

Abstract
The serine/threonine protein kinase, TBK1, plays a crucial role as the hub for many innate immune signaling pathways that lead to the induction of type I interferon (IFN) and interferon-stimulated genes (ISGs). Due to its key function in maintaining homeostasis of the immune system, cell survival and proliferation, TBK1 activity is tightly regulated. Dysregulation of TBK1 activity is often associated with autoimmune diseases and cancer, implicating the potential therapeutic benefit for targeting TBK1. Tremendous effort from both academic institutions and private sectors during the past few years has led to the development of many potent and selective TBK1 inhibitors, many of which have shown great promise in disease models in vivo. This review summarizes recent advance on the pharmacological inhibition of TBK1 and its potential for treating autoimmune diseases and interferonopathies.
Keywords: Autoimmune disease, Interferonopathy, TBK1, Type I interferon, Lupus
1. General overview of TBK1 signaling
The serine/threonine protein kinase, TBK1, is ubiquitously expressed and it belongs to the IκB kinase (IKK) family. Canonical IKK family members include IKKα and IKKβ. They are key molecules in the NF-κB pathway involved in the clearance of incoming pathogen, cell survival and proliferation. Given the importance of IKKα/β, collective effort from several research groups led to the discovery of two IKK-related kinases [1]. One of which is TBK1 (TANK-binding kinase), originally identified by yeast two-hybrid screen using TANK as the bait. TBK1 is also known as NAK (NF-κB-activating kinase), discovered in parallel based on sequence similarity to IKKα/β and the ability to activate NF-κB. The other IKK-related kinase is IKKε (also known as IKKi), also discovered through homology search and functional screens. IKKε shares 60% sequence identity to TBK1. IKKε is the only IKK family member that is inducible, in response to LPS, viral infection and several cytokines. Its expression is also limited to pancreas, thymus, spleen and peripheral blood leukocytes. In contrast, IKKα, IKKβ, and TBK1 are all constitutively expressed in all tissue and cell types. Both TBK1 and IKKε are also essential player of the NF-κB pathway; MEFs deficient for either enzyme would abrogate NF-κB target gene expression in response to classical inducers [2,3]. In addition, TBK1-deficienct mice are embryonic lethal and arrest development at embryonic stage E14.5 due to liver degeneration and apoptosis, a phenotype similar to IKKβ- or p65-deficient mice [3].
The unique function that sets TBK1 and IKKε apart from IKKα/β is their ability to phosphorylate interferon regulator factor (IRF) 3 and 7, both of which are transcription factors critical for activation of type I interferon (IFN-I) genes and interferon-stimulated genes (ISGs) in response to microbial infection [4,5]. Upon phosphorylation, IRF3 and IRF7 homo- or hetero-dimerize and translocate into the nucleus, where they activate IFN-I genes, ISGs and proinflammatory genes [6]. A variety of pattern recognition receptors (PRRs), such as endosomal TLR3, 7, 9, cytosolic RNA and DNA sensors, once engaged with a ligand, could activate TBK1/IKKε, leading to both NF-κB and IRF3/7 signaling. These innate immune signaling pathways are crucial for establishing an immediate antiviral state during acute infection. On the other hand, these signaling pathways also need to be carefully regulated, because their chronic activation could be detrimental to the host and cause autoimmune diseases. A schematic overview of TBK1-mediated signaling as well as downstream IFN-I receptor (IFNAR) and the JAK/STAT pathway is shown in Fig. 1.
Fig. 1.

TBK1 is the innate immune signaling hub mediating the induction of type I interferon response.
Besides innate immune signaling, TBK1 also mediates crosstalk between innate immunity and autophagy [7]. TBK1 contains an N-terminal kinase domain, an ubiquitin-like domain (ULD) in the middle and an α-helical scaffold dimerization domain on the C-terminus. TBK1 is required for autophagic clearance of salmonella enterica, through binding and phosphorylation of autophagsome adaptor proteins optineurin (OPTN) and NDP52 [8,9]. Phosphorylation enhances OPTN interaction with LC3 and facilitates the recruitment of salmonella to autophagosomes. This TBK1-mediated autophagy function is also important in maintaining motor neuron homeostasis; genetic mutations that truncate TBK1 C-terminus OPTN-binding domain while preserving the kinase activity were associated with a neurodegenerative syndrome called familial amyotrophic lateral sclerosis (ALS) [10]. Thus, it is also possible that TBK1 acts as a scaffolding protein in autophagy in neurons.
2. Role of TBK1 in autoimmune diseases, cancers and beyond
Canonical IKKs such as IKKα/β are clearly important for broad NF-κB signaling governing cell survival, proliferation and cancer development. TBK1 and IKKε are better known for activating NF-κB and IRF signaling in infection and autoimmune disease settings. Recent findings also revealed a role for TBK1 and IKKε in Rasinduced oncogenesis [11]. TBK1 acts as an effector downstream of Ras, through RalGEF-RalB-Sec5 pathway, where it directly phosphorylates AKT to promote pro-survival signaling in transformed cells [12]. TBK1 also binds mTORC1 and inhibits its activity to regulate prostate cancer dormancy, although it is unclear whether kinase activity or scaffolding function is involved in regulating mTORC1 [13]. In certain cancers like lung, breast, pancreatic, and colon cancers, TBK1 is highly expressed and provides pro-survival signals [14]. It is remarkable that the same kinase is involved in both antiviral immune response and oncogenic transformation. This may be due to the fact that TBK1 can act on different targets in different pathological settings.
In the settings of autoimmune disease, TBK1-mediated signaling contributes to production of IFN-I, autoantibodies and chemokines such as CXCL10/IP-10. These molecules then initiate and amplify immune attack on tissues leading to organ damage and disease. Immune cell activation (e.g. T cells, B cells and neutrophils) and subsequent migration to peripheral organs also play important role in tissue destructions including the nervous system in autoimmune disease. One example is Aicardi-Goutières syndrome (AGS), a several neurological autoinflammatory disease that is a genetic mimic of congenital viral infection, with elevated levels of IFN-I in the serum and cerebrospinal fluid [15]. Seven genes are associated with AGS, including RNASEH2A, RNASEH2B, RNASEH2C, TREX1, SAMHD1, ADAR1, and IFIH1 (encodes MDA5), all of which are nucleases or nucleic acids sensor. Accumulating evidence suggest that the cytosolic innate immune response to self-DNA or RNA, both require TBK1 kinase activity, is likely a common molecular cause of AGS [16–21]. Pharmacological inhibition of TBK1 in the most well-studied AGS mouse model, Trex1−/−, showed clear therapeutic benefit, such as reduced inflammation and mortality caused by the disease [22].
Recent technological advance on the next-generation sequencing (NGS) allowed rapid discovery of causal mutations in more monogenic autoimmune diseases like AGS. Many of these patient cells exhibit elevated IFN-I or ISG gene expression (also known as the “IFN signature”), and these diseases are collectively called interferonopathy [23]. Although underlying causes and immune pathways leading to elevated IFN signature may be diverse amongst these diseases, given the critical role of TBK1 in activating IFN-I gene expression, pharmacological inhibition of TBK1 is an attractive avenue of therapeutic intervention that could have a broad impact on many interferonopathies.
TBK1-mediated immune signaling has also been implicated in complex autoimmune disease such as systemic lupus erythematosus (SLE) [22,24]. TBK1 mRNA expression, but not other IKK mRNAs, is elevated in several key immune cell populations isolated from SLE patient blood compared to healthy controls [22]. Many common genetic variants associated with SLE encode proteins associated with molecular pathways involved in TBK1-mediated induction of IFN-I. In lupus patients, elevated IFN signature is associated with the presence of RNA/protein-complex autoantibodies [25,26]. Recent studies of monogenic SLE associated with chronic activation of cytoplasmic nucleic acid receptors also demonstrate that IFN-I is induced through TBK1-mediated activation of IRF3. Collective evidence from both murine and human systems suggest IFN-I pathway as a rational therapeutic target for patients with SLE. As a proof-of-concept, we recently showed that lymphoblasts derived from SLE patients with high IFN signature responded well to TBK1 inhibition in vitro [22].
Autoimmune diseases such as rheumatoid arthritis (RA) and multiple sclerosis (MS) may also benefit from inhibiting TBK1-mediated immune signaling. Innate immunity plays an important role in inflammatory cell activation and pathogenesis of RA and MS. Rheumatoid fibroblast-like synoviocytes (FLS) express TLRs and produce inflammatory mediators such as cytokines and matrix metalloproteinases that contribute to the destruction of rheumatoid joints [27,28]. One study highlighted the importance of TLR3-TBK1 signaling pathway in FLS, leading to IRF7 activation and CXCL10/IP-10 expression, which is closely related to the pathogenesis of RA [28]. TBK1 also controls the migration of autoreactive T cells to the brain and is involved in the pathogenesis of MS-like autoimmune manifestations [29]. TBK1 also interacts with the master regulator of cellular metabolism, mTORC1, and suppresses its function [13]. Metabolic dysregulation is becoming more commonly detected in autoimmune diseases such as SLE and AGS [30–32]. Thus, it is logical to speculate that TBK1 may play a broader role than previously thought, coordinating the crosstalk between immune response and metabolism.
3. Potential benefits of targeting TBK1 in autoimmune disease
Autoimmune disease such as SLE is a complex immune disorder caused by a combination of genetic and environmental factors. Although the underlying causes of this disease is heterogeneous, the majority of SLE patients share a prominently common feature, which is increased serum levels of IFN-I and increased expression of ISGs. Accumulating evidence from animal studies and human clinical trials implicate an important ethiopathogenic role of dysregualted IFN-I system in SLE [25]. Therapeutic targeting of IFN-I pathway thus is becoming an exciting option for treating SLE.
Current clinical studies have focused on targeting IFN-I downstream signaling, including IFN-I proteins, IFNAR and the JAK/STAT signaling pathway [25]. Sifalimumab, an anti-IFNα antibody, partially reduced IFN signature in whole blood cells in moderate SLE patients, while may be less effective in severe SLE patients. The anti-IFNAR antibody anifrolumab was also tested in a phase IIb study with encouraging results, and will progress to phase III studies. Ruxolitinib and tofacitinib are JAK inhibitors approved by the FDA to treat psoriasis and rheumatoid arthritis. Clinical studies are just beginning to evaluate these drugs in SLE patients. These important clinical studies bring exciting new hope to SLE treatment. However, one important consideration is that blocking downstream IFN-I signaling would only temper IFN-I target cell function, and have little or no affect on IFN-I producing cells, thus allowing endogenous pathogenic stimulus continue to induce IFN-I and ISGs (Fig. 1).
In comparison, upstream signaling pathways initiated by many PRRs signal through TBK1/IKKε to induce IFN-I expression; thus therapeutic targeting of these two closely related kinases could be effective at blocking IFN-I induction by a variety of stimuli. The rationales for targeting TBK1/IKKε are: 1) Correct the “root cause”. Inhibiting TBK1 would eliminate endogenous ligand-stimulated induction of IFN-I, thus correcting the “root cause” of the problem. This can be done as a single therapy in moderate SLEs, which would leave the downstream IFNAR and JAK/STAT pathway operational for maintaining important cell-to-cell communication and immune hemostasis. TBK1 can also be targeted in combination with IFNAR and/or JAK inhibitors for more complete block of IFN-I signaling in severe SLEs. 2) Kinase activity is amendable to small molecule targeting. TBK1/IKKε are kinases that can be conveniently targeted with small molecule inhibitors that are easy to deliver and pharmacologically more stable, compared to large bulky antibodies currently being used to target downstream IFN-Is and IFNAR. In fact, many potent and selective TBK1/IKKε inhibitors have already been developed to treat cancer and inflammatory diseases ([33], see below). Re-purposing these inhibitors for treating SLE could drastically expedite therapeutic development. 3) Also effective at blocking IFN-independent pathways. Some aspect of SLE pathology may be caused independent of or only partially dependent on IFN-I. For example, CXCL10 plays an important role in systemic vasculopathy, and it can be activated by TBK1-IRF3 signaling pathway in a cell-intrinsic manner [17,34]. 4) “On-target” for TBK1-dependent monogenic SLE. Many monogenic SLE and interferonopathies are clearly caused by TBK1-dependent signaling, such as AGS and STING-associated vasculopathy with onset in infancy (SAVI) [35]. These patients could potentially benefit more from direct therapeutic targeting of TBK1. We recently demonstrated that inhibiting TBK1 with a small molecular inhibitor, Compound II, ameliorated autoimmune disease phenotypes in an AGS disease model Trex1−/− mouse. 5) TBK1 expression is elevated in SLE. TBK1-mediated immune signaling has been implicated in SLE [22,24]. In addition, TBK1 mRNA expression, but not other related family member IKK mRNAs, is elevated in several key immune cell populations isolated from SLE patient blood compared to healthy controls [22]. We recently also showed that lymphoblast cell lines derived from SLE patients with high IFN-I signatures responded well to TBK1 inhibition in vitro [22]. Taken together, TBK1 is an attractive target for therapeutic inhibition of IFN-I induction in autoimmune diseases such as SLE.
4. Viral antagonism of TBK1-mediated induction of interferon response
The immunological profiles of SLE patient bear striking similarity to patient with chronic viral infection, especially the IFN signature. Many viruses have evolved mechanisms to antagonize the IFN-I pathway, which is highly similar to our ongoing therapeutic effort in developing inhibitors for the IFN-I pathway for treating autoimmune diseases. In the majority of cases of viral antagonism, virus usually targets intermediate steps of the upstream signaling pathway to prevent induction of IFN-I, as oppose to targeting downstream IFN-I signaling. Part of this may be because virus directly encounters components of the upstream signaling pathway, thus providing an opportunity for direct host:pathogen interaction. These lessons from virus nonetheless implicate that it may be advantageous or more efficient to target upstream signaling pathway that induces IFN-I expression. Taking herpesvirus (an DNA virus) for an example [36], innate immune cGAS/IFI16-STING-TBK1-IRF3/7 signaling cascade is critical for inducing IFN-I response to herpesvirus infection. To counteract at the PRR level, KSHV encodes ORF52 to block cytosolic DNA sensor cGAS activity; HSV-1 ICP0 causes degradation of another DNA sensor IFI16. The intermediate steps, especially TBK1-mediated signaling, are more extensively targeted by herpesviruses, including KSHV ORF45 and vIRF1, MHC68 ORF11, HSV-1 ICP34.5 and ICP0, that either block TBK1 interaction with STING or with IRF3/7, or cause its degradation [36]. A unified theme of these viral evasion mechanisms is to prevent IFN-I induction, which is most effectively achieved by targeting multiple steps of the upstream signaling cascade including the signaling hub TBK1.
5. TBK1 inhibitors and therapeutic potential
During the past few years, many academic institutions and private sectors have developed inhibitors against TBK1 [33]. The most well-known and widely used compound, BX795, is a potent TBK1 inhibitor, but non-selective. Thus, BX795 has served as the basic structure for further modification to achieve more potency and selectivity. We summarize below major groups of TBK1 inhibitors from published literatures and patents (Table 1).
Table 1.
Existing TBK1 inhibitors reported in the literature and patents.
| Inhibitor Name | Structure | Pharmacological class |
IC50 for TBK1 (nM) |
In vivo efficacy | FDA approval |
Refs. |
|---|---|---|---|---|---|---|
| Compound II |
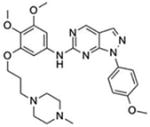
|
Pyrazolo-pyrimidine derivatives | 13 | Yes | No | [12,22] |
| Amlexanox |
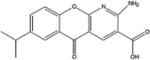
|
Benzopyrano-pyridine derivatives | 1000–2000 | Yes | Yes | [41–43] |
| BX795 |
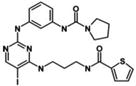
|
Amino-pyrimidine derivatives | 1000 | No | No | [44] |
| MRT67307 |
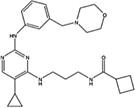
|
Amino-pyrimidine derivatives | 19 | Yes | No | [29] |
| AZ13102909 |
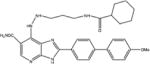
|
Azabenzimidazole derivatives | 5 | No data available | No | [38] |
| SR8185 (TBK1/IKKε dual) |
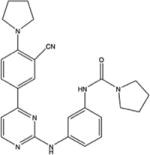
|
Phenyl-pyrimidine derivatives | 1 | Yes | No | [39] |
| TBK1/IKKε dual inhibitor (Domainex) |
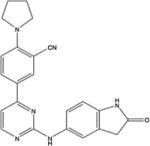
|
Pyrimidine derivatives | 1–2 | Yes | No | [39] |
| MPI-0485520 (Myrexis) | No data available | Amino-pyrimidine derivatives | 0.5 | Yes | No | [40] |
5.1. MRT67307
MRT67307 was developed by University of Dundee, and is similar to BX795 in chemical structure [37]. Both compounds are Aminopyrimidine derivatives. The potency and specificity of MRT67307 is drastically improved over BX795. The IC50 of MRT67307 against TBK1 is 19 nM, compared to 1 µM for BX795. MRT67307 also does not inhibit related kinases such IKKα/β, JAK and p38 MAPK up to 10 µM.
5.2. AZ13102909
During the past few years, AstraZeneca has reported more than 44 compounds as part of an azabenzimidazole derivative series. Several of these compounds inhibit TBK1 at or below 10 nM [37]. The best compound, AZ13102909, inhibits TBK1 with IC50 of 5 nM. Treatment of AZ13102909 together with MEK inhibitors demonstrated clear efficacy at enhancing cancer cell apoptosis in 3D tissue culture models [38].
5.3. SR8185-related compounds
The Scripps Research Institute developed SR8185 with phenylpyrimidine scaffold as an JAK inhibitor [39]. Modification of SR8185 generated SR8185-related compounds with IC50 against TBK1 below 1 nM. These compounds have low molecular weight and high metabolic stability. They were also tested in tissue culture and xenograft and allograft mouse models to have excellent efficacy at inhibiting tumor development.
5.4. Domainex compounds
Domainex is an advanced drug discovery company that developed the first-in-class small molecule inhibitors of TBK1/IKKε. Subsequently, Domainex developed more than 80 pyrimidinyl compounds that show high potency and selectivity against TBK1. These Domainex TBK1 inhibitors have IC50 of 1–2 nM, and inhibitions to related kinases such as IKKβ and JAK were more than 200 times higher. These compounds have been tested in inflammatory disease animal models, where they inhibited production of inflammatory cytokines without toxicity.
5.5. MPI-0485520
Myrexis has developed many amino-pyrimidine compounds that inhibit TBK1 with IC50 in the range of 10 nM or lower. The best compound, MPI-0485520, has IC50 against TBK1 at 0.5 nM [40]. Several of these compounds were tested in vitro and in vivo using inflammatory disease models with excellent inhibition of RANTES, IP-10 and IFNβ.
5.6. Amlexanox
Amlexanox is an anti-inflammatory drug approved by the FDA to treat recurrent aphthous ulcers of the mouth in the US. Its mechanism of action is unknown. Recently, amlexanox has been shown to inhibit TBK1 and IKKε, both of which are induced in the liver and fat by high-fat diet-mediated NF-κB activation [41]. Treatment of obese mice with amlexanox increased thermogenesis, reduced weight, improved insulin sensitivity and decreased steatosis.
5.7. Compound II
Compound II is a pyrazolo-pyrimidine derivative. Amgen developed it in collaboration with Michael White’s group at University of Texas Southwestern Medical Center [12]. Compound II was first shown as a potent and selective TBK1 inhibitor in cancer cells with IC50 of 13 nM. Compound II inhibition of TBK1 impairs AKT signaling and promotes apoptosis of several non-small-cell lung cancer cell lines [12]. More recently, our group tested Compound II in autoimmune disease settings. We showed that Compound II inhibited poly(I:C)-induced immune activation in vitro and in vivo [22]. Interestingly, we found that Compound II potently inhibited TBK1 self-phosphorylation whereas BX795 did not, although both compounds inhibited downstream IRF phosphorylation. Compound II treatment also ameliorated autoimmune disease phenotypes of Trex1−/− mice, increased mouse survival, and dampened the IFN signature in TREX1 mutant patient lymphoblasts [22]. SLE cells with high IFN signature also responded well to Compound II treatment [22]. Thus, Compound II is an excellent candidate for therapeutic development in treating AGS, SLE and interferonopathies.
6. Possible risk of TBK1 inhibition
Although several studies convincingly demonstrated that pharmacological inhibition of TBK1 improved autoimmune and inflammatory disease phenotypes in mice, prolonged inhibition of TBK1 kinase activity may also increase the risk of viral infections. It is possible that the human immune system has evolved redundant processes to cope with infection during short or long term inhibition of TBK1 signaling. Antiviral drugs should be considered when infections do occur. Haploinsuficiency of TBK1 causes neurodegenerative diseases due to loss of TBK1 protein scaffolding function [10]. Thus identification and development of novel drugs that selectively target the kinase function of TBK1 without affecting total protein abundance will minimize the risk of potential adverse effects. Another potential side effect of targeting TBK1 may be dysregulated NF-κB pathway that is critical for cell proliferation and survival, although several potent TBK1 inhibitors mentioned above have shown excellent safety profile and were well tolerated in animals.
7. Conclusions and perspectives
TBK1 is critically involved in diverse biology including innate immune signaling, Ras-mediated tumorgenesis, autophagy and diet-induced obesity. There are strong interests in inhibiting TBK1 kinase activity as a novel avenue of therapeutics for autoimmune disease and cancer. Significant development has been made over the past few years for small molecule inhibitors of TBK1 by the combined efforts from academia and industries. Many of these TBK1 inhibitors are potent and selective against the kinase activity, and are effective at ameliorating autoimmune and inflammatory diseases in animal models with minimal toxicity. Some of these compounds are already in or about to enter clinical trials. As described earlier, prolonged inhibition of TBK1 may increase the risk of viral infections; thus, such treatments should proceed with caution or be used in combination with appropriate antiviral therapies. For severe life-threatening monogenic autoimmune and autoinflammatory diseases such as AGS, SAVI and interferonopathies that presently have no effective treatment, inhibiting TBK1 could potentially be an exciting life saving treatment option. For chronic and complex autoimmune diseases such as lupus, rheumatoid arthritis and multiple sclerosis, TBK1 inhibitors will provide much needed alternatives that can be used alone or in combination with the current standard of care.
Acknowledgments
We thank A. Mir, R. Hersperger and D. Patel for the discussion and helpful suggestions relating to the manuscript. Research in Nan Yan’s lab is supported by the Rita C. and William P. Clements, Jr. Endowed Scholar Award from UT Southwestern, the US National Institute of Health (AI98569, AR067135), Alliance for Lupus Foundation, Welch Foundation and Burroughs Wellcome Fund.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Clément J-F, Meloche S, Servant MJ. The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 2008;18:889–899. doi: 10.1038/cr.2008.273. http://dx.doi.org/10.1038/cr.2008.273. [DOI] [PubMed] [Google Scholar]
- 2.Kravchenko VV, Mathison JC, Schwamborn K, Mercurio F, Ulevitch RJ. IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. J. Biol. Chem. 2003;278:26612–26619. doi: 10.1074/jbc.M303001200. http://dx.doi.org/10.1074/jbc.M303001200. [DOI] [PubMed] [Google Scholar]
- 3.Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, et al. 2000, Deficiency of T2 K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. Embo J. 2000;19:4976–4985. doi: 10.1093/emboj/19.18.4976. http://dx.doi.org/10.1093/emboj/19.18.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003;4:491–496. doi: 10.1038/ni921. http://dx.doi.org/10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 5.Sharma S, Tenoever BR, Grandvaux N, Zhou G-P, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. http://dx.doi.org/10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 6.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. http://dx.doi.org/10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 7.Weidberg H, Elazar Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 2011;4:pe39–pe39. doi: 10.1126/scisignal.2002355. http://dx.doi.org/10.1126/scisignal.2002355. [DOI] [PubMed] [Google Scholar]
- 8.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. http://dx.doi.org/10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thurston TLM, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. http://dx.doi.org/10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 10.Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015;18:631–636. doi: 10.1038/nn.4000. http://dx.doi.org/10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- 11.Chien Y, Kim S, Bumeister R, Loo Y-M, Kwon SW, Johnson CL, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157–170. doi: 10.1016/j.cell.2006.08.034. http://dx.doi.org/10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 12.Ou Y-H, Torres M, Ram R, Formstecher E, Roland C, Cheng T, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol. Cell. 2011;41:458–470. doi: 10.1016/j.molcel.2011.01.019. http://dx.doi.org/10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JK, Jung Y, Wang J, Joseph J, Mishra A, Hill EE, et al. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia. 2013;15:1064–1074. doi: 10.1593/neo.13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen RR, Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2011;30:631–641. doi: 10.1038/onc.2010.493. http://dx.doi.org/10.1038/onc.2010.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015;15:429–440. doi: 10.1038/nri3850. http://dx.doi.org/10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 16.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. http://dx.doi.org/10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, et al. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat. Immunol. 2013;14:61–71. doi: 10.1038/ni.2475. http://dx.doi.org/10.1038/ni.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, et al. RNase H2 catalytic core Aicardi-Goutières syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J. Exp. Med. 2016 doi: 10.1084/jem.20151464. http://dx.doi.org/10.1084/jem.20151464 (jem.20151464) [DOI] [PMC free article] [PubMed]
- 19.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-Editing enzyme ADAR1 independently control nucleic acid sensor MDA5-Driven autoimmunity and multi-organ development. Immunity. 2015;43:933–944. doi: 10.1016/j.immuni.2015.11.001. http://dx.doi.org/10.1016/j.immuni.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014;9:1482–1494. doi: 10.1016/j.celrep.2014.10.041. http://dx.doi.org/10.1016/j.celrep.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao D, Li T, Li X-D, Chen X, Li Q-Z, Wight-Carter M, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. U. S. A. 2015;1646 doi: 10.1073/pnas.1516465112. 201516465 5, http://dx.doi.org/10.1073/pnas.1516465112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasan M, Dobbs N, Khan S, White MA, Wakeland EK, Li Q-Z, et al. Cutting edge: inhibiting TBK1 by compound II ameliorates autoimmune disease in mice. J. Immunol. 2015 doi: 10.4049/jimmunol.1500162. http://dx.doi.org/10.4049/jimmunol.1500162. [DOI] [PMC free article] [PubMed]
- 23.Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann. N. Y. Acad. Sci. 2011;1238:91–98. doi: 10.1111/j.1749-6632.2011.06220.x. http://dx.doi.org/10.1111/j.1749-6632.2011.06220.x. [DOI] [PubMed] [Google Scholar]
- 24.Becker AM, Dao KH, Han BK, Kornu R, Lakhanpal S, Mobley AB, et al. SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLoS One. 2013;8:e67003. doi: 10.1371/journal.pone.0067003. http://dx.doi.org/10.1371/journal.pone.0067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crow MK, Olferiev M, Kirou KA. Targeting of type I interferon in systemic autoimmune diseases. Transl. Res. 2015;165:296–305. doi: 10.1016/j.trsl.2014.10.005. http://dx.doi.org/10.1016/j.trsl.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003;197:711–723. doi: 10.1084/jem.20021553. http://dx.doi.org/10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Migita K, Nakamura T. TBK1: a potential therapeutic target in RA. Rheumatology (Oxford) 2012;51:588–589. doi: 10.1093/rheumatology/ker207. http://dx.doi.org/10.1093/rheumatology/ker207. [DOI] [PubMed] [Google Scholar]
- 28.Hammaker D, Boyle DL, Firestein GS. Synoviocyte innate immune responses: TANK-binding kinase-1 as a potential therapeutic target in rheumatoid arthritis. Rheumatology (Oxford) 2012;51:610–618. doi: 10.1093/rheumatology/ker154. http://dx.doi.org/10.1093/rheumatology/ker154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu J, Zhou X, Chang M, Nakaya M, Chang J-H, Xiao Y, et al. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat. Commun. 2015;6:6074. doi: 10.1038/ncomms7074. http://dx.doi.org/10.1038/ncomms7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu T, Xie C, Han J, Ye Y, Weiel J, Li Q, et al. Metabolic disturbances associated with systemic lupus erythematosus. PLoS One. 2012;7:e37210. doi: 10.1371/journal.pone.0037210. http://dx.doi.org/10.1371/journal.pone.0037210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnerias C, Giurgea I, Hertz-Pannier L, Bahi-Buisson N, Boddaert N, Rustin P, et al. Respiratory chain deficiency in a female with Aicardi-Goutières syndrome. Dev. Med. Child Neurol. 2006;48:227–230. doi: 10.1017/S001216220600048X. http://dx.doi.org/10.1017/S001216220600048X. [DOI] [PubMed] [Google Scholar]
- 32.Haaxma CA, Crow YJ, van Steensel MAM, Lammens MMY, Rice GI, Verbeek MM, et al. A de novo p.Asp18Asn mutation in TREX1 in a patient with Aicardi-Goutières syndrome. Am. J. Med. Genet. A. 2010;152A:2612–2617. doi: 10.1002/ajmg.a.33620. http://dx.doi.org/10.1002/ajmg.a.33620. [DOI] [PubMed] [Google Scholar]
- 33.Yu T, Yang Y, Yin DQ, Hong S, Son Y-J, Kim J-H, et al. TBK1 inhibitors: a review of patent literature (2011–2014) Expert Opin. Ther. Pat. 2015;25:1385–1396. doi: 10.1517/13543776.2015.1081168. http://dx.doi.org/10.1517/13543776.2015.1081168. [DOI] [PubMed] [Google Scholar]
- 34.Hasan M, Fermaintt CS, Gao N, Sakai T, Miyazaki T, Jiang S, et al. Cytosolic nuclease TREX1 regulates oligosaccharyltransferase activity independent of nuclease activity to suppress immune activation. Immunity. 2015;43:1–13. doi: 10.1016/j.immuni.2015.07.022. http://dx.doi.org/10.1016/j.immuni.2015.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum. Mol. Genet. 2009;18:R130–6. doi: 10.1093/hmg/ddp293. http://dx.doi.org/10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christensen MH, Paludan SR. Viral evasion of DNA-stimulated innate immune responses. Cell. Mol. Immunol. 2016 doi: 10.1038/cmi.2016.06. http://dx.doi.org/10.1038/cmi.2016.06. [DOI] [PMC free article] [PubMed]
- 37.Wang T, Block MA, Cowen S, Davies AM, Devereaux E, Gingipalli L, et al. Discovery of azabenzimidazole derivatives as potent, selective inhibitors of TBK1/IKKε kinases. Bioorg, Med. Chem. Lett. 2012;22:2063–2069. doi: 10.1016/j.bmcl.2012.01.018. http://dx.doi.org/10.1016/j.bmcl.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 38.Vu HL, Aplin AE. Targeting TBK1 inhibits migration and resistance to MEK inhibitors in mutant NRAS melanoma. Mol. Cancer Res. 2014;12:1509–1519. doi: 10.1158/1541-7786.MCR-14-0204. http://dx.doi.org/10.1158/1541-7786.MCR-14-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Huang J, Jeong J-H, Park S-J, Wei R, Peng J, et al. Selective TBK1/IKKi dual inhibitors with anticancer potency. Int. J. Cancer. 2014;134:1972–1980. doi: 10.1002/ijc.28507. http://dx.doi.org/10.1002/ijc.28507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richards B, Carlson R. Cellular and in vivo properties of MPI-0485520, a novel and potent small molecule inhibitor of IKKe. FASEB J. 2010;24(Supplement 753.6) http://www.fasebj.org/content/24/1_Supplement/753.6.short. [Google Scholar]
- 41.Reilly SM, Chiang S-H, Decker SJ, Chang L, Uhm M, Larsen MJ, et al. An inhibitor of the protein kinases TBK1 and IKK-ε improves obesity-related metabolic dysfunctions in mice. Nat. Med. 2013 doi: 10.1038/nm.3082. http://dx.doi.org/10.1038/nm.3082. [DOI] [PMC free article] [PubMed]
- 42.Mowers J, Uhm M, Reilly SM, Simon J, Leto D, Chiang S-H, et al. Inflammation produces catecholamine resistance in obesity via activation of PDE3 B by the protein kinases IKKε and TBK1. Elife. 2013;2:e01119. doi: 10.7554/eLife.01119. http://dx.doi.org/10.7554/eLife.01119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Guan H, Li J, Fang Z, Chen W, Li F. Amlexanox suppresses osteoclastogenesis and prevents ovariectomy-induced bone loss. Sci. Rep. 2015;5:13575. doi: 10.1038/srep13575. http://dx.doi.org/10.1038/srep13575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feldman RI, Wu JM, Polokoff MA, Kochanny MJ, Dinter H, Zhu D, et al. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem. 2005;280:19867–19874. doi: 10.1074/jbc.M501367200. http://dx.doi.org/10.1074/jbc.M501367200. [DOI] [PubMed] [Google Scholar]