

Abstract
Chromatin is the natural form of DNA in the eukaryotic nucleus and is the substrate for diverse biological phenomena. The functional analysis of these processes ideally would be carried out with nucleosomal templates that are assembled with customized core histones, DNA sequences, and chromosomal proteins. Here we report a simple, reliable, and versatile method for the ATP-dependent assembly of evenly spaced nucleosome arrays. This minimal chromatin assembly system comprises the Drosophila nucleoplasmin-like protein (dNLP) histone chaperone, the imitation switch (ISWI) ATP-driven motor protein, core histones, template DNA, and ATP. The dNLP and ISWI components were synthesized in bacteria, and each protein could be purified in a single step by affinity chromatography. We show that the dNLP-ISWI system can be used with different DNA sequences, linear or circular DNA, bulk genomic DNA, recombinant or native Drosophila core histones, native human histones, the linker histone H1, the non-histone chromosomal protein HMGN2, and the core histone variants H3.3 and H2A.V. The dNLP-ISWI system should be accessible to a wide range of researchers and enable the assembly of customized chromatin with specifically desired DNA sequences, core histones, and other chromosomal proteins.
Keywords: chromatin, chromatin structure, histone, histone chaperone, nucleosome
Introduction
In the eukaryotic nucleus, DNA is packaged into a nucleoprotein complex known as chromatin (1). The chromatin fiber consists of repeating units of nucleosomes, which comprise nucleosome cores (∼147 bp of DNA wrapped around a core histone octamer of two copies each of histones H2A, H2B, H3, and H4) that are connected by about 20–50 bp of linker DNA. Chromatin is involved not only in the compaction and organization of the genome, but it is also the substrate for DNA-utilizing processes such as transcription, DNA replication, and DNA repair.
Chromatin is fascinating yet challenging because of its multidimensional nature. In addition to the core histones, chromatin contains the linker histone H1 as well as non-histone chromosomal proteins such as the high-mobility-group (HMG)2 proteins. There are also different types of core histones: the canonical S phase–regulated histones as well as S phase–independent histone variants with specialized functions. Furthermore, there are many different covalent modifications of the histones.
A functional analysis of the multifarious components of chromatin requires the assembly of customized histones and non-histone chromosomal proteins into nucleosomes (for reviews, see Refs. 2 and 3). For the assembly of periodic arrays of nucleosomes onto natural DNA sequences, there are purified and defined enzyme-driven systems that comprise an ATP-dependent motor protein, such as ACF (ATP-utilizing chromatin assembly and remodeling factor), Chd1 (chromodomain helicase DNA-binding protein 1), or RSF (remodeling and spacing factor), and a core histone chaperone such as NAP1 (nucleosome assembly protein 1) (4, 5). These enzyme-driven systems have been successfully used for the assembly of chromatin, but they also have some limitations that restrict their more widespread use. For instance, the ACF and RSF motor proteins are synthesized in insect cells by using baculovirus vectors, and it would be easier and less expensive to use bacterially synthesized proteins. On the other hand, Chd1, which can be made in bacteria, cannot assemble histone H1–containing chromatin (6). NAP1 is widely used as a histone chaperone, but bacterially synthesized NAP1 is often contaminated with a nuclease that can degrade the DNA template (7).
To circumvent these problems, we have developed and characterized a simple and reliable method for the ATP-dependent assembly of periodic arrays of nucleosomes with a bacterially synthesized ISWI (imitation switch) motor protein and dNLP (Drosophila nucleoplasmin-like protein) histone chaperone. Importantly, we tested the versatility of this dNLP-ISWI assembly system and found that it can function with circular or linear DNA templates, human or Drosophila core histones, histone H1, an HMGN (high-mobility-group, nucleosome-binding) protein, and core histone variants. Thus, this system should be useful for the preparation of many different types of customized chromatin for a wide range of specific applications.
Results
ATP-dependent assembly of periodic nucleosome arrays with dNLP and ISWI
In this work, we sought to establish and characterize a simple, reliable, and versatile system for the ATP-dependent assembly of periodic nucleosome arrays. The minimal chromatin assembly process is mediated by a combination of an ATP-utilizing motor protein, such as ACF, Chd1, or RSF, and a core histone chaperone, such as NAP1 or dNLP. In our simplified system, we chose to use the Drosophila ISWI motor protein (8), which is the ATPase subunit of the ACF assembly factor (9). ACF comprises ISWI and the Acf1 protein (10), and ISWI alone can assemble chromatin in the absence or presence of histone H1 in conjunction with the NAP1 histone chaperone (6, 10, 11). Moreover, ISWI can be synthesized in bacteria and purified by one-step affinity chromatography (11), and thus, the purified protein can be obtained quickly and inexpensively. Hence, ISWI is ideally suited for a simplified assembly system because it can easily be produced in bacteria and can mediate the assembly of chromatin in the presence or absence of histone H1.
For the core histone chaperone, we chose to use the Drosophila nucleoplasmin-like protein, dNLP (12) (also known as p22 or CRP1 (13, 14)), which has been found to function in chromatin assembly in conjunction with purified ACF (15). In the past, we extensively used dNAP1 that had been synthesized in Sf9 insect cells with a baculovirus vector (4). In contrast, we found that bacterially synthesized dNAP1 could be contaminated with a bacterial nuclease (7) and could also lose its activity upon long-term storage (e.g. several months) at −80 °C. The use of dNLP circumvents these problems. Like Xenopus nucleoplasmin, dNLP is a heat-stable protein (14, 16), and hence, it can be heat-treated under conditions that inactivate contaminating nucleases. In addition, we have found that purified bacterially synthesized dNLP retains its activity upon long-term storage at −80 °C.
We therefore tested whether purified bacterially synthesized ISWI could assemble chromatin in conjunction with purified bacterially synthesized dNLP. To this end, we purified ISWI by a variation of the method of Corona et al. (11), and we generated and purified an N-terminally His6-tagged version of dNLP (termed dNLP). Each of the proteins was purified by a single step of affinity chromatography (Fig. 1A). We then carried out chromatin assembly reactions with ISWI and dNLP along with purified recombinant canonical (S phase–regulated) Drosophila core histones, relaxed circular plasmid DNA, and ATP. It should also be noted that the reactions additionally contained an ATP regeneration system (pyruvate kinase and phosphoenolpyruvate), purified topoisomerase I to relax DNA superhelical tension, and bovine serum albumin (BSA) as a stabilizing agent.
Figure 1.

ATP-dependent assembly of periodic nucleosome arrays with the dNLP-ISWI system. A, purification of recombinant Drosophila dNLP, ISWI, and S phase–regulated core histones. The proteins were synthesized in E. coli, purified, and analyzed by 10% (left) or 15% (right) polyacrylamide-SDS gel electrophoresis and staining with Coomassie Brilliant Blue R-250. The dNLP is the N-terminally His6-tagged version of the protein. The sizes of molecular mass markers (in kDa) and the positions of the individual histones are indicated. B, DNA supercoiling analysis reveals efficient nucleosome assembly by dNLP and ISWI. Complete chromatin assembly reactions were carried out with dNLP, ISWI, core histones, ATP, and relaxed plasmid DNA in the presence of purified topoisomerase I, an ATP regeneration system, and BSA as a stabilizing agent. Reactions lacking individual components are indicated. The reaction products were deproteinized, subjected to 0.8% agarose gel electrophoresis, and stained with ethidium bromide. Samples of supercoiled DNA and relaxed DNA were included as references. The positions of nicked DNA, relaxed DNA, and supercoiled DNA are shown. The black dot corresponds to a minor unknown contaminant, which may be supercoiled dimeric plasmid DNA. C, partial MNase digestion analysis indicates the assembly of periodic nucleosome arrays by dNLP and ISWI. Chromatin assembly reactions were performed as described in B. The reaction products were partially digested with two different concentrations of MNase, deproteinized, and subjected to 1.3% agarose gel electrophoresis. The resulting DNA fragments were detected by staining with ethidium bromide. The DNA bands that correspond to mono-, di-, tri-, and tetranucleosomes are shown. The DNA size markers (M) are the 123-bp ladder (Invitrogen).
Chromatin assembly was monitored by the DNA supercoiling and micrococcal nuclease (MNase) digestion assays. During nucleosome assembly, the wrapping of the DNA around the core histone octamer results in a change in the linking number of about −1 (17, 18). The DNA supercoiling assay monitors the formation of negative supercoiling in the plasmid DNA as nucleosomes are assembled in the presence of topoisomerase I. The extent of nucleosome assembly is revealed by the formation of negative supercoils in the DNA from the deproteinized reaction products. As seen in Fig. 1B, we observed the most efficient nucleosome assembly in the complete reaction containing dNLP, ISWI, core histones, and ATP. More detailed analyses of different concentrations of dNLP and ISWI in the chromatin assembly reactions are shown in supplemental Figs. S1 and S2.
The partial MNase digestion assay reveals whether there are extended periodic arrays of nucleosomes in the reconstituted chromatin (19). MNase catalyzes the double-stranded cleavage of DNA in the linker region between the nucleosome cores. If the nucleosomes are evenly spaced, then the different DNA fragments derived from the mono- and oligonucleosomes generated by partial MNase cleavage will form a ladder that corresponds to mononucleosomes, dinucleosomes, trinucleosomes, and increasingly larger oligonucleosomes. As seen in Fig. 1C and supplemental Figs. S1 and S2, we observed the most distinct pattern of MNase digestion products (“MNase ladder”) with the complete reaction containing dNLP, ISWI, core histones, and ATP. These experiments revealed that purified bacterially synthesized dNLP, ISWI, and core histones can be assembled efficiently into periodic nucleosome arrays.
The dNLP-ISWI system can assemble chromatin with either human or Drosophila core histones
We next sought to test the versatility of the dNLP-ISWI chromatin assembly system. We initially addressed the following two issues. First, because the dNLP and ISWI are Drosophila proteins, it was important to test whether the factors would work with human histones, which would be more widely studied than Drosophila histones. Second, bacterially synthesized recombinant histones are homogeneous polypeptides, whereas native histones consist of S phase–regulated histones and their variants with a variety of different covalent modifications. We therefore tested the ability of the dNLP-ISWI system to assemble chromatin with native core histones. To this end, we purified native core histones from Drosophila embryos as well as from human HeLa cells (Fig. 2A) and then carried out chromatin assembly reactions with dNLP and ISWI. As shown in Fig. 2, B and C, both native and recombinant Drosophila core histones as well as native human core histones are efficiently assembled into extended periodic nucleosome arrays with dNLP and ISWI. The core histones are highly conserved among eukaryotes. It is thus likely that the dNLP-ISWI system will function with core histones from many different organisms. In addition, it is useful to know that the dNLP-ISWI system can be used with native core histones as well as with recombinant core histones, which can be modified specifically as desired.
Figure 2.
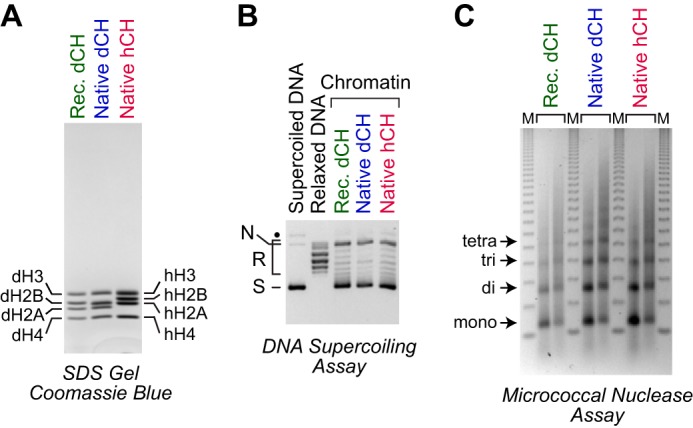
Assembly of native Drosophila and human histones with dNLP and ISWI. A, purification of core histones from Drosophila embryos and human HeLa cells. The proteins were analyzed by 15% polyacrylamide-SDS gel electrophoresis and staining with Coomassie Brilliant Blue R-250. The positions of the individual Drosophila (d) and human (h) core histones (CH) are indicated. B, DNA supercoiling analysis. Chromatin assembly reactions were carried out and analyzed, as described in the legend for Fig. 1B, with the indicated core histones. The positions of nicked DNA (N), relaxed DNA (R), and supercoiled DNA (S) are shown. C, partial MNase digestion analysis. Chromatin was assembled and analyzed, as described in the legend for Fig. 1C, with the indicated core histones.
The dNLP-ISWI system can assemble chromatin onto different DNA templates
We then examined the ability of the dNLP-ISWI system to assemble chromatin onto different DNA templates. First, as shown in Fig. 3, A and B, we found that the dNLP-ISWI system is able to assemble chromatin onto relaxed circular DNAs containing sequences from the adenovirus E4 promoter (3 kb), bacteriophage T4 (7 kb), and the Drosophila Antennapedia gene (8 kb). Thus, the assembly factors function with both prokaryotic and eukaryotic DNA sequences. Second, we observed that dNLP and ISWI can assemble chromatin efficiently onto linearized plasmid DNA (Fig. 3C). Third, we also tested the ability of the dNLP-ISWI system to assemble chromatin with high-molecular-weight (mostly >12 kb; see supplemental Fig. S3) bulk genomic DNA. These experiments revealed efficient assembly of extended periodic nucleosome arrays with the high-molecular-weight genomic DNA from calf thymus (Fig. 3D). It thus appears that the dNLP-ISWI system can be used for the assembly of chromatin with a wide range of linear and circular DNAs.
Figure 3.

Assembly of various DNA templates with the dNLP-ISWI system. A, DNA supercoiling analysis shows the efficient assembly of three different plasmid DNAs. Chromatin assembly reactions were carried out with three plasmid DNAs: pGIE-0 (3 kb) (50), which contains sequences from the adenovirus E4 promoter; pJH187 (8 kb), which contains sequences from the Drosophila Antennapedia gene; and pLA4 (7 kb) (51), which contains sequences from bacteriophage T4. Chromatin assembly and analysis were performed as described in the legend for Fig. 1B, except that native Drosophila core histones were used instead of recombinant Drosophila core histones. B, partial MNase digestion analysis reveals the assembly of periodic nucleosome arrays on three different plasmid DNAs. Reactions were performed as described in A, and the samples were analyzed as described in the legend for Fig. 1C. C, chromatin can be assembled with either circular or linear DNA. Reactions were performed as described in A, with relaxed circular or linearized pLA4 (7 kb), and the resulting samples were subjected to partial MNase digestion analysis. D, the dNLP-ISWI system can be used for the assembly of genomic DNA into periodic nucleosome arrays. Chromatin assembly reactions were carried out as described in A with native calf thymus genomic DNA that is mostly greater than 10 kb in length (supplemental Fig. S3). The chromatin (right) and genomic DNA (left) were subjected to partial MNase digestion analysis in parallel.
Assembly of histone H1–containing chromatin with the dNLP-ISWI system
In metazoa, histone H1 is a major component of chromatin with a stoichiometry that generally ranges from 0.5 to 0.8 molecules per nucleosome (20) (for reviews, see Refs. 21–23). It is thus important to include histone H1 in the analysis of chromatin structure and function. We therefore tested whether the dNLP-ISWI system could be used to assemble histone H1–containing chromatin. To this end, we purified native histone H1 from Drosophila embryos as well as recombinant human histone H1 variant H1.0 (Fig. 4A). We then carried out chromatin assembly reactions in the absence or presence of histone H1. Because dNAP1 and ACF can assemble histone H1–containing chromatin (4), we used these factors as a reference. As seen in Fig. 4, B and C, chromatin is assembled in the presence of histone H1, and the incorporation of histone H1 into chromatin results in an increase in the nucleosome repeat length, as seen previously in the assembly of histone H1–containing chromatin with crude or purified systems (4, 6, 24, 25). We further analyzed the histone H1–containing chromatin by extensive MNase digestion into mononucleosomes followed by native gel electrophoresis (Fig. 4D). In these experiments, we found that the H1-containing chromatin yields chromatosomes (mononucleosomes + H1), as seen previously with chromatin isolated from cells or assembled in vitro with ACF and dNAP1 (6, 26, 27). Some differences can be seen between chromatin that is assembled with Drosophila H1 relative to human H1.0, effects, which are likely due to the different properties of the different H1 molecules. Importantly, histone H1–containing chromatin that is assembled with dNAP1-ACF is similar to that assembled with dNLP-ISWI. These results indicate that histone H1 is incorporated into chromatin during assembly with the dNLP-ISWI system. This property of the dNLP-ISWI system contrasts with that of the dNAP1-Chd1 system, which is unable to mediate the assembly of H1-containing chromatin (6).
Figure 4.
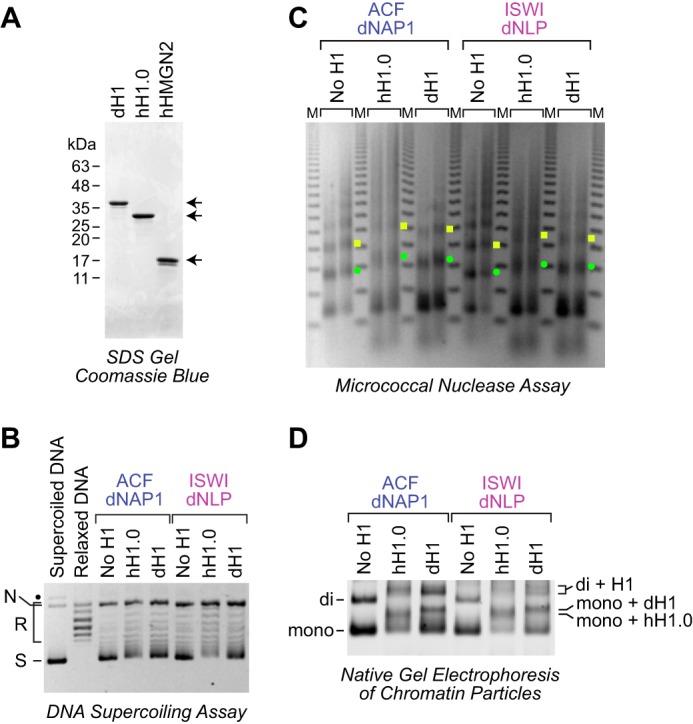
The dNLP-ISWI system can be used to assemble histone H1–containing chromatin. A, purification of native Drosophila histone H1, recombinant human H1.0, and recombinant human HMGN2. The proteins were subjected to 15% polyacrylamide-SDS gel electrophoresis and staining with Coomassie Brilliant Blue R-250. B, DNA supercoiling analysis of chromatin assembled with histone H1. Reactions were performed either with the dNAP1-ACF system (4) or with the dNLP-ISWI system, as described in the legend for Fig. 3. Where indicated, Drosophila histone H1 and human H1.0 were included at a ratio of 0.75 molecules of H1/nucleosome. The positions of nicked DNA (N), relaxed DNA (R), and supercoiled DNA (S) are shown. The black dot corresponds to a minor unknown contaminant. C, partial MNase analysis shows an increase in the nucleosome repeat length upon incorporation of histone H1 into chromatin. Reactions were performed as described in B, and the resulting samples were subjected to partial MNase digestion analysis. The green dots correspond to the DNA bands derived from dinucleosomes, and the yellow squares designate DNA fragments from trinucleosomes. D, native gel electrophoresis of mono- and dinucleosomes obtained by extensive MNase digestion of chromatin either lacking or containing histone H1. The chromatin particles were detected by staining of the DNA with SYBR Gold. The positions of mononucleosomes (mono), dinucleosomes (di), H1-containing mononucleosomes (chromatosomes; mono + H1), and H1-containing dinucleosomes (di + H1) are indicated.
Assembly of HMGN2-containing chromatin with the dNLP-ISWI system
Beyond the core histones and H1, there are other abundant components of chromatin, such as the high-mobility-group (HMG) proteins (for reviews, see Refs. 28–30). We therefore tested whether the dNLP-ISWI system could assemble chromatin that contains the HMGN proteins, which are abundant nuclear proteins (31) that bind with high affinity to two sites on nucleosomes (32–34). We purified recombinant human HMGN2 (Fig. 4A), and then carried out chromatin assembly reactions in the absence or presence of different concentrations of this protein. The DNA supercoiling and partial MNase digestion assays indicated that periodic arrays of nucleosomes are assembled in the presence of HMGN2 (Fig. 5, A and B). The incorporation of HMGN2 into chromatin was examined by digestion of the chromatin into mononucleosomes with MNase followed by native gel electrophoresis of the resulting chromatin particles (Fig. 5C). These experiments revealed the incorporation of up to two molecules of HMGN2 per nucleosome. Hence, we concluded that the dNLP-ISWI system can be used for the assembly of periodic nucleosome arrays containing the HMGN proteins.
Figure 5.

The dNLP-ISWI system can be used to assemble HMGN2-containing chromatin. A, DNA supercoiling analysis of chromatin assembled with HMGN2. Chromatin assembly reactions were performed as described in the legend for Fig. 3, in the absence or presence of purified human HMNG2 (shown in Fig. 4A) at the indicated molar ratios of HMGN2 to nucleosomes. B, partial MNase analysis of HMGN2-containing chromatin assembled as described in A. C, native gel electrophoresis of mono- and dinucleosomes obtained by extensive MNase digestion of chromatin either lacking or containing HMGN2. The chromatin particles were detected by staining of the DNA with SYBR Gold. The positions of mononucleosomes (mono), dinucleosomes (di), mononucleosomes containing one molecule of HMGN2 (mono + 1 HMGN2), and mononucleosomes containing two molecules of HMGN2 (mono + 2 HMGN2) are indicated.
The dNLP-ISWI system can be used to assemble chromatin that contains histone variants H3.3 and H2A.V (Drosophila H2A.X/H2A.Z)
In the study of chromatin structure and function, it is important to analyze the S phase–independent histone variants in addition to the canonical S phase–regulated histones (for reviews, see Refs. 35 and 36). For instance, nucleosomes containing the histone variants H3.3 and H2A.Z have been found at active promoters, enhancers, and insulator regions in humans (37). We therefore investigated whether the dNLP-ISWI system could be used to assemble chromatin that contains histone variants. To this end, we synthesized and purified Drosophila histone H3.3 and H2A.V (Fig. 6A). Drosophila H3.3 has the same amino acid sequence as human H3.3, whereas histone H2A.V is a Drosophila H2A variant (38) with the combined features of mammalian H2A.X and H2A.Z. By using the dNLP-ISWI system, we performed chromatin assembly reactions with H2A.V and H3.3 along with canonical H2B and H4. DNA supercoiling and partial MNase digestion analyses revealed the formation of evenly spaced arrays of nucleosomes containing H2A.V and H3.3 (Fig. 6, B and C). Thus, the dNLP-ISWI system has considerable promise for the assembly of a wide range of histone substrates into periodic nucleosome arrays.
Figure 6.
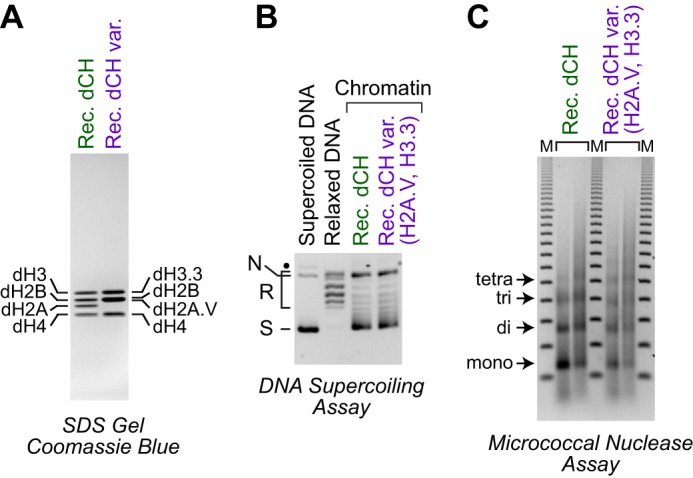
The dNLP-ISWI system can assemble containing histone variants H3. 3 and H2A.V. A, purification of Drosophila core histones with the H3.3 and H2A.V variants. The core histone proteins were synthesized in E. coli, purified, and analyzed by 15% polyacrylamide-SDS gel electrophoresis and staining with Coomassie Brilliant Blue R-250. The positions of the individual histones are shown. B, DNA supercoiling analysis of chromatin assembled with histones H3.3 and H2A.V. Chromatin assembly reactions were performed as described in the legend for Fig. 1B except that the H3.3 and H2A.V variants were used instead of the S phase–regulated H3 and H2A histones. The positions of nicked DNA (N), relaxed DNA (R), and supercoiled DNA (S) are shown. The black dot corresponds to a minor unknown contaminant. C, partial MNase digestion analysis of chromatin assembled with histones H3.3 and H2A.V. The samples were analyzed as described in the legend for Fig. 1C.
A minimal dNLP-ISWI chromatin assembly system that lacks an ATP regeneration system, topoisomerase I, and bovine serum albumin
Many applications may require the simplest possible system for the assembly of chromatin. We therefore examined the effects of the removal of the potentially nonessential protein components in our standard chromatin assembly reactions. First, we tested the omission of the ATP regeneration system (pyruvate kinase and phosphoenolpyruvate), which converts the ADP that is produced during chromatin assembly back into ATP. As shown in Fig. 7, A and B, the absence of the ATP regeneration system has little effect on the efficiency of assembly of periodic nucleosome arrays under our standard reaction conditions. It should be noted, however, that the ATP regeneration system might be essential for efficient chromatin assembly if a higher concentration of the ISWI ATPase or a longer reaction time were employed.
Figure 7.

A simplified minimal chromatin assembly system. A, an ATP-regeneration system is not essential for chromatin assembly by dNLP and ISWI. Chromatin assembly reactions were performed in the presence or absence of the ATP regeneration system, which consists of pyruvate kinase and phosphoenolpyruvate. The reactions were carried out as described in the legend for Fig. 3. DNA supercoiling analysis revealed that the efficiency of nucleosome assembly is not affected by the inclusion of the ATP regeneration system. The positions of nicked DNA (N), relaxed DNA (R), and supercoiled DNA (S) are shown. The black dot corresponds to a minor unknown contaminant. B, partial MNase digestion analysis of the chromatin assembled in A shows that the ATP regeneration system is not essential for the assembly of periodic nucleosome arrays. The samples were analyzed as described in the legend for Fig. 1C. C, topoisomerase I and bovine serum albumin (BSA) are not essential for the assembly of periodic nucleosome arrays with supercoiled plasmid DNA. Reactions were carried out in the absence of the ATP regenerating system, as in A, with supercoiled plasmid DNA in the presence or absence of topoisomerase I and BSA, as indicated. The samples were analyzed by the partial MNase digestion assay. The minimal system that consists only of dNLP, ISWI, core histones, DNA, and ATP is highlighted in red.
Then, in the absence of the ATP regeneration system, we further investigated the omission of topoisomerase I. Under normal reaction conditions, topoisomerase I serves two functions: first, it is used to relax the negative supercoils in plasmid DNA prior to chromatin assembly; and second, it functions to relax the positive supercoils that are formed as the DNA wraps around the histone octamer during chromatin assembly. The second function is the basis for the DNA supercoiling assay. The first function is relevant to the energy that drives the formation of nucleosomes. In this regard, we preferred to use relaxed DNA for chromatin assembly because negatively supercoiled DNA is “spring-loaded” for chromatin assembly, as the formation of nucleosomes relieves the stress of the negative supercoiling. If chromatin is assembled from relaxed DNA in the presence of topoisomerase I, then the energy that drives the formation of nucleosomes is provided only by the ATP-dependent motor protein (in this case, ISWI) without any contribution from superhelical tension in the DNA. Hence, the use of relaxed DNA is useful for the study of the mechanism of chromatin assembly. However, if the objective of the experiment is to make chromatin without regard to the specific mechanism, then negatively supercoiled DNA could be used as a substrate for chromatin assembly. Thus, as shown in Fig. 7C, we carried out chromatin assembly reactions with negatively supercoiled plasmid DNA in the absence or presence of topoisomerase I and observed the assembly of periodic arrays of nucleosomes in the absence of topoisomerase I. (Note that without topoisomerase I, it is not possible to perform the DNA supercoiling assay.) Thus, it should be possible to perform chromatin assembly reactions with the dNLP-ISWI system with negatively supercoiled plasmid DNA in the absence of topoisomerase I. In these reactions, the negative supercoils in the DNA are relieved by the wrapping of the DNA around the histones. It is also important to note that topoisomerase I is not necessary for the assembly of linear DNA, as shown in Fig. 3, C and D.
Lastly, we tested whether BSA is essential for chromatin assembly with the dNLP-ISWI system. BSA is commonly added to biochemical reactions to minimize adsorption of proteins to plastic as well as to maintain the activity of enzymes in vitro. However, in the absence of the ATP regeneration system and topoisomerase I, we observed that the omission of BSA has no discernable effect on chromatin assembly (Fig. 7C). Thus, taken together, the efficient assembly of periodic nucleosome arrays can be achieved with the minimal dNLP-ISWI system, which consists only of dNLP, ISWI, core histones, and ATP, in conjunction with supercoiled or linear DNA. Nevertheless, when possible, we recommend the inclusion of the ATP regeneration system and BSA to provide additional assurance of the consistent success of the reaction. In addition, topoisomerase I would be required if the chromatin is to be analyzed by the DNA supercoiling assay.
Discussion
We have described here a simple, reliable, and versatile method for the ATP-dependent assembly of periodic nucleosome arrays. The dNLP-ISWI system can be used with different DNA sequences, linear or circular DNA, recombinant or native core histones from Drosophila or humans, histone H1, HMGN2, and core histone variants H3.3 and H2A.V (H2A.Z/H2A.X). We have also described a minimal assembly system that does not involve the inclusion of an ATP regeneration system, topoisomerase I, or BSA. Notably, dNLP and ISWI are synthesized in bacteria, and each protein can be purified in a single step by affinity chromatography. The dNLP-ISWI system is accessible to a wide range of researchers, enabling the assembly of customized chromatin with specifically desired DNA sequences, core histones, and other chromosomal proteins. Because there are many different possible approaches to the use of the dNLP-ISWI system, some specific comments and recommendations are as follows.
dNLP and ISWI
For all applications, it is necessary to purify dNLP and ISWI.
Core histones from various organisms
The core histones are highly conserved proteins, and it is likely that the dNLP-ISWI system will work with core histones from many different organisms. For any given organism, if the core histones (especially histones H3 and H4) have a strong similarity to the human histones, it is likely that they will function in the dNLP-ISWI system.
Recombinant versus native core histones
We have generally found that native core histones yield slightly better (i.e. more evenly spaced) chromatin than recombinant histones (for example, see Fig. 2C and Ref. 39). However, native core histones, a mixture of S phase–regulated histones and histone variants, have a variety of different covalent modifications. Thus, one key advantage of recombinant core histones is that each individual histone is homogeneous. In addition, a second important advantage of recombinant histones is their ability to use mutant histones, histone variants, or specifically modified histones (for recent review, see Ref. 40). Such customized chromatin would allow the investigation of specific biological phenomena.
Core histone octamer to DNA mass ratio
This is the single most critical component of the assembly reaction. It is essential to determine the optimal core histone octamer to DNA mass ratio for chromatin assembly because there may be some error in the measurements of the concentrations of DNA and histones. A 10% error in either of these measurements could lead to a significant reduction in the quality of the chromatin. Therefore, for each new preparation of histones and DNA, it is necessary to perform a series of reactions with a range of core histone octamer to DNA mass ratios each varying by only about 10% (for example, octamer:DNA mass ratios of 0.7, 0.8, 0.9, 1.0, and 1.1). In this manner, the optimal ratio would be identified. As a reference, in the absence of histone H1, the optimal core histone octamer to DNA mass ratio is about 0.9:1 (which corresponds to ∼1 core histone octamer/180 bp DNA), whereas in the presence of histone H1, the longer repeat length results in a lower optimal core histone octamer to DNA mass ratio of about 0.8:1.
Purification of the DNA template
We normally purify the DNA by two successive CsCl density gradient centrifugation steps. This strategy yields DNA with minimal contamination by other species that might inhibit the assembly reaction or affect the accuracy of the determination of the DNA concentration. It is important to note, however, that high quality chromatin can be assembled with DNA purified by some other methods (supplemental Fig. S4).
DNA supercoiling assay
This assay provides an indication of the efficiency of chromatin assembly. Moreover, if the different topoisomers are analyzed by two-dimensional agarose gel electrophoresis (41), it is possible to obtain quantitative data on the efficiency of chromatin assembly (see, for example, Refs. 6 and 42). In general, we recommend analyzing the chromatin by one-dimensional agarose gel electrophoresis, as done in this work, to estimate the efficiency of nucleosome assembly.
Partial MNase digestion assay
This assay reveals the periodicity of the nucleosomes. In addition, if a distinct MNase ladder is achieved, it is likely that the chromatin is efficiently assembled because inefficiently assembled nucleosomes would yield a diffuse and smeared digestion pattern. However, the DNA supercoiling assay gives more reliable information on the efficiency of nucleosome assembly.
Topoisomerase I
Because topoisomerase I is needed only for the DNA supercoiling assay, one might be tempted to bypass the purification of the topoisomerase I and the use of the DNA supercoiling assay. We recommend, however, that initial work on the use of the dNLP-ISWI system should include the DNA supercoiling analysis, as it provides an indication of the overall success of the assembly reactions.
Linear versus circular DNA
The dNLP-ISWI system works with linear as well as circular DNA. In the initial experiments, we recommend using circular DNA so that the success of the assembly reaction can be monitored with the DNA supercoiling assay. Then, once the assembly system is established, linear templates could be assembled and analyzed with the partial MNase digestion assay. Also, as noted above, topoisomerase I is not needed for the assembly of linear DNA templates.
ATP regeneration system and BSA
We recommend that initial work with the dNLP-ISWI system include these components, which might increase the efficiency of chromatin assembly under some conditions. After the dNLP-ISWI system is successfully established, then the omission of these components could be tested.
The minimal assembly system, lacking the ATP regeneration system, topoisomerase I, and BSA
If the standard assembly system with all of the components is established, then the omission of the ATP regeneration system, topoisomerase I, and BSA could be tested. It is important to remember that this minimal system would work best with either linear DNA or negatively supercoiled plasmid DNA, as positive supercoils are generated during the assembly process. One potential advantage of the minimal system is that the resulting chromatin is of higher purity, as there are fewer components in the reaction medium, which could be useful for some applications.
Conclusion and perspectives
We are entering an exciting new era of chromatin research with the advent of new technologies such as techniques for the synthesis of customized histones (40). The dNLP-ISWI assembly system could be used in conjunction with conventional or customized histones for the assembly of many different types of chromatin that could be used for a diverse range of potential applications.
Experimental procedures
Purification of recombinant His6-tagged dNLP
The full-length dNLP coding sequence (12) was subcloned into a pET21-based vector to give pET21-His6dNLP, which encodes N-terminally His6-tagged dNLP (termed dNLP). The coding sequence, which was reconfirmed by DNA sequence analysis, is provided in supplemental Table S1. For expression, freshly transformed Escherichia coli strain BL21(DE3) was grown in LB medium (0.5-liter volume, 40 μg/ml ampicillin) at 37 °C to A600 nm of ∼0.6, and the synthesis of dNLP was induced by the addition of isopropyl-β-thiogalactopyranoside (IPTG) to 1 mm final concentration. The culture was incubated for an additional 3 h at 37 °C, and the cells were pelleted by centrifugation (Fiberlite F14S-6x250y rotor; 6000 rpm, 5 min at 4 °C). Unless stated otherwise, all subsequent operations were performed at 4 °C. The cells were resuspended in 20 ml of buffer N (50 mm phosphate (Na+), pH 7.0, containing 0.5 m NaCl, 1.5 mm MgCl2, 0.1% (v/v) Nonidet P-40, 15% (v/v) glycerol, 0.2 mm phenylmethylsulfonyl fluoride, 0.5 mm benzamidine-HCl, and 10 mm sodium metabisulfite) containing 15 mm imidazole, and the bacteria were lysed by sonication (Branson Sonifier 450 with a 0.25-inch microtip; 20% output, 4–8 cycles of sonication of 45 s “on” and 60 s “off”). The mixture was immersed in water at 80 °C for 15 min, and the insoluble material was removed by centrifugation (Fiberlite F21S-8x50y rotor; 10,000 rpm, 10 min). The lysate was combined with 2 ml (50% slurry in buffer N containing 15 mm imidazole) of nickel-nitrilotriacetic acid-agarose (Qiagen) and incubated for 3 h on a nutator. The dNLP-bound nickel-nitrilotriacetic acid beads were transferred into a Poly-Prep chromatography column (Bio-Rad, catalog no. 731-1550) and washed with 30 ml of buffer N containing 15 mm imidazole. Then, the protein was eluted with a step gradient of 2 ml each of buffer N containing 50, 100, 200, and 400 mm imidazole. The protein fractions were analyzed by 10% polyacrylamide-SDS gel electrophoresis and staining with Coomassie Brilliant Blue R-250. Nuclease activity in the fractions was detected by incubating plasmid DNA with the samples at 37 °C for 1 h in the presence of 10 mm MgCl2 followed by 1% agarose gel electrophoresis and staining with ethidium bromide. Peak dNLP-containing fractions lacking nucleases were dialyzed (molecular weight cutoff: 10 kDa) against 2 liters of buffer R (10 mm Hepes (K+), pH 7.6, containing 10 mm KCl, 1.5 mm MgCl2, 10% glycerol, 10 mm β-glycerophosphate, 1 mm dithiothreitol, 0.2 mm phenylmethylsulfonyl fluoride, 1 mm benzamidine-HCl, and 1 mm sodium metabisulfite) for 2 h, frozen in liquid nitrogen, and stored at −80 °C. A typical yield of dNLP from 0.5 liter of bacterial culture is ∼21 mg of purified protein. In the course of the purification and use of dNLP, the frozen protein should not be subjected to slow thawing such as on wet ice. Instead, the frozen dNLP should be thawed quickly in room temperature water.
Purification of recombinant Drosophila ISWI
The coding sequence of full-length Drosophila ISWI fused to an intein and chitin-binding domain (11) was subcloned into a pET24-based vector to give the plasmid pET24-ISWI-iCBD (the coding sequence, which was reconfirmed by DNA sequence analysis, is provided in supplemental Table S1). For expression, freshly transformed E. coli strain Rosetta(DE3) was grown in LB medium (1-liter volume, 40 μg/ml kanamycin, 34 μg/ml chloramphenicol) at 37 °C to an A600 nm of ∼0.6, and the synthesis of the ISWI fusion protein was induced by the addition of IPTG to 1 mm final concentration. The culture was incubated for an additional 16–18 h at 16 °C, and the bacteria were pelleted by centrifugation (Fiberlite F14S-6x250y rotor; 6000 rpm, 5 min at 4 °C). The cells were resuspended in cold (4 °C) PBS (250 ml; 137 mm NaCl, 2.7 mm KCl, 3.0 mm Na2HPO4, and 1.5 mm KH2PO4) and repelleted by centrifugation (Fiberlite F14S-6x250y rotor; 6000 rpm, 5 min at 4 °C). Unless stated otherwise, all subsequent operations were performed at 4 °C. The cells were resuspended in 50 ml of buffer M (20 mm Hepes (K+), pH 7.6, containing 1 m NaCl, 1 mm EDTA, 0.1% (v/v) Nonidet P-40, 15% (v/v) glycerol, 0.2 mm phenylmethylsulfonyl fluoride, and 0.5 mm benzamidine-HCl), subjected to two freeze-thaw cycles (frozen in liquid nitrogen and quickly thawed with room temperature water), and then lysed by sonication on ice (Branson Sonifier 450 with a 0.25-inch microtip; 20% output, 8 cycles for 15 s each) (it is important to keep the cells cold during sonication). The insoluble material was removed by centrifugation (Fiberlite F21S-8x50y rotor; 16,000 rpm for 20 min). The lysate was then combined with 2 ml (50% slurry in buffer M) of chitin resin (New England Biolabs) and incubated for 1 h on a nutator. The chitin beads were washed twice with 35 ml each of buffer M (for each wash, the resin should be allowed to settle by force of gravity followed by the removal of the supernatant by decanting), resuspended in 2 ml of buffer M, and transferred into a Poly-Prep chromatography column (Bio-Rad, catalog no. 731-1550). The column was washed four times each with 8 ml of buffer M. This was followed by a quick wash (∼1 min total time) with 1.5 ml of buffer E (buffer M containing 50 mm dithiothreitol). The bottom of the column was sealed, 0.5 ml of buffer E was added, and the beads were incubated for 16–20 h. (In this step, the dithiothreitol mediates the cleavage and release of ISWI from the chitin resin-bound ISWI-iCBD fusion protein.) The bottom of the column was opened, and the ISWI-containing eluate was collected. Three additional fractions of 0.25 ml (in buffer E) were collected. The ISWI-containing fractions were dialyzed (molecular weight cutoff: 10 kDa) against 2 liters of buffer D (20 mm Hepes (K+), pH 7.6, containing 200 mm NaCl, 2 mm MgCl2, 1 mm EDTA, 15% (v/v) glycerol, 0.02% (v/v) Nonidet P-40, 0.5 mm dithiothreitol, 0.2 mm phenylmethylsulfonyl fluoride, and 0.5 mm benzamidine-HCl) for 4 h, frozen in liquid nitrogen, and stored at −80 °C. A typical yield of ISWI is 0.1–0.2 mg of purified protein from 1 liter of bacterial culture.
Purification of recombinant Drosophila core histones
The coding sequences of Drosophila melanogaster S phase–regulated histones H2A and H2B were each individually subcloned into pET11a. In the H2B expression plasmid, an additional Ile codon (ATA) was inserted after the initiating Met codon. This modification was found to increase the level of expression of H2B and to have no adverse effects on chromatin assembly (43). The S phase–regulated histones H3 and H4 were co-synthesized from a single pET11a-based expression plasmid. Whereas histone H3 alone as well as histone H4 alone is each individually insoluble in aqueous solution, the co-synthesized histones H3 and H4 are soluble in standard aqueous buffers and can be purified as H3-H4 tetramers (39). For expression, freshly transformed E. coli strain BL21(DE3) was grown in LB medium (40 μg/ml ampicillin) at 37 °C to an A600 nm of ∼0.6, and protein synthesis was induced by the addition of IPTG to 0.4 mm final concentration. The cultures were incubated for an additional 3 h at 37 °C, and the cells were pelleted by centrifugation (Fiberlite F14S-6x250y rotor; 6000 rpm, 5 min at 4 °C).
Histones H3 and H4
Histones H3 and H4 were purified using a modification of the method of Levenstein and Kadonaga (39). Unless stated otherwise, all operations were performed at 4 °C. The pelleted cells (from 4 × 0.5 liters of culture) were resuspended in 40 ml of buffer D (10 mm Hepes (K+), pH 7.6, 1 mm EDTA, 10% (v/v) glycerol, 1 mm dithiothreitol, 0.1 mm phenylmethylsulfonyl fluoride, 1 mm sodium metabisulfite, and 2 mm benzamidine-HCl) containing 0.1 m NaCl and then lysed by sonication on ice (Branson Sonifier 450 with a 0.25-inch microtip; 20% output, 3 cycles for 30 s each). The insoluble material (including the histones) was pelleted by centrifugation (Fiberlite F21S-8x50y rotor; 13,200 rpm for 20 min), and the supernatant was discarded. The histone-containing pellet was suspended in 20 ml of 0.25 N HCl, and the dissolution of the material was facilitated by dispersion of the sample with a Wheaton Dounce homogenizer (A pestle). The mixture was incubated at −20 °C for 30 min and then subjected to centrifugation (Fiberlite F21S-8x50y rotor; 13,200 rpm for 20 min). The supernatant was neutralized with 0.125 volumes of 2 m Tris base and dialyzed (molecular weight cutoff: 3500 Da) for 2–3 h against buffer D containing 0.1 m NaCl. The insoluble material was removed by centrifugation (Fiberlite F21S-8x50y rotor; 13,200 rpm for 20 min), and the supernatant was applied to a Source 15S column (GE Healthcare; column dimensions (length × diameter) = 5.0 × 0.5 cm, column volume = 1 ml, flow rate = 1 ml/min) that was equilibrated with buffer D containing 0.1 m NaCl. The column was washed with 15 ml of buffer D containing 0.1 m NaCl, and the H3-H4 tetramers were eluted with a linear gradient of 0.1–2 m NaCl in buffer D over 10 column volumes. The peak fractions containing H3-H4 tetramers, which typically elute at about 1 m NaCl, were identified by 15% polyacrylamide-SDS gel electrophoresis and staining with Coomassie Brilliant Blue R-250. The H3-H4 peak fractions were pooled, and 2 m Tris-HCl, pH 7.6, was added to a final concentration of 10 mm. Then, 3 volumes of 8 m urea (deionized immediately before use by gentle mixing for 1 h at room temperature with Bio-Rad AG 501-X8 mixed bed resin) was added to the H3-H4 peak fractions to a final urea concentration of 6 m. The sample was incubated with nutation for 1–2 h at 22 °C before being dialyzed (molecular weight cutoff: 3500 Da) extensively against three changes of 2 liters of water containing 5 mm 2-mercaptoethanol. The first and third dialysis steps were performed for 2 h, and the second dialysis step was performed overnight. The insoluble material was removed by centrifugation (Fiberlite F21S-8x50y rotor; 13,000 rpm for 20 min). The absorbance at 276 nm was measured, and the concentration of H3-H4 was determined by using the molar extinction coefficient of 10,430/cm·m at 276 nm and a molecular mass of 26,719 g/mol. The H3-H4 was lyophilized to a dry pellet with medium heat and then stored at −80 °C. A typical yield of H3-H4 from 2 liters of bacterial culture is about 8 mg of purified protein.
Histones H2A and H2B
Histones H2A and H2B were each synthesized and purified separately by using the same method, which is a modified version of the protocol described by Luger et al. (44). (It is also useful to note that histones H3, H4, H2A.V, and H3.3 can be purified individually by this same method.) Unless stated otherwise, all operations were performed at 4 °C. The pelleted cells (from 4 × 0.5 liters of culture) were resuspended in 20 ml of buffer W (50 mm Tris-HCl, pH 7.5, 0.1 m NaCl, 1 mm EDTA, 5 mm 2-mercaptoethanol, 0.2 mm phenylmethylsulfonyl fluoride, and 1 mm benzamidine-HCl) and then frozen in liquid nitrogen. (If desired, the cells could be stored at −80 °C at this point.) The sample was thawed in warm (42 °C) water and lysed by sonication on ice (Branson Sonifier 450 with a 0.25-inch microtip; 20% output, 6–10 cycles for 15 s each). The insoluble material was pelleted by centrifugation (Fiberlite F21S-8x50y rotor; 13,000 rpm for 20 min). The resulting pellet was resuspended in 50 ml of buffer W containing 1% (v/v) Triton X-100, dispersed and washed by using a Wheaton Dounce homogenizer (A pestle), and repelleted by centrifugation (Fiberlite F21S-8x50y rotor; 13,000 rpm for 20 min). This process of dispersion, washing, and recentrifugation of the histone-containing pellet was repeated two to four times until the appearance of the pellet lightened to an off-white color. At this stage, the pellet can be frozen in liquid nitrogen and stored at −80 °C. The further purification of H2A or H2B was carried out as follows. The pellet was sliced into smaller pieces with a spatula and immersed in 0.7 ml of DMSO for 30 min at 22 °C. To extract the histones from the pellet, 15 ml of buffer UM (10 mm Tris-HCl, pH 8.0, 7 m urea (before use, freshly deionized as an 8 m urea solution in water with Bio-Rad AG 501-X8), 0.1 m NaCl, 1 mm EDTA, 0.2 mm phenylmethylsulfonyl fluoride, and 5 mm 2-mercaptoethanol) was added, and the mixture was dispersed with a Wheaton Dounce homogenizer (A pestle) and then incubated with nutation at room temperature for 1 h. The insoluble material was pelleted by centrifugation (Fiberlite F21S-8x50y rotor; 13,000 rpm, 22 °C for 20 min). The histone-containing supernatant was saved, and the pellet was subjected to an additional round of extraction with buffer UM (15 ml), incubation, and centrifugation. The two histone-containing supernatants were combined, and small particulate matter was removed by ultracentrifugation (Beckman SW41 rotor; 35,000 rpm, 22 °C for 35 min). The resulting sample was filtered through a Millipore Millex-HN nylon syringe filter (0.45 μm) and then applied to tandem Source 15Q (GE Healthcare; column dimensions (length × diameter) = 5.0 × 0.5 cm, volume = 1 ml, flow rate = 1 ml/min) followed by Source 15S (GE Healthcare; column dimensions (length × diameter) = 10 × 1 cm, volume = 4 ml, flow rate = 1 ml/min) chromatography columns that were equilibrated with buffer UD (10 mm Tris-HCl, pH 8.0, 7 m urea (before use, freshly deionized as a 8 m urea solution in water with Bio-Rad AG 501-X8), 1 mm EDTA, 0.2 mm phenylmethylsulfonyl fluoride, and 1 mm dithiothreitol) containing 0.1 m NaCl. After the tandem columns were washed with 1 volume (5 ml) of buffer UD containing 0.1 m NaCl, the Source 15Q column was removed. The remaining Source 15S column was washed with 5 column volumes of buffer UD containing 0.1 m NaCl, and protein was eluted with a 0.1–0.35 m NaCl gradient in buffer UD over 7.5 column volumes followed by a 0.35–0.4 m NaCl gradient in buffer UD over 2.5 column volumes and a step to 1.0 m NaCl in buffer UD for 1 column volume. H2A and H2B typically elute from the column at ∼0.3 M NaCl. Peak fractions, identified by 15% polyacrylamide-SDS gel electrophoresis and staining with Coomassie Brilliant Blue R-250, were then combined and dialyzed (molecular weight cutoff: 3500 Da) extensively against three changes of 2 liters of water containing 5 mm 2-mercaptoethanol. The first and third dialysis steps were performed for 2 h, and the second dialysis step was performed overnight. The insoluble material was removed by centrifugation (Fiberlite F21S-8x50y rotor; 13,000 rpm for 20 min). The absorbance at 276 nm was measured, and the histone concentration was determined by using the molar extinction coefficients at 276 nm and molecular masses as follows: H2A, 4470/cm·m, 12,231 g/mol; H2B with additional Ile, 7450/cm·m, 13,678 g/mol; H3, 4470/cm·m, 15,356 g/mol; H4, 5960/cm·m, 11,363 g/mol. The histone was lyophilized to a dry pellet and then stored at −80 °C. A typical yield of H2A or H2B from 2 liters of bacterial culture is about 20–40 mg of purified protein.
Reconstitution of the core histone octamer
The core histone octamer was reconstituted by using a modified version of the protocol described by Luger et al. (44). Each of the lyophilized histones (∼2 mg of each individual histone) was dissolved in 0.1 ml of buffer G (10 mm Hepes (K+), pH 7.6, 6 m guanidine-HCl, 1 mm EDTA, 10% (v/v) glycerol, and 1 mm dithiothreitol) at 22 °C for 30 min. The concentrations of the dissolved histones were determined by the absorbance at 276 nm as described above. Then, equimolar amounts of histones H3 and H4 were combined with a 25% molar excess of each of H2A and H2B to give a 1:1:1.25:1.25 molar ratio of H3:H4:H2A:H2B. If necessary, buffer G was added to give a final volume of 0.4 ml. The mixture was dialyzed (molecular weight cutoff: 3500 Da) against 1 liter of buffer RF (10 mm Tris-HCl, pH 7.5, 2 m NaCl, 1 mm EDTA, and 5 mm 2-mercaptoethanol) overnight at 4 °C. The insoluble material was removed by centrifugation (Eppendorf 5415R; 13,200 rpm, 10 min at 4 °C), and the supernatant was loaded onto a Superose 12 column (GE Healthcare; column dimensions (length × diameter) = 30 × 1 cm, volume = 24 ml, flow rate = 0.2 ml/min) that was equilibrated in buffer RF. The core histone octamer elutes with a Kav of ∼0.25, and the excess histones H2A and H2B elute with a Kav of ∼0.4. Fractions containing equimolar ratios of core histones were identified by 15% polyacrylamide-SDS gel electrophoresis and staining with Coomassie Brilliant Blue R-250. The peak fractions were combined and dialyzed (molecular weight cutoff: 3500 Da) against 1 liter of buffer CH (10 mm Hepes (K+), pH 7.6, containing 10 mm KCl, 1 mm EDTA, 10% (v/v) glycerol, and 1 mm dithiothreitol) overnight at 4 °C. The concentration of the histones was determined by the BCA assay (Pierce) with bovine serum albumin as the reference. The histones were frozen in liquid nitrogen and stored at −80 °C.
Purification of native core histones, recombinant human HMGN2, and recombinant Drosophila topoisomerase ND423 catalytic domain
Native Drosophila core histones were purified from Drosophila embryos as described (45, 46) and dialyzed into buffer CH, frozen in liquid nitrogen, and stored at −80 °C. Native human histones were purified by the method of Bulger and Kadonaga (45) and Fyodorov and Levenstein (46), except that HeLa cell nuclei were used instead of Drosophila embryo nuclei, and then dialyzed into buffer CH, frozen in liquid nitrogen, and stored at −80 °C. The human HMGN2 bacterial expression construct from Rattner et al. (47) (phHMGN2) was transformed into E. coli strain Rosetta(DE3), and HMGN2 protein was purified as described in Paranjape et al. (48). The purified protein was dialyzed into buffer CH, frozen in liquid nitrogen, and stored at −80 °C. The ND423 catalytic domain of Drosophila topoisomerase I was synthesized and purified as described by Fyodorov and Kadonaga (4).
Purification of recombinant human histone H1.0
The full-length sequence of human histone H1.0 was the kind gift of Dr. Woojin An (USC School of Medicine). It was slightly modified and subcloned into a pET24 vector to give pET24-H1.0. The coding sequence, which was confirmed by DNA sequence analysis, is provided in supplemental Table S1. Recombinant human H1.0 was purified by using a modified version of a method for the purification of native histone H1 (49). For expression, freshly transformed E. coli strain Rosetta(DE3) was grown in LB medium (40 μg/ml ampicillin) at 37 °C to an A600 nm of ∼0.6, and protein synthesis was induced by the addition of IPTG to 1 mm final concentration. The culture was incubated for an additional 2 h at 30 °C, and the cells were pelleted by centrifugation (Fiberlite F14S-6x250y rotor; 6000 rpm, 5 min at 4 °C). Unless stated otherwise, all subsequent operations were performed at 4 °C. The bacterial pellet was resuspended in 20 ml of buffer L (20 mm Tris-HCl, pH 7.9, containing 0.5 m NaCl, 0.2 mm EDTA, 10%(v/v) glycerol, 0.2 mm phenylmethylsulfonyl fluoride, 10 mm 2-mercaptoethanol, 1 μg/ml pepstatin, 1 μg/ml leupeptin, and 1 μg/ml aprotinin) and lysed by sonication on ice (Branson Sonifier 450 with a 0.25-inch microtip; 20% output, 6–8 cycles, 45 s each). The insoluble material was pelleted by centrifugation (Fiberlite F21S-8x50y rotor; 11,000 rpm for 20 min). Pulverized ammonium sulfate was slowly added to the lysate to 2.1 m final concentration, and the mixture was incubated with nutation for 15 min. The insoluble material was pelleted by centrifugation (Fiberlite F21S-8x50y rotor; 15,000 rpm for 20 min), and the supernatant was applied to a phenyl-Sepharose CL-4B chromatography column (GE Healthcare; column dimensions (length × diameter) = 10 × 1.6 cm, column volume = 20 ml, flow rate = 0.5 ml/min). H1.0 was eluted with a linear gradient from 2.1 to 0.1 m ammonium sulfate in HEMG buffer (25 mm Hepes (K+), pH 7.6, 0.1 mm EDTA, 12.5 mm MgCl2, 10% (v/v) glycerol, and 1 mm dithiothreitol) containing 0.5 mm phenylmethylsulfonyl fluoride over 7.5 column volumes. Fractions containing H1.0 were identified by 15% polyacrylamide-SDS gel electrophoresis and staining with Coomassie Brilliant Blue R-250. The peak fractions were combined and dialyzed against HEG buffer (25 mm Hepes (K+), pH 7.6, 0.1 mm EDTA, 10% (v/v) glycerol, and 1 mm dithiothreitol) containing 0.1 m KCl until the conductivity of the sample was identical to that of HEG buffer containing 0.15 m KCl. Nonidet P-40 was added to a final concentration of 0.01% (v/v), and HEG buffer lacking KCl was added until the conductivity of the sample was identical to that of HEG buffer containing 0.1 m KCl. The sample was subjected to centrifugation (Fiberlite F21S-8x50y rotor; 10,000 rpm for 10 min) and then applied to a Source 15S column (GE Healthcare; column dimensions (length × diameter) = 5 × 0.5 cm, volume = 1 ml, flow rate = 1 ml/min). H1.0 was eluted with a linear gradient of 0.1 to 1 m KCl in HEG buffer containing 0.01% (v/v) Nonidet P-40 and 0.5 mm phenylmethylsulfonyl fluoride over 15 column volumes. H1.0-containing fractions were identified by 15% polyacrylamide-SDS gel electrophoresis and staining with Coomassie Brilliant Blue R-250. The peak fractions were combined and dialyzed (molecular weight cutoff: 3500 Da) for 2 h against 2 liters of HEMG buffer containing 0.1 m KCl, 0.01% (v/v) Nonidet P-40, and 0.5 mm phenylmethylsulfonyl fluoride, frozen in liquid nitrogen, and stored at −80 °C. A typical yield from 1 liter of bacterial culture was about 0.1 mg of purified protein. Histone H1 proteins adhere to glass and plastic; hence, it is generally useful to include 0.01% (v/v) Nonidet P-40 in buffers containing histone H1.
Chromatin assembly reactions
Chromatin assembly reactions and DNA supercoiling and partial MNase digestion analyses were performed generally as described by Fyodorov and Levenstein (46) and Fyodorov and Kadonaga (4) with ACF and dNAP1. Here, with the dNLP-ISWI system, we included different variations of the chromatin assembly method that could be used to assemble chromatin for different specific applications. The different steps should be performed in the order listed below. This protocol is sufficient for six chromatin assembly reactions in which 0.54 μg of DNA is assembled into chromatin per reaction.
Determination of the correct amount of topoisomerase I to use for the relaxation of supercoiled plasmid DNA (optional for analysis of the chromatin by the DNA supercoiling assay)
If the chromatin is to be analyzed using the DNA supercoiling assay, then the amount of topoisomerase I to use for the relaxation of supercoiled plasmid DNA should be determined. Otherwise, the topoisomerase I step can be omitted. (Note also that topoisomerase I should not be included in chromatin assembly reactions with linear DNA.) The purified ND423 topoisomerase I (4) should have a concentration of about 1–2 mg/ml. Before use, the enzyme should be diluted about 100-fold into buffer S (10 mm Hepes (K+), pH 7.6, 0.1 mm EDTA, 50% (v/v) glycerol, 50 mm NaCl, 0.2 mg/ml recombinant insulin, 0.01% (v/v) Nonidet P-40, 10 mm 2-mercaptoethanol, 0.2 mm phenylmethylsulfonyl fluoride, 0.5 mm benzamidine-HCl, and 1 μg/ml leupeptin). In the context of this work, we defined 1 unit to be the amount of topoisomerase I that is sufficient to relax 5 μg of template DNA completely in 10 min at 30 °C in 1× Topo I buffer (50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 0.1 mm EDTA, 50 μg/ml BSA, and 0.5 mm dithiothreitol). By analyzing the activity of different dilutions of the topoisomerase I stock, it should be possible to determine the appropriate dilution that would give a concentration of 0.5 unit/μl. It is also useful to note that excess topoisomerase I can inhibit the assembly reaction. We therefore recommend the addition of only 1 unit of topoisomerase I to the DNA relaxation reactions described below.
Dilution of dNLP, ISWI, and core histones
Before use, the proteins should be diluted to the following working concentrations: dNLP to 2.0 mg/ml (22 μm of dNLP pentamers) in HEG buffer (12 μl of 2.0 mg dNLP/ml is sufficient for six reactions); ISWI to 0.12 mg/ml (1.0 μm) in HEG buffer (18 μl of 0.12 mg ISWI/ml is sufficient for six reactions); and core histones to 0.35 mg/ml (3.3 μm of histone octamers) in buffer CH (12 μl of 0.35 mg core histones/ml is sufficient for six reactions). Note that frozen stocks of dNLP, ISWI, and core histones should be thawed quickly in room temperature (22 °C) water immediately before use, kept on ice during use, and then frozen in liquid nitrogen before storage at −80 °C.
Preparation of ATP mix (two versions)
An ATP regeneration system (phosphoenolpyruvate and pyruvate kinase) is usually not necessary for chromatin assembly, but it is typically included to ensure that sufficient ATP is available for the reaction. Two versions of the ATP mix are as follows. Version 1 (7× scale ATP mix with ATP regeneration system) consists of the following: 63 μl of water, 37.5 μl of 100 mm MgCl2, 4.5 μl of 0.5 m ATP, pH 7.0, 4.5 μl of 0.5 m phosphoenolpyruvate, pH 7.0 (Millipore Sigma, catalog no. 860077), and 3 μl of 5000 units/ml pyruvate kinase (Millipore Sigma, catalog no. P9136) in 10 mm Hepes (K+), pH 7.6, containing 10 mm KCl and 50% (v/v) glycerol) to a final volume of 112.5 μl (use 15.8 μl/reaction as indicated below). Version 2 (7× ATP mix without ATP regeneration system) consists of the following: 70.5 μl of water, 37.5 μl of 100 mm MgCl2, and 4.5 μl of 0.5 m ATP, pH 7.0, to a final volume of 112.5 μl (use 15.8 μl/reaction as indicated below). The ATP mixes are prepared on ice and stored on ice before use.
Preparation of the dNLP mix
A 7× scale dNLP mix is prepared with the following components: 290 μl of HEG buffer, 123 μl of 600 mm KCl, 147 μl of a solution of 5% (w/v) polyethylene glycol (Millipore Sigma, catalog no. P2139) and 5% (w/v) polyvinyl alcohol (Millipore Sigma, catalog no. P8136), 8 μl of 2 mg/ml BSA (Pierce, catalog no. 23209), and 9.1 μl of 2 mg/ml dNLP to a final volume of 577 μl (use 82.5 μl/reaction). Mix the tube by gentle flicking, and consolidate the liquid by low speed centrifugation (2000 rpm or less). It should be noted that BSA is not required for the chromatin assembly reaction but is included to maximize the consistency and reproducibility of the reactions. If desired, the 8 μl of BSA could be replaced with 8 μl of water. The dNLP mix is prepared on ice and store on ice before use.
Formation of dNLP–core histone complexes
For each reaction (1× scale), 1.5 μl of the core histones (0.35 mg/ml) was added to 82.5 μl of the dNLP mix on ice. The tube was mixed by gentle flicking or vortexing and kept on ice for 20 min.
Preparation of the DNA mix (two versions)
Version 1 involves relaxation of supercoiled DNA with topoisomerase I. If DNA supercoiling analysis is to be performed, it would be necessary to relax supercoiled plasmid DNA with topoisomerase I. While the dNLP–core histone mixture is incubating on ice, relax the plasmid DNA by combining the following components (9× scale): 18 μl of water, 3 μl of 10× Topo I buffer, 6 μl of DNA (0.90 μg DNA/μl in water or Tris-EDTA buffer), and 3 μl of topoisomerase I (0.5 units/μl, as defined above) to 30 μl final volume. Mix and incubate the mixture at 30 °C for 10 min. Store at room temperature (22 °C) before use. Version 2 involves the use of DNA without treatment with topoisomerase I by combining the following components: 21 μl water, 3 μl 10× Topo I buffer, and 6 μl of DNA (0.90 μg DNA/μl in water or Tris-EDTA buffer) to 30 μl final volume. Mix and incubate the mixture at 30 °C for 10 min. Store at room temperature (22 °C) before use.
Chromatin assembly reactions
For each reaction (1× scale), place the tube containing the dNLP–core histone complexes (84 μl) in a 22 °C water bath. (Note that the samples will remain at 22 °C until the 27 °C incubation described below.) Add 15.8 μl of the ATP mix and 3.3 μl of the DNA mix to the tube. Blend the components by gentle vortexing for about 3 s. Then, initiate the chromatin assembly reaction by the addition of 2.1 μl of the 0.12 mg/ml ISWI. Mix the sample by gentle flicking, and incubate the tube at 27 °C in a water bath for 1.5 h. Terminate the reactions by the addition of 11.4 μl of 0.5 m EDTA (Na+), pH 8.0, to 50 mm final concentration of EDTA. Further analyses, such as DNA supercoiling or MNase digestion assays, may be performed as described by Fyodorov and Levenstein (46) and Fyodorov and Kadonaga (4). In the case of MNase digestion assays, the chromatin assembly reactions should not be terminated with 50 mm EDTA, as this would inhibit MNase activity.
Additional notes on chromatin assembly conditions
Some additional notes regarding the chromatin assembly reactions are as follows. First, the core histone to DNA mass ratio is a critical factor in the successful assembly of chromatin. For each new preparation of histones and DNA, it is necessary to determine the optimal histone:DNA ratio empirically by careful titration, such as by varying the ratio from 0.7 to 1.2 in increments of 0.1. Second, in the assembly of chromatin with histone H1, reduce the histone:DNA mass ratio by about 10–20% to adjust for the longer repeat length of the nucleosomes. Third, in reactions containing histone H1 or HMGN2, add 0.01% (v/v) Nonidet P-40 and 1 mm dithiothreitol to the reaction medium.
Native gel electrophoresis of mononucleosomes
In this assay, the newly assembled chromatin is digested to mononucleosomes by MNase, and the resulting particles are analyzed by native gel electrophoresis. For each sample, an aliquot from the assembly reaction (32 μl, from the step prior to the addition of EDTA to 50 mm final concentration) is combined with 100 mm CaCl2 (1 μl). MNase (0.1 unit/μl) is added to a final concentration of about 0.003 unit of MNase/ml. The reaction is carried out for 1 min at 22 °C and then terminated by the addition of 0.5 m EDTA (Na+), pH 8.0, to a final concentration of 50 mm. The sample is applied to a 5% nondenaturing polyacrylamide gel (height × width × thickness: 21 cm × 19 cm × 1 mm) made from freshly prepared 0.8:30 bisacrylamide:acrylamide and 0.5 × Tris-borate-EDTA buffer. The gel (pre-run for 1 h prior to the application of the samples) should be run at 60 V, 4 °C for about 5.5 h.
Author contributions
This study was conceived by M. T. K., J. F., and J. T. K. The experiments were performed mostly by M. T. K. J. F. developed and optimized the expression and purification of the recombinant histones and reconstitution of the core histone octamer. G. C. tested the reproducibility of the assembly system and performed the studies on the different methods of DNA purification. All authors analyzed the results, contributed to the writing of the manuscript, and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Long Vo ngoc and George Kassavetis for critical reading of the manuscript.
This work was supported by National Institutes of Health Grant R35 GM 118060 (to J. T. K.). The authors declare potential competing financial interests with the contents of this article. The chromatin assembly system described in this article could potentially be sold commercially as a research reagent. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S4 and Table S1.
- HMG
- high mobility group
- ACF
- ATP-utilizing chromatin assembly and remodeling factor
- Chd1
- chromodomain helicase DNA-binding protein 1
- dNAP1
- Drosophila nucleosome assembly protein 1
- dNLP
- Drosophila nucleoplasmin-like protein
- HMGN
- high-mobility-group, nucleosome-binding
- ISWI
- imitation switch
- MNase
- micrococcal nuclease
- RSF
- remodeling and spacing factor.
References
- 1. Craig N. L., Cohen-Fix N., Green R., Greider C., Storz G., and Wolberger C. (2014) Molecular Biology: Principles of Genome Function, 2nd Ed., Oxford University Press, Oxford, UK [Google Scholar]
- 2. Stein A. (1989) Reconstitution of chromatin from purified components. Methods Enzymol. 170, 585–603 [DOI] [PubMed] [Google Scholar]
- 3. Lusser A., and Kadonaga J. T. (2004) Strategies for the reconstitution of chromatin. Nat. Methods 1, 19–26 [DOI] [PubMed] [Google Scholar]
- 4. Fyodorov D. V., and Kadonaga J. T. (2003) Chromatin assembly in vitro with purified recombinant ACF and NAP-1. Methods Enzymol. 371, 499–515 [DOI] [PubMed] [Google Scholar]
- 5. Loyola A., and Reinberg D. (2003) Histone deposition and chromatin assembly by RSF. Methods 31, 96–103 [DOI] [PubMed] [Google Scholar]
- 6. Lusser A., Urwin D. L., and Kadonaga J. T. (2005) Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat. Struct. Mol. Biol. 12, 160–166 [DOI] [PubMed] [Google Scholar]
- 7. Ito T., Bulger M., Kobayashi R., and Kadonaga J. T. (1996) Drosophila NAP-1 is a core histone chaperone that functions in ATP-facilitated assembly of regularly spaced nucleosomal arrays. Mol. Cell. Biol. 16, 3112–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elfring L. K., Deuring R., McCallum C. M., Peterson C. L., and Tamkun J. W. (1994) Identification and characterization of Drosophila relatives of the yeast transcriptional activator SNF2/SWI2. Mol. Cell. Biol. 14, 2225–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ito T., Bulger M., Pazin M. J., Kobayashi R., and Kadonaga J. T. (1997) ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell 90, 145–155 [DOI] [PubMed] [Google Scholar]
- 10. Ito T., Levenstein M. E., Fyodorov D. V., Kutach A. K., Kobayashi R., and Kadonaga J. T. (1999) ACF consists of two subunits, Acf1 and ISWI, that function cooperatively in the ATP-dependent catalysis of chromatin assembly. Genes Dev. 13, 1529–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corona D. F., Längst G., Clapier C. R., Bonte E. J., Ferrari S., Tamkun J. W., and Becker P. B. (1999) ISWI is an ATP-dependent nucleosome remodeling factor. Mol. Cell 3, 239–245 [DOI] [PubMed] [Google Scholar]
- 12. Ito T., Tyler J. K., Bulger M., Kobayashi R., and Kadonaga J. T. (1996) ATP-facilitated chromatin assembly with a nucleoplasmin-like protein from Drosophila melanogaster. J. Biol. Chem. 271, 25041–25048 [DOI] [PubMed] [Google Scholar]
- 13. Kawasaki K., Philpott A., Avilion A. A., Berrios M., and Fisher P. A. (1994) Chromatin decondensation in Drosophila embryo extracts. J. Biol. Chem. 269, 10169–10176 [PubMed] [Google Scholar]
- 14. Crevel G., Huikeshoven H., Cotterill S., Simon M., Wall J., Philpott A., Laskey R. A., McConnell M., Fisher P. A., and Berrios M. (1997) Molecular and cellular characterization of CRP1, a Drosophila chromatin decondensation protein. J. Struct. Biol. 118, 9–22 [DOI] [PubMed] [Google Scholar]
- 15. Khuong M. T., Fei J., Ishii H., and Kadonaga J. T. (2015) Prenucleosomes and active chromatin. Cold Spring Harb. Symp. Quant. Biol. 80, 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Namboodiri V. M., Dutta S., Akey I. V., Head J. F., and Akey C. W. (2003) The crystal structure of Drosophila NLP-core provides insight into pentamer formation and histone binding. Structure 11, 175–186 [DOI] [PubMed] [Google Scholar]
- 17. Germond J. E., Hirt B., Oudet P., Gross-Bellark M., and Chambon P. (1975) Folding of the DNA double helix in chromatin-like structures from simian virus 40. Proc. Natl. Acad. Sci. U.S.A. 72, 1843–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Simpson R. T., Thoma F., and Brubaker J. M. (1985) Chromatin reconstituted from tandemly repeated cloned DNA fragments and core histones: A model system for study of higher order structure. Cell 42, 799–808 [DOI] [PubMed] [Google Scholar]
- 19. Noll M., and Kornberg R. D. (1977) Action of micrococcal nuclease on chromatin and the location of histone H1. J. Mol. Biol. 109, 393–404 [DOI] [PubMed] [Google Scholar]
- 20. Bates D. L., and Thomas J. O. (1981) Histones H1 and H5: one or two molecules per nucleosome? Nucleic Acids Res. 9, 5883–5894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Woodcock C. L., Skoultchi A. I., and Fan Y. (2006) Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 14, 17–25 [DOI] [PubMed] [Google Scholar]
- 22. Happel N., and Doenecke D. (2009) Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene 431, 1–12 [DOI] [PubMed] [Google Scholar]
- 23. Kalashnikova A. A., Rogge R. A., and Hansen J. C. (2016) Linker histone H1 and protein-protein interactions. Biochim. Biophys. Acta 1859, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rodríguez-Campos A., Shimamura A., and Worcel A. (1989) Assembly and properties of chromatin containing histone H1. J. Mol. Biol. 209, 135–150 [DOI] [PubMed] [Google Scholar]
- 25. Becker P. B., and Wu C. (1992) Cell-free system for assembly of transcriptionally repressed chromatin from Drosophila embryos. Mol. Cell. Biol. 12, 2241–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Varshavsky A. J., Bakayev V. V., and Georgiev G. P. (1976) Heterogeneity of chromatin subunits in vitro and location of histone H1. Nucleic Acids Res. 3, 477–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Albright S. C., Wiseman J. M., Lange R. A., and Garrard W. T. (1980) Subunit structures of different electrophoretic forms of nucleosomes. J. Biol. Chem. 255, 3673–3684 [PubMed] [Google Scholar]
- 28. Reeves R. (2010) Nuclear functions of the HMG proteins. Biochim. Biophys. Acta 1799, 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Postnikov Y., and Bustin M. (2010) Regulation of chromatin structure and function by HMGN proteins. Biochim. Biophys. Acta 1799, 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu N., and Hansen U. (2010) Transcriptional regulation by HMGN proteins. Biochim. Biophys. Acta 1799, 74–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuehl L., Salmond B., and Tran L. (1984) Concentrations of high-mobility-group proteins in the nucleus and cytoplasm of several rat tissues. J. Cell Biol. 99, 648–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sandeen G., Wood W. I., and Felsenfeld G. (1980) The interaction of high mobility proteins HMG14 and 17 with nucleosomes. Nucleic Acids Res. 8, 3757–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mardian J. K., Paton A. E., Bunick G. J., and Olins D. E. (1980) Nucleosome cores have two specific binding sites for nonhistone chromosomal proteins HMG 14 and HMG 17. Science 209, 1534–1536 [DOI] [PubMed] [Google Scholar]
- 34. Alfonso P. J., Crippa M. P., Hayes J. J., and Bustin M. (1994) The footprint of chromosomal proteins HMG-14 and HMG-17 on chromatin subunits. J. Mol. Biol. 236, 189–198 [DOI] [PubMed] [Google Scholar]
- 35. Weber C. M., and Henikoff S. (2014) Histone variants: Dynamic punctuation in transcription. Genes Dev. 28, 672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maze I., Noh K. M., Soshnev A. A., and Allis C. D. (2014) Every amino acid matters: Essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 15, 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jin C., Zang C., Wei G., Cui K., Peng W., Zhao K., and Felsenfeld G. (2009) H3.3/H2A.Z double variant-containing nucleosomes mark “nucleosome-free regions” of active promoters and other regulatory regions. Nat. Genet. 41, 941–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van Daal A., White E. M., Gorovsky M. A., and Elgin S. C. (1988) Drosophila has a single copy of the gene encoding a highly conserved histone H2A variant of the H2A.F/Z type. Nucleic Acids Res. 16, 7487–7497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Levenstein M. E., and Kadonaga J. T. (2002) Biochemical analysis of chromatin containing recombinant Drosophila core histones. J. Biol. Chem. 277, 8749–8754 [DOI] [PubMed] [Google Scholar]
- 40. David Y., and Muir T. W. (2017) Emerging chemistry strategies for engineering native chromatin. J. Am. Chem. Soc. 139, 9090–9096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peck L. J., and Wang J. C. (1983) Energetics of B-to-Z transition in DNA. Proc. Natl. Acad. Sci. U.S.A. 80, 6206–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fyodorov D. V., and Kadonaga J. T. (2002) Dynamics of ATP-dependent chromatin assembly by ACF. Nature 418, 897–900 [DOI] [PubMed] [Google Scholar]
- 43. Hamiche A., Kang J. G., Dennis C., Xiao H., and Wu C. (2001) Histone tails modulate nucleosome mobility and regulate ATP-dependent nucleosome sliding by NURF. Proc. Natl. Acad. Sci. U.S.A. 98, 14316–14321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luger K., Rechsteiner T. J., and Richmond T. J. (1999) Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 119, 1–16 [DOI] [PubMed] [Google Scholar]
- 45. Bulger M., and Kadonaga J. T. (1994) Biochemical reconstitution of chromatin with physiological nucleosome spacing. Methods Mol. Genet. 5, 241–262 [Google Scholar]
- 46. Fyodorov D. V., and Levenstein M. E. (2002) Chromatin assembly using Drosophila systems. Curr. Protoc. Mol. Biol. 21, unit 21.7 [DOI] [PubMed] [Google Scholar]
- 47. Rattner B. P., Yusufzai T., and Kadonaga J. T. (2009) HMGN proteins act in opposition to ATP-dependent chromatin remodeling factors to restrict nucleosome mobility. Mol. Cell 34, 620–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Paranjape S. M., Krumm A., and Kadonaga J. T. (1995) HMG17 is a chromatin-specific transcriptional co-activator that increases the efficiency of transcription initiation. Genes Dev. 9, 1978–1991 [DOI] [PubMed] [Google Scholar]
- 49. Croston G. E., Lira L. M., and Kadonaga J. T. (1991) A general method for the purification of H1 histones that are active for repression of basal RNA polymerase II transcription. Protein Expr. Purif. 2, 162–169 [DOI] [PubMed] [Google Scholar]
- 50. Pazin M. J., Kamakaka R. T., and Kadonaga J. T. (1994) ATP-dependent nucleosome reconfiguration and transcriptional activation from preassembled chromatin templates. Science 266, 2007–2011 [DOI] [PubMed] [Google Scholar]
- 51. Albright L. M., Kassavetis G. A., and Geiduschek E. P. (1988) Bacteriophage T4 late transcription from plasmid templates is enhanced by negative supercoiling. J. Bacteriol. 170 1279–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.