

SUMMARY
Many adult stem cells display prolonged quiescence, promoted by cues from their niche. Upon tissue damage, a coordinated transition to the activated state is required, as non-physiological breaks in quiescence often lead to stem cell depletion and impaired regeneration. Here, we identify cadherin-mediated adhesion and signaling between muscle stem cells (satellite cells; SCs) and their myofiber niche as a mechanism that orchestrates the quiescence-to-activation transition. Conditional removal of N-cadherin and M-cadherin in mice leads to a break in SC quiescence with long-term expansion of a regeneration-proficient SC pool. These SCs have an incomplete disruption of the myofiber-SC adhesive junction, and maintain niche residence and cell polarity, yet show properties of SCs in a state of transition from quiescence towards full activation. Among these is nuclear localization of β-catenin, which is necessary for this phenotype. Injury-induced perturbation of niche adhesive junctions is therefore a likely first step in the quiescence-to-activation transition.
Keywords: Stem cell, niche, muscle, satellite cell, cell adhesion, quiescence, regeneration, cadherin, β-catenin
eTOC Blurb
Using muscle- and muscle stem cell (SC)-specific mutants, Goel et al. demonstrate that N- and M-cadherins are niche-based regulators of SC quiescence. SCs from mice lacking these cadherins (dKO SCs) exist in a state between quiescence and full activation. β-catenin signaling is required for adoption of this state.
INTRODUCTION
Adult stem cells are critical to homeostatic maintenance and regeneration of many tissues. They inhabit a specialized microenvironment, or niche, that imparts signals to support their behavior (Morrison and Spradling, 2008; Scadden, 2014). Some types of stem cells, such as long-term hematopoietic stem cells and skeletal muscle stem cells, are characterized by prolonged periods of quiescence. They are “activated” and enter the cell cycle principally upon injury, thereby providing progenitor cells for tissue repair. In these cases, the niche provides cues that promote stem cell quiescence; such signals must be overridden upon injury, to permit activation. The identity of the niche signals required for long-term stem cell quiescence, and the mechanisms that underlie the transition from quiescence to activation, are poorly understood.
Cell-cell adhesion participates in niche-regulated stem cell behavior, with cadherin-based adhesive junctions (AJs) anchoring stem cells to niche cells (Chen et al., 2013; Marthiens et al., 2010). Genetic removal of E-cadherin from Drosophila germline stem cells leads to their departure from the niche, alteration of cell polarity, and loss of long-term self-renewal (Chen et al., 2013; Yamashita, 2010). Similarly, in mice, N-cadherin is required to maintain normal architecture between neural stem cells and niche cells and, consequently, neural stem cell quiescence (Porlan et al., 2014). It is likely that in addition to physical anchorage of stem cells, niche AJs provide direct signaling cues essential to stem cell behavior (Chen et al., 2013). However, as these examples illustrate, it has been difficult to study direct signaling mechanistically because, when AJs are disrupted, stem cells depart the niche and are deprived of all niche functions that regulate proliferation, polarity, and differentiation.
Skeletal muscle stem cells, or satellite cells (SCs), are the source of this tissue’s regenerative capacity (Brack and Rando, 2012; Dumont et al., 2015). SCs display long-term quiescence and express Pax7, a transcription factor required for this property (von Maltzhan et al., 2013). Following muscle injury, quiescent SCs are activated, a process that involves expression of the myogenic transcription factors Myf5 and MyoD and proliferation of these cells as transit-amplifying myoblasts. Myoblasts subsequently differentiate and fuse to each other and to existing myofibers to repair the injury. At least a subset of SCs self-renew, and muscles are capable of multiple rounds of SC-dependent regeneration. When SC quiescence is broken in a non-physiological manner (e.g., during old age or via genetic manipulation in mice), it generally leads to loss of a functional SC pool and impaired regeneration (Boonsanay et al., 2016; Bjornson et al., 2012; Chakkalakal et al., 2012; Cheung et al., 2012; Gopinath et al., 2014; Mourikis et al., 2012; Rozo et al., 2016; von Maltzahn et al., 2013; Yamaguchi et al., 2015; Yue et al., 2017; Zhang et al., 2015). Similar observations have been made with long-term hematopoietic stem cells (Orford and Scadden, 2008). Therefore, it is thought that quiescence is a critical property of these stem cells for long-term function (Dumont et al., 2015; Orford and Scadden, 2008). Mechanisms whereby stem cells transit normally from a quiescent to a fully activated state and preserve function remain largely unknown.
The myofiber is a source of quiescence-promoting signals to its associated SCs (Bischoff, 1990), but the identity of these is obscure. SCs reside between myofibers and the surrounding basal lamina and have polarized adhesive contacts that tether them to this immediate niche (Yin et al., 2013). Basally, they express integrins (e.g., integrin α7β1) that bind laminins present in the basal lamina, and this interaction is important for maintenance of SC quiescence (Rozo et al., 2016). Apically, M-cadherin (Mcad, encoded by Cdh15) contributes to adhesive interactions with the myofiber.
Cadherins are a major class of cell adhesion molecule (Niessen et al., 2011). Extracellularly, they bind in a homophilic manner across the intercellular space. Cadherin intracellular regions bind directly to β- and γ-catenin (also called plakoglobin), which in turn bind α-catenin. α-catenin interacts directly and indirectly with the F-actin cytoskeleton, providing cortical tension and dynamic stability of cell-cell junctions. Several cadherins are expressed in developing skeletal muscle, including N-cadherin (Ncad, encoded by Cdh2) and Mcad (Krauss et al., 2005). Ncad is present throughout development of the muscle lineage, from uncommitted progenitor cells to proliferative myoblasts and nascent myofibers (Cifuentes-Diaz et al., 1993). It is down-regulated in adult myofibers, but strongly re-expressed during muscle regeneration, in a pattern similar to that seen in development (Cifuentes-Diaz et al., 1993). Mcad expression begins at the myoblast stage and is nearly exclusive to the skeletal muscle lineage (Moore and Walsh, 1993). In adult muscle, Mcad is localized to the apical SC membrane that is in direct contact with the myofiber plasma membrane (Irintchev et al., 1994). Ncad and Mcad have been proposed to play roles in lineage determination, myoblast differentiation, and myoblast fusion (Krauss et al., 2005). However, mice with a targeted, germline mutation in Cdh15 (Mcad KO) had no apparent defects in muscle development or regeneration (Hollnagel et al., 2002). Mice with a germline mutation in Cdh2 died by E10, but cultured somites from mutant embryos formed elongated cells that expressed muscle markers and had intact AJs (Radice et al., 1997). Therefore, neither Ncad nor Mcad is essential for myogenesis, and they may have compensatory functions during muscle development and regeneration.
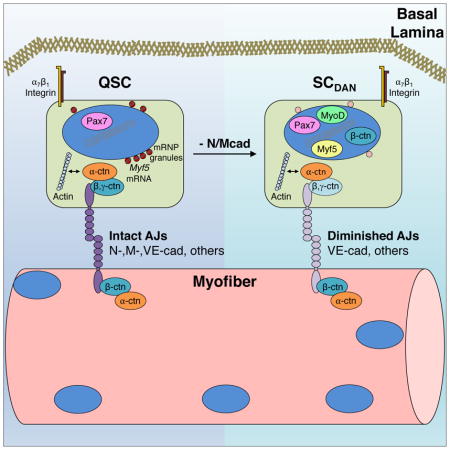
Here, we identify Ncad and Mcad as components of the quiescence-promoting SC niche. In adult muscle, Ncad and Mcad are expressed at sites of direct contact between SCs and myofibers. Genetic removal of these cadherins from SCs led to a break in quiescence but, in contrast to previous observations, resulted in long-term expansion of a regeneration-proficient SC pool. Ncad/Mcad-deficient SCs displayed a partial disruption of the myofiber-SC AJ but remained in the SC niche and maintained apical-basal polarity due to expression of additional AJ components. Removal of these niche cadherins resulted in: 1) multiple characteristics associated with the earliest stages of SC activation; and 2) a stem cell state that falls between quiescence and complete activation. In this manner, SC niche localization and polarity could be dissociated from initial signaling events in SC activation. Together, our findings argue that partial disruption of AJs in response to injury is a first step in the transition from quiescence to activation.
RESULTS
Ncad and Mcad are Dispensable for Skeletal Muscle Development
To assess the roles of Ncad and Mcad at different stages of myogenesis, a conditional mutagenesis approach was taken with Ncad, plus or minus germline removal of Mcad. Mice with a conditional Cdh2f/f allele were crossed to three different Cre driver mice: Pax7cre (active at the pre-commitment muscle progenitor stage), MyoDiCre (active at the developmental myoblast stage), and Pax7CreERT2 (for temporal removal from adult SCs). Removal of Ncad with Pax7Cre resulted in embryonic lethality, likely due to defects in neural crest development (Luo et al., 2006) (not shown). Mice in which Ncad was removed with MyoDiCre (Ncad cKO mice) were crossed with Mcad KO mice to generate double mutants (dKO mice). Ncad cKO, Mcad KO, and dKO mice were born in Mendelian ratios and were outwardly indistinguishable from control mice through at least one year of age. Additionally, muscle wet weights and fiber cross-sectional areas of all three mutants were similar to controls over this period (Figure S1A–E). MyoDiCre-mediated loss of Ncad was efficient, as seen by Western blot and immunohistochemistry of regenerating whole muscle (Figure S1F, G), and the Mcad KO is a protein null (Figure S1H). Therefore, Ncad and Mcad, singly or together, are not essential for muscle development.
Primary myoblasts were isolated from control and dKO animals and allowed to differentiate and fuse into multinucleated myotubes in culture. Previous studies showed that individual loss of Ncad or Mcad had no effect on this process (Charlton et al., 1997; Hollnagel et al., 2002). Myoblasts from control and dKO mice expressed similar levels of muscle differentiation markers, including Myosin Heavy Chain (MyHC) and Troponin T (TNNT-1) (Figure S1I). However, while myoblasts from control mice fused efficiently into myotubes, myoblasts from dKO mice failed to fuse (Figure S1J, K). Therefore, the combined absence of Ncad and Mcad led to a specific fusion defect in vitro. We suspect that this reflects the predicted function for these factors in myoblast fusion, but this role may be masked in vivo by the expression of additional cadherins that may not be maintained in culture.
SC Quiescence is Broken in Ncad cKO and dKO Mice
In adult muscle, Mcad was localized exclusively at the myofiber-SC junction (Figure 1A). As loss of Mcad does not lead to a SC phenotype, Ncad may be a compensatory factor. Ncad was difficult to observe in sections of uninjured adult muscle, possibly due to relatively low expression (not shown). To explore further, primary myofibers were isolated from the Extensor Digitorum Longus (EDL) muscle of wild-type animals, and analyzed by IF for Ncad and the SC marker Syndecan-4 (Sdc4). As with Mcad, Ncad was localized specifically to the myofiber-SC interface (Figure 1B). Ncad expression was detectable in >70% of EDL SCs.
Figure 1. SC Quiescence is Broken in Ncad cKO and dKO Mice.

(A) IF of a TA section stained for Mcad and laminin.
(B) IF of a single EDL fiber stained for Ncad and the SC marker, Sdc4.
(C) IF of EDL fibers from control and dKO mice stained for Ncad and Sdc4. Arrows indicate SCs. See also Figure S1F–H.
(D) IF of TA sections from control and dKO mice stained for the SC marker Pax7 and the basal lamina marker Laminin. Arrows indicate SCs. Insets show higher magnification. See also Figure S1L–O.
(E) Quantification of Pax7+ SCs in TA sections over a 12-month (MO) time course. Ncad cKO and dKO mice are different from control and Mcad KO mice after 1MO.
(F) IF of EDL fibers from control and dKO mice. Arrows indicate SCs.
(G) Quantification of Pax+ SCs on EDL fibers at 6MO.
(H) BrdU dosage protocol.
(I) IF of a TA section from dKO mouse stained for Pax7, BrdU, and Laminin. Arrows indicate a Pax7+/BrdU+ SC.
(J) Quantification of Pax7+/BrdU+ SCs in TA sections from control and dKO mice.
Scale bars: (A, B) 5μm; (C) 10μm; (D, I) 20μm; (F) 50μm. *, p <0.05, means±SD.
Virtually all SCs express MyoD in their history (Kanisicak et al., 2009). Consistent with this observation, and with the high efficiency of the MyoDiCre allele (Kanisicak et al., 2009), SCs from Ncad cKO mice did not display Ncad protein (Figure 1C); >1200 Ncad cKO SCs were scored and no Ncad+ cells were observed. The numbers of SCs in the Tibialis Anterior (TA) from control, Ncad cKO, Mcad KO, and dKO mice were then quantified at 1, 2, 6, and 12 months of age by staining for the SC marker Pax7 and the basal lamina marker laminin. At one month of age, about the time that SCs achieve adult numbers and quiescence (White et al., 2010), mice of all four genotypes had similar numbers of Pax7+ cells. However, beginning at two months of age, Ncad cKO and, to a greater extent, dKO mice showed an age-dependent increase in the number of SCs relative to control and Mcad mice (Figure 1D, E). All SCs remained within their niche, between the fiber and basal lamina (Figure 1D). Elevated numbers of SCs were seen in all muscles examined, including the TA, quadriceps, EDL, and diaphragm (Figure 1D–G, Figure S1L–O). Furthermore, this phenotype was fully penetrant and remarkably uniform between animals. Therefore, loss of Ncad resulted in an age-dependent expansion of SC numbers; loss of Mcad was without effect, but it enhanced the effects of Ncad mutation.
To validate that SCs in dKO mice had broken quiescence, three-month-old control and dKO mice were given a single dose of BrdU, and 12 hours later muscles were harvested (Figure 1H–J). BrdU incorporation was not seen in SCs from control animals, confirming their quiescent nature. In contrast, 4% of SCs from dKO mice incorporated BrdU, demonstrating that some dKO SCs were actively in the cell cycle.
Ncad cKO and dKO Muscles Display a Pseudo-Regenerative Response
Nuclei in adult myofibers reside in a peripheral location. Central nucleation is associated with regenerated fibers following acute injury or with muscle diseases (Folker and Baylies, 2013). We noticed that some dKO myofibers had centrally-located nuclei. To examine this further, control, Mcad KO, Ncad cKO, and dKO muscles were sectioned and stained with H&E at 1, 2, 6, and 12 months of age. TA muscles in all Ncad cKO and dKO mice, but not control or Mcad KO mice, displayed fibers with central nuclei at 2 months of age. Moreover, the percentage of fibers with at least one central nucleus increased over 12 months (Figure 2A, B, Figure S2A). dKO mice had a greater percentage of centrally-nucleated fibers than Ncad cKO mice. Serial sections showed that each TA fiber had only one or a few central nuclei, and, overall, six-month-old dKO fibers did not have an increased number of myonuclei (Figure S2B). Furthermore, fibers with long strings of central nuclei, which are characteristic of fibers arising anew from SC-mediated regeneration, were not observed. Central nucleation was also observed in the quadriceps and diaphragm of Ncad cKO and dKO mice (Figure S2C, D).
Figure 2. Ncad cKO and dKO Muscles Display a Pseudo-Regenerative Response.

(A) H&E-stained sections of uninjured TA muscle from 6MO-old control, McadKO, Ncad cKO, and dKO mice. Arrows indicate fibers with central nuclei. See also Figure S2A–D.
(B) Quantification of centrally-nucleated fibers in serial TA muscle sections.
(C) BrdU dosage protocol.
(D) IF of a TA section stained for Pax7, BrdU, and Laminin. Arrows indicate a Pax7+/BrdU+ SC; arrowheads, a Pax7+/BrdU− SC.
(E) Quantification of Pax7+/BrdU+ SCs in TA sections from control and dKO mice.
(F) IF of BrdU and Laminin in TA sections from control and dKO mice. Arrows indicate BrdU+ interstitial cells, arrowheads indicate BrdU+ central nuclei.
(G) Quantification of BrdU+ central nuclei in control and dKO mice.
(H) IF of BrdU and PDGFRα in TA sections from dKO mice. Arrows indicate BrdU+/PDGFRα+ interstitial cells, arrowhead a BrdU+ central nucleus. The dashed line indicates outline of a myofiber. See also Figure S2H.
(I) Quantification of BrdU+/PDGFRα+ interstitial cells in sections from control and dKO mice in areas with and without central nuclei.
Scale bars: (A) 50μm; (D, F, H) 20μm. *, p <0.05, means±SD.
As centrally-nucleated fibers arose in Ncad cKO and dKO mice in the absence of acute injury, their muscles were evaluated for evidence of disease. Ncad cKO and dKO muscles did not show expression of the early regeneration marker embryonic Myosin Heavy Chain (eMyHC), often seen in muscular dystrophies (Figure S2E). Similarly, they did not display other markers of muscle disease or degeneration, including fibrosis, lipid accumulation, muscle fiber-type switching, or inflammation (Figure S2E, F). Western blots on whole muscle extracts from these mice revealed similar levels of the muscle markers, MyHC and TNNT-1 (Figure S2G). A modest increase in MyoD expression was seen in Ncad cKO and dKO mice (Figure S2G), likely from expression in mutant SCs themselves (see below). Accumulation of central nuclei therefore occurred in the absence of obvious myopathy.
The increase in SC numbers and the accumulation of centrally-nucleated fibers had similar time courses, suggesting that the central nuclei arose from proliferative SCs which subsequently differentiated and fused with fibers, depositing their nuclei centrally. To address this possibility, two-month old control and dKO mice were labeled with BrdU for five days, followed by a three-week chase period. We reasoned that so long as proliferative dKO SCs did not divide to such an extent that the BrdU label was completely diluted prior to fusion, central nuclei should also be labeled. Consistent with this notion, 14% of dKO SCs and 62% of central nuclei in dKO TA fibers were BrdU+ with this protocol (Figure 2C–G). Importantly, BrdU+ SCs remained within their niche, under the basal lamina (Figure 2F).
During SC-mediated regeneration, PDGFRα+/Sca1+ interstitial cells called fibro-adipogenic progenitor cells (FAPs) also proliferate, and SCs and FAPs regulate each other’s numbers and differentiation during this process (Joe et al., 2010; Murphy et al., 2011). We noted that, while the increase in SC numbers in dKO mice was seen throughout the TA muscle, centrally-nucleated fibers were more regionally localized, accumulating largely at the superficial edge. Interstitial cells also incorporated BrdU, and in dKO mice BrdU+ cells were much more numerous in areas of central nucleation (Figure 2F, I). Regions of dKO muscle without central nucleation had similar numbers of BrdU+ interstitial cells as muscles of control mice. These interstitial cells expressed PDGFRα and Sca1, identifying them as FAPs (Figure 2H, Figure S2H). Elevated numbers of CD45+ cells were not found in regions of central nucleation. Together, these results argue that in specific areas of Ncad cKO and dKO muscle, resident SCs and FAPs communicated in a manner that led to proliferation of both cell types, with some SCs differentiating and fusing to their associated fibers. This is therefore a pseudo-regenerative process, in that it involved multiple muscle-resident cells that participate in regeneration, but was not associated with acute injury or signs of inflammation or muscle disease.
dKO Mice are Capable of Repetitive Muscle Regeneration
Loss of SC quiescence is usually associated with a reduction in the SC pool and impaired regeneration, yet dKO mice have an increase in SC numbers that persists for at least one year. To test regenerative potential, control and dKO mice were subjected to either one or three consecutive acute freeze injuries, with a 30 day recovery period after each injury (Figure 3A). dKO mice repaired both the single and triple injuries indistinguishably from control mice (Figure 3B). Even after the triple injury, the cross-sectional area of fibers in the regenerated region of the dKO mice was similar to control mice (Figure 3C). dKO Pax7+ SCs were found in their normal niche, between the basal lamina and myofiber, even after the third injury (Figure 3B). In previous studies, it was shown that within days of acute injury the number of Pax7+ SCs was strongly elevated and then tapered off over months (Hardy et al., 2016; Murphy et al., 2014). One month after a single freeze injury, control mice had ~4–fold more Pax7+ SCs than uninjured mice (Figure 3D). dKO mice displayed a similar ~4-fold-elevation of SC numbers one month following injury, relative to uninjured dKO animals (Figure 3D). Therefore, dKO mice maintained ~2.5–fold more SCs than controls after regeneration (Figure 3E). The same was true one month after the third injury (Figure 3D, E). Therefore, despite the break in quiescence by dKO SCs, expansion of SCs in dKO mice was stable through three cycles of regeneration.
Figure 3. dKO Mice are Capable of Repetitive Muscle Regeneration.

(A) Protocol for assessing injury-induced regeneration.
(B) H&E-stained sections from control and dKO TA muscles subjected to one or three injuries; IF of Pax7 and laminin in TA sections. Arrows indicate SCs. Insets show higher magnification. Scale bar, 20μm.
(C) Box and whisker plots for cross-sectional areas of fibers from the regenerated region of TA after triple injury.
(D) Quantification of Pax7+ cells in TA sections from control and dKO mice prior to injury and after single and triple injury. *, p<0.005, means±SD.
(E) Ratio of Pax7+ cells in dKO vs. control mice prior to injury and after single and triple injury.
dKO SCs are in a State of Transition from Quiescence to Activation
To study dKO SCs in further detail, we analyzed single fiber preparations that were freshly isolated or cultured under conditions in which SCs undergo activation and progression through discernable stages. Control SCs on single fibers are initially Pax7+/MyoD−/Myf5−. When cultured in medium supplemented with chick embryo extract (CEE), they become Pax7+/MyoD+/Myf5+, subsequently express the differentiation factor myogenin (MyoG), and later yet lose expression of Pax7. As expected, no control SCs on freshly isolated fibers expressed MyoD or Myf5; in contrast, 42% of dKO SCs were Pax7+/MyoD+ (Figure 4A, B), and 43% were Pax7+/Myf5+ (Figure S3A, B). When these fibers were incubated an additional three hours in medium without CEE, control SCs remained Pax7+/MyoD−, whereas 91% of dKO SCs were now Pax7+/MyoD+. After culture in CEE medium for 24 hours, 87% of control and 91% of dKO SCs were Pax7+/MyoD+ (Figure 4B). These results indicate that a large fraction of dKO SCs were MyoD+ in vivo, and, ex vivo, most of the rest expressed MyoD precociously without the need for CEE medium. Despite these findings, we were unable to detect MyoD by IF on sections of dKO muscles, even though MyoD was easily visualized in sections of control muscles at five days post-injury, a time when myoblasts are plentiful (not shown). However, we noticed that the intensity of the MyoD signal in dKO SCs on freshly isolated fibers was weaker than that seen in control or dKO SCs on fibers cultured for 24–48 hours with CEE medium. To quantify this, we measured corrected total cell fluorescence (CTCF) of MyoD in SCs of freshly isolated fibers (time 0) and fibers cultured with CEE medium for 24 and 48 hours. Control SCs at time 0 had a CTCF signal that represents background, and this signal rose substantially at 24 and 48 hours (Figure 4C). dKO SCs had significant MyoD CTCF signal at time 0, but this was less than one-third the signal achieved by 48 hours of culture. In fact, control and dKO SCs were similar at the 24- and 48-hour time points. These results argue that some dKO SCs were MyoD+ in vivo, but their MyoD levels were lower than in fully activated SCs or myoblasts, rendering it below the IF detection level in sections. To prove this point, and to rule out that MyoD expression was induced during fiber isolation, we dissected dKO muscles and isolated fibers in cycloheximide (CHX)-containing media (Figure 4D). CHX did not alter the percentage of dKO SCs that were Pax7+/MyoD+ at time 0. However, after 24 hours of culture in CEE medium, only the SCs without CHX progressed to >90% Pax7+/MyoD+, showing that CHX was effective in blocking SC activation. Therefore, consistent with their incorporation of BrdU in vivo, >40% of dKO SCs expressed the activation marker MyoD+ in vivo, albeit at levels below those seen in myoblasts.
Figure 4. dKO SCs Display Properties of an Early State of Activation.

(A) IF of freshly isolated single EDL fibers from control and dKO mice stained for Pax7 and MyoD. Arrows indicate SCs. Insets, same SCs at higher magnification.
(B) Quantification of Pax7+/MyoD− and Pax7+/MyoD+ SCs on freshly isolated fibers (0hr), after 3hrs in collection buffer, and after 24hr of culture with CEE medium.
(C) Corrected total cell fluorescence (CTCF) of MyoD per SC on control and dKO fibers at 0hr, and after 24 and 48hrs of culture in CEE medium. AU, arbitrary units.
(D) Quantification of Pax7+/MyoD− and Pax7+/MyoD+ SCs on fibers from dKO mice at 0hr and after 24hrs of culture with CEE medium, ± cycloheximide (CHX).
(E) Quantification of MyoD+/MyoG− and MyoD+/MyoG+ cells on fibers from control and dKO mice after 24, 48, and 72hrs of culture with CEE medium. See also Figure S3D.
(F) Quantification of Pax7+/MyoD−, Pax7+/MyoD+ and Pax7−/MyoD+ cells on fibers from control and dKO mice at 0hrs and after 24, 48, and 72hrs of culture with CEE medium. See also Figure S3C.
(G) IF of Pax7 and Ddx6 in SCs on freshly isolated fibers from control and dKO mice.
(H) Beeswarm plot with violin plot overlaid of number of mRNP granules per SC in control and dKO mice. Scale bars: (A) 20μm; (G) 10μm. *, p<0.0001, means±SD.
To assess later stages of SC activation and differentiation, control and dKO fibers were cultured in CEE medium for up to 72 hours and then stained for Pax7, MyoD, and MyoG. No differences between control and dKO SCs were seen in the percentages or kinetics of MyoD+/MyoG+ and Pax7−/MyoD+ cells (Figure 4E, F; Figure S3C, D). Together, these data indicate that dKO SCs were in a partial state of activation characterized by low levels of MyoD, but once they received a full activation stimulus, they proceeded through activation and began differentiation similarly to controls. The findings further suggest that loss of Ncad/Mcad may mimic mechanisms whereby wild type SCs enter the activation process. We therefore examined dKO SCs for one of the earliest known steps to occur during SC activation.
In quiescent SCs, full length Myf5 mRNA is sequestered within cytoplasmic mRNP granules, so that its translation is prevented by miR-31 (Crist et al., 2012). Upon SC activation, mRNP granules dissociate, and Myf5 mRNA is rapidly translated. Freshly isolated fibers from control and dKO mice were stained for Pax7 and Ddx6, a component of the mRNP granules (Figure 4G, H). Similar to a previous report (Crist et al., 2012), control SCs had 6 to 16 Ddx6+ cytoplasmic puncta per cell, corresponding to intact mRNP granules. However, dKO SCs separated into two discrete populations: 1) cells with a number of cytoplasmic granules similar to control SCs; and 2) cells showing fewer granules and diffuse cytoplasmic Ddx6 staining, consistent with the granule dissociation seen early after wild type SC activation (Crist et al., 2012). The percentage of dKO SCs in the second population is similar to the percentage of dKO SCs that are positive for Myf5 and MyoD. Therefore, dKO SCs displayed properties associated with the early stages of SC activation. Following muscle injury, expression of Ncad and Mcad increases in the myoblast progeny of SCs (Krauss et al., 2005). Consistent with these reports, Ncad levels increased in SCs on fibers cultured for 6–48 hours (Figure S3E), likely related to its roles in differentiation and fusion. Therefore, a simple loss of Ncad expression is not a mechanism of early SC activation (see Discussion).
Removal of Ncad from Adult SCs Phenocopies Its Removal in Development
Use of MyoDiCre leads to loss of Ncad in developing myoblasts, yet the earliest phenotype seen in Ncad cKO and dKO mice was in SCs at two months of age. Additionally, both myofibers and SCs lacked Ncad in these mice. To prove this was a SC-specific phenotype, Ncad was removed with the Pax7CreERT2 allele (these mice were also germline mutant for Mcad and are designated dKOSC). Mice were treated with tamoxifen at one month of age, a time when control and dKO mice have similar numbers of SCs, and studied one month after completion of tamoxifen treatment (Figure 5A). Based on scoring Ncad+ SCs, recombination frequency was 87% (Figure S4A). dKOSC animals had a two-fold elevation in Pax7+ SC numbers in vivo and on single fibers (Figure 5B–D), similar to two-month-old dKO mice. Furthermore, 29% of SCs on single fibers from dKOSC mice were Pax7+/MyoD+ (Figure 5E, Figure S4B). Finally, dKOSC mice displayed fibers with central nuclei (Figure 5F) and an elevated number of FAPs that incorporated BrdU in regions of central nucleation (Figure 5G, H), also similar to dKO mice. Therefore, removal of Ncad at either the developmental myoblast stage or cell-autonomously from SCs resulted in very similar phenotypes.
Figure 5. Removal of Ncad from Adult SCs Phenocopies Removal During Development.

(A) Strategy for inducing recombination in dKOSC mice.
(B) IF of TA sections and EDL fibers from control and dKOSC mice stained for Pax7 and Laminin (sections, left) or Pax7 (fibers, right).
(C, D) Quantification of Pax7+ SCs from such sections and fibers. *, p<0.05, means±SD.
(E) Quantification of Pax7+/MyoD− and Pax7+/MyoD+ SCs on freshly isolated single fibers from control and dKOSC mice. See also Figure S4B.
(F) H&E-stained sections of uninjured TA muscle from control and dKOSC mice. Arrows indicate fibers with central nuclei.
(G) IF of BrdU and PDGFRα in TA sections from dKOSC mice. Arrows indicate BrdU+/PDGFRα+ interstitial cells, arrowhead indicates a central nucleus. Dashed lines indicate outlines of myofibers. BrdU labeling was as in Figure 2C.
(H) Quantification of BrdU+/PDGFRα+ interstitial cells in sections from control and dKOSC mice in areas with and without central nuclei.
Scale bars: (B, G) 20μm; (F) 50μm.
dKO SCs Have Partially Disrupted Adhesive Junctions
Classical cadherins exert their effects by assembling intracellular proteins, notably the catenins, at sites of homophilic adhesion between two cells (Niessen et al., 2011). We used IF to assess localization of AJ-associated proteins in control and dKO SCs on freshly isolated fibers and quantified the ratio of apical to basal signal for each factor. Confocal images revealed β-catenin was localized at the apical membrane in all control SCs, similar to Ncad and Mcad (Figure 6A, G). In contrast, in fibers from dKO mice, 76% of SCs displayed β-catenin at both the cell periphery and in the nucleus (Figure 6B, G). dKOSC SCs also had nuclear-localized β-catenin (Figure S5A). Although nuclear β-catenin is associated with the transcriptional response to canonical Wnt signaling, it can also be derived via perturbation of the junctional pool (McCrea et al., 2015). We suspect that this is the case in dKO SCs (see Discussion). To begin to assess whether nuclear β-catenin promoted a transcriptional response in dKO SCs, fibers were stained for Axin2, a direct transcriptional target of β-catenin/TCF signaling. Control SCs were negative for cytoplasmic Axin2. In contrast, 22% of dKO SCs were strongly positive for cytoplasmic Axin2 (Figure S5B, C), indicating that some dKO SCs responded transcriptionally to nuclear β-catenin.
Figure 6. dKO SCs Have Partially Disrupted Adhesive Junctions.

(A) IF of freshly isolated, single EDL fibers from control and dKO mice stained for Sdc4 and β-catenin. See also Figure S5A.
(B) Quantification of SCs with nuclear β-catenin.
(C–F) IF of freshly isolated, single EDL fibers from control and dKO mice stained for (C) Sdc4 and αT-catenin;(D) Sdc4 and γ-catenin; (E) Sdc4 and VE-cadherin; and (F) α7-integrin and αT-catenin.
(G) Quantification of the ratio of apical to basal IF signal intensity for the indicated proteins in SCs from control and dKO mice. Data are plotted as log2 of the apical-to-basal ratio of signal intensity. Positive values indicate apical localization, negative values indicate basal localization. Note that polarized expression of β-catenin was lost in dKO SCs, whereas it was maintained for the other factors.
Scale bars: 5μm. *, p<0.001, means±SD.
α-catenin is encoded by three isoforms (αE, αT, and αN); αE and αT are expressed in SCs at the RNA level (Ryall et al., 2015). αT-catenin localized mainly to the SC apical membrane in both control and dKO SCs (Figure 6C, G). This finding, combined with the mislocalization of β-catenin, suggests that additional cadherins and β-catenin-like adhesive function remain in dKO SCs. SCs express VE-cadherin (Fukada et al., 2007), and we found that both control and dKO SCs displayed VE-cadherin enriched at apical membranes (Figure 6E, G). Similarly, γ-catenin is expressed by SCs (Cohen et al., 2014), and it too was enriched at apical membranes of control and dKO SCs (Figure 6D, G). Furthermore, the basal membrane SC marker, α7-integrin, was basally localized in both control and dKO SCs (Figure 6F, G). Therefore, despite disruption of β-catenin localization, dKO SCs appeared to maintain normal apical-basal polarity. Together, these results argue that SC AJs are diminished in the absence of Ncad/Mcad, but not so compromised as to lead to a loss of cell polarity.
β-catenin Haploinsufficiency Rescues SC phenotypes in dKO mice
To address whether deregulation of β-catenin was required for the break in quiescence in SCs lacking Ncad/Mcad, we crossed mice carrying a conditional β-catenin null allele (Ctnnb1f/f) to dKO mice to generate dKO animals lacking one copy of β-catenin (dKO;β-ctnf/+ mice). Two-month-old control, dKO, and dKO;β-ctnf/+ mice were compared for SC numbers in vivo, SC numbers and percentage of Pax7+/MyoD+ SCs on freshly isolated fibers, and presence of centrally nucleated fibers in vivo. Strikingly, dKO;β-ctnf/+ mice had a similar number of SCs as control mice (Figure 7A, B; Figure S6A). Also included as a control were mice heterozygous for the Ncadf, Mcad−, MyoDiCre, and Ctnnb1f alleles (4H mice), so as to assess the effects of reduced β-catenin levels on a genetic background not associated with elevated SC numbers. 4H mice had similar numbers of SCs as control mice, indicating that β-catenin heterozygosity itself had no effect. Furthermore, 17% of SCs on single fibers from dKO;β-ctnf/+ mice were Pax7+/MyoD+, as compared to 42% in dKO mice (Figure 7C; Figure S6B). Additionally, there were very few centrally-nucleated fibers in dKO;β-ctnf/+ mice (Figure 7D), demonstrating that the normalized numbers of SCs in these mice were not a consequence of enhanced differentiation and fusion to fibers. Consistent with the rescue of the dKO phenotype, we did not observe dKO;β-ctnf/+ SCs with strong cytoplasmic Axin2 staining (not shown).
Figure 7. β-catenin Haploinsufficiency Rescues SC phenotypes in dKO mice.

(A) IF of TA sections from control, dKO, and dKO;β-ctnf/+ mice stained for Pax7 and laminin. Arrows indicate SCs. Arrowhead indicates fiber with central nucleus.
(B) Quantification of Pax7+ SCs in TA sections from control, 4H, dKO, and dKO;β-ctnf/+ mice. *, p<0.01, means±SD.
(C) Quantification Pax7+/MyoD− and Pax7+/MyoD+ SCs on freshly isolated single fibers from control, dKO and dKO;β-ctnf/+ mice. See also Figure S6B.
(D) H&E-stained sections of uninjured TA muscle from dKO and dKO;β-ctnf/+ mice. Arrows indicate fibers with central nuclei.
(E) Model in which SCDAN arises from loss of N/Mcad and may represent a transition state that occurs as quiescent SCs become activated. The deepening color of the different cell states represents progression from quiescence to transit-amplifying myoblast. Dashed lines indicate the possibilities that SCDAN cells can either return to the quiescent state or self-renew.
Scale bars: 50μm.
Together, our findings indicate that genetic removal of the niche cadherins Ncad and Mcad diminishes, but does not fully abrogate, AJ function, leading to a state that mimics the earliest stages of SC activation. These results further suggest that partial disruption of AJs is an initial signal in the transition from quiescence to activation.
DISCUSSION
Many adult stem cells are characterized by prolonged states of quiescence. Mechanisms must exist for both maintenance of quiescence and, following injury, a well-orchestrated transition into the activation process. Perturbations in this process are often associated with loss of long-term stem cell function (Dumont et al., 2015; Orford and Scadden, 2008). Cues from the stem cell niche are involved in maintaining quiescence but the identity of these signals is still emerging. Additionally, how the exit from quiescence is integrated with entry to the activated state is largely unknown. We have addressed this issue with skeletal muscle SCs. Our results demonstrate that cadherin-mediated adhesion is required for SC quiescence. Furthermore, removal of specific cadherins from SCs engenders a state resembling an early stage of SC activation. Adhesion of stem cells to neighboring cells is a common theme of stem cell-niche interactions, and these findings may be of general relevance to how stem cells undergo quiescence-to-activation transitions.
The Myofiber-SC AJ: A Critical Component of the SC Niche
SCs are polarized, with cadherins present on their apical membrane and integrins on their basal membrane (Yin et al., 2013). Mcad, a largely muscle-specific cadherin, was known to reside specifically at the myofiber-SC interface, but the absence of apparent defects in Mcad KO mice raised the possibility that cadherin-based AJs were not key regulators of SC behavior (Hollnagel et al., 2002). Our findings demonstrate that AJs are required for maintenance of SC quiescence, but Ncad plays a primary role with Mcad functioning in a compensatory manner. This study also showed several other factors to be localized to the myofiber-SC AJ, including at least one additional cadherin (VE-cadherin), both β- and γ-catenin, and αT-catenin. dKO SCs maintained a polarized distribution of VE-cadherin, γ-catenin, αT-catenin, and α7-integrin, despite their loss of Ncad/Mcad and a consequent partial relocalization of β-catenin to the nucleus. Therefore, even in the absence of Ncad/Mcad and an incomplete complement of junctional β-catenin, dKO AJs had sufficient levels of other cadherins, γ-catenin, and α-catenins to preserve key aspects of niche function, including apical attachment of SCs to the fiber (i.e., residence in the niche) and apical-basal SC polarity.
The nuclear β-catenin seen in dKO and dKOSC SCs likely represents β-catenin that would normally be localized at AJs, but translocated to the nucleus as an outcome of fewer binding sites being available at the AJ due to loss of Ncad/Mcad. Alterations of cadherin levels can have this effect, although the “liberated” β-catenin must still avoid degradation (McCrea et al., 2015). An alternative possibility is that loss of Ncad/Mcad led to activation of canonical Wnt signaling. However, this seems less likely, as in the case of dKOSC SCs, Ncad removal is cell-autonomous to a very small population of isolated single cells in muscle.
Cadherin-Mediated Adhesion and the Quiescence-to-Activation Transition
Removal of Ncad/Mcad led to a break in quiescence and a stable increase in Pax7+ SCs that persisted for at least one year. Furthermore, dKO mice were regeneration-proficient and maintained a 2.5 – 3-fold elevation of Pax7+ SCs relative to control mice, through multiple injuries. This was therefore distinct from the breaks in SC quiescence observed upon genetic removal of many other factors, including Pax7, RBP-J, FOXO3, Dicer, Pten, β1 integrin, calcitonin receptor, Suv4-20h1, and Prmt5, since removal of any of these factors led to a loss of SCs and an inability to repair an acute injury (Boonsanay et al., 2016; Bjornson et al., 2012; Cheung et al., 2012; Gopinath et al., 2014; Mourikis et al., 2012; Rozo et al., 2016; von Maltzahn et al., 2013; Yamaguchi et al., 2015; Yue et al., 2017; Zhang et al., 2015). This distinction may derive from whether the break in quiescence occurs in a way that mirrors what occurs normally in response to injury (see below).
Several events occur rapidly as SCs transition from quiescence into activation. Among these are: 1) dissociation of Ddx6-containing mRNP granules, resulting in liberation and translation of Myf5 mRNA; 2) posttranscriptional stabilization and translation of MyoD mRNA; and 3) β-catenin/TCF-dependent reporter activity and Axin2 expression (Brack et al., 2008; Crist et al., 2012; Hausburg et al., 2015; Murphy et al., 2014). More than 40% of dKO SCs constitutively displayed these properties; therefore, they appear comparable to SCs in an early stage of activation. Consistent with this conclusion, the amount of MyoD protein expressed by dKO SCs on freshly isolated single fibers was significantly lower than the amount expressed by control or dKO SCs on fibers cultured for 48 hours in activation medium. Similarly, while 76% of dKO SCs displayed nuclear β-catenin, only 22% of these cells were positive for cytoplasmic Axin2. We suspect that nuclear β-catenin may accumulate with time, and only cells that have reached a threshold level transcribe Axin2. It should be noted that the β-catenin/TCF-dependent reporter activity seen early after muscle injury has been viewed as indicative of canonical Wnt signaling. However, some of this activity may arise from β-catenin translocation from the SC AJ to the nucleus. This is reminiscent of the results of Sieiro et al., who showed that some β-catenin/TCF-dependent activation of Myf5 expression in somitic muscle progenitors was driven by AJ-derived β-catenin (Sieiro et al., 2016).
Once dKO SCs in single fiber cultures were placed in activation medium, they behaved similarly to control SCs in expression kinetics of later markers. Therefore, dKO SCs have likely accomplished the early stages of activation in a manner resembling that which occurs normally as a physiological response to injury, allowing them to progress normally to full activation. This then suggests that the SC loss associated with genetic removal of the factors listed above may be linked to an entry into the activation process that is temporally out of sequence.
Niche-based, quiescence-inducing cues must be overridden when SCs respond to muscle injury. We hypothesize that AJs maintain the quiescent SC state by providing structural integrity, mechanosensation, cell polarity, and juxtacrine signaling. Our findings argue that signals generated by cadherin-based AJs are abrogated as part of the initial activation process, promoting an orderly entry into the activated state, including dissociation of mRNP granules, post-transcriptional production of MyoD and Myf5, and liberation of β-catenin from AJs to allow nuclear translocation. Localization of β-catenin in the nucleus, rather than in AJs, provides one mechanism whereby the break in quiescence can be linked directly to the transition to activation. Removal of one copy of β-catenin efficiently rescued dKO SC phenotypes, providing genetic evidence that its nuclear function in dKO cells was required for the quiescence-to-activation transition. However, it is very unlikely to be sufficient, as SCs in mice that were engineered to have constitutively nuclear β-catenin did not break quiescence, at least not in the time frame exhibited by dKO or dKOSC SCs (Parisi et al., 2015; Rudolf et al., 2016). Therefore, additional mechanisms seen in dKO SCs (e.g., posttranscriptional events associated with Myf5 and MyoD protein synthesis) are also required, as would be expected for a highly controlled exit from quiescence and entry into the activation process.
Several potential mechanisms exist for dismantling myofiber-SC junctions upon muscle injury. One likely possibility is that injury-induced physical disruption of myofibers is immediately sensed by AJs, which are then altered, physically and/or functionally. Cadherins are components of the junctional fluid shear stress mechanoreceptor in endothelial cells (Baeyens and Schwartz, 2016), and SC AJs may also have mechanoreception properties. Following an immediate, injury-induced physical response, SC cadherins might continue to have their quiescence-promoting functions limited by additional mechanisms. Cadherins can undergo protease-mediated ectodomain shedding, and their intracellular regions are subject to phosphorylation (as is β-catenin); these events diminish cell-cell adhesion (Daugherty and Gottardi, 2007; Saftig and Lichtenthaler, 2015). These are non-mutually exclusive possibilities and each may occur as SCs proceed through the activation process. We note, however, that Ncad and Mcad expression is not down-regulated upon injury, as the myoblast progeny of SCs show higher expression levels than SCs (Krauss et al., 2005; this study), likely due to the cadherins’ later participation in myoblast differentiation and fusion.
Incomplete Disruption of the Myofiber-SC AJ Reveals the SCDAN State
Adult stem cells have generally been thought to exist in either quiescent or activated states. Our findings, and the recent work of others, suggest the existence of states that fall between quiescence and full activation. Incomplete disruption of AJs in dKO SCs allowed these cells to enter an early stage of activation but held them back from complete activation. Presumably, all dKO SCs achieve some minimal phenotype associated with loss of Ncad/Mcad, and then proceed in a stochastic manner through various additional steps (e.g., nuclear β-catenin localization, expression of Myf5, MyoD, and Axin2, etc.). Some dKO SCs in this state appeared to progress to full activation, even in the absence of injury, as demonstrated by their incorporating BrdU, proliferating, communicating with local FAPs, and differentiating to fuse with fibers. Their residence in an apparently normal early stage of activation in turn allowed them to proceed into full activation when confronted with an acute injury in vivo or activation medium in single fiber cultures. We have named this the SCDAN state, for “diminished adhesive niche”. Although we identified the SCDAN state via genetic removal of specific niche AJ cadherins, it may represent a transition state that SCs normally traverse in response to injury, when emerging from quiescence and on the way to a fully activated state (Figure 7E). If so, it is not clear whether this state may exist as a relatively stable one, perhaps dependent on the severity of the injury, or one that is traversed quickly without “pause”. A distinct SC state, GAlert, was recently described as an adaptive response associated with SCs in muscles contralateral to an injured muscle (Rodgers et al., 2014). GAlert SCs entered the first division more rapidly than quiescent SCs and displayed enhanced regenerative properties upon injury; however, they did not cycle efficiently in vivo and did not express MyoD, distinguishing them from SCDAN cells as being closer to G0 quiescent SCs. GAlert SCs could give rise to fully activated cells or return to G0 (Rodgers et al., 2014). SCDAN cells also gave rise to fully activated cells and, based on the ability of dKO mice to repair multiple injuries and maintain elevated numbers of Pax7+ SCs, we infer that they are capable of self-renewal. It is possible that SCDAN cells themselves may be able to self-renew; alternatively, they may need to return to a quiescent state to do so (Figure 7E). Both the GAlert and SCDAN states can be distinguished from yet another possible state, seen in mice lacking the cell surface SC proteoglycan syndecan-3 (Sdc3). Sdc3−/− mice have a normal number of Pax7+ SCs that generate many Pax7−/Myf5+ progeny in the absence of injury, yet retain regenerative potential (Pisconti et al., 2016). The notion of multiple SC states may be analogous to the evolving view of pluripotency, in which naïve, formative, and primed states exist in a progression towards ultimate lineage commitment (Smith, 2017).
In summary, this study uncovered mechanisms whereby stem cell niche components both promote long-term quiescence and coordinate the transition from quiescence to activation. We used skeletal muscle SCs as a model system but it is likely that analogous mechanisms underlie these processes in other quiescent stem cell types. Furthermore, it is possible that our findings can be exploited in a therapeutic manner. The impressive regenerative potential of SCs has suggested that they may be useful in cell-based therapies for regenerative medicine (Cosgrove et al., 2014; Hall et al., 2010; Tedesco et al., 2010). Alternatively, it has been proposed that targeting stem cell niches may enhance the therapeutic potential of endogenous stem cell populations (Dimmeler et al., 2014; Lane et al., 2014). Agents that target the SC niche to promote the progression of quiescent SCs into the SCDAN state may be useful in this regard. As niche-based regulators of SC quiescence, Ncad and Mcad are potential targets in such strategies.
EXPERIMENTAL PROCEDURES
Mice
This study was carried out in accordance with recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH. The protocol was approved by the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee. The Mount Sinai animal facility is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Cdh2f/f and Cdh15−/− mice were as described (Hollnagel et al., 2002; Kostetskii et al., 2005). All other mouse lines were obtained from Jackson Laboratories (Bar Harbor, ME). For muscle regeneration assays, the TA muscle of anesthetized mice was subjected to a freeze injury (Hardy et al., 2016). Dissected muscles were frozen, sectioned, and analyzed by IF, as described (Joseph et al., 2017). Male and female mice had similar phenotypes and were studied in equal numbers at all stages studied, which ranged from one to twelve months of age.
Single Fiber and Primary Myoblast Isolation and Culture
Single muscle fibers were isolated from EDL muscles of adult mice and cultured and analyzed by IF as described elsewhere (Vogler et al., 2016; Zammit et al., 2004). Primary myoblasts were isolated from hind limbs of P14-P21 mice and cultured as described (Cole et al., 2004; Rando and Blau, 1994).
Imaging and Post-Imaging Analyses
Wide-field images were acquired using Zeiss Axioplan2 and AxioImager Z2 microscopes with a Zeiss Axiocam 503 monochrome camera for imaging fluorescence. Confocal images were acquired using a Leica SP5 DMI inverted quad laser scanning confocal microscope with Leica Application Suite software. Images were exported to ImageJ and FIJI.
Statistical analysis
Data are expressed as means±SD. Means, SD, 95% confidence intervals, and unpaired Student’s t tests, were calculated using Microsoft Excel and GraphPad Software. Boxplots, beeswarm, and violin plots were generated using R software. Three to six mice were used per condition, and each experiment was repeated at least twice. For quantifications using sections, 6–15 random sections were analyzed per mouse. For quantifications using primary EDL myofibers, 15–30 fibers were analyzed per mouse. For quantification of central nuclei, 2×30 serial sections were analyzed per mouse, with groups of 110–160 myofibers analyzed per section. Ncad recombination efficiency using the Pax7CreERT2 allele was determined by scoring Ncad+ SCs in myofibers from control and dKOSC mice (100 SCs scored from 3 mice per genotype).
Supplementary Material
Highlights.
Niche cadherins regulate muscle stem cell (SC) quiescence
Expansion of a regeneration-proficient SC pool associated with loss of cadherins
Cadherin-deficient SCs enter a state of partial activation
Nuclear β-catenin signaling is a necessary downstream driver of these phenotypes
Acknowledgments
We thank D. Cornelison, C. Crist, A. Brack, and P. Gilbert for gifts of reagents and helpful advice, N. Beckmann for help with statistics and graphing, I. Cheng for technical support, I. Kos tetskii and E. Barton for assistance with the initial analysis of the Ncad cKO model, and P. Soriano and P. Wassarman for critical reading of the manuscript. Microscopy was performed at the Mount Sinai Microscopy CoRE. IVF rederivation was performed at the Mount Sinai Mouse Genetics Facility. This work was supported by NIH grants AR046207 and AR070231 to R.S.K. and DFG support to H.-H.A. This research was supported in part by the Tisch Cancer Institute at Mount Sinai P30 CA196521 – Cancer Center Support Grant. No authors of this manuscript have a conflict of interest.
Footnotes
AUTHOR CONTRIBUTIONS
A.J.G. conceived and performed experiments, acquired and analyzed data, and wrote the manuscript. M.K.R. acquired data and provided technical assistance. H.-H.A. and G.L.R. provided critical reagents and scientific input. R.S.K. conceived experiments, wrote the manuscript, and managed the project.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baeyens N, Schwartz MA. Biomechanics of vascular mechanosensation and remodeling. Mol Biol Cell. 2016;27:7–11. doi: 10.1091/mbc.E14-11-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff R. Interaction between satellite cells and skeletal muscle fibers. Development. 1990;109:943–952. doi: 10.1242/dev.109.4.943. [DOI] [PubMed] [Google Scholar]
- Bjornson C, Cheung T, Liu L, Tripathi P, Steeper K, Rando T. Notch Signaling Is Necessary to Maintain Quiescence in Adult Muscle Stem Cells. Stem Cells. 2012;30:232–242. doi: 10.1002/stem.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonsanay V, Zhang T, Georgieva A, Kostin S, Qi H, Yuan X, Zhou Y, Braun T. Regulation of Skeletal Muscle Stem Cell Quiescence by Suv4-20h1-Dependent Facultative Heterochromatin Formation. Cell Stem Cell. 2016;18:229–242. doi: 10.1016/j.stem.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell. 2008;2:50–59. doi: 10.1016/j.stem.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Brack AS, Rando TA. Tissue-Specific Stem Cells: Lessons from the Skeletal Muscle Satellite Cell. Cell Stem Cell. 2012;10:504–514. doi: 10.1016/j.stem.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakkalakal JV, Jones KM, Basson MA, Brack AS. The aged niche disrupts muscle stem cell quiescence. Nature. 2012;490:355–360. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton CA, Mohler WA, Radice GL, Hynes RO, Blau HM. Fusion competence of myoblasts rendered genetically null for N-cadherin in culture. J Cell Biol. 1997;138:331–336. doi: 10.1083/jcb.138.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Lewallen M, Xie T. Adhesion in the stem cell niche: biological roles and regulation. Development. 2013;140:255–265. doi: 10.1242/dev.083139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TH, Quach NL, Charville GW, Liu L, Park L, Edalati A, Yoo B, Hoang P, Rando TA. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature. 2012;482:524–528. doi: 10.1038/nature10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Nicolet M, Goudou D, Rieger F, Mège R. N-cadherin and N-CAM-mediated adhesion in development and regeneration of skeletal muscle. Neuromuscular Disorders. 1993;3:361–365. doi: 10.1016/0960-8966(93)90078-x. [DOI] [PubMed] [Google Scholar]
- Cohen S, Lee D, Zhai B, Gygi SP, Goldberg AL. Trim32 reduces PI3K–Akt–FoxO signaling in muscle atrophy by promoting plakoglobin–PI3K dissociation. J Cell Biol. 2014;204:747–758. doi: 10.1083/jcb.201304167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole F, Zhang W, Geyra A, Kang JS, Krauss RS. Positive regulation of myogenic bHLH factors and skeletal muscle development by the cell surface receptor CDO. Dev Cell. 2004;7:843–854. doi: 10.1016/j.devcel.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med. 2014;20:255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crist CG, Montarras D, Buckingham M. Muscle Satellite Cells Are Primed for Myogenesis but Maintain Quiescence with Sequestration of Myf5 mRNA Targeted by microRNA-31 in mRNP Granules. Cell Stem Cell. 2012;11:118–126. doi: 10.1016/j.stem.2012.03.011. [DOI] [PubMed] [Google Scholar]
- Daugherty RL, Gottardi CJ. Phospho-regulation of β-Catenin Adhesion and Signaling Functions. Physiology. 2007;22:303–309. doi: 10.1152/physiol.00020.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Ding S, Rando TA, Trounson A. Translational strategies and challenges in regenerative medicine. Nat Med. 2014;20:814–821. doi: 10.1038/nm.3627. [DOI] [PubMed] [Google Scholar]
- Dumont NA, Wang YX, Rudnicki MA. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development. 2015;142:1572–1581. doi: 10.1242/dev.114223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folker E, Baylies M. Nuclear positioning in muscle development and disease. Front Physiol. 2013;4 doi: 10.3389/fphys.2013.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada S, Uezumi A, Ikemoto M, Masuda S, Segawa M, Tanimura N, Yamamoto Hi, Miyagoe-Suzuki Y, Takeda S. Molecular Signature of Quiescent Satellite Cells in Adult Skeletal Muscle. Stem Cells. 2007;25:2448–2459. doi: 10.1634/stemcells.2007-0019. [DOI] [PubMed] [Google Scholar]
- Gopinath S, Webb A, Brunet A, Rando T. FOXO3 Promotes Quiescence in Adult Muscle Stem Cells during the Process of Self-Renewal. Stem Cell Reports. 2014;2:414–426. doi: 10.1016/j.stemcr.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JK, Banks GB, Chamberlain JS, Olwin BB. Prevention of Muscle Aging by Myofiber-Associated Satellite Cell Transplantation. Sci Transl Med. 2010;2:57ra83–57ra83. doi: 10.1126/scitranslmed.3001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy D, Besnard A, Latil M, Jouvion G, Briand D, Thépenier C, Pascal Q, Guguin A, Gayraud-Morel B, Cavaillon JM, et al. Comparative Study of Injury Models for Studying Muscle Regeneration in Mice. PLOS ONE. 2016;11:e0147198. doi: 10.1371/journal.pone.0147198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausburg MA, Doles JD, Clement SL, Cadwallader AB, Hall MN, Blackshear PJ, Lykke-Andersen J, Olwin BB. Post-transcriptional regulation of satellite cell quiescence by TTP-mediated mRNA decay. eLife. 2015;4:e03390. doi: 10.7554/eLife.03390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollnagel A, Grund C, Franke WW, Arnold HH. The cell adhesion molecule M-cadherin is not essential for muscle development and regeneration. Mol Cell Biol. 2002;22:4760–4770. doi: 10.1128/MCB.22.13.4760-4770.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irintchev A, Zeschnigk M, Starzinski-Powitz A, Wernig A. Expression pattern of M-cadherin in normal, denervated, and regenerating mouse muscles. Developmental Dynamics. 1994;199:326–337. doi: 10.1002/aja.1001990407. [DOI] [PubMed] [Google Scholar]
- Joe AWB, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FMV. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010;12:153–163. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph GA, Lu M, Radu M, Lee JK, Burden SJ, Chernoff J, Krauss RS. Group I Paks Promote Skeletal Myoblast Differentiation In Vivo and In Vitro. Mol Cell Biol. 2017;37:e00222–00216. doi: 10.1128/MCB.00222-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Mulieri PJ, Miller C, Sassoon DA, Krauss RS. CDO, a Robo-related cell surface protein that mediates myogenic differentiation. J Cell Biol. 1998;143:403–413. doi: 10.1083/jcb.143.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanisicak O, Mendez JJ, Yamamoto S, Yamamoto M, Goldhamer DJ. Progenitors of skeletal muscle satellite cells express the muscle determination gene, MyoD. Dev Biol. 2009;332:131–141. doi: 10.1016/j.ydbio.2009.05.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD, Radice GL. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res. 2005;96:346–354. doi: 10.1161/01.RES.0000156274.72390.2c. [DOI] [PubMed] [Google Scholar]
- Krauss RS, Cole F, Gaio U, Takaesu G, Zhang W, Kang JS. Close encounters: regulation of vertebrate skeletal myogenesis by cell-cell contact. J Cell Sci. 2005;118:2355–2362. doi: 10.1242/jcs.02397. [DOI] [PubMed] [Google Scholar]
- Lane SW, Williams DA, Watt FM. Modulating the stem cell niche for tissue regeneration. Nat Biotech. 2014;32:795–803. doi: 10.1038/nbt.2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, High FA, Epstein JA, Radice GL. N-cadherin is required for neural crest remodeling of the cardiac outflow tract. Dev Biol. 2006;299:517–528. doi: 10.1016/j.ydbio.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthiens V, Kazanis I, Moss L, Long K, ffrench-Constant C. Adhesion molecules in the stem cell niche – more than just staying in shape? J Cell Sci. 2010;123:1613–1622. doi: 10.1242/jcs.054312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea PD, Maher MT, Gottardi CJ. Nuclear Signaling from Cadherin Adhesion Complexes. Curr Top Dev Biol. 2015;112:129–196. doi: 10.1016/bs.ctdb.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R, Walsh FS. The cell adhesion molecule M-cadherin is specifically expressed in developing and regenerating, but not denervated skeletal muscle. Development. 1993;117:1409–1420. doi: 10.1242/dev.117.4.1409. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Spradling AC. Stem Cells and Niches: Mechanisms That Promote Stem Cell Maintenance throughout Life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourikis P, Sambasivan R, Castel D, Rocheteau P, Bizzarro V, Tajbakhsh S. A Critical Requirement for Notch Signaling in Maintenance of the Quiescent Skeletal Muscle Stem Cell State. Stem Cells. 2012;30:243–252. doi: 10.1002/stem.775. [DOI] [PubMed] [Google Scholar]
- Murphy MM, Keefe AC, Lawson JA, Flygare SD, Yandell M, Kardon G. Transiently Active Wnt/β-Catenin Signaling Is Not Required but Must Be Silenced for Stem Cell Function during Muscle Regeneration. Stem Cell Reports. 2014;3:475–488. doi: 10.1016/j.stemcr.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 2011;138:3625–3637. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen CM, Leckband D, Yap AS. Tissue Organization by Cadherin Adhesion Molecules: Dynamic Molecular and Cellular Mechanisms of Morphogenetic Regulation. Physiol Rev. 2011;91:691–731. doi: 10.1152/physrev.00004.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9:115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- Parisi A, Lacour F, Giordani L, Colnot S, Maire P, Le Grand F. APC is required for muscle stem cell proliferation and skeletal muscle tissue repair. J Cell Biol. 2015;210:717–726. doi: 10.1083/jcb.201501053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisconti A, Banks GB, Babaeijandaghi F, Betta ND, Rossi FMV, Chamberlain JS, Olwin BB. Loss of niche-satellite cell interactions in syndecan-3 null mice alters muscle progenitor cell homeostasis improving muscle regeneration. Skelet Muscle. 2016;6:34. doi: 10.1186/s13395-016-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porlan E, Martí-Prado B, Morante-Redolat JM, Consiglio A, Delgado AC, Kypta R, López-Otín C, Kirstein M, Fariñas I. MT5-MMP regulates adult neural stem cell functional quiescence through the cleavage of N-cadherin. Nat Cell Biol. 2014;16:629–638. doi: 10.1038/ncb2993. [DOI] [PubMed] [Google Scholar]
- Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M, Hynes RO. Developmental defects in mouse embryos lacking N-cadherin. Dev Biol. 1997;181:64–78. doi: 10.1006/dbio.1996.8443. [DOI] [PubMed] [Google Scholar]
- Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated therapy. J Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, Brunson C, Mastey N, Liu L, Tsai CR, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to GAlert. Nature. 2014;510:393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozo M, Li L, Fan CM. Targeting [beta]1-integrin signaling enhances regeneration in aged and dystrophic muscle in mice. Nat Med. 2016;22:889–896. doi: 10.1038/nm.4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf A, Schirwis E, Giordani Lo, Parisi A, Lepper C, Taketo MM, Le Grand F. β-Catenin Activation in Muscle Progenitor Cells Regulates Tissue Repair. Cell Rep. 2016;15:1277–1290. doi: 10.1016/j.celrep.2016.04.022. [DOI] [PubMed] [Google Scholar]
- Ryall JG, Dell’Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez-Cruz G, Fulco M, et al. The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. Cell Stem Cell. 2015;16:171–183. doi: 10.1016/j.stem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P, Lichtenthaler SF. The alpha secretase ADAM10: A metalloprotease with multiple functions in the brain. Prog Neurobiol. 2015;135:1–20. doi: 10.1016/j.pneurobio.2015.10.003. [DOI] [PubMed] [Google Scholar]
- Scadden DT. Nice Neighborhood: Emerging Concepts of the Stem Cell Niche. Cell. 2014;157:41–50. doi: 10.1016/j.cell.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieiro D, Rios AC, Hirst CE, Marcelle C. Cytoplasmic NOTCH and membrane-derived β-catenin link cell fate choice to epithelial-mesenchymal transition during myogenesis. Elife. 2016;24:e14847. doi: 10.7554/eLife.14847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. Formative pluripotency: the executive phase in a developmental continuum. Development. 2017;144:365–373. doi: 10.1242/dev.142679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedesco FS, Dellavalle A, Diaz-Manera J, Messina G, Cossu G. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J Clin Invest. 2010;120:11–19. doi: 10.1172/JCI40373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler TO, Gadek KE, Cadwallader AB, Elston TL, Olwin BB. Isolation, Culture, Functional Assays, and Immunofluorescence of Myofiber-Associated Satellite Cells. In: Kyba M, editor. Skeletal Muscle Regeneration in the Mouse: Methods and Protocols. New York, NY: Springer New York; 2016. pp. 141–162. [DOI] [PubMed] [Google Scholar]
- von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci U S A. 2013;110:16474–16479. doi: 10.1073/pnas.1307680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R, Bierinx A, Gnocchi V, Zammit P. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev Biol. 2010;10:21. doi: 10.1186/1471-213X-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Watanabe Y, Ohtani T, Uezumi A, Mikami N, Nakamura M, Sato T, Ikawa M, Hoshino M, Tsuchida Ko, et al. Calcitonin Receptor Signaling Inhibits Muscle Stem Cells from Escaping the Quiescent State and the Niche. Cell Rep. 2015;13:302–314. doi: 10.1016/j.celrep.2015.08.083. [DOI] [PubMed] [Google Scholar]
- Yamashita YM. Cell adhesion in regulation of asymmetric stem cell division. Curr Opin Cell Biol. 2010;22:605–610. doi: 10.1016/j.ceb.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Price F, Rudnicki MA. Satellite Cells and the Muscle Stem Cell Niche. Physiological Reviews. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Bi P, Wang C, Shan T, Nie Y, Ratliff TL, Gavin TP, Kuang S. Pten is necessary for the quiescence and maintenance of adult muscle stem cells. Nat Commun. 2017;8:14328. doi: 10.1038/ncomms14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zammit PS, Golding JP, Nagata Y, Hudon V, Partridge TA, Beauchamp JR. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J Cell Biol. 2004;166:347–357. doi: 10.1083/jcb.200312007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Günther S, Looso M, Künne C, Krüger M, Kim JH, Zhou Y, Braun T. Prmt5 is a regulator of muscle stem cell expansion in adult mice. Nat Commun. 2015;6:7140. doi: 10.1038/ncomms8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.