

Abstract
Pictilisib, a weakly basic compound, is an orally administered, potent, and selective pan‐inhibitor of phosphatidylinositol 3‐kinases for oncology indications. To investigate the significance of high‐fat food and gastric pH on pictilisib pharmacokinetics (PK) and enable label recommendations, a dedicated clinical study was conducted in healthy volunteers, whereby both top‐down (population PK, PopPK) and bottom‐up (physiologically based PK, PBPK) approaches were applied to enhance confidence of recommendation and facilitate the clinical development through scenario simulations. The PopPK model identified food (for absorption rate constant (Ka)) and proton pump inhibitors (PPI, for relative bioavailability (Frel) and Ka) as significant covariates. Food and PPI also impacted the variability of Frel. The PBPK model accounted for the supersaturation tendency of pictilisib, and gastric emptying physiology successfully predicted the food and PPI effect on pictilisib absorption. Our research highlights the importance of applying both quantitative approaches to address critical drug development questions.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Many anticancer drugs or candidates are susceptible to absorption‐related DDI risk when coadministered with ARAs due to pH‐dependent solubility, including pictilisib, a weak base. The applications of PBPK model to investigate the impact of food and gastric pH on drug absorptions have been reviewed and reported.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study used both top‐down (PopPK) and bottom‐up (PBPK) approaches to quantitatively and mechanistically understand the food and PPI effect on pictilisib PK. It addresses the question as to how exactly food and PPI exert their effects and how strong the effects are.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ This is the first research report to use combined modeling approaches to systemically investigate the food and PPI effect on drug absorptions, incorporating a deep understanding of the role of gastric emptying physiology.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This study highlighted an area with considerable potential to identify and mechanistically understand DDI liability and sources of PK variability that can be integrated in clinical trial designs.
A key objective of the clinical pharmacology plan is to build an integrated understanding of pharmacokinetics (PK), efficacy, and safety, as well as assessing the need of dose adjustment based on intrinsic (e.g., genetics) and extrinsic factors (e.g., drug–drug interactions (DDI)). The assessment of DDI risk is especially important for oncology drugs, where the therapeutic windows are often narrow,1 and cancer patients may be taking multiple concomitant prescribed drugs for comorbidities.2, 3 In addition to the metabolic‐related DDI,4 there may be other PK‐related DDIs depending on the rate‐determining step of the absorption, distribution, metabolism, and excretion (ADME) property of the drugs.5, 6, 7 In particular, for orally administered drugs, tablet disintegration, dissolution, and membrane permeability are critical for drug exposure. For weakly basic drugs, drug dissolution process can be drastically impacted by coadministration with acid‐reducing agents (ARAs), such as proton pump inhibitors (PPI), which are recognized as some of the most commonly prescribed and utilized drugs globally.8 We recently reported that many molecular‐targeted anticancer drugs and drug candidates are susceptible to absorption‐related DDI risk when coadministered with ARAs due to the pH‐dependent solubility.9, 10
Recently, there is growing recognition of the value of physiologically based PK (PBPK) modeling and simulation in predicting human PK, especially regarding DDI risk.11, 12, 13 The PBPK approach integrates drug‐specific (i.e., ADME and physicochemical properties) and system‐specific information (e.g., human physiology, demographics, and heterogeneity), and is thus generally recognized as a “bottom‐up” approach. This “bottom‐up” approach has been recently used in the clinical development to evaluate how food, formulation, and ARAs impact drug absorption.14, 15 The population PK (PopPK) modeling based on clinical PK observation is generally recognized as a “top‐down” approach to characterize the impact of intrinsic and extrinsic factors (covariates) on PK.16, 17 These modeling approaches are complementary in nature and may provide unique or confirmatory insights from different angles. The value of combining both approaches was demonstrated in the assessment of how ethnic difference impacts bitopertin clearance.18
Pictilisib (GDC‐0941) is an orally administered potent, selective pan‐inhibitor of phosphatidylinositol 3‐kinases (PI3Ks) with good preclinical antitumor activity in xenograft models and favorable PK and tolerability in phase I anticancer trials.19 However, as recently reported in two randomized phase II studies, pictilisib did not meet its primary endpoint when combined with paclitaxel or fulvestrant for patients with hormone receptor‐positive, HER2‐negative locally recurrent or metastatic breast cancer.20, 21
Based on in vitro, nonclinical and clinical healthy volunteer investigations, we have previously reported that pictilisib displays marked pH‐dependent solubility and its systemic concentrations can be significantly decreased by PPIs such as rabeprazole.22 Using pictilisib as an example, this study provides a real‐life case in clinical drug development, where both “top‐down” and “bottom‐up” approaches are applied using all the available data (in vitro, nonclinical, and healthy volunteer) to elucidate how food and PPI coadministration will impact the PK of a weakly basic compound.23 Although same label recommendations could be made by the statistical evaluation of the healthy volunteer data for pictilisib, the integrated approaches can enhance confidence of recommendation and facilitate the clinical development through scenario simulations.
MATERIALS AND METHODS
Clinical pharmacology study
Due to the steep pH‐dependent solubility profile (Figure 1 a), and the pH‐dependent absorption in hypochlorhydric dog,24 a phase I randomized study was conducted in healthy volunteers to investigate the effect of food and PPI on pictilisib PK. This was a single‐center, four‐period, two‐sequence, open‐label, randomized, crossover study conducted using a 40‐mg dose of pictilisib (NCT00999128). Standard high‐fat food (referred to as “food” hereafter) was administered within 30 min of pictilisib dosing. Patients were pretreated with 20 mg rabeprazole daily for 4 days before coadministrating with a single dose of pictilisib. Thirty‐two subjects were randomized equally to two sequences as follows, with four periods for each sequence. Thirty‐one subjects received study treatment.
Figure 1.
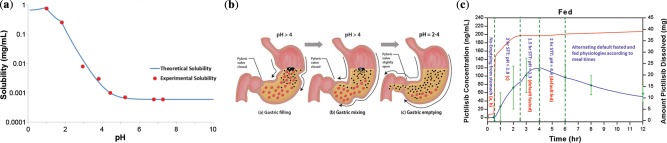
The impact of pH on the solubility and absorption of pictilisib: illustrated from the in vitro solubility assessment of pictilisib and the mechanistic understanding of stomach emptying process. (a) In vitro solubility vs. pH profile for pictilisib. (b) Stomach emptying physiology. (c) The impact of stomach emptying physiology on the absorption of pictilisib in fed state ((a,b), gastric filling and gastric mixing; (c), gastric emptying).
Sequence 1: fasted → fed → fasted/PPI → fed/PPI
Sequence 2: fed → fasted → fed/PPI → fasted/PPI
PopPK model development
PopPK analysis and covariate selection was conducted based on 1,202 plasma samples from 31 subjects in all periods using NONMEM v. 7.3. Natural log‐transformed data were used for modeling. Interindividual variability was modeled as log‐normal distribution. An additive error model on the log‐transformed data was applied. The same subjects in different periods were assumed to have the same elimination and distribution properties, but different absorption properties. The effect of food (fasted/fed) and PPI (yes/no) on the fixed effect parameters (relative bioavailability, Frel, absorption rate constant, Ka, and absorption lag time, Tlag), as well as random effect parameters (interindividual variability for Frel) were tested. EVID = 4 was set between periods of the same subject to represent complete PK washout. Covariate effects were judged for their significance on the basis of a likelihood ratio test at a P‐value of 0.01 for forward inclusion and 0.001 for backward deletion. The PopPK model was evaluated with goodness‐of‐fit diagnostics and Visual Predictive Checks (VPC).
PBPK model development
The PBPK models were developed for pictilisib using GastroPlus v. 8.5 (Simulations Plus, Lancaster, CA)25 for the average and representative subjects from fasted, fed, and fasted state with PPI. The model development process included prospective prediction using the default settings in GastroPlus and the model refinement. The refinement was driven by the mechanistic understanding of the stomach emptying process and the physicochemical properties for pictilisib. The final parameters in the refined model were fine‐tuned by the clinical data.
Physicochemical and biopharmaceutical properties
Key physicochemical and biopharmaceutical properties of pictilisib are provided in Supplemental Table S1. Pictilisib is a weakly basic compound with pKa values of 4.2 and 1.5.22 Given the high permeability and poor solubility at physiologically relevant pH values (Figure 1 a), pictilisib is classified as a BCS class II drug.26 Pictilisib also demonstrates a supersaturation tendency, a common characteristic for weak bases given the substantially higher solubility in fasted gastric fluid than in intestinal fluid.27, 28, 29, 30 The default precipitation time in GastroPlus (900 sec) was used for prospective prediction, and it was prolonged in the model refinement to account for the supersaturation tendency.
Intestinal absorption and pharmacokinetics of pictilisib
Intestinal absorption was described by the Advanced Compartmental Absorption and Transit (ACAT) model, which is an enhanced version of the absorption and transit model originally developed by Yu et al.31 The ACAT model considers the local solubility, dissolution, precipitation, and absorption in each region of the intestinal tract. The Johnson model32 was used to describe the drug dissolution process in individual intestinal compartments.
The default human fasted and fed physiology in GastroPlus were used for the prospective model predictions.25, 33 The stomach transit time (STT) and stomach pH were adjusted in the model refinement to account for the stomach emptying physiology34, 35 (Figure 1 b). The effect of PPI was also refined based on the literature.36
A PBPK model with all perfusion‐limited tissues described the systemic distribution of pictilisib. Human organ weights, volumes, and tissue perfusion rates were generated by GastroPlus' internal Population Estimates for Age‐Related (PEAR) Physiology Module according to the gender, age, and body weight. A volume of distribution (Vss) of 191 L (2.7 L/kg) for the typical subject (32 years, 71 kg male) was obtained. Hepatic clearance of 5 mL/min/kg parameterized from in vitro measurements in human liver microsomes was used in simulations.
PBPK model verification
To warrant the reliability of the PBPK model for pictilisib, we used the average subject data from fed state with PPI to verify the model. The model for fed with PPI was based on the refined model for fed state, with modifications considering the effect of PPI. The predictions for fed with PPI were compared directly to the clinical data. No further parameter fine‐tune was conducted.
RESULTS
Results of the clinical pharmacology study
The average PK profiles following a single dose of 40 mg pictilisib with or without food and PPI are presented in Figure 2 . The PBPK model predictions were overlaid.
Figure 2.
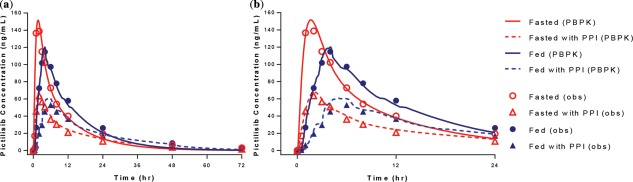
Influence of high‐fat meal and rabeprazole on the PK of pictilisib (observation and PBPK model prediction). (a) Average plasma PK profile of pictilisib (72 h). (b) Average plasma PK profile of pictilisib (24 h).
The median AUCinf for pictilisib were 1,760, 2,070, 906, and 1,180 ng/mL*h for fasted, fed, fasted with PPI, and fed with PPI, respectively.22 The presence of PPI, regardless of food, decreased the AUC significantly with similar Tmax. Regardless of PPI, the presence of food slightly increased AUC, with Tmax increased significantly. The median Tmax were 2, 4, 2, and 6 h for fasted, fed, fasted with PPI, and fed with PPI, respectively. The interindividual variabilities for AUCinf were 40, 23, 83, and 46%, respectively. For all periods, the elimination half‐life was unchanged.
Results of the PopPK analysis
Pictilisib PK was best described by a two‐compartment model with first‐order absorption with lag time, and first‐order elimination from the central compartment. The goodness‐of‐fit plots showed good agreement between model prediction and observations (Supplemental Figure S1). VPC plots indicated reasonable description of the central tendency and variability in different periods (Figure 3).
Figure 3.
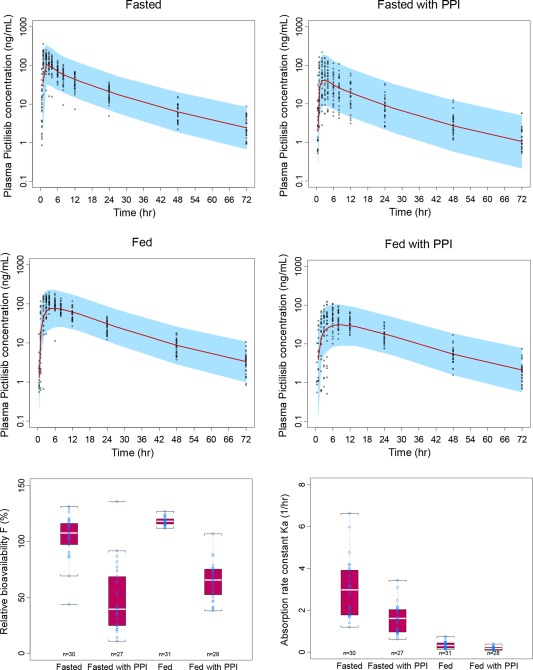
Visual predictive check (VPC) for the plasma PK profiles of pictilisib and the boxplots for the relative bioavailability (Frel) and Ka for each period.
The parameter estimates are shown in Table 1 and the covariate screening process is shown in Table S3. Briefly, PPI and food were the most influential covariates impacting Ka, where Ka decreased by ∼80% with food, regardless of PPI, and decreased by ∼50% with PPI, regardless of food. PPI and food were also identified as the key covariates impacting Frel. Although the impact of food on Frel was not statistically significant (P = 0.0017) during the backward elimination, it was kept in the model given the clinical relevance. The Frel decreased by 50–60% with PPI, regardless of food, and increased by 20–40% with food, regardless of PPI. The between‐subject variability around Frel was also impacted significantly by PPI and food, with 27, 8.9, 64, and 32% for fasted, fed, fasted with PPI and fed with PPI, respectively (Figure 3).
Table 1.
PopPK estimation and covariate identification for pictilisib in healthy volunteers
| Parameter | Unit | Typical value | BSV | |
|---|---|---|---|---|
| Absorption rate constant (Ka) | PPI=0, Fasted | 1/h | 2.63 | 51.9% |
| PPI=0, Fed | 0.297 | |||
| PPI=1, Fasted | 1.361 | |||
| PPI=1, Fed | 0.154 | |||
| Absorption lag time (Tlag) | H | 0.47 | 3.00% | |
| Clearance (CL/F)a* | L/h | 26.0 | 23.0% | |
| Distribution clearance (Q/F) | L/h | 13.9 | — | |
| Central volume (Vc/F) | L | 304.9 | 37.7% | |
| Peripheral volume (Vp/F) | L | 175.9 | — | |
| Relative bioavailability (F) | PPI=0, Fasted | % | 100 | 27% |
| PPI=1, Fasted | 42.9 | 64% | ||
| PPI=0, Fed | 118.4 | 8.9% | ||
| PPI=1, Fed | 61.3 | 32% | ||
| Residual error (proportional) | % | 56.1% | ||
*The apparent clearance for fasted state without PPI.
Results of the PBPK analysis
Fasted state
The observed and PBPK model predicted average PK profiles for pictilisib under fasted state are shown in Figure 4 a. The prospective model significantly underpredicted the Cmax, Tmax, and AUC with the default fasted physiology (0.25 h STT and pH 1.3),25, 33 and default precipitation time (Tprecip) of 900 sec.25, 30 The default Tprecip led to the overprediction of the precipitation potential for pictilisib, which was illustrated by the sharp dip shown in the profile of accumulative amount of dissolved compound shortly after dosing. This was the main reason for underprediction in fasted state. Given the supersaturation tendency for pictilisib, the Tprecip was refined to 90,000 sec (fine‐tuned based on average PK profile) to prevent any precipitation during absorption. The default STT of 0.25 h for fasted state was adjusted to 1 h, given the slower dissolution from tablet. In the refined model, pictilisib is predicted to be completely dissolved and absorbed from the small intestine at 40 mg (Table S2).
Figure 4.
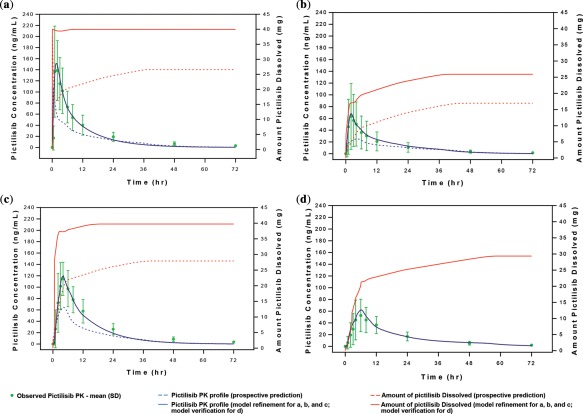
PBPK model predicted plasma PK profiles of pictilisib for each period. (a) Fasted state; (b) fasted state with PPI; (c) fed state; (d) fed state with PPI (a–c for model development; d for model verification).
The relatively large between‐subject variability in fasted state could be due to the differences in stomach pH, STT, and Tprecip among individuals.37
Fasted state with PPI
The observed data indicated that PPI decreases pictilisib exposure significantly. For fasted state with PPI, a stomach pH of 4.5 was used for the prospective prediction, resulting in a much stronger PPI effect (Figure 4 b). A closer look at different PPIs and the degree of stomach pH elevation revealed that pH 4.5 is the high boundary of the stomach pH for the fasted state in the presence of a PPI.38, 39 Quite often, PPIs increase the fasted stomach pH to values between 2 and 3.40 The default STT (0.25 h) and Tprecip (900 sec) for the fasted state also contributed to the underprediction. The average PK profile could be reproduced at a stomach pH 2.9, 0.7 h STT, and 90,000 sec Tprecip. In the refined model, the predicted fraction absorbed for the average subject is 64.5% (Table S2).
The effect of PPIs may vary significantly across individuals,41 leading to the increased PK variability for pictilisib (largest among the four states).
Fed state
Food has been shown to slightly increase pictilisib exposure. However, the prospective model with default fed physiology (mainly, 1 h STT, stomach pH 4.9) and default Tprecip (900 sec) only predicted half of the observed data (Figure 4 c).
Of note, the stomach emptying process slowly transits between fed and fasted physiologies after taking a meal. By considering the time‐ and pH‐dependent stomach emptying physiology (Figure 1 b),34, 35 the prediction can be significantly improved. The low pH window during the stomach emptying process provided the absorption opportunity for pictilisib. In the refined model, STT and stomach pH were programmed as the segmental changes over time to represent the stomach emptying physiology (Appendix S1, Table 2). Supersaturation tendency was also considered (90,000 sec Tprecip). The enlarged PK profile is shown in Figure 1 c with the impact of stomach emptying physiology highlighted.
Table 2.
The segmental changes for STT and stomach pH for pictilisib in fed state and fed state with PPI in healthy volunteers
| State | Time (h) | STT (h) | Stomach pH | Description |
|---|---|---|---|---|
| Fed | 0 | 100 | 4.9 | Absorption delay |
| 0.5 | 3 | 2.8 | Fed physiology (gastric emptying) | |
| 2.5 | 1.5 | 1.3 | Fasted physiology | |
| 4 | 2 | 4.9 | Fed physiology | |
| 6 | 1 | 1.3 | Fasted physiology | |
| Fed with PPI | 0 | 100 | 5.0 | Absorption delay |
| 0.5 | 3 | 5.0 | Fed physiology under PPI | |
| 2.5 | 1.5 | 2.9 | Fasted physiology under PPI | |
| 4 | 2 | 5.0 | Fed physiology under PPI | |
| 6 | 1 | 2.9 | Fasted physiology under PPI |
Nearly complete dissolution and absorption (99.4%) can be achieved in the refined model for the average subject (Table S2), which otherwise cannot be achieved with the default model. The individual subject profiles under the fed state can be well predicted by adjusting the segmental changes for STT and stomach pH.
Fed state with PPI
The PPI effect on pictilisib exposure appeared to be slightly attenuated by food. The average subject data for fed state with PPI were used for model verification. The parameter settings were adapted from the refined model for fed state, with the following rationales: PPI will elevate stomach pH to 2.9 for fasted physiology,40 and to 5 for fed physiology. The segmental changes for STT and stomach pH are provided in Table 2 and Appendix S1.
The model prediction is in very good agreement with the average subject data for fed state with PPI (Figure 4 d). The predicted fraction absorbed was 74.2% (Table S2).
Predicting the impact of PPI at a clinically relevant dose in breast cancer patients
The PBPK model developed at the 40 mg dose was used to predict pictilisib PK at the clinically relevant dose (340 mg) in breast cancer patients. As a sensitivity analysis, the predictions were conducted with varying STT and stomach pH values. Briefly, the STTs were tested at the same settings for healthy volunteers and the settings double the values in all four states; stomach pH was tested at 2, 2.9, and 5 in fasted state with PPI. Doubling the STT had no impact on the fraction absorbed for fasted state regardless of PPI; however, it slightly increased the value for fed state from 61 to 74% without PPI, and 19 to 21% with PPI. The fraction bioavailable for fed and fasted state alone were predicted to be comparable (56% and 57%) under the extended STT. For fasted state alone, the model prediction at 340 mg revealed incomplete absorption (78%) due to the solubility limitations. Increasing the stomach pH from 2 to 5 decreased the fraction absorbed from 50 to 12%. The detailed results are shown in Table S4.
DISCUSSION
Given the identified PPI/DDI risk from in vitro and preclinical evidence, a clinical pharmacology study was conducted for pictilisib in healthy volunteers at an earlier point in oncology drug development to inform the design of phase II and pivotal studies. Food effect was also assessed.
Being the first modeling initiative, the PopPK covariate analysis identified food and PPI as significant covariates for Ka (food, PPI) and Frel (food). Food increased Frel by about 20% (P > 0.001). Slower Ka was estimated for food and PPI. Ka was about 6% of the fasted state value when both PPI and food were present. Food and PPI also altered the variability of Frel significantly, with an increasing effect for PPI and decreasing effect for food. The magnitude and ranking order for the estimated variability of Frel in the four states (low to high: fed, fasted, fed state with PPI, and fasted state with PPI) are consistent with the observed data and the mechanisms. Based on the statistical comparisons of PK parameters in the four states, pictilisib may be taken without regard to the timing of meals and the recommendation is to take it with a meal to reduce PK variability. By analyzing the totality of the data, the popPK model provided comprehensive support for this recommendation.
As the follow‐up modeling initiative, the PBPK models were implemented to elucidate the source of the PK interactions and to perform scenario simulations. Conducting separate PBPK and popPK analysis can be a viable alternative to the population PBPK approach,42 given that it is less technical challenging and might provide similar interpretations. The PBPK models were developed for average and representative subjects (results not shown) from fasted, fed, and fasted state with PPI. The prospective predictions with the default fasted and fed state physiologies, default precipitation time, and the assumed PPI effect significantly underpredicted the average subject profiles. The models were refined by accounting for the stomach emptying physiology, supersaturation tendency, and the ideal PPI effect. The prospective model predictions are purely “bottom‐up.” Although the clinical data were involved in the model refinement, it is still considered “bottom‐up,” because the refinements were driven by the mechanistic understanding of the system‐ and drug‐specific properties.
Specifically, in fasted state the drastic precipitation in small intestine under the default Tprecip (900 sec) was the main reason for the underprediction of the prospective model. The 900 sec precipitation time originated from Kostewicz et al.'s in vitro experiment.30 Without properly considering the disappearance of the compound that occurred in vivo due to gut absorption, the in vitro experiments typically overpredicted the precipitation potential, especially for the highly permeable compounds. The proposed Tprecip of 90,000 sec may not reflect the exact in vivo conditions, but the sensitivity analysis indicated that the exposure difference under fasted state was only 1.1% for 15,000 sec and 90,000 sec Tprecip. The Tprecip of 90,000 sec was applied for all the four states. For the fasted state with PPI, the profiles for the average and representative subjects could be captured by adjusting the stomach pH to different levels (pH 2.9 for an average subject), given the variability around PPI effect across individuals.41 PPI has also been shown to delay gastric emptying.43, 44, 45 Compared to the consistent delaying effect for solid meals, the effect on liquids is inconsistent,45 which might indicate the possibility of having similar or slightly lower STT for pictilisib when PPI present.
The prediction for the fed state presented challenges because the application of default fed physiology with constant pH value (pH 4.9) and transit time could not describe the average profile at all. The stomach emptying physiology (Figure 1 b) offers a physiological explanation for the higher than expected pictilisib exposure in the fed state. After solid food is ingested, stomach pH increases above 4 (Figure 1 b(a)), the pyloric valve closes, and food digestion begins.34 During this phase, liquids and small suspended particles (<1 mm in diameter) can flow into the duodenum, whereas the viscous and solid mass is retained in the stomach and further mixed with gastric juice (Figure 1 b(b)). The pyloric valve opens when the pH drops below 4, and the emptying process will be completed usually 4 to 6 h after a meal with transition to a fasted state. The dissolution of pictilisib accelerates when stomach pH drops below 3. The delayed gastric emptying of pictilisib could be due to the lack of a substantial dissolution at pH >4 right after food is taken. The formation of a film of precipitated food components around the tablets might be another reason.46
The impact of food and ARAs on drug absorption is generally considered an area of PBPK application associated with low confidence in prediction. To enhance the confidence, the average subject data from fed state with PPI was used for model verification. Similar to the fasted state, PPI decreased pictilisib exposure by half in the fed state. The average PK profile for fed with PPI can be well predicted by the fed state model, with elevated stomach pH to 2.9 for fasted physiology and to 5 for fed physiology. Both pH 5 and 7 were tested for the fed physiology, and showed no difference in prediction. The ability to predict a new scenario by mechanistically adjusting the stomach pH and STT indicated that the PBPK model with gastric empty physiology was implemented successfully for pictilisib and can be used for further simulations. It also provides a flexible platform for evaluating the impact of food and PPI on the absorption of week bases in general.
Given the strong PPI effect observed in the healthy volunteer study at the 40 mg dose, the concomitant use of ARAs for pictilisib should be restricted. The PBPK models were used to further investigate whether the effect of PPI at the clinically relevant dose (340 mg) in breast cancer patients are consistent with healthy volunteers. Cancer patients in general have delayed gastric empting given the cancer itself and the complication of its treatment, such as surgery,47 radiation,48 and chemotherapy,49 or other mechanisms that lead to gastrointestinal dysmotility, such as paraneoplastic syndromes (mostly associated with small cell lung cancer and breast cancer50). Given all the causes, extending the STT to twice of the healthy volunteer settings is a reasonable assumption for breast cancer patients. Sensitivity analysis indicated that doubling the STT increased the fraction absorbed slightly for fed state but had no impact on fasted state, regardless of PPI. Under the extended STT, the breast cancer patients were predicted to have a comparable fraction bioavailable under fed and fasted state at the 340 mg dose. As compared to the complete absorption at 40 mg in fasted state, incomplete absorption (78%) was predicted for 340 mg due to solubility limitations. An oncology phase I dose‐escalation study indicated dose‐proportionality from 15 to 450 mg given in fasted state.19 There appeared to be some discrepancy between the 340 mg prediction and the dose‐proportionality assessment. Different formulations were used for the phase I dose‐escalation study and the healthy volunteer study. The relative bioavailability study indicated a slightly better absorption for the phase I formulation (data on file), which provided a rational explanation for the discrepancy. In the phase I expansion cohort, pictilisib PK following a 340 mg single dose before and after rabeprazole was determined in nine patients under fasted state. The results indicated a strong (45–50% decrease) but highly variable PPI effect (data on file), which is in line with the PBPK sensitivity simulations with varying stomach pH values. So, in general, the recommendations will stay the same for cancer patients taking the clinically relevant dose.
Given the high prevalence of ARA in cancer populations,10 mitigation strategies were also assessed to temporarily reacidify the gastric environment at the time of treatment to allow for maximum absorption of pictilisib and ensure continuous PPI therapy (results not shown). This was another PBPK scenario simulation to facilitate the clinical development of pictilisib.
For the case of pictilisib, the dedicated healthy volunteer study and integrated PBPK/popPK approach (Figure 5 , middle panel) presented an informative way to assess the effect of PPI and food and enable dose recommendations. Although the same recommendations might be derived without modeling and simulation, the integrated analysis can enhance confidence of recommendations and facilitate the clinical development of pictilisib through quantitative assessment (ascertain food effect by PopPK analysis) and scenario simulations (PBPK simulation for food and PPI effect at a clinically relevant dose, and for mitigation strategies to avoid PPI/DDI).
Figure 5.
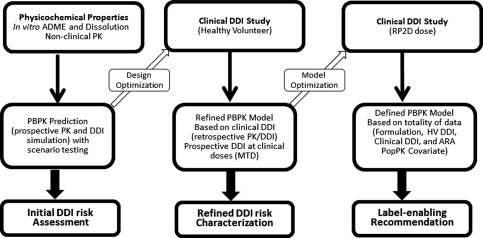
High‐level strategy for the use of bottom‐up PBPK model for drug candidates with pH‐dependent solubility.
Figure 5 represents the most comprehensive workflow to reach label‐enabling recommendations for drug candidates with pH‐dependent solubility. Besides the healthy volunteer study, the clinical DDI might also be assessed in patients at the recommended phase II dose (RP2D) given the potential changes in formulation and dose for patients as compared to healthy volunteers. For pictilisib, a clinical PPI/DDI study was conducted in the phase I expansion cohort at the 340 mg dose. On a case‐by‐case basis, the DDI assessment can be conducted in either healthy volunteers or patients or both. The PBPK model based on a healthy volunteer study can be further improved using data from a clinical DDI study in patients, if available, to account for the changes such as formulation and dose, which is referred to as “defined PBPK model” in the right panel of Figure 5.
When the dedicated healthy volunteer study or a controlled clinical study in patients is not available for DDI assessment, the PBPK model built from the in vitro and nonclinical PK can be refined, to a less extent, by the clinical trial data, given the limitations such as larger PK variability, sparse PK sampling, and the uncontrolled use of comedications or food. Similar limitations will apply for the popPK analysis to assess the potential covariates for DDI using clinical trial data.16
CONFLICT OF INTEREST/DISCLOSURE
During the preparation of this article, T.L., L.S., N.B., G.D., G.S.S., K.M.M., J.D.D., J.A.W. and J.Y.J. are full‐time employees of Genentech, Inc., and own Roche stock. G.F. is an employee of Simulations Plus, and served in a consultant/advisory role to Genentech.
AUTHOR CONTRIBUTIONS
J.W., T.L., G.F., N.B., G.D., G.S., K.M., J.D.D., and J.Y.J. wrote the article; J.W. designed the research; J.W., T.L., G.F., L.S., N.B., G.D., G.S., K.M., J.D.D., and J.Y.J. performed the research; T.L., G.F., N.B., and G.D. analyzed the data.
Supporting information
Figure S1: Goodness of Fit plot for the PopPK model of pictilisib in healthy volunteers
Table S1: Key physicochemical and biopharmaceutical parameters for pictilisib used in GastroPlus simulations
Table S2: Summary of simulated amount dissolved, fraction of absorbed and fraction bioavailable for pictilisib at 40 mg in healthy volunteers
Table S3: Covariate screening and final model identification for the PopPK model of pictilisib in healthy volunteers
Table S4: Summary of simulated fraction of absorbed and fraction bioavailable for Pictilisib at clinical relevant dose (340 mg) in breast cancer patients using the PBPK model developed at 40 mg in healthy volunteers (HV)
Appendix S1: Detail descriptions for the segmental changes of STT and stomach pH for pictilisib in fed state and fed state with PPI in healthy volunteers
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors thank the investigators, healthy volunteers, and patients who participated in the clinical development of pictilisib. The authors acknowledge the pictilisib development team and Genentech Leadership from the Departments of Clinical Pharmacology, DMPK, and Global Pharmaceutical Sciences for their feedback support. Special thanks to Dr. Bert Lum for critical review and feedback on the article. The authors thank Christine (Yuying) Gao from Certara Strategic Consulting for assistance with popPK model development, and Anshin BioSolutions for editorial support of the article. The analysis was funded by Genentech, Inc., a member of the Roche group.
References
- 1. Lu, D. et al A survey of new oncology drug approvals in the USA from 2010 to 2015: a focus on optimal dose and related postmarketing activities. Cancer Chemother. Pharmacol. 77, 459–476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beijnen, J.H. & Schellens, J.H. Drug interactions in oncology. Lancet Oncol. 5, 489–496 (2004). [DOI] [PubMed] [Google Scholar]
- 3. van Leeuwen, R.W. , van Gelder, T. , Mathijssen, R.H. & Jansman, F.G. Drug‐drug interactions with tyrosine‐kinase inhibitors: a clinical perspective. Lancet Oncol. 15, e315–326 (2014). [DOI] [PubMed] [Google Scholar]
- 4. Ito, K. et al Prediction of pharmacokinetic alterations caused by drug‐drug interactions: metabolic interaction in the liver. Pharmacol. Rev. 50, 387–412 (1998). [PubMed] [Google Scholar]
- 5. Hochman, J. , Tang, C. & Prueksaritanont, T. Drug‐drug interactions related to altered absorption and plasma protein binding: theoretical and regulatory considerations, and an industry perspective. J. Pharm. Sci. 104, 916–929 (2015). [DOI] [PubMed] [Google Scholar]
- 6. Guidance for industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations . US Food and Drug Administration <http://wwwfdagov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362pdf> 2012.
- 7. Guideline on the investigation of drug interactions . European Medicines Agency <http://wwwemaeuropaeu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606pdf> 2012.
- 8. Zhang, L. , Wu, F. , Lee, S.C. , Zhao, H. & Zhang, L. pH‐dependent drug‐drug interactions for weak base drugs: potential implications for new drug development. Clin. Pharmacol. Ther. 96, 266–277 (2014). [DOI] [PubMed] [Google Scholar]
- 9. Budha, N.R. et al Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH‐dependent solubility the Achilles heel of targeted therapy? Clin. Pharmacol. Ther. 92, 203–213 (2012). [DOI] [PubMed] [Google Scholar]
- 10. Smelick, G.S. , et al Prevalence of acid‐reducing agents (ARA) in cancer populations and ARA drug‐drug interaction potential for molecular targeted agents in clinical development. Mol. Pharm. 10, 4055–4062 (2013). [DOI] [PubMed] [Google Scholar]
- 11. Zhao, P. , Rowland, M. & Huang, SM. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 92, 17–20 (2012). [DOI] [PubMed] [Google Scholar]
- 12. Jones, H.M. et al Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin. Pharmacol. Ther. 97, 247–262 (2015). [DOI] [PubMed] [Google Scholar]
- 13. Wagner, C. et al Application of Physiologically Based Pharmacokinetic (PBPK) Modeling to Support Dose Selection: Report of an FDA Public Workshop on PBPK. CPT Pharmacometrics Syst. Pharmacol. 4, 226–230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parrott, N.J. , Yu, L.J. , Takano, R. , Nakamura, M. & Morcos, P.N. Physiologically based absorption modeling to explore the impact of food and gastric pH changes on the pharmacokinetics of alectinib. AAPS J. 18, 1464–1474 (2016). [DOI] [PubMed] [Google Scholar]
- 15. Parrott, N. , Lukacova, V. , Fraczkiewicz, G. & Bolger, M.B. Predicting pharmacokinetics of drugs using physiologically based modeling—application to food effects. AAPS J. 11, 45–53 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bonate, P.L. et al Methods and strategies for assessing uncontrolled drug‐drug interactions in population pharmacokinetic analyses: results from the International Society of Pharmacometrics (ISOP) Working Group. J. Pharmacokinet. Pharmacodyn. 43, 123–135 (2016). [DOI] [PubMed] [Google Scholar]
- 17. Joerger, M. Covariate pharmacokinetic model building in oncology and its potential clinical relevance. AAPS J. 14, 119–132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feng, S. et al Combining 'bottom‐up' and 'top‐down' methods to assess ethnic difference in clearance: bitopertin as an example. Clin. Pharmacokinet. 55, 823–832 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sarker, D. et al First‐in‐human phase I study of pictilisib (GDC‐0941), a potent pan‐class I phosphatidylinositol‐3‐kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 21, 77–86 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vuylsteke, P. et al Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3‐kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor‐positive, HER2‐negative, locally recurrent, or metastatic breast cancer: interim analysis of the multicentre, placebo‐controlled, phase II randomised PEGGY study. Ann. Oncol. 27, 2059–2066 (2016). [DOI] [PubMed] [Google Scholar]
- 21. Krop, I.E. et al Pictilisib for oestrogen receptor‐positive, aromatase inhibitor‐resistant, advanced or metastatic breast cancer (FERGI): a randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Oncol. 17, 811–821 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ware, J.A. et al Impact of food and the proton pump inhibitor rabeprazole on the pharmacokinetics of GDC‐0941 in healthy volunteers: bench to bedside investigation of pH‐dependent solubility. Mol. Pharm. 10, 4074–4081 (2013). [DOI] [PubMed] [Google Scholar]
- 23. Lu, T. et al Where top‐down meets bottom‐up: combined population PK (Poppk) and PBPK approaches to evaluate the impact of food and gastric pH on the pharmacokinetics of pictilisib. American Conference on Pharmacometrics 2014, Poster Abstracts (T‐041).
- 24. Pang, J. , Dalziel, G. , Dean, B. , Ware, J.A. & Salphati, L. Pharmacokinetics and absorption of the anticancer agents dasatinib and GDC‐0941 under various gastric conditions in dogs—reversing the effect of elevated gastric pH with betaine HCl. Mol. Pharm. 10, 4024–4031 (2013). [DOI] [PubMed] [Google Scholar]
- 25. Simulations Plus I. GastroPlus User Manual. V. 8.5 In: Simulations Plus I, editor. Lancaster, CA; 2013. [Google Scholar]
- 26. Amidon, G.L. , Lennernas, H. , Shah, V.P. & Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12, 413–420 (1995). [DOI] [PubMed] [Google Scholar]
- 27. Box, K.J. & Comer, J.E. Using measured pKa, LogP and solubility to investigate supersaturation and predict BCS class. Curr. Drug Metab. 9, 869–878 (2008). [DOI] [PubMed] [Google Scholar]
- 28. Carlert, S. et al Predicting intestinal precipitation—a case example for a basic BCS class II drug. Pharm. Res. 27, 2119–2130 (2010). [DOI] [PubMed] [Google Scholar]
- 29. Psachoulias, D. et al Precipitation in and supersaturation of contents of the upper small intestine after administration of two weak bases to fasted adults. Pharm. Res. 28, 3145–3158 (2011). [DOI] [PubMed] [Google Scholar]
- 30. Kostewicz, E.S. et al Predicting the precipitation of poorly soluble weak bases upon entry in the small intestine. J. Pharm. Pharmacol. 56, 43–51 (2004). [DOI] [PubMed] [Google Scholar]
- 31. Yu, L.X. , Crison, J.R. & Amidon, G.L. Compartmental transit and dispersion model analysis of small intestinal transit flow in humans. Int . J. Pharm. 140, 111–118 (1996). [Google Scholar]
- 32. Lu, A.T. , Frisella, M.E. & Johnson, K.C. Dissolution modeling: factors affecting the dissolution rates of polydisperse powders. Pharm. Res. 10, 1308–1314 (1993). [DOI] [PubMed] [Google Scholar]
- 33. Parrott, N. , Lukacova, V. , Fraczkiewicz, G. & Bolger, M.B. Predicting pharmacokinetics of drugs using physiologically based modeling—application to food effects. AAPS J. 11, 45–53 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ehrlein, H.J. & Schemann, M. Gastrointestinal motility. <http://humanbiology.wzw.tum.de/motvid01/tutorial.pdf> [DOI] [PubMed]
- 35. Hellstrom, P.M. , Gryback, P. & Jacobsson, H. The physiology of gastric emptying. Best Pract. Res. Clin. Anaesthesiol. 20, 397–407 (2006). [DOI] [PubMed] [Google Scholar]
- 36. Shin, J.M. & Sachs, G. Pharmacology of proton pump inhibitors. Curr. Gastroenterol. Rep. 10, 528–534 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van Den Abeele, J. , Rubbens, J. , Brouwers, J. & Augustijns, P. The dynamic gastric environment and its impact on drug and formulation behaviour. Eur. J. Pharm. Sci. 96, 207–231 (2017). [DOI] [PubMed] [Google Scholar]
- 38. Hoogerwerf, W.A. P.P. Pharmacotherapy of gastric acidity peptic ulcers, and gastroesophageal reflux disease Goodman & Gilman's The Pharmacological Basis of Therapeutics. 11. McGraw‐Hill; New York, 2006, pp. 967–981. [Google Scholar]
- 39. Pace, F. , Pallotta, S. , Casalini, S. & Porro, G.B. A review of rabeprazole in the treatment of acid‐related diseases. Ther. Clin. Risk Manag. 3, 363–379 (2007). [PMC free article] [PubMed] [Google Scholar]
- 40. Aubert, C.M. , Schror, K. & Vavricka, S. Omeprazole MUPS®: An advanced formulation offering flexibility and predictability for self medication. SelfCare 2, 1–14 (2011). [Google Scholar]
- 41. Kirchheiner, J. et al Relative potency of proton‐pump inhibitors‐comparison of effects on intragastric pH. Eur. J. Clin. Pharmacol. 65, 19–31 (2009). [DOI] [PubMed] [Google Scholar]
- 42. Tsamandouras, N. , Rostami‐Hodjegan, A. & Aarons, L. Combining the 'bottom up' and 'top down' approaches in pharmacokinetic modelling: fitting PBPK models to observed clinical data. Br. J. Clin. Pharmacol. 79, 48–55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parkman, H.P. et al Effect of gastric acid suppressants on human gastric motility. Gut 42, 243–250 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sud, D. , Joseph, I.M.P. & Kirschner, D. Predicting efficacy of proton pump inhibitors in regulating gastric acid secretion. J. Biol. Syst. 12, 1–34 (2004). [Google Scholar]
- 45. Sanaka, M. , Yamamoto, T. & Kuyama, Y. Effects of proton pump inhibitors on gastric emptying: a systematic review. Dig. Dis. Sci. 55, 2431–2440 (2010). [DOI] [PubMed] [Google Scholar]
- 46. Brouwers, J. , Tack, J. & Augustijns, P. Parallel monitoring of plasma and intraluminal drug concentrations in man after oral administration of fosamprenavir in the fasted and fed state. Pharm. Res. 24, 1862–1869 (2007). [DOI] [PubMed] [Google Scholar]
- 47. Kelly, D. , Moran, C. , Maher, M. & O'Mahony, S. Malignancy‐associated gastroparesis: an important and overlooked cause of chronic nausea and vomiting. BMJ Case Rep. bcr2013201815 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Otterson, M.F. Effects of radiation upon gastrointestinal motility. World J. Gastroenterol. 13, 2684–2692 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nelson, K. , Walsh, D. & Sheehan, F. Cancer and chemotherapy‐related upper gastrointestinal symptoms: the role of abnormal gastric motor function and its evaluation in cancer patients. Support. Care Cancer 10, 455–461 (2002). [DOI] [PubMed] [Google Scholar]
- 50. DiBaise, J.K. Paraneoplastic gastrointestinal dysmotility: when to consider and how to diagnose. Gastroenterol. Clin. N. Am. 40, 777–786 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Goodness of Fit plot for the PopPK model of pictilisib in healthy volunteers
Table S1: Key physicochemical and biopharmaceutical parameters for pictilisib used in GastroPlus simulations
Table S2: Summary of simulated amount dissolved, fraction of absorbed and fraction bioavailable for pictilisib at 40 mg in healthy volunteers
Table S3: Covariate screening and final model identification for the PopPK model of pictilisib in healthy volunteers
Table S4: Summary of simulated fraction of absorbed and fraction bioavailable for Pictilisib at clinical relevant dose (340 mg) in breast cancer patients using the PBPK model developed at 40 mg in healthy volunteers (HV)
Appendix S1: Detail descriptions for the segmental changes of STT and stomach pH for pictilisib in fed state and fed state with PPI in healthy volunteers
Supporting Information
Supporting Information