

CONSPECTUS
C–H activation and functionalization are on the forefront of modern synthetic chemistry. Imagine if any C–H bond of a molecule could be converted to a C–X bond, where X is target functionality. Collaborations between many experimental and computational groups have led to rapid developments of new C–H functionalization methods. Our groups represent an example of this; we were brought together as part of the NSF-supported Center for Selective C–H Functionalization. Many examples of experimental-computational synergy for selective Pd(II)-catalyzed C–H activation of aryl and alkyl groups are described in this Account. We describe computations by the Houk group made in response to experimental stimuli by the Yu group. The first section discusses the experimental and computational investigations of oxazoline-directed, stereoselective Pd(II)-catalyzed C(sp3)–H bond activation that occurs through the concerted metalation-deprotonation (CMD) pathway involving a monomeric Pd functionality. The second section involves two types of bidentate ligands, mono-N-protected amino acid (MPAA) and acetyl-protected aminoethyl quinoline (APAQ) ligands that facilitate the C–H activation reactions in an internal base mechanism. In the MPAA-assisted remote C–H bond activation, the basic dianionic amidate ligand participates in the deprotonation of a specific C–H bond. This mechanism accounts for the improved reactivity and selectivity in C–H activation reactions with MPAA ligands. The chiral APAQ ligands enable the asymmetric palladium insertion into prochiral C–H bonds on a single methylene carbon center. The origins of the dramatic differences of 5-membered (relatively inactive) and 6-membered chelation (highly active) in the β-methylene C(sp3)–H activation reactions by Pd(II) catalyst were explained with density functional theory (DFT) calculations. This is mainly due to the steric repulsions between the ArF group of substrate and the quinoline group of the ligand. The steric repulsion between the ArF group of substrate and the quinoline group of the acetyl-protected aminomethyl quinoline ligand destabilizes the 5-membered chelate transition structure, increasing the energy of transition state. The third section discusses a mechanism involving a Pd-Ag heterodimeric complex intermediate in the template-directed, Pd(II)-catalyzed meta-functionalization of toluene derivatives and benzoic acid derivatives. The nitrile directing group of the template coordinates with Ag while the Pd is placed adjacent to the meta-C–H bond in the transition state, leading to the observed high meta-selectivity. The dual role of AgOAc as both an oxidant and part of the heteronuclear active species in the mechanism involving PdAg(OAc)3 was determined by DFT calculation. The interaction between the experimental and computational groups, and the interplay that led to these discoveries, are highlighted in this Account.
Graphical abstract
1. INTRODUCTION
Transition-metal-catalyzed C–H activation and functionalization have been extraordinary subjects of interest in recent years.1,2,3, 4 Among the most challenging aspects of developing robust C–H functionalization methodologies are identifying catalyst and reagent combinations capable of site-selective as well as diastereo- and enantioselective reactions.5 Among the plethora of transition-metal catalysts, palladium are incredibly powerful for the construction of carbon-carbon and carbon-heteroatom bonds by C–H activation of aryl and alkyl groups.6,7,8
The understanding of the mechanism and the stereoselectivity of palladium-catalyzed C–H activation reactions is key to the progress of this research field. Kinetics and isolation of intermediates provides key information of mechanistic pathways.9 Computations can also provide details of both structures and energetics in specific steps in the whole catalytic cycle.10,11,12 Computations have become essential to elucidate structures and properties of molecules, and mechanisms and selectivities of reactions.13 Thanks to the rapid development of hardware, software, and theoretical methods, computational chemistry has evolved to a very powerful and routine tool to study the mechanisms and selectivities of more complex reactions. In this Account, we present several examples of collaboration between experimental and computational researchers that led to advances in understandings and innovative new methods.
In our computational studies of organometallic reactions, the B3LYP method14,15 was used normally for geometry optimization, and the M06 method16 was used for single-point solvation energy calculations with the SMD (solvation model based on density) model.17 The B3LYP method has been in use since 1995,18 and it has known limitations. Nevertheless, it was proved to be a rapid way to explore mechanisms and to obtain structures of intermediates and transition state that can then be evaluated with higher accuracy functionals.
2. PALLADIUM ACETATE-CATALYZED DIASTEREOSLECTIVIE C–H BOND ACTIVATION OF OXAZOLINES
Transition-metal-catalyzed C–H functionalization is a powerful tool for the construction and modification of complex molecules, but many difficulties need to be overcome to make it a routine tool. Asymmetric induction through installation of chiral auxiliaries is a well-established procedure in organic synthesis,19 but the possibility of a robust transition-metal catalyst for stereoselective C–H functionalization is most appealing. Encouraged by many successful asymmetric catalysis reactions using chiral oxazolines as ligands,20 we explored diastereoselective C–H iodination with oxazolines (Scheme 1).21 Surprisingly, the transformations with the 4-t-Bu-substituted oxazolines are very effective, while the 4-i-Pr-substituted oxazolines lack reactivity and stereoselectivity. Intrigued by this dramatic change in reactivity and stereoselectivity with subtle changes in substituents on the oxazoline ring, the mechanism and origin of reactivity and diastereoselectivity was investigated.21 Yu Lan and Peng Liu, the postdocs in the Houk group who worked on this problem, are now professors at Chongqing University and the University of Pittsburgh, respectively.
Scheme 1.

C–H Iodinations with t-Bu- and i-Pr-Substituted Oxazolines
Computations revealed that the reactions with 4-i-Pr- and 4-t-Bu-substituted oxazolines involve different catalyst resting states prior to the C–H bond activation.21 The Pd3(OAc)6 trimer is the catalyst resting state for reactions with 4-t-Bu-substituted oxazolines. For the reaction with 4-i-Pr-substituted oxazolines, the monomeric [bis(oxazoline)]Pd(OAc)2 6 in Figure 1, which is 10.4 kcal/mol more stable than Pd3(OAc)6 trimer. Because of the greater stability of the catalyst resting state 6, the reaction has a higher activation barrier (38.4 kcal/mol from 6 to 8 in Figure 2) for C–H activation. The 4-t-Bu-substituted oxazoline complex gives C–H activation with an overall barrier of 26.2 kcal/mol; the 5-membered palladacycle intermediate associates with another molecule and Pd(OAc)2 to form a stable trinuclear Pd metallacycle that was confirmed by the X-ray crystallographic study.21
Figure 1.

Optimized geometries of [bis(oxazoline)]Pd(OAc)2 complexes 5 and 6. Gibbs free energies are with respect to Pd3(OAc)6.
Figure 2.

Optimized geometries of C–H activation transition states 7, 8, 9 and 10 and the Newman Projections along the highlighted C–C Bond. The distances are shown in Ångströms.
The stereoselectivity (>99:1 dr) with the 4-t-Bu-substituted oxazoline was also investigated.21 C–H activation transition structures 7, 8, 9 and 10 and the Newman projections along the C–C Bond (highlighted in red) are shown in Figure 2. Structures 9 and 10 have the bulky t-Bu substituents cis; they lead to the minor diastereomer. The preferred transition state for C–H activiation is 8. Here the t-Bu substituent gauche interacting with the breaking C–H bond is less than with the C–H bonds in 7.
3. PALLADIUM-CATALYZED C–H BOND ACTIVATION WITH BIDENDATE LIGANDS
3.1 Palladium-Catalyzed C–H Bond Activation with MPAA Ligands
The Yu group has discovered a variety of ways that mono-N-protected amino acid (MPAA) ligands can influence Pd-catalyzed C-H activations. These additions increase yields and selectivities of both C(sp2)–H and C(sp3)–H bonds.22 The performance of the Yu group’s templates that direct the activation of meta-C–H bond of tethered arenes,23 are also improved with MPAA ligands (Scheme 2a). Chiral MPAA ligands have also been employed to achieve high enantioselectivity of C–H bond activation reactions (Scheme 2b).24
Scheme 2.

Pd/MPAA-Catalyzed C–H Activation Reactions
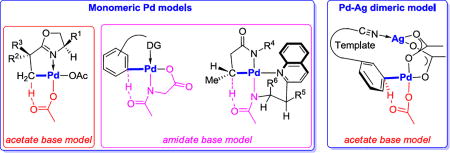
The deprotonated dianionic MPAA ligand binds to Pd(II) center through a bidentate mode.25 Transition structure 15 in Figure 3 is the external base model for Pd-catalyzed C–H activation reactions with directing group (DG) involving what is often called a concerted metalation-deprotonation (CMD) mechanism. In this model, an acetate anion, which is not coordinated to Pd(II) center, acts as the external base for deprotonation through an outer-sphere mechanism.26 Transition structure 16 in Figure 3 is the internal base CMD model, in which the amidate O acts as an internal base to deprotonate the target C–H bond. In this model, the MPAA ligand has dual roles, stabilizing monomeric Pd complexes as a dianion amidate and deprotonating the C–H bond with the amidate O.27 The internal base model is lower in energy and gives results consistent with the experimental meta-selectivity.
Figure 3.

External and internal base models for Pd/MPAA-catalyzed C–H activation reactions.
The internal base transition state is lower in energy than the external base transition state, and explains the enantioselectivity of the C–H activation reactions with chiral MPAA ligands as well. The transition structure 17 (Figure 3) for C–H activation reactions with the Boc-Val-OH ligand leads to the experimental major enantiomer 14. Pd(II) coordinates with the dianionic ligand and the directing group of the substrate in a square-planar coordination.28 The side chain R1 (e.g. i-Pr group) of the MPAA ligand pushes the N-protecting group (e.g. Boc group) downward below the Pd coordination plane. With the other phenyl to react, the methyl group at the pro-chiral carbon would be forced onto the proximity with Boc.
3.2 Palladium-Catalyzed C–H Bond Activation with APAQ Ligands
It is more difficult to activate methylene C–H bonds than arene C–H bonds, but the chiral acetyl-protected aminoethyl quinoline (APAQ) ligands developed by the Yu group enable activation of the β-methylene C(sp3)–H bonds with high enantiomeric ratios reaching up to 96:4, as shown in Scheme 3.29 MPAA ligands were ineffective for this transformation. The deprotonated APAQ ligand forms a 6-membered chelate with the Pd(II) center. Yu’s group had attempted such reactions for 14 years, but the APAQ ligands finally made this enantioselective reaction of an unactivated methylene feasible.
Scheme 3.

Enantioselective Methylene C–H Activation Reactions with APAQ Ligands
The optimized geometries of the transition states for C(sp3)–H bond activation with the APAQ ligand are shown in Figure 4. Eight transition structures were located, four leading to the (R)-product and four leading to the (S)-product. The two most favorable are shown in Figure 4. In both, the Pd(II) center is chelated by N of the quinoline group and the N of the amidate group of the APAQ ligand. The N of the substrate amidate group coordinates and delivers the CH2 in proximity to undergo the CMD process. The amidate O of the APAQ ligand serves as an internal base to deprotonate the methylene C(sp3)–H bond. Transition structure 20 is 1.2 kcal/mol more favorable than transition structure 21, in agreement with the experimentally observed preference for 19. In both TSs, the large aryl group (R) is in an axial-position that is pointing perpendicular to the square-planar Pd(II) coordination plane. The methyl group of the substrate is anti to R in 20 while but syn in 21. The distortion of the palladacycle is reflected in the N-C-C-N dihedral angle (marked with green circle in Figure 4) as 49°.
Figure 4.

Optimized geometries of C(sp3)–H bond activation transition states 20 and 21. R group is 3,5-di-tert-butylphenyl group.
3.3 5-Membered vs. 6-Membered Chelation of Pd(II) on Efficiency of C(sp3)–H Bond Activation
The chiral APAQ ligands in Scheme 3 form 6-membered chelates with Pd. The achiral analog, 22, is also effective. By contrast, the deprotonated acetyl-protected aminomethyl quinoline ligand (23) forms a 5-membered chelate with the Pd(II) catalyst and it is inactive in this transformation. The dramatic difference between ligands forming 5-membered or 6-membered chelates with Pd(II) was found to be due to unusual circumstances.30
Figure 5 shows the TS geometries and energies for the C(sp3)–H bond activation involving 6- and 5-membered chelates. The C(sp3)–H bond activation with a 5-membered chelate 27 is unfavorable by 7.7 kcal/mol compared to the corresponding 6-membered chelate 26. Two factors cause the difference: (1) the dimeric Pd species with 5-membered chelation square-planar structure is more stable than that with 6-membered chelation by 2 kcal/mol; (2) steric repulsion between the ArF group of substrate and the quinoline group of the acetyl-protected aminomethyl quinoline ligand destabilizes the 5-membered chelate transition structure by 5.7 kcal/mol. The 6-membered chelate of Pd(II) with acetyl-protected aminoethyl quinoline ligand orients the ligand away from the ArF group of substrate and alleviates the steric repulsion. This occurs because of the longer link between the chelates Ns, allowing rotation of the quinoline group.
Figure 5.

Optimized geometries of C(sp3)–H bond activation transition states 26, 27, 28 and 29. The values not in italics are with respect to the separated reactants. The values in italics are with respect to the resting state dimeric palladium complexes. The distances are shown in Ångströms.
The steric clash caused by the ArF group of substrate and the quinoline group of the ligand accounts for the higher C(sp3)–H bond activation barrier in 27. Replacing the quinoline group with the pyridine group is expected to alleviate the steric repulsion interaction. In the C(sp3)–H bond activation transition structure, 29, shown in Figure 5, the 5-membered chelation is even closer to planar coordination, and results greater stability of its catalyst resting state and higher C–H activation barrier. For the 6-membered chelation, the barrier for C–H activation with the pyridine ligand (transition structure 28) is 0.7 kcal/mol slightly higher than that with the quinoline ligand (transition structure 26). The computational studies predict that the 6-membered pyridine containing complex will likely undergo reaction. The experiments also showed low reactivity of the 6-membered chelate pyridine ligand that it gives 28% yield of product. The computational prediction was confirmed by experiments and demonstrated the power of computational-experimental synergy.
4. PALLADIUM-SILVER HETERODIMERIC MODEL FOR META C–H BOND ACTIVATION
4.1 Palladium-Catalyzed Meta C–H Olefination of Toluene Derivatives
Controlling site-selectivity, especially meta-selective functionalization of electron-rich aromatic rings, is a very usefull approach for the synthesis of meta-substituted aromatics. Scheme 5 shows an easily removable nitrile-containing template attached to benzyl alcohol. This directs the activation of the meta-C–H bonds of electron-rich mono-substituted arenes.23 This template overrides any electronic preferences and leads selectively to cleavage of the meta-C–H bonds.
Scheme 5.

Template-Mediated, Palladium-Catalyzed Meta C–H Olefination of Toluene Derivatives
Computational explorations showed that the commonly proposed Pd monomeric model with nitrile directing group coordinating with Pd(II) center, 32, is ortho-selective (Scheme 6) and cannot explain the experimental observations.31 Distortion/Interaction analysis of the Pd monomeric transition states indicates the origins of differences of activation energies in the ortho, meta and para transition states results from the differences of distortion energies of the template in the transition states. The meta and para transition states bearing 11-membered and 12-membered rings, outlined in color in Scheme 6, are highly distorted due to the ring strain.
Scheme 6.

Pd Monomeric Model and Pd-Pd/Pd-Ag Dimeric Models
We also investigated possibilities how the Pd-Pd or Pd-Ag complexes might be involved. In both, two acetates act as bridges for two metal centers and lengthen the tether by 4 atoms.31 In these two models 33 and 34, the nitrile directing groups of the template binds to one Pd or Ag in the dimeric complex, while the arene meta-C–H bond is placed adjacent to another Pd to be cleaved. Such dimeric activation models relieve the ring strain of the meta transition state by forming a larger macrocycle. A systematic conformational search was performed to obtain the low-energy conformer due to the increased conformational space for the meta-transition structures involving 16-membered ring. Computational results showed that the two dimeric mechanisms require lower barriers for C–H bond activation than the Pd monomeric mechanism, and both are meta-selective. The Pd-Ag heterodimeric mechanism was found to be the most favorable one, with the distortion of the meta-transition structure of Pd-Ag heterodimeric mechanism is the smallest. The silver salt plays a dual role in this reaction, besides acting as an oxidant to effectively turn over the catalytic cycle; it also binds to the nitrile directing group. The preference for the heterodimeric pathway is highly depending on the unique structure of the templates. The Pd-Ag herterodimeric mechanism has been applied in other C–H bond activation reactions.32,33
4.2 Palladium-Catalyzed Meta C–H Olefination of Benzoic Acid Derivatives
An efficient, practical, and selective method with a surprisingly flexible 2-(2-(methylamino)ethoxy)benzonitrile templates (Scheme 7), was developed for the meta-olefination of benzamide derivatives with yields up to 98% and meta-regioselectivity up to 92%.34
Scheme 7.

Template-Mediated, Palladium-Catalyzed Meta C–H Olefination of Benzoic Acid Derivatives
Unlike the previously reported templates for meta-C–H activation that have sterically bulky group at specific positions on the template, this template is rather flexible. Considering the similarity to the template for hydrocinnamic acid derivatives shown in Scheme 2a, monomeric palladium ligated by a dianionic N-acetylglycine ligand (37 in Scheme 8) was studied computationally. However, the para-selective transition states were favored by this mechanism.
Scheme 8.

Pd-MPAA monomeric model and Pd-Ag Heterodimeric Model
While meta-selective C–H activation involves the Pd-Ag heterodimeric transition states were favored, with a barrier of only 23.2 kcal/mol. The meta-olefination product is also favored by the palladium homodimer mechanism, Pd-Pd, but this is higher in energy compared to the Pd-Ag mechanism. The origin of the meta-selectivity was also investigated by a distortion/interaction analysis. The meta-transition state, 38, has a low (ΔE‡template distortion=1.4 kcal/mol) energy to distort the template into the transition-state geometry. By contrast, greater conformational changes are necessary in the ortho- and para-transition states, with distortion energies of 3.3 kcal/mol and 4.8 kcal/mol, respectively.
Kinetics studies showed that the MPAA ligand produce a 4-fold rate increase on the rates of reaction compared to those without this ligand. The computational results showed that MPAA stabilizes the Pd-Ag heterodimer prior to C–H activation step and lowers the barrier to coordination to the nitrile of the template (Scheme 9). In the absence of MPAA ligand the rate-determining step is formation of the intermediate prior to C–H activation step, but the MPAA ligand facilitates formation of 40, and the rate-determining transition state becomes C–H activation 38.
Scheme 9.

MPAA Stabilize Pd-Ag Heterodimeric Complex
5. CONCLUSION
Collaborations between our groups have involved computational and experimental investigations of a variety of selective Pd(II)-catalyzed C–H activations of aryl and alkyl groups. The mechanisms and selectivities are found to be highly dependent on the ligands, substrates, additives, oxidants and solvents. The CMD mechanism is very common in base/ligand-assisted, Pd(II)-catalyzed C–H activation, but we have shown many variations that depend on ligands and condition. The mechanistic insights from these studies have forged an expanding synergism between experiment and computations for future development of C–H activations.
Scheme 4.

5-Membered vs. 6-Membered Chelate Ligands with Pd(II)
Acknowledgments
This Account is dedicated to Professor Yun-Dong Wu on the occasion of his 60th birthday. We are grateful to all researchers who contributed to these studies. This work was funded by the National Science Foundation (CHE-1361104 to K.N.H.), the National Institutes of Health (NIGMS, 2R01GM084019 to J.-Q.Y.) and the CCI Center for Selective C–H Functionalization (CHE-1205646). This center made the extensive collaborations described in this Account possible. The Chinese “Thousand Youth Talents Plan” (to X.H.) also facilitated this research.
Biographies
Yun-Fang Yang received her Ph.D. in chemistry from Peking University (P. R. of China) in 2013 under the guidance of Professor Yun-Dong Wu. In 2014, she joined the group of Professor K. N. Houk as a postdoctoral scholar in the Department of Chemistry and Biochemistry at University of California, Los Angeles (USA). Her current research focuses on computational studies of mechanisms and stereoselectivity of organic and organometallic reactions.
Xin Hong received his Ph.D. in chemistry from University of California, Los Angeles (USA) in 2014 under the guidance of Professor K. N. Houk. He then worked as a postdoctoral scholar in Professor Houk’s group focusing on computational studies of organometallic reactions. In 2015, he conducted postdoctoral research in Professor Jens K. Nørskov’s group in the Department of Chemical Engineering at Stanford University (USA). He is a 2016 Fellow of The Thousand Talents Plan for Young Scientists (P. R. of China). In 2016, he joined the Department of Chemistry at Zhejiang University (P. R. of China) as an assistant professor.
Jin-Quan Yu received his Ph.D. degree at the University of Cambridge with Prof. Jonathan B. Spencer. After some time as a Junior Research Fellow at Cambridge, he joined the laboratory of Prof. E. J. Corey at Harvard University as a postdoctoral fellow. He then began his independent career at Cambridge (2003–2004), before moving to Brandeis University (2004–2007), and then to The Scripps Research Institute, where he is currently the Frank and Bertha Hupp Professor of Chemistry. His group studies transition-metal-catalyzed C–H activation.
K. N. Houk is the Saul Winstein Chair in Organic Chemistry at University of California, Los Angeles. He is a computational organic chemist. He received his Ph.D. at Harvard University with R. B. Woodward in 1968, doing experimental work in orbital symmetry-predicted chemistry. He has published extensively on pericyclic reactions, stereoselectivity, molecular recognition, and enzyme design. He is a Fellow of the American Academy of Arts and Sciences and a member of the National Academy of Sciences.
Footnotes
The authors declare no competing financial interest.
References
- 1.Shilov AE, Shul'pin GB. Activation of C–H Bonds by Metal Complexes. Chem. Rev. 1997;97:2879–2932. doi: 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]
- 2.Crabtree RH. Introduction to Selective Functionalization of C–H Bonds. Chem. Rev. 2010;110:575-575. doi: 10.1021/cr900388d. [DOI] [PubMed] [Google Scholar]
- 3.Wencel-Delord J, Droge T, Liu F, Glorius F. Towards Mild Metal-Catalyzed C–H Bond Activation. Chem. Soc. Rev. 2011;40:4740–4761. doi: 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]
- 4.Smith KT, Berritt S, González-Moreiras M, Ahn S, Smith MR, Baik M-H, Mindiola DJ. Catalytic Borylation of Methane. Science. 2016;351:1424–1427. doi: 10.1126/science.aad9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giri R, Shi B-F, Engle KM, Maugel N, Yu J-Q. Transition Metal-Catalyzed C–H Activation Reactions: Diastereoselectivity And Enantioselectivity. Chem. Soc. Rev. 2009;38:3242–3272. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]
- 6.Yu J-Q, Giri R, Chen X. σ-Chelation-Directed C-H Functionalizations Using Pd(II) And Cu(II) Catalysts: Regioselectivity, Stereoselectivity and Catalytic Turnover. Org. Biomol. Chem. 2006;4:4041–4047. doi: 10.1039/b611094k. [DOI] [PubMed] [Google Scholar]
- 7.Daugulis O, Do H-Q, Shabashov D. Palladium- and Copper-Catalyzed Arylation of Carbon–Hydrogen Bonds. Acc. Chem. Res. 2009;42:1074–1086. doi: 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lyons TW, Sanford MS. Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blackmond DG. Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. J. Am. Chem. Soc. 2015;137:10852–10866. doi: 10.1021/jacs.5b05841. [DOI] [PubMed] [Google Scholar]
- 10.Cheng G-J, Zhang X, Chung LW, Xu L, Wu Y-D. Computational Organic Chemistry: Bridging Theory and Experiment in Establishing the Mechanisms of Chemical Reactions. J. Am. Chem. Soc. 2015;137:1706–1725. doi: 10.1021/ja5112749. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Chung LW, Wu Y-D. New Mechanistic Insights on the Selectivity of Transition-Metal-Catalyzed Organic Reactions: The Role of Computational Chemistry. Acc. Chem. Res. 2016;49:1302–1310. doi: 10.1021/acs.accounts.6b00093. [DOI] [PubMed] [Google Scholar]
- 12.Sperger T, Sanhueza IA, Schoenebeck F. Computation and Experiment: A Powerful Combination to Understand and Predict Reactivities. Acc. Chem. Res. 2016;49:1311–1319. doi: 10.1021/acs.accounts.6b00068. [DOI] [PubMed] [Google Scholar]
- 13.Houk KN, Liu F. Holy Grails for Computational Organic Chemistry and Biochemistry. Acc. Chem. Res. 2017;50:539–543. doi: 10.1021/acs.accounts.6b00532. [DOI] [PubMed] [Google Scholar]
- 14.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti Correlation-Energy Formula into A Functional of the Electron Density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 15.Becke A. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993;98:1372–1377. [Google Scholar]
- 16.Zhao Y, Truhlar D. The M06 Suite Of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]
- 17.Marenich AV, Cramer CJ, Truhlar DG. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 18.Del Bene JE, Person WB, Szczepaniak K. Properties of Hydrogen-Bonded Complexes Obtained from the B3LYP Functional with 6-31G(d,p) and 6-31+G(d,p) Basis Sets: Comparison with MP2/6-31+G(d,p) Results and Experimental Data. J. Phys. Chem. 1995;99:10705–10707. [Google Scholar]
- 19.Meyers AI. Asymmetric Carbon-Carbon Bond Formation From Chiral Oxazolines. Acc. Chem. Res. 1978;11:375–381. [Google Scholar]
- 20.Hargaden GC, Guiry PJ. Recent Applications of Oxazoline-Containing Ligands in Asymmetric Catalysis. Chem. Rev. 2009;109:2505–2550. doi: 10.1021/cr800400z. [DOI] [PubMed] [Google Scholar]
- 21.Giri R, Lan Y, Liu P, Houk KN, Yu J-Q. Understanding Reactivity and Stereoselectivity in Palladium-Catalyzed Diastereoselective sp3 C–H Bond Activation: Intermediate Characterization and Computational Studies. J. Am. Chem. Soc. 2012;134:14118–14126. doi: 10.1021/ja304643e. [DOI] [PubMed] [Google Scholar]
- 22.Shi B-F, Maugel N, Zhang Y-H, Yu J-Q. PdII-Catalyzed Enantioselective Activation of C(sp2)–H and C(sp3)–H Bonds Using Monoprotected Amino Acids as Chiral Ligands. Angew. Chem., Int. Ed. 2008;47:4882–4886. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]
- 23.Leow D, Li G, Mei T-S, Yu J-Q. Activation of Remote Meta-C-H bonds Assisted by an End-on Template. Nature. 2012;486:518–522. doi: 10.1038/nature11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi B-F, Zhang Y-H, Lam JK, Wang D-H, Yu J-Q. Pd(II)-Catalyzed Enantioselective C–H Olefination of Diphenylacetic Acids. J. Am. Chem. Soc. 2010;132:460–461. doi: 10.1021/ja909571z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baxter RD, Sale D, Engle KM, Yu J-Q, Blackmond DG. Mechanistic Rationalization of Unusual Kinetics in Pd-Catalyzed C–H Olefination. J. Am. Chem. Soc. 2012;134:4600–4606. doi: 10.1021/ja207634t. [DOI] [PubMed] [Google Scholar]
- 26.Musaev DG, Kaledin A, Shi B-F, Yu J-Q. Key Mechanistic Features of Enantioselective C–H Bond Activation Reactions Catalyzed by [(Chiral Mono-N-Protected Amino Acid)–Pd(II)] Complexes. J. Am. Chem. Soc. 2011;134:1690–1698. doi: 10.1021/ja208661v. [DOI] [PubMed] [Google Scholar]
- 27.Cheng G-J, Yang Y-F, Liu P, Chen P, Sun T-Y, Li G, Zhang X, Houk KN, Yu J-Q, Wu Y-D. Role of N-Acyl Amino Acid Ligands in Pd(II)-Catalyzed Remote C–H Activation of Tethered Arenes. J. Am. Chem. Soc. 2014;136:894–897. doi: 10.1021/ja411683n. [DOI] [PubMed] [Google Scholar]
- 28.Cheng G-J, Chen P, Sun T-Y, Zhang X, Yu J-Q, Wu Y-D. A Combined IM-MS/DFT Study on [Pd(MPAA)]-Catalyzed Enantioselective C–H Activation: Relay of Chirality through a Rigid Framework. Chem. Eur. J. 2015;21:11180–11188. doi: 10.1002/chem.201501123. [DOI] [PubMed] [Google Scholar]
- 29.Chen G, Gong W, Zhuang Z, Andrä MS, Chen Y-Q, Hong X, Yang Y-F, Liu T, Houk KN, Yu J-Q. Ligand-Accelerated Enantioselective Methylene C(sp3)–H Bond Activation. Science. 2016;353:1023–1027. doi: 10.1126/science.aaf4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y-F, Hong X, Chen G, Yu J-Q, Houk KN. The Origins of Dramatic Differences in Five-Membered vs Six-Membered Chelation of Pd(II) on Efficiency of C(sp3)–H Bond Activation. J. Am. Chem. Soc. 2017;139:8514–8521. doi: 10.1021/jacs.7b01801. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y-F, Cheng G-J, Liu P, Leow D, Sun T-Y, Chen P, Zhang X, Yu J-Q, Wu Y-D, Houk KN. Palladium-Catalyzed Meta-Selective C–H Bond Activation with a Nitrile-Containing Template: Computational Study on Mechanism and Origins of Selectivity. J. Am. Chem. Soc. 2014;136:344–355. doi: 10.1021/ja410485g. [DOI] [PubMed] [Google Scholar]
- 32.Anand M, Sunoj RB, Schaefer HF. Non-innocent Additives in a Palladium(II)-Catalyzed C–H Bond Activation Reaction: Insights into Multimetallic Active Catalysts. J. Am. Chem. Soc. 2014;136:5535–5538. doi: 10.1021/ja412770h. [DOI] [PubMed] [Google Scholar]
- 33.Anand M, Sunoj RB, Schaefer HF. Palladium–Silver Cooperativity in an Aryl Amination Reaction through C–H Functionalization. ACS Catal. 2016;6:696–708. [Google Scholar]
- 34.Fang L, Saint-Denis TG, Taylor BLH, Ahlquist S, Hong K, Liu SS, Han L, Houk KN, Yu J-Q. Experimental and Computational Development of a Conformationally Flexible Template for the meta-C–H Functionalization of Benzoic Acids. 2017;139:10702–10714. doi: 10.1021/jacs.7b03296. [DOI] [PMC free article] [PubMed] [Google Scholar]