed a calmodulin-like fold (19, 20), with two lobes, each composed of four α-helices, connected by a flexible linker (Fig. 3c), consistent with the well-established evolutionary relationship of myosin light chains to calmodulin proteins (21, 22). Notably, however, MLC1 lacks functional calcium-binding motifs, and no role for calcium was identified from previous studies with the plasmodium homologue PfMTIP (11, 23). The two lobes of MLC1 completely encompass the MyoA peptide, resulting in an extensive buried surface area (1186.1 Å2). The complex is stabilized via an apical salt bridge (MLC1, Asp145–MyoA, Arg808) (Fig. 2c, inset 1) and a basal, interdomain clamp, which secures MLC1 in a closed conformation around MyoA with a pair of hydrogen bonds (MLC1, Glu179–backbone amides of Tyr114 and Ala115) (Fig. 2c, inset 2). Overall, the buried interface between MLC1 and MyoA is largely electrostatic in nature (Fig. 2c, right panels). Although residues 801–818 of MyoA adopt an ordered α-helix, consistent with the HDX results (Fig. 2a), the C-terminal extension (819–831) lacks secondary structure, and its conformation appears to be a by-product of the crystal-packing interface. A comparison of the binary complex with the homologous MTIP-MyoA peptide structure (PDB code 4AOM) (18) from P. falciparum yielded a root mean square deviation of 0.83 Å (114 Cαs) and a comparable buried surface of 1131.5 Å2.
Figure 3.

ELC1 binds calcium using a classical EF-hand motif and interacts with MLC1, increasing its affinity for MyoA. a, putative EF-hands 1 and 2 of ELC1, based on consensus sequences. Ca2+-interacting residues are numbered, and mutated aspartates are boxed in red. b, bar graphs of representative peptides from ELC1 WT and mutants, showing changes in backbone deuterium exchange induced by Ca2+ binding. Sequences of peptides are shown above corresponding graphs. The data are from a single time point of exchange (300 s at 23 °C). The error bars (S.D.) are from independent triplicate experiments. Refer to supplemental Table S2 for the full HDX-MS data set relating to this figure. c, representative ITC binding isotherms of CaCl2 titrated into ELC1 wild-type (left panel), ELC1 D15A (middle panel), or ELC1 D80A (right panel). d, representative ITC binding isotherms of ELC1 titrated into MyoA (775–818) without (left panel) and with (right panel) calcium. e, representative ITC binding isotherms ELC1 titrated into MyoA (775–818), prebound with MLC1 without (left panel) or with (right panel) calcium.
A discrete ELC1–calcium complex shows enhanced affinity for MyoA
Myosin light chains are evolutionarily related to calmodulin proteins, which employ EF-hand motifs (Fig. 3a) to coordinate calcium (19–22). Two putative EF-hand motifs were identified in ELC1 using the program PROSITE (24) consistent with a previous prediction (17). Notably, calcium was previously observed to enhance the thermostability of T. gondii ELC1 (17), although no effect was observed on the in vitro velocity generated by the MyoA complex (16, 25). To assess the ability of ELC1 to directly coordinate Ca2+, we generated D15A and D80A mutants, corresponding to key Ca2+-coordinating residues of putative EF hands 1 and 2, respectively, based on the EF-hand consensus motif (Fig. 3a). HDX-MS revealed significant decreases in exchange in the N-terminal lobes (residues ∼1–58) of ELC1 WT and D80A in the presence of calcium; however, no decreases in exchange were observed in the D15A mutant (Fig. 3b). ITC measurement revealed low-micromolar affinity of Ca2+ binding to ELC1 WT and D80A but no binding to D15A (Fig. 3c). These data definitively show that ELC1 possesses a single functional EF-hand motif capable of directly binding Ca2+ ions. It is of note that ELC1 was also shown to bind Mg2+, albeit with an affinity at least 6-fold weaker (Table 1). We next reasoned that an ELC1–Ca2+ complex might display enhanced binding to the MyoA neck. ITC experiments revealed greater than a 7-fold (Kd = 591 nm to 83 nm) stronger binding affinity of ELC1 to MyoA (775–818) when titrations were carried out in the presence of CaCl2 (Fig. 3d). Together, these results are consistent with a model where calcium functions as a regulator for ELC1 binding to the MyoA neck.
ELC1 is cooperatively recruited to the MyoA neck by MLC1
Through mapping of the molecular interfaces, we established that MLC1 and ELC1 bind adjacent sites on the MyoA neck (Fig. 2a). The close proximity of the two light chain-binding sites suggests the possibility of direct interactions between light chains, as observed in canonical myosins (22, 26). This, coupled with the observation that MLC1 indirectly orders the ELC1 binding region in absence of ELC1 (Fig. 2a), led us to speculate that ELC1 may be cooperatively recruited to the MyoA neck when MLC1 is already present. We reasoned that if such an interaction were to occur, the affinity for ELC1 should be greater for the preformed MyoA–MLC1 complex. To test this, ITC experiments were performed assessing the affinity of ELC1 binding to the MyoA neck peptide (775–818), with the added step of prebinding MLC1 to the peptide (Fig. 3e). These experiments showed significantly stronger binding of ELC1 to MyoA when MLC1 is prebound (Kd = 591 nm to 72 nm). It is worth noting that the reverse experiment, binding MLC1 to the preformed MyoA–ELC1 complex, was attempted but could not be completed due to limited solubility of the MyoA–ELC1 complex. The enhanced binding supports an assembly mechanism that relies on cooperative binding. Repeating these experiments in the presence of CaCl2 further strengthened binding of ELC1 to MyoA–MLC1 (Kd = 12 nm). These data support a model where ELC1 binds weakly on its own, but calcium and MLC1-based cooperative recruitment of ELC1 combine to yield a highly stable ternary complex.
ELC1 interacts with MyoA converter domain, increasing binding affinity and further stabilizing the MyoA neck
To control for the fact that our previous binding studies were performed with truncated neck peptides and with the C775S mutation, we next used ITC to measure the affinity of ELC1 for the full-length, WT MyoA (1–831) prebound with MLC1 (Fig. 4a). Full-length recombinant MyoA was produced in insect cells by co-expressing with MLC1 and the recently identified myosin chaperone, UNC (16). Notably, co-expression with MLC1 was required to stabilize the neck region of full-length MyoA, and thus we were unable to perform ITC studies with the apo form of the motor. These experiments revealed significantly stronger binding of ELC1 to full-length MyoA (1–831), compared with the truncated MyoA neck peptide (775–818) (Kd = 72 nm to 1.9 nm). Although our previous experiments suggest it is likely that the addition of Ca2+ would further enhance binding affinity, a Kd of 1 nm is near the detection limit of ITC, and thus, enhanced binding could not be accurately measured.
Figure 4.

ELC1 interacts with the MyoA converter subdomain, increasing its affinity for the MyoA neck and stabilizing the myosin lever arm. Schematics shown are colored as previously. a, representative ITC binding isotherm of ELC1 titrated into MyoA (1–831) prebound with MLC1. b, HDX difference map showing reduction in total deuteron incorporation of full-length MyoA, prebound with MLC1, when ELC1 is added. The decreases in amide exchange between conditions are summed over three time points of HDX (3, 30, and 300 s at 23 °C), and the error bars (S.D.) represent independent triplicate. Peptide # corresponds to the centroid amino acid of the peptide from which a data point is obtained. Inset, expanded view of HDX difference map showing order induced in MyoA converter domain (residues ∼710–740) by ELC1 binding. c, representative deuterium incorporation time course for MyoA peptide in the converter subdomain (716–720). Refer to supplemental Table S3 for the full HDX-MS data set relating to this figure.
We reasoned that the increase in affinity was likely due to interactions between ELC1 and additional regions of full-length MyoA not represented in the MyoA peptide-based experiments. Previous genetic studies have shown that MyoA residues 706–763 may form the “converter subdomain” (11), a discrete region of the myosin responsible for transducing small conformational changes in the motor domain, that are then amplified into larger steps by the swinging motion of the neck region. This led us to hypothesize that ELC1 could form contacts with the converter subdomain of MyoA, in addition to its contacts with the MyoA neck. To test this hypothesis, HDX-MS was performed on the MyoA (1–831)–MLC1 complex in the presence and absence of ELC1–Ca2+ (Fig. 4, b and c). These results showed the expected reduction in deuterium incorporation in the MyoA neck (775–818) as observed previously (Fig. 2a). Notably, however, an additional region of reduced deuterium incorporation on MyoA (∼710–740) was observed that corresponds to the predicted converter subdomain (Fig. 4, b and c). Thus, it appears that ELC1 engages two distinct regions on MyoA (710–740 and 775–795), resulting in an exceptionally stable complex that is optimized for force transduction. Interestingly, an isolated region of the MyoA N terminus also showed reduced deuteration in response to ELC1 binding consistent with a stabilizing interaction (Fig. 4b). Notably, the N terminus of MyoA has been shown to contain a regulatory phosphorylation site important for parasite egress (27), and ELC1 was shown to interact with the N terminus of smooth muscle myosin type II (28). However, the variability in the N termini of myosins makes it difficult to draw firm conclusions regarding the role of an interaction between the N termini of MyoA and ELC1. The structure of the full-length MyoA ternary complex will likely be required to ultimately validate this putative interaction.
Discussion
Apicomplexan parasites rely on a unique form of “gliding motility” powered by a divergent class XIV myosin motor. Previous studies have identified key components of the apicomplexan motor complex composed of MyoA and its light chains, MLC1 and ELC1/ELC2 (7, 13, 15, 17, 29–33). Here we sought to interrogate the dynamic interface underlying complex formation and establish the mechanisms governing assembly and overall architecture of this intriguing motor complex.
Solution-binding studies were initially used to define the binding regions of both ELC1 and MLC1 on the MyoA neck, which showed a clear 1:1:1 stoichiometry for the ternary complex. Complementary structural data yielded a high-resolution blueprint of the binding interface between MLC1 and its cognate MyoA neck peptide. Notably, the MLC1–MyoA complex showed significant homology to the PfMTIP-MyoA complex (18). In particular, the basal clamping region of MLC1 is conserved with that of PfMTIP, which was shown to be crucial for high-affinity binding to MyoA, and a key interaction hot spot for a peptide-fragment chimera that showed enhanced affinity for PfMTIP (18, 34). Thus, our MLC1–MyoA structure, which is only the second from an apicomplexan parasite, and the first from T. gondii, may prove valuable in refining engineered peptidomimetics capable of disrupting assembly of the class XIV myosin complexes. Notably, the potential for disrupting the light chains was recently demonstrated with the MLC1-binding small molecule tachypleginA-2, which caused impaired motility and invasion of T. gondii tachyzoites (35, 36).
Analysis of the calcium-binding properties of ELC1 definitively shows that ELC1 employs a single EF-hand motif to bind a Ca2+ ion and that this significantly enhances the affinity of ELC1 for the MyoA neck. The low-micromolar affinity of ELC1 for Ca2+ measured here supports the possibility that ELC1 may act as a Ca2+-activated “switch,” induced by the rapidly oscillating levels of intracellular calcium observed during T. gondii motility (37–40). These data are consistent with a recent study that showed a role for Ca2+ in increasing thermostability of ELC1 and maximizing invasion and egress of host cells (17). It is also noteworthy that ELC1 binds to the full-length MyoA–MLC1 complex with a Kd of 1–2 nm in the absence of calcium, which suggests a limited role for calcium in motor function. This observation is consistent with in vitro motility data that do not show a clear effect for calcium in enhancing motor velocity (16, 25). It is possible that, through enhancing ELC1 binding to MyoA, calcium effectively reduces the amount of ELC1 in the cell required to saturate MyoA rather than calcium directly enhancing motor velocity. This explanation would effectively reconcile our own calcium-binding data with that of the aforementioned motility assay, because this assay was performed in the presence of a molar excess of ELC1, as well as a buffer containing magnesium, which we show can also bind ELC1, albeit with a significantly lower affinity than calcium (Table 1).
Our results strongly support the hypothesis that ELC1 is cooperatively recruited to the myosin neck by MLC1. However, the mechanistic basis for cooperativity remains enigmatic because this phenomenon could result from the preordering of the MyoA neck, induced by MLC1 or additional interactions between MLC1 and ELC1 as they assemble on the MyoA neck. The HDX data support the former scenario (Fig. 2a), whereas structural studies of canonical myosins support the latter and suggest that binding of Ca2+ may alter the conformation of ELC1 so as to generate more extensive contacts with MLC1 (22, 26). Notably, we did not observe any direct interactions between MLC1 and ELC1 in the absence of the MyoA neck. Ultimately, the structure of the full MyoA–MLC1–ELC1 ternary complex will likely be required to define the complement of supporting interactions.
Finally, we performed solution-binding studies with the full-length MyoA, which yielded the first evidence that the MyoA converter subdomain may constitute an additional binding interface for ELC1. The additional ELC1 binding surface is likely to provide greater rigidity, which is necessary when force transduction is mediated by the coordinated light chains rather than simply through the neck or tail regions as observed in canonical myosins.
In this study, we have established a detailed model of the assembly of the MyoA, MLC1, and ELC1 complex, which relies on calcium coordination and a prescribed order of binding to support cooperativity. Structural and binding data of the MLC1–MyoA complex may enable the expansion and refinement of current efforts to develop peptidomimetic and small-molecule inhibitors that interfere with assembly of the apicomplexan MyoA complex (34, 41).
Experimental procedures
Cloning, protein production, and purification
For ELC1–MLC1–MyoA (801–831), clones encoding ELC1, the C-terminal domain of MLC1 (Ala66–Glu210), and the C-terminal region of the MyoA neck (Lys801–Phe831) were codon optimized for expression in Escherichia coli and synthesized by GenScript. Light chain genes were subcloned into an engineered vector encoding a TEV protease cleavable N-terminal hexahistidine tag, whereas the MyoA (801–831) vector encoded an additional N-terminal GB1 fusion tag. ELC1, MLC1, and MyoA (801–831) were expressed in BL21 cells overnight at 30 °C and purified with nickel-affinity chromatography. For crystallization, MLC1 was combined with a 2-fold molar excess of the MyoA peptide to ensure saturation of MLC1 and cleaved overnight with TEV protease. The MLC1–MyoA (801–831) complex was further purified using size exclusion chromatography (SEC) in 20 mm Hepes, pH 8.0, 150 mm NaCl, and 1 mm DTT. The final, purified sample was concentrated to 8 mg/ml for crystallization. For ITC, nickel-purified ELC1 and MLC1 were cleaved overnight with TEV protease and purified with SEC in 20 mm Hepes, pH 7.5, 150 mm NaCl, and 1 mm TCEP.
Full-length MyoA (1–831) was expressed with a C-terminal hexahistidine tag in Hi5 cells along with MLC1 and the co-chaperone UNC, according to protocols established by Bookwalter et al. (16). After 72 h, the cells were lysed by sonication in 30 mm imidazole, 20 mm Hepes, pH 8.0, and 1.0 m NaCl; purified with nickel affinity chromatography; and cleaved overnight with TEV protease. The MyoA–MLC1 complex was further purified with SEC in 20 mm Hepes, pH 7.5, 150 mm NaCl, and 1 mm TCEP.
In vitro motility assays
FLAG-tagged MyoA was co-expressed in Sf9 cells with UNC and purified by FLAG affinity chromatography as previously described (16). It was further purified using His-tagged ELC1, as follows: 500 μg of cobalt-derivatized magnetic beads (Dynabeads His tag pulldown and isolation; Life Technologies) were placed in a 1.5-ml microcentrifuge tube on a DynaMag-2 magnet (Life Technologies) and washed four times with 250 μl of B/W buffer (50 mm sodium phosphate, pH 8.0, 300 mm NaCl, 0.01% Tween 20). The beads were resuspended in 500 μl of B/W buffer containing 7.5 μg of bacterially expressed His-tagged ELC1 (16) and gently rotated for 15 min at 4 °C. The ELC1-coupled beads were washed four times with 1.0 μl of B/W buffer and then resuspended with 15 μg of recombinant MyoA in 500 μl 3.25 mm sodium phosphate, pH 7.4, 70 mm NaCl, 0.01% Tween 20, and rotated for 30 min at 4 °C. The beads were washed four times on the magnet with 1000 μl of B/W buffer, and the MyoA–ELC1 binary complex was eluted by gentle rotation for 15 min at 4 °C in 115 μl of 300 mm imidazole, 50 mm sodium phosphate, pH 8.0, 300 mm NaCl, 0.01% Tween 20. Tubes were again placed on the magnet; the eluate was recovered and analyzed by SDS-PAGE.
In vitro motility assays were performed using the eluted MyoA–ELC1 as previously described (16) with the following modifications: (a) neutravidin (40 μg/ml, Molecular Probes) in buffer B was added to the flow cells first, followed by three washes with BSA (in buffer B) and three washes with buffer B; (b) 85 ng of ELC1 affinity-purified MyoA was used per flow cell; (c) buffer C was in all cases supplemented with oxygen scavengers (3 mg/ml glucose, 0.125 mg/ml glucose oxidase, and 0.05 mg/ml catalase); (d) all washes with Buffer C were done three times; and (e) bacterially expressed ELC1, MLC1, and truncated MLC1 were each added to the flow cells at a concentration of 25 μg/ml. The data were collected on an Eclipse Ti-U inverted microscope (Nikon) equipped with a 100× Plan Apo objective lens (1.49 NA) and a XR/Turbo-Z camera (Stanford Photonics) running Piper Control software (v2.3.39). The data were collected at 10 frames/s. Actin filament tracking and analysis was done using Dia Track 3.04 (Semasopht).
Isothermal titration calorimetry
Purified ELC1, MLC1, and MyoA (1–831) were dialyzed separately against 20 mm Hepes, pH 7.5, 150 mm NaCl, and 1 mm TCEP (±1 mm CaCl2) at 4 °C overnight. All MyoA neck peptide constructs used for ITC were synthesized by Genscript, N-terminally acetylated, and C-terminally amidated. All ITC experiments were carried out at 25 °C on a MicroCal iTC200 instrument (GE Healthcare). The sample cell contained MyoA (1–831) or MyoA neck peptides, with or without prebound MLC1 (20–40 μm), and ELC1 (200–400 μm) was added in 19 injections of 2 μl each. The data were processed using Origin software (MicroCal), and the dissociation constants (Kd) were determined using a one-site model. Figures are of a single experiment but are representative of at least three independent experiments.
Crystallization and data collection
Crystals of MLC1–MyoA (801–831) complex were grown at 18 °C by mixing the preformed, purified complex in a 1:1 ratio with reservoir solution containing 0.1 m Bis-Tris, pH 5.5, and 25% PEG3350. Crystals were cryoprotected in reservoir solution supplemented with 12.5% glycerol and flash cooled in liquid nitrogen. Diffraction data were collected on Beamline 7-1 at the Stanford Synchrotron Radiation Lightsource at 1.127 Å.
Data processing, structure solution, and refinement
Diffraction data for MLC1–MyoA (801–831) were processed to 1.85 Å resolution using Imosflm (42) and Aimless (43). The structure of MLC1–MyoA (801–831) was determined by molecular replacement in Phaser (44) using PDB code 2QAC as the search model. COOT (45) was used for model building and selection of solvent atoms, and the model was refined in Phenix.refine (46). Structural validation was performed with MolProbity (47), including analysis of the Ramachandran plots, with greater than 98% of residues in the most favored conformations. 5% of reflections were set aside for calculation of Rfree. The data collection and refinement statistics are presented in Table 2. The atomic coordinates and structure factors have been deposited in the PDB with code 5VT9.
Table 2.
MLC1/MyoA (801–831) data collection and refinement statistics
The crystals contained two complexes in the asymmetric unit. Complex 1 (chains A and C for MLC1 and MyoA, respectively) was used for all structural evaluation. The values in parentheses are for the highest resolution shell. 5% of reflections were set aside for calculation of Rfree.
| Data collection statistics | |
| Space group | P21 |
| a, b, c (Å) | 40.99, 64.39, 65.15 |
| α, β, γ (°) | 90, 97.72, 90 |
| Wavelength (Å) | 1.127 |
| Resolution range (Å) | 64.56–1.85 (1.89–1.85) |
| Measured reflections | 172,371 (10,604) |
| Unique reflections | 28,665 (1,749) |
| Redundancy | 6.0 (6.1) |
| Completeness (%) | 99.6 (99.8) |
| I/σ(I) | 15.3 (2.9) |
| Rmerge | 0.076 (0.627) |
| Refinement statistics | |
| Resolution (Å) | 40.63–1.85 |
| Rwork/Rfree | 0.191/0.229 |
| No. of atoms | |
| Protein (A-C/B-D) | 1057–238/1093–220 |
| Solvent | 147 |
| B-values (Å2) | |
| Protein (A-C/B-D) | 26.6–24.0/28.4–25.1 |
| Solvent | 27.2 |
| Root mean square deviation from ideality | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.95 |
| Ramachandran statistics (%) | |
| Most favored | 98.5 |
| Allowed | 1.5 |
| Disallowed | 0.0 |
HDX-MS
HDX reactions were conducted in 50-μl reactions with a final concentration of 2.0 μm of each protein per sample. The reactions were initiated by the addition of 45 μl of D2O buffer (10 mm Hepes, pH 7.5, 50 mm NaCl, 0.5 mm TCEP, 97% D2O) to 5 μl of protein solution to give a final concentration of 87% D2O. Exchange was carried out for 3 s at 0 and 23 °C for MyoA neck experiments, at 300 s at 23 °C for ELC1 calcium experiments, and at all three time points for MyoA (1–831) experiments. Exchange was terminated by the addition guanidine HCl (final concentration, 0.6 m) and 0.8% formic acid. The experiments were carried out in triplicate. The samples were immediately frozen in liquid nitrogen and stored at −80 °C until mass analysis. Protein samples were rapidly thawed and injected onto an ultra-performance liquid chromatography system at 2 °C. The protein was run over two immobilized pepsin columns (Applied Biosystems; Porosyme, 2-3131-00) at 10 and 2 °C at 200 μl/min for 3 min, and the peptides were collected onto a VanGuard Precolumn trap (Waters). The trap was subsequently eluted in line with an ACQUITY 1.7 μm particle, 100 × 1 mm2 C18 ultra-performance liquid chromatography column (Waters), using a gradient of 5–36% B (buffer A 0.1% formic acid, buffer B 100% acetonitrile) over 16 min. Mass spectrometry experiments were performed on an Impact II QTOF (Bruker) acquiring over a mass range from 150 to 2,200 m/z using an electrospray ionization source operated at a temperature of 200 °C and a spray voltage of 4.5 kV. Peptides were identified using data-dependent acquisition methods following tandem MS/MS experiments (0.5-s precursor scan from 150 to 2200 m/z; twelve 0.25-s fragment scans from 150 to 2200 m/z). MS/MS data sets were analyzed using PEAKS7 (PEAKS), and a false discovery rate was set at 1% using a database of purified proteins and known contaminants.
HDExaminer Software (Sierra Analytics) was used to calculate deuterium incorporation into each peptide. All peptides were manually inspected for the correct charge state and the presence of overlapping peptides. Deuteration levels were calculated using the centroid of the experimental isotope clusters. The average error of all time points and conditions for each HDX project was less than 0.2 Da. Refer to supplemental Tables S1–S3 for full HDX-MS data sets.
Availability of supporting data
The data set supporting the conclusions of this article is available in the Protein Data Bank with identifier 5VT9.
Author contributions
C. J. P., M. L. P., G. E. W., and M. J. B. designed the study. C. J. P., M. L. J., A. K., and M. L. P. performed the experiments. C. J. P., M. L. J., M. L. P., R. R., A. K., D. M. W., G. E. W., J. E. B., and M. J. B. analyzed the data. C. J. P. and M. J. B. wrote the paper with editorial support from R. R., D. M. W., and G. E. W.
Supplementary Material
Acknowledgments
We thank Samantha Previs and Guy Kennedy for technical support and the staff at the Stanford Synchrotron Radiation Lightsource.
This work was supported by Canadian Institutes of Health Research Grants MOP82916 and 148596 (to M. J. B.), U. S. Public Health Service Grants AI054961 (to G. E. W.) and GM094229 (to D. M. W.), and Natural Sciences and Engineering Research Council of Canada Grant NSERC-2014-05218 (to J. E. B.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1–S3.
The atomic coordinates and structure factors (code 5VT9) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ELC1
- essential light chain 1
- HDX-MS
- hydrogen-deuterium exchange mass spectrometry
- ITC
- isothermal titration calorimetry
- MLC1
- myosin light chain 1
- MTIP
- myosin tail interacting protein
- Pf
- P. falciparum
- SEC
- size exclusion chromatography
- PDB
- Protein Data Bank.
References
- 1. World Health Organization (2015) Guidelines for the treatment of malaria, 3rd Ed., World Health Organization Press, Geneva, Switzerland [Google Scholar]
- 2. Shirley D. A., Moonah S. N., and Kotloff K. L. (2012) Burden of disease from cryptosporidiosis. Curr. Opin. Infect. Dis. 25, 555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Checkley W., White A. C. Jr., Jaganath D., Arrowood M. J., Chalmers R. M., Chen X. M., Fayer R., Griffiths J. K., Guerrant R. L., Hedstrom L., Huston C. D., Kotloff K. L., Kang G., Mead J. R., Miller M., et al. (2015) A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for Cryptosporidium. Lancet Infect. Dis. 15, 85–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Torgerson P. R., and Mastroiacovo P. (2013) The global burden of congenital toxoplasmosis: a systematic review. Bull. World Health Organ. 91, 501–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luft B. J., and Remington J. S. (1992) Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis. 15, 211–222 [DOI] [PubMed] [Google Scholar]
- 6. Tenter A. M., Heckeroth A. R., and Weiss L. M. (2000) Toxoplasma gondii: from animals to humans. Int. J. Parasitol. 30, 1217–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heintzelman M. B. (2015) Gliding motility in apicomplexan parasites. Semin. Cell Dev. Biol. 46, 135–142 [DOI] [PubMed] [Google Scholar]
- 8. Foth B. J., Goedecke M. C., and Soldati D. (2006) New insights into myosin evolution and classification. Proc. Natl. Acad. Sci. U.S.A. 103, 3681–3686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frenal K., Foth B. J., and Soldati-Favre D. (2008) Myosin class XIV and other myosins in protists. In Myosins: A superfamily of Molecular Motors (Coluccio L. M., ed) pp. 421–440, Springer, Dordrecht, The Netherlands [Google Scholar]
- 10. Heintzelman M. B., and Schwartzman J. D. (2001) Myosin diversity in apicomplexa. J. Parasitol. 87, 429–432 [DOI] [PubMed] [Google Scholar]
- 11. Herm-Götz A., Weiss S., Stratmann R., Fujita-Becker S., Ruff C., Meyhöfer E., Soldati T., Manstein D. J., Geeves M. A., and Soldati D. (2002) Toxoplasma gondii myosin A and its light chain: a fast, single-headed, plus-end-directed motor. EMBO J. 21, 2149–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meissner M., Reiss M., Viebig N., Carruthers V. B., Toursel C., Tomavo S., Ajioka J. W., and Soldati D. (2002) A family of transmembrane microneme proteins of Toxoplasma gondii contain EGF-like domains and function as escorters. J. Cell Sci. 115, 563–574 [DOI] [PubMed] [Google Scholar]
- 13. Baum J., Richard D., Healer J., Rug M., Krnajski Z., Gilberger T. W., Green J. L., Holder A. A., and Cowman A. F. (2006) A conserved molecular motor drives cell invasion and gliding motility across malaria life cycle stages and other apicomplexan parasites. J. Biol. Chem. 281, 5197–5208 [DOI] [PubMed] [Google Scholar]
- 14. Tardieux I., and Baum J. (2016) Reassessing the mechanics of parasite motility and host-cell invasion. J. Cell Biol. 214, 507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frénal K., Polonais V., Marq J. B., Stratmann R., Limenitakis J., and Soldati-Favre D. (2010) Functional dissection of the apicomplexan glideosome molecular architecture. Cell Host Microbe 8, 343–357 [DOI] [PubMed] [Google Scholar]
- 16. Bookwalter C. S., Kelsen A., Leung J. M., Ward G. E., and Trybus K. M. (2014) A Toxoplasma gondii class XIV myosin, expressed in Sf9 cells with a parasite co-chaperone, requires two light chains for fast motility. J. Biol. Chem. 289, 30832–30841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Williams M. J., Alonso H., Enciso M., Egarter S., Sheiner L., Meissner M., Striepen B., Smith B. J., and Tonkin C. J. (2015) Two essential light chains regulate the MyoA lever arm to promote Toxoplasma gliding motility. MBio 6, e00845–e00815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Douse C. H., Green J. L., Salgado P. S., Simpson P. J., Thomas J. C., Langsley G., Holder A. A., Tate E. W., and Cota E. (2012) Regulation of the Plasmodium motor complex: phosphorylation of myosin A tail-interacting protein (MTIP) loosens its grip on MyoA. J. Biol. Chem. 287, 36968–36977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rhoads A. R., and Friedberg F. (1997) Sequence motifs for calmodulin recognition. FASEB J. 11, 331–340 [DOI] [PubMed] [Google Scholar]
- 20. Denessiouk K., Permyakov S., Denesyuk A., Permyakov E., and Johnson M. S. (2014) Two structural motifs within canonical EF-hand calcium-binding domains identify five different classes of calcium buffers and sensors. PLoS One 9, e109287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Debreczeni J. E., Farkas L., Harmat V., Hetényi C., Hajdú I., Závodszky P., Kohama K., and Nyitray L. (2005) Structural evidence for non-canonical binding of Ca2 to a canonical EF-hand of a conventional myosin. J. Biol. Chem. 280, 41458–41464 [DOI] [PubMed] [Google Scholar]
- 22. Houdusse A., Silver M., and Cohen C. (1996) A model of Ca2+-free calmodulin binding to unconventional myosins reveals how calmodulin acts as a regulatory switch. Structure 4, 1475–1490 [DOI] [PubMed] [Google Scholar]
- 23. Bosch J., Turley S., Daly T. M., Bogh S. M., Villasmil M. L., Roach C., Zhou N., Morrisey J. M., Vaidya A. B., Bergman L. W., and Hol W. G. (2006) Structure of the MTIP-MyoA complex, a key component of the malaria parasite invasion motor. Proc. Natl. Acad. Sci. U.S.A. 103, 4852–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sigrist C. J., de Castro E., Cerutti L., Cuche B. A., Hulo N., Bridge A., Bougueleret L., and Xenarios I. (2013) New and continuing developments at PROSITE. Nucleic Acids Res. 41, D344–D347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bookwalter C. S., Tay C. L., McCrorie R., Previs M. J., Krementsova E. B., Fagnant P. M., Baum J., and Trybus K. M. (2017) Binding of a newly identified essential light chain to expressed Plasmodium falciparum class XIV myosin enhances actin motility. BioRXiv, 10.1101/127118 [DOI] [Google Scholar]
- 26. Houdusse A., and Cohen C. (1996) Structure of the regulatory domain of scallop myosin at 2 A resolution: implications for regulation. Structure 4, 21–32 [DOI] [PubMed] [Google Scholar]
- 27. Gaji R. Y., Johnson D. E., Treeck M., Wang M., Hudmon A., and Arrizabalaga G. (2015) Phosphorylation of a myosin motor by TgCDPK3 facilitates rapid initiation of motility During Toxoplasma gondii egress. PLoS Pathog. 11, e1005268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dominguez R., Freyzon Y., Trybus K. M., and Cohen C. (1998) Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell 94, 559–571 [DOI] [PubMed] [Google Scholar]
- 29. Keeley A., and Soldati D. (2004) The glideosome: a molecular machine powering motility and host-cell invasion by Apicomplexa. Trends Cell Biol. 14, 528–532 [DOI] [PubMed] [Google Scholar]
- 30. Heintzelman M. B. (2006) Cellular and molecular mechanics of gliding locomotion in eukaryotes. Int. Rev. Cytol. 251, 79–129 [DOI] [PubMed] [Google Scholar]
- 31. Green J. L., Rees-Channer R. R., Howell S. A., Martin S. R., Knuepfer E., Taylor H. M., Grainger M., and Holder A. A. (2008) The motor complex of Plasmodium falciparum: phosphorylation by a calcium-dependent protein kinase. J. Biol. Chem. 283, 30980–30989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soldati-Favre D. (2008) Molecular dissection of host cell invasion by the apicomplexans: the glideosome. Parasite 15, 197–205 [DOI] [PubMed] [Google Scholar]
- 33. Boucher L. E., and Bosch J. (2015) The apicomplexan glideosome and adhesins: structures and function. J. Struct. Biol. 190, 93–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Douse C. H., Vrielink N., Wenlin Z., Cota E., and Tate E. W. (2015) Targeting a dynamic protein-protein interaction: fragment screening against the malaria myosin A motor complex. ChemMedChem 10, 134–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carey K. L., Westwood N. J., Mitchison T. J., and Ward G. E. (2004) A small-molecule approach to studying invasive mechanisms of Toxoplasma gondii. Proc. Natl. Acad. Sci. U.S.A. 101, 7433–7438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leung J. M., Tran F., Pathak R. B., Poupart S., Heaslip A. T., Ballif B. A., Westwood N. J., and Ward G. E. (2014) Identification of T. gondii myosin light chain-1 as a direct target of TachypleginA-2, a small-molecule inhibitor of parasite motility and invasion. PLoS One 9, e98056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lovett J. L., and Sibley L. D. (2003) Intracellular calcium stores in Toxoplasma gondii govern invasion of host cells. J. Cell Sci. 116, 3009–3016 [DOI] [PubMed] [Google Scholar]
- 38. Wetzel D. M., Chen L. A., Ruiz F. A., Moreno S. N., and Sibley L. D. (2004) Calcium-mediated protein secretion potentiates motility in Toxoplasma gondii. J. Cell Sci. 117, 5739–5748 [DOI] [PubMed] [Google Scholar]
- 39. Nagamune K., Beatty W. L., and Sibley L. D. (2007) Artemisinin induces calcium-dependent protein secretion in the protozoan parasite Toxoplasma gondii. Eukaryotic Cell 6, 2147–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Borges-Pereira L., Budu A., McKnight C. A., Moore C. A., Vella S. A., HortuaTriana M. A., Liu J., Garcia C. R., Pace D. A., and Moreno S. N. (2015) Calcium signaling throughout the Toxoplasma gondii lytic cycle: a study using gentically encoded calcium indicators. J. Biol. Chem. 290, 26914–26926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kortagere S., Mui E., McLeod R., and Welsh W. J. (2011) Rapid discovery of inhibitors of Toxoplasma gondii using hybrid structure-based computational approach. J. Comput. Aided Mol. Des. 25, 403–411 [DOI] [PubMed] [Google Scholar]
- 42. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.